 Kristen B. Long1
Kristen B. Long1 Regina M. Young2,3
Regina M. Young2,3 Alina C. Boesteanu2
Alina C. Boesteanu2 Megan M. Davis2,4J. Joseph Melenhorst2,3,4
Megan M. Davis2,4J. Joseph Melenhorst2,3,4 Simon F. Lacey2,3,4David A. DeGaramo1Bruce L. Levine2,4
Simon F. Lacey2,3,4David A. DeGaramo1Bruce L. Levine2,4 Joseph A. Fraietta2,3,4*
Joseph A. Fraietta2,3,4*- 1Department of Biology, Mansfield University, Mansfield, PA, United States
- 2Center for Cellular Immunotherapies, Abramson Cancer Center, University of Pennsylvania, Philadelphia, PA, United States
- 3Parker Institute for Cancer Immunotherapy, University of Pennsylvania, Philadelphia, PA, United States
- 4Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, United States
Chimeric antigen receptor (CAR)-engineered T cells represent a breakthrough in personalized medicine. In this strategy, a patient's own T lymphocytes are genetically reprogrammed to encode a synthetic receptor that binds a tumor antigen, allowing T cells to recognize and kill antigen-expressing cancer cells. As a result of complete and durable responses in individuals who are refractory to standard of care therapy, CAR T cells directed against the CD19 protein have been granted United States Food and Drug Administration (FDA) approval as a therapy for treatment of pediatric and young adult acute lymphoblastic leukemia and diffuse large B cell lymphoma. Human trials of CAR T cells targeting CD19 or B cell maturation antigen in multiple myeloma have also reported early successes. However, a clear and consistently reproducible demonstration of the clinical efficacy of CAR T cells in the setting of solid tumors has not been reported to date. Here, we review the history and status of CAR T cell therapy for solid tumors, potential T cell-intrinsic determinants of response and resistance as well as extrinsic obstacles to the success of this approach for much more prevalent non-hematopoietic malignancies. In addition, we summarize recent strategies and innovations that aim to augment the potency of CAR T cells in the face of multiple immunosuppressive barriers operative within the solid tumor microenvironment. Advances in the field of CAR T cell biology over the coming years in the areas of safety, reliability and efficacy against non-hematopoietic cancers will ultimately determine how transformative adoptive T cell therapy will be in the broader battle against cancer.
Introduction
The use of genetically engineered T cells as a form of cancer therapy heralds a new era of synthetic biology and medicine. Within the past few years, clinical trials using chimeric antigen receptor (CAR) T cells to recognize and eliminate hematopoietic malignancies have demonstrated high rates of response as well as durability of remission that are unprecedented in ALL (1–3), chronic lymphocytic leukemia (CLL) (4, 5), and refractory B cell lymphomas (6, 7). This culminated in the recent United States Food and Drug Administration approvals of CD19-directed CAR T cells for relapsed/refractory pediatric and young adult ALL and diffuse large B cell lymphoma (DLBCL). While CAR T cell therapy is poised to revolutionize the treatment of leukemias and lymphomas, the field awaits a clear demonstration of efficacy against non-hematopoietic malignancies. The key challenges for these immunotherapies are how to: (I) safely enhance the potency and sustain the function of CAR T cells in vivo and (II) develop mechanism-based strategies to increase the resistance of CAR T cells to intrinsic and extrinsic dysfunction. Advances in basic and translational research aimed at improving the safety, consistency and effectiveness of CAR T cells against tumors of non-hematopoietic origin will ultimately determine whether this approach can find wider applications in cancer as well as other diseases.
Adoptive cellular immunotherapy involves expanding T cells from a patient or donor in vitro, followed by reinfusion of tumor-specific lymphocytes as cancer therapy. Transfer of expanded tumor infiltrating lymphocytes (TILs) from a subset of individuals with metastatic melanoma has shown potent anti-tumor effects (8, 9). It is likely that TILs target neoantigens within the broad landscape of mutant peptides encoded by de novo somatic mutations (10–14). In rare instances, adoptive transfer of autologous T cells targeting antigens encoded by somatically mutated genes has also resulted in clinically meaningful regressions of colon, metastatic bile duct, cervical and breast cancers (15–19). However, this strategy has little effect on other common epithelial malignancies that have lower mutation rates.
Transfer of genetically-redirected T cells bypasses many of the mechanisms involved in immunological tolerance by the creation of antigen-specific lymphocytes independently of intrinsic tumor immunogenicity that is driven at least in part by a high mutational burden. T cells can be directed to novel tumor antigens by introducing genes encoding new antigen receptors, including natural T cell receptors (TCRs) and CARs. CARs are synthetic molecules that combine the effector functions of T cells with the ability of antibodies to detect pre-defined antigens with a high degree of specificity in a non-major histocompatibility complex (MHC) restricted manner (20). These receptors can therefore recognize intact proteins and do not rely on endogenous antigen processing and presentation. CARs are typically comprised of an extracellular domain for tumor recognition and an intracellular signaling domain that mediates T cell activation [reviewed in 21–24)]. The antigen-binding function of a CAR is usually conferred by a single chain variable fragment (scFv) containing the variable heavy (VH) and variable light (VL) chains of an antibody fused to peptide linker (20, 25, 26). This extracellular portion of the receptor is fused to a transmembrane domain followed by intracellular signaling modules. First-generation chimeric receptors bearing CD3ζ alone were not sufficient to elicit proliferation or cytokine production in peripheral T cells (27), which likely explains their failure to consistently expand and persist in some of the earliest clinical trials of CAR T cells (28, 29). However, the incorporation of co-stimulatory endodomains into CARs can recapitulate natural co-stimulation (30–32). We and others have demonstrated remarkable rates of complete and durable remission in patients with CLL (4, 5, 33), ALL (1–3), and Non-Hodgkin lymphomas (6, 7, 34) treated with second-generation CD19-directed CARs incorporating 4-1BB or CD28 co-stimulation. Early clinical trials of CAR T cells for the treatment of multiple myeloma have also demonstrated promising results (35–37). Thus, in the setting of hematopoietic malignancies, CAR T cells are emerging as a powerful therapy with the curative potential of allogeneic stem cell transplantation, but without the acute and chronic toxicity of graft-vs.-host disease and conditioning regimens. In contrast, CAR modified T cells are less effective than immune checkpoint blockade and in some cases TIL-based immunotherapy in treating patients with solid tumors to date. In this review, we will discuss the history and current status of CAR T cell therapy for non-hematopoietic malignancies, outline intrinsic mechanisms of T cell potency, describe extrinsic barriers operative in the setting of treating solid tumors, and suggest strategies to enhance the effectiveness of this approach for a variety of these incurable cancers.
History and Current Status of Car T Cell Therapy for Non-hematopoietic Cancers
Initial Clinical Trials of Car T Cell Therapy in Solid Tumors
In early clinical trials of first-generation CAR T cells for solid tumors, safety and therapeutic efficacy were difficult to determine because of the aforementioned poor in vivo expansion and persistence of the transferred lymphocytes. These studies included patients with advanced epithelial ovarian cancer or metastatic renal cell carcinoma and targeted the folate receptor or carbonic anhydrase IX (CAIX), respectively (28, 29). A clinical trial of L1-cell adhesion molecule-specific (CD171) CAR T cells for the treatment of metastatic neuroblastoma demonstrated similar results of short-persisting (1–7 days) CAR T cells in individuals with bulky disease, but significantly longer persistence (42 days) in a single patient with limited tumor burden (38). Later trials of first-generation GD2-targeted CAR T cells administered to children with advanced neuroblastoma were more encouraging, with 3 of 11 patients experiencing complete remission, no substantial toxicity observed and sustained therapeutic benefit reported for several subjects (39, 40). Although the results of these trials were encouraging and provided the impetus to incorporate co-stimulatory signaling motifs in addition to CD3ζ, a third-generation CAR specific to the tumor antigen Her2 and integrating CD28, 4-1BB, and CD3ζ signaling moieties resulted in death of a patient with metastatic colon cancer (41). In this case, toxicity was caused by on-target, off-tumor reactivity of the CAR T cells with Her2 on normal lung and/or cardiac tissue (41). This serious adverse event was likely attributed to the infusion of substantially higher numbers of CAR T cells following lymphodepleting chemotherapy compared to most other trials. A second-generation Her2 CAR was also tested in patients with sarcoma without evidence of toxicity (42). Although there were some indications of anti-tumor activity in this trial, T cell persistence was limited, similar to earlier clinical studies.
Recent Clinical Studies of Car T Cell Therapy in Non-hematopoietic Malignancies
Less dramatic clinical responses have also been observed in recently conducted clinical trials designed for the treatment of solid tumors with CAR T lymphocytes. Although evaluable data are not yet available from many of these studies, there is enough proof-of-concept from successful human studies of CAR T cells in leukemia and lymphoma to establish a concrete platform to treat these other indications. A complete response to CAR T cell therapy of recurrent multifocal glioblastoma was achieved using autologous T cells genetically-redirected to the tumor-associated antigen interleukin-13 receptor alpha 2 (IL13Rα2) (43). Interestingly, multiple intracavitary and intraventricular administrations of IL13Rα2 CAR T cells induced increases in the frequencies and absolute numbers of endogenous immune cells (i.e., CD3+ T cells, CD14+ CD11b+ HLA-DR+ mature myeloid populations, CD19+ B cells, and few CD11b+ CD15+granulocytes) in association with the elaboration of inflammatory cytokines. This case underscores the possible role of the endogenous immune system in potentiating the anti-tumor activity of engineered CAR T cells and the potential of this approach to safety and dramatically increase quality of life in patients with malignant brain tumors (43).
We have recently generated CARs directed against the epidermal growth factor receptor variant III (EGFRvIII) and used them to gene engineer glioblastoma multiforme (GBM)-specific T cells. We found that we can redirect GBM patient T cells to target glioma tumors via lentiviral transduction with a CAR recognizing EGFRvIII in vitro, as well as in vivo in murine models (44) and in 10 patients (45) without the systemic toxicity associated with current standard-of-care treatments. In our first-in-human trial of EGFRvIII CAR T cells, we were able to confirm that a single intravenous infusion of these modified lymphocytes resulted in T cell engraftment in the peripheral blood, trafficking to the brain and antigen-directed activity (45). However, we observed that the inhibitory tumor microenvironment ultimately hampers clinical efficacy: following CAR T cell administration, several immunosuppressive factors were upregulated in the tumor environment including programmed death-ligand 1 (PD-L1), tryptophan 2,3-dioxygenase, indoleamine 2,3-dioxygenase, and IL-10. The lack of CAR T cell anti-tumor activity was accompanied by the presence of immunosuppressive regulatory T cells (TREGS) based on their expression of CD4, CD25, and FoxP3. Furthermore, the heterogeneity of EGFRvIII expression was a clear barrier to ongoing clinical responses in this study (45). Thus, adoptive cell therapies for non-hematopoietic malignancies will need to address how to increase both the potency and persistence of CAR T cells in the face of antigen heterogeneity and a strongly suppressive tumor microenvironment (Figure 1). This clinical report (45) presents several known obstacles to CAR T cell therapy for solid tumors which are described below in detail.
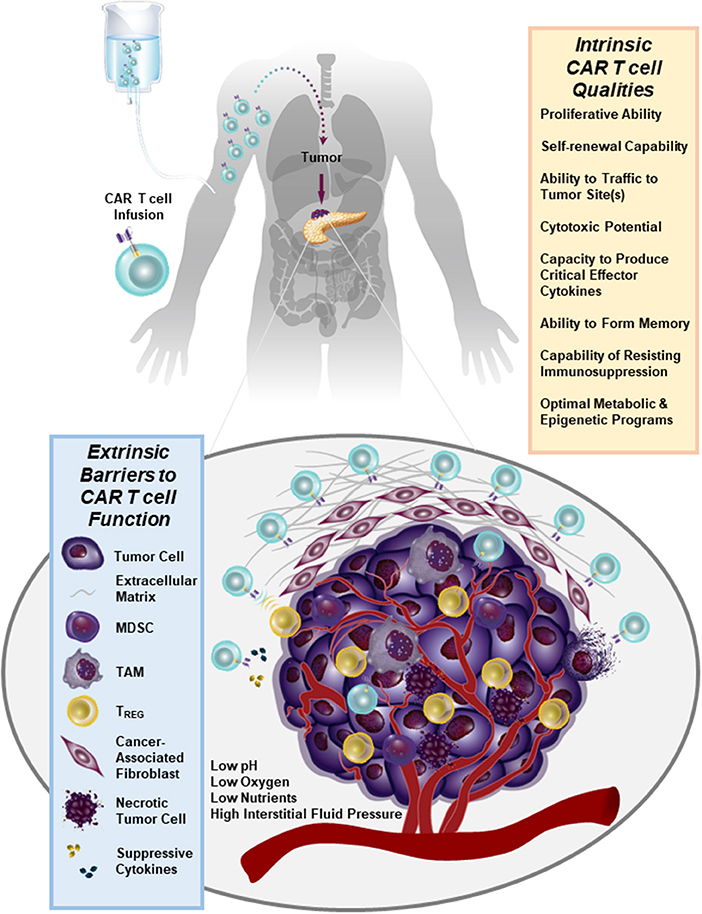
Figure 1. Numerous immunosuppressive barriers present in the solid tumor microenvironment that can hamper the efficacy of CAR T cell therapy are schematically depicted. Several intrinsic qualities of CAR T cells that may impact the anti-tumor potency of these lymphocytes are also listed.
Tuning Car T Cell Specificity and Intrinsic Fitness for Immunotherapy of Solid Tumors
Tumor Antigen Expression and Heterogeneity
Despite the fact that antigens such as CD19 and B-cell maturation antigen (BCMA) have been successfully targeted by CARs in the setting of hematopoietic cancer, there is an unmet need to identify similarly ideal antigens expressed by solid tumors. A major barrier to the development of CARs for solid tumor indications is, indeed, the identification of tumor antigens that can be targeted safely and effectively [reviewed in 46). In an optimal setting, CAR T cells should be directed against a tumor-restricted antigen to avoid on-target, off-tumor reactivity with healthy tissues. The proposed target antigen should be differentially expressed on tumor cells relative to essential normal tissues. In addition, the chimeric receptor must be highly specific for an antigen that is broadly expressed on the majority of cancer cells (46, 47). A variety of tumor-specific and tumor-associated antigens that can be targeted using CAR T cell therapy in non-hematopoietic malignancies have been identified (e.g., EGFR/EGFRvIII, IL13Rα2, Her2, CD171, mesothelin (MSLN), folate receptor alpha, GD2, carcinoembryonic antigen (CEA), chondroitin sulfate proteoglycan 4, c-Met, etc.). Antigens that display high constitutive expression that is tumor-restricted (e.g., chondroitin sulfate proteoglycan 4) may permit the application of CAR T cell therapy to higher proportions of patients and reduce the likelihood of tumor escape (48). However, because most tumor-associated antigens are heterogeneously expressed in tumor tissue, the efficacy of CAR T cells is often limited. Thus, combination therapies incorporating CARs that target multiple antigens will likely be required. There is progress in more safely and specifically targeting non-hematopoietic tumors with CAR T cells, either through creating CAR T cells specific for RNA splice variants or tumor-specific glycans (49, 50), or by generating CAR T cells that are conditionally specific for solid tumors. The latter is achieved by employing sensing and switching strategies in the tumor microenvironment (51–54). In addition to selectively replicating in and killing tumor cells directly, oncolytic viruses armed with payloads (e.g., bispecific T cell engagers, cytokines) may further synergize with CAR T cells to overcome tumor heterogeneity, while simultaneously bolstering anti-tumor activity (55, 56) (Figure 2).
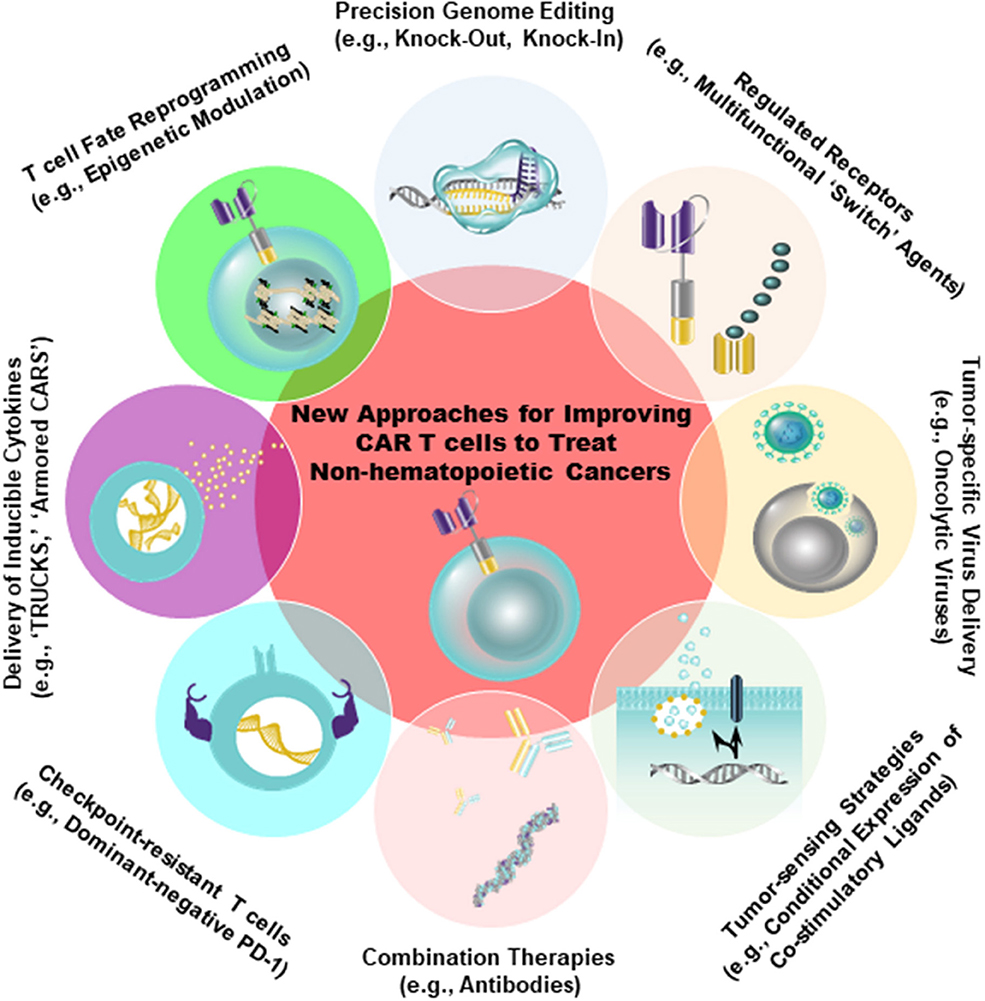
Figure 2. Strategies to improve the safety (e.g., tumor-sensing strategies) as well as to augment the anti-tumor efficacy of CAR T cells are shown. Genetic engineering can be accomplished using viral (e.g., lentiviruses, retroviruses) and non-viral (e.g., CRISPR/Cas9) approaches to endow CAR T cells with gain-of-function or loss-of-function alterations. The overall aim of these approaches is to improve intrinsic T cell fitness and allow these cells to elicit optimal effector activity in the setting of several extrinsic barriers operative within solid tumors, as shown in Figure 1.
Car T Cell Trafficking to Solid Tumors
Following infusion of CAR T cells targeting an appropriate antigen into patients, these lymphocytes are faced with the immediate obstacle of having to successfully localize to the tumor bed. This process is critically dependent on chemokine receptors expressed by the transferred cells and the chemokine gradient produced by the tumor. This presents a challenge because T cells often do not express the cognate receptors for the chemokines produced by tumors. In addition to this chemokine/chemokine receptor mispairing, tumors produce very small amounts of the chemokines needed for successful trafficking of T cells to the lesion. For example, melanoma cells do not produce sufficient amounts of CXCR3 ligands and this results in inefficient localization of CXCR3 receptor-bearing effector CD8+ T cells to metastatic sites 57. We and others have co-expressed better matched chemokine receptors with CARs which resulted in improved trafficking of CAR T cells and enhanced tumor elimination (58, 59).
Characteristics of Intrinsic Car T Cell Potency
Systematic evaluations of patients with hematologic malignancies responding or not responding to CAR T cell therapy has yielded insights into key determinants of T cell potency that may inform treatment of solid tumors. In CLL, CAR T cells that were particularly effective exhibited robust proliferative capacity as well as long-term persistence in vivo. Transcriptomic profiling of patient-derived cell products revealed that CAR T cells from complete-responding patients were enriched in memory related genes, including IL-6/STAT3 signatures, whereas products from non-responding patients upregulated programs involved in effector T cell differentiation, glycolysis, exhaustion, and apoptosis (33). Unexpectedly, there was no association with typical patient-(e.g., age, sex, prior therapy) or disease-related (prior therapies, genetic and other risk profile, tumor burden, etc.) factors with likelihood of response. This makes the important point that cell-intrinsic properties are major determinants of success and failure in CAR T cell therapy (Figure 1).
Generation of Quality Car T Cells
The optimal “seed” population of T cells needed for the generation of CAR T cells that can sustain durable responses against cancer is still a matter of debate. One school of thought is that effector CD8+ T cells producing high amounts of interferon-gamma are most effective at eliminating tumors, while other investigators believe that naïve or early memory CD8+ T cells which differentiate and expand at the tumor site are superior for eliciting long-lasting anti-tumor immunity (60–62). If one assumes a linear model of CD8+ T cell differentiation, naïve T lymphocytes (TN) are programmed into the earliest identifiable memory T cell stage, stem cell memory (TSCM). This population is thought to give rise to the successive stages of differentiation: central memory (TCM), effector memory (TEM), terminally differentiated effector memory RA (TEMRA), and effector (TEFF) cells (63). Many studies have supported the idea that early memory CD8+ T cells generate the most potent CAR T cells against both liquid and solid tumors. For example, CAR-engineered TSCM cells directed to mesothelin were significantly more effective at regressing established solid tumors compared to TEM and TEFF cells (63). Retrospective profiling of ex vivo CD4+ and CD8+ T cells from CLL patients treated with anti-CD19 CAR T cells revealed that responding and non-responding patients did not differ in their frequencies of TN, TCM, TEM, or TEFF cells at the time of T cell collection. However, responding patients did exhibit a modest increase in TSCM cells compared to non-responders (33). More significantly, unbiased biomarker analysis revealed that the frequency of apheresed CD27+ CD45RO− CD8+ T cells from patients responding to CAR T cell therapy was significantly higher compared to non-responder T cells. Notably, this subpopulation of CD8+ T cells possessed functional characteristics of early memory as well as effector T cells (33).
Based on growing pre-clinical and clinical evidence of less-differentiated cells mediating superior anti-tumor efficacy, there is interest in developing ways to conduct large-scale T cell expansion, while simultaneously preserving the functional features of early-memory T cells. Human T cells undergo a series of profound changes with successive rounds of division in vitro and in vivo. Among these changes are the loss of certain co-stimulatory receptors (e.g., CD28, CD27) and the erosion of telomeres. Depending on the molecular design, co-stimulatory endodomains from these receptors may or may not be incorporated into the CAR. Therefore, culture systems that can prevent telomere loss or potentiate the maintenance of endogenous co-stimulatory receptor expression could restore proliferative potential to conventional effector T cells and presumably increase the functional lifespan of these cells following re-infusion into patients (64, 65). We have recently described a culture system for the production of CAR T cells in 3–5 days, relative to a traditional 9-day process (66). This process allowed us to generate CD19-directed CAR T cells that were less differentiated and, at limited cell doses, significantly more potent against leukemia in an in vivo animal model (66). Alternative approaches for reducing CAR T cell differentiation during in vitro expansion include inhibition of signaling mediators downstream of the IL-2 pathway such as subunits of Glycogen synthase kinase 3β (60), Protein kinase B (AKT) (67), and Phosphoinositide 3-kinase (68). In addition, replacement of IL-2 with other cytokines such as IL-7 and IL-15 that signal through the γ-common chain receptor (69), but regulate survival and homeostatic T cell proliferation independently of TCR stimulation (70–72) may enhance the in vivo expansion and persistence of CAR T cells (73, 74). Genetic reprogramming of induced pluripotent stem cells derived from somatic cells could also be used to generate more naïve-like CAR T lymphocytes for adoptive transfer (75). Finally, in a “bedside-to-bench” study, we demonstrated that unintentional disruption of the gene encoding the methylcytosine dioxygenase TET2 resulted in the massive clonal expansion of CAR T cells that were all derived from a single cell. Furthermore, TET2-disrupted lymphocytes exhibited a predominantly TCM phenotype at the peak of the anti-tumor response (76). These findings, along with other recent reports (77–81), underscore the power of epigenetic modulation in effectively re-programming T lymphocyte fate for the generation of CAR T cells with optimal anti-tumor potency (Figure 2).
Surmounting Tumor-mediated Barriers to Car T Cell Therapy of Non-hematopoietic Cancers
A major issue to be addressed for improving the efficacy of CAR T cells against non-hematopoietic malignancies is determining how to effectively enhance the persistence and function of these lymphocytes in toxic tumor microenvironments. CAR T cells are vulnerable to both immunological and metabolic checkpoints as well as other suppressive factors present in the tumor bed. In pre-clinical mouse models, both CAR and TCR transgenic T cells cease to function or die shortly after entering the tumor microenvironment (82, 83). Although repeated infusions of freshly engineered T cells may help to improve engraftment, this approach is not always clinically feasible. Tumor-imposed extrinsic barriers as well as strategies to overcome several of these hurdles for the generation of efficacious CAR T cells to treat solid cancers are described below.
Overcoming Physical Barriers in Solid Tumors
Unlike liquid tumors which do not typically possess physical barriers that would prevent their interactions with CAR T cells, many solid tumors have a formidable barricade that renders these masses inaccessible to invasion by immune cells. This landscape includes stromal cells, immune cells, cancer cells and extracellular matrix (ECM) components (i.e., proteins and glycans). Collagens, fibronectin, laminin, hyaluronan, and proteoglycans heavily contribute to the proliferation of fibrous or connective tissue (desmoplasia). The fibrotic tumor stroma of many solid malignancies, including pancreatic, breast and ovarian cancer is thought to impede effective drug delivery (84–86) and may also prevent infiltration by CAR T cells (Figure 1). Accordingly, diffusion of the CAR T cells into tumor tissue was shown to be blocked by the ECM are therefore often trapped (87) and unable to deeply penetrate tumor tissue (88). Desmoplasia combined with high interstitial fluid pressure and rapid tumor cell proliferation also contributes to the collapse of vasculature, which may further impede CAR T cell infiltration from vessels into tumor tissue (89). Tumor vessels may also not possess the receptors necessary for T cell homing and extravasation, including E- and P-selectins, VCAM-1, and ICAM-1 (87). Furthermore, following in vitro culture, CAR T cells often lack normal expression of the enzyme heparanase which degrades matrix proteoglycans and potentiates extravasation (90).
Administration of collagenases or hyaluronidase into solid tumors has been shown to enhance ECM breakdown, rendering the tumor more penetrable and thus susceptible to drug and cell-based therapies. Collagenase or hyaluronidase treatment has aided in increased antibody diffusion and chemotherapy uptake in pre-clinical in vivo and in vitro models of disease (91–94). Alternatively, reprogramming of myeloid cells, which naturally traffic and infiltrate into solid tumors, can effect anti-fibrotic activity and ECM breakdown (95). Depletion of ECM-producing cells (e.g., cancer-associated fibroblasts) can also render solid tumors more susceptible to therapy (96). In this regard, targeting stromal fibroblasts with anti-fibroblast activation protein (FAP) CAR T cells significantly stalls the growth of multiple types of solid tumors (97). In addition, administration of CAR T cells engineered to overexpress heparanase leads to partial ECM degradation, enhanced T cell infiltration and anti-tumor activity (90). Although these strategies seem promising, the potential negative impact of tumor ECM depletion should not be overlooked. In some studies, ECM reduction can paradoxically accelerate disease progression (98, 99). To avoid this potential negative outcome, direct intracavitary or intratumoral injection relative to intravenous infusion of CAR T cells may circumvent many of the physical barriers described above. In this vein, Klampatsa et al. used intracavitary methods to eliminate mesothelioma cell lines with some success (100), and Adusumilli and colleagues demonstrated that intrapleural administration of CAR T cells was significantly more successful at eliciting anti-tumor activity than the intravenous route (101).
Targeting the Tumor Vasculature and Immune Stimulatory Car T Cell Modifications
In addition to tumor antigens, CARs can be targeted to the tumor vasculature in an effort to restrict blood flow and nutrient supplies to the tumor, which impedes malignant growth and simultaneously increases T cell localization (102). A strategy based on regional infusion of IL-12 secreting CAR T cells directed against VEGFR-2 which is expressed on angiogenic endothelial cells resulted in enhanced accumulation of these lymphocytes and tumor regression in multiple pre-clinical models (103). “Armored CARs” or “TRUCKs” (T cells Redirected for Universal Cytokine Killing) delivering other cytokines such as IL-15 (104, 105) or IL-18 (106) to the tumor microenvironment have also demonstrated superior anti-tumor activity compared to conventional CAR T cells (Figure 2). Furthermore, echistatin CARs targeting the angiogenic integrin αvβ3, which is commonly expressed on vascular endothelium of solid tumors (107), increased nanoparticle deposition in tumors (108). These findings indicate that the use of vasculature-targeted CAR T cells may be a potential “lead-in” strategy to enhance delivery of drugs or other adoptively transferred immune cells.
Overcoming Cell-Mediated Immunosuppression in the Solid Tumor Microenvironment
Along with physical barriers, the tumor microenvironment is composed of multiple cellular components and molecular factors that can abrogate the elicitation of effective endogenous anti-tumor immune responses. This immunosuppressive milieu can also severely inhibit the effector functions of adoptively transferred CAR T cells. However, CAR T cell hypofunction is tightly dependent on the tumor microenvironment and in some instances removal of engineered T cells from the tumor restores their functional activity (109). This report as well as other studies (110–112) suggest that favorably altering the toxic tumor microenvironment by directly targeting immunosuppressive cells or engineering T cells to resist tumor-specific inhibitory mechanisms may provide new opportunities to improve CAR T cell function.
Tumor associated macrophages (TAMs) are an immunosuppressive cell type commonly found in solid tumors, and these cells aid in tumor cell survival and growth. While the phenotype of macrophages is pliable and these cells can be programmed to be either tumor-promoting or tumor-suppressive, macrophage function is ultimately dictated by signals from the surrounding tissue-specific niche (113). The tumor microenvironment often pushes macrophages toward a tumor-promoting phenotype (114), and this aids in angiogenesis, growth, immune evasion and metastasis. Therefore, targeting TAMs may improve the efficacy of CAR T cells against solid tumors. Ruella and colleagues recently devised a strategy to deplete tumor-promoting macrophages with macrophage-targeted CAR T cells. This approach was efficacious in a mouse model of Hodgkin lymphoma and led to the establishment of long-term immunological memory (115).
Myeloid derived suppressor cells (MDSCs) are another immunosuppressive cell type found in solid tumors that can dampen CAR T cell function. MDSCs express arginase and indoleamine, which metabolize amino acids that are essential for effector T cell activation and proliferation (116). Accordingly, Burga et al. demonstrated that depletion of GR1+ cells (targeting immunosuppressive tumor-associated neutrophils and MDSCs) augmented the ability anti-carcinoembryonic antigen CAR T cells to reduce colorectal cancer liver metastases (117). MDSCs also produce high levels of reactive oxygen species, which may impair the cytotoxic ability and proliferative capacity of CAR T cells (118). To overcome this oxidative stress, CAR T cells have been modified to express the anti-oxidant enzyme catalase into the local environment and this modification significantly improves their anti-tumor activity (119).
TREGS are well-documented suppressors of T cell function capable of inhibiting anti-tumor activity through multiple mechanisms, including cell-cell contact inhibition, sequestration of IL-2 and the production of immunosuppressive cytokines such as TGF-β and IL-10 (120). Although these cells promote the growth and metastasis of tumors, they are difficult to directly deplete due to the lack of specificity of targeting agents, and the potential to induce autoimmune diseases when global disruption approaches are used (121). Given the high level of TGF-β produced by TREGS, MDSCs, and tumor cells, blocking TGF-β signaling through overexpression of a dominant-negative TGF-β receptor on adoptively-transferred T cells may improve their anti-tumor potency (122, 123). Overexpression of dominant-negative TGF-β receptor II on CAR T cells results in enhanced T cell proliferation, cytokine production, in vivo persistence and ability to eradicate tumors in mouse models of aggressive human prostate cancer (124).
Many types of cells including tumor cells, fibroblasts, endothelial cells and immune cells produce the lipid-signaling molecule prostaglandin E2 (PGE2) by activation of cyclooxygenase (COX)-2 and prostaglandin E synthase. PGE2 enhances tumor progression by stimulating multiple pathways, including those that mediate angiogenesis and immunosuppression (125). For example, PGE2 plays a significant role in the suppression of effector T cells and the attraction of TREGS and MDSCs. PGE2 and adenosine activate protein kinase A (PKA), which then inhibits antigen receptor –triggered T cell activation. PGE2 is also known to cooperate with adenosine in the dampening of immune responses mediated by TREGS (126). Recently, Newick et al. engineered CAR T cells to produce a small peptide that inhibits the association of PKA with ezrin, thus reducing the negative effects of PKA on TCR activation (127). This PKA inhibitor ameliorated the immunosuppressive actions of both adenosine and PGE2, resulting in increased CAR T cell trafficking, tumor cell cytotoxicity, and pro-inflammatory cytokine production (127).
Enhancing the Metabolic Fitness of Car T Cells
Immune cell function and metabolism are impacted by the solid tumor microenvironment. Glucose utilization is heterogeneous within the tumor and associated with perfusion, with lesser-perfused regions of the tumor displaying higher glucose metabolism (128). Both proliferating tumors and effector T cells responding to antigen challenge rely primarily on aerobic glycolysis to fuel expansion, creating competing demands for metabolites within nutrient-poor regions of the tumor (129). This competition for nutrients, metabolites and oxygen (O2) is thought to impact T cell metabolism, limit T cell-mediated anti-tumor efficacy and contribute to T cell exhaustion and cancer progression (130–132). Stabilization of HIF-1α drives glucose uptake, induces production of S-2-hydroxyglutarate (S-2HG) and consequential epigenetic remodeling as well as increased expression of IL-2, which potentiates CD8+ T cell mediated anti-tumor activity (133, 134). However, under O2 and glucose limiting conditions, reduction of HIF-1α expression may enhance T cell function (135). In a recent study, CD8+ TILs isolated from clear cell renal cell carcinoma (ccRCC) were shown to exhibit an impaired ability to consume glucose, mitochondrial fragmentation and hyperpolarization, as well as increased production of ROS (136). Because ccRCC develops a unique pathological pseudo-hypoxic response [reviewed in 137), with increased aerobic glycolysis and vascularization, it is tempting to speculate that the altered tumor microenvironment in ccRCC may have contributed to these observed defects in ccRCC CD8 TIL metabolism (136). Likewise, hypoxic areas within solid tumors are often negatively correlated with patient survival and thought to promote tumor metastasis and resistance to radiotherapy (138–140). Another metabolic checkpoint in the tumor microenvironment regulating immune modulation is amino acid limitation (129). For example, degradation of L-arginine by MDSCs in the tumor microenvironment can lead to reduced expression of CD3ζ and impaired T cell responses (141). In contrast, increased levels of arginine shift T cell metabolism to oxidative phosphorylation and increase central memory differentiation (142).
Activation, growth, proliferation, effector and memory function, and return to homeostasis are linked to the metabolic profile of the T cell (131). T cell subsets differently metabolize nutrients and regulation of nutrient availability can influence T cell differentiation as well as fate (129). Naïve T cells are metabolically quiescent and rely on glucose, fatty acids and amino acids as fuel sources for oxidative phosphorylation (143, 144). TCM cells maintain spare respiratory capacity through oxidation of fatty acids in mitochondria which allows for a rapid recall of the memory response upon antigen re-challenge (145, 146). In contrast, effector T cells, like tumor cells, rely on aerobic glycolysis to provide energy, metabolic intermediates for rapid cell growth and NAD+/NADH to maintain redox balance (147); although under metabolically challenging conditions CD8+ TILs can partially preserve effector function by catabolizing fatty acids (135). Glutamine is also essential for effector function (148). After conversion to α-ketoglutarate, glutamine can serve as a TCA intermediate or contribute to the citrate pool. Similarly, altering metabolism can impact T cell phenotype; restraining glycolysis, AKT, and mTOR activity or enhancing STAT3 or Wnt/β catenin signaling can arrest T cell development and retain TCM differentiation, which are associated with enhanced T cell persistence and may promote the efficacy of adoptive cell therapy (60, 149–152).
Different types of co-stimulatory endodomains incorporated into a CAR can differentially program T cell metabolism and mitochondrial biogenesis (153). This indicates that the fate of CAR T cells toward memory or effector differentiation can be directed, as cells expressing CARs with 4-1BB signaling domains have enhanced mitochondrial biogenesis and fatty acid oxidation, while CARs with CD28 signaling domains have enhanced aerobic glycolysis (i.e., Warburg metabolism) (153). Therefore, in addition to being able to direct CARs to virtually any cell surface structure on tumor cells, we also have the potential to engineer these lymphocytes to be resistant to the tumor microenvironment by specifying their metabolic program. Alternatively, host pre-conditioning strategies involving the treatment of tumors with HIF blocking agents or metabolic enzymes may represent a promising strategy to limit the metabolic flexibility of tumors as well as the localization of inhibitory immune cells (154). This would allow CAR T cells to function in a more nutrient replete and less suppressive tumor microenvironment.
Engineering Car T Cell Resistance to Immune Checkpoint Inhibitors
Tumors cells can also directly modulate effector T cell activation by expression of inhibitory signals that block T lymphocyte activation and function, thus preventing immune control of tumor growth (155). In addition to secreting immunosuppressive cytokines, tumor cells or other cells in the tumor microenvironment express a number of proteins on their surface that are capable of inactivating CAR T cells. These include PD-1 ligands, PD-L1 (B7–H1), and PD-L2 (B7-DC), all belonging to the B7 receptor superfamily. Other B7 family members, such as B7–H3 and B7–H4, and the unrelated receptors herpes virus entry mediator (HVEM), inhibitory receptor Ig-like transcript-3 and−4 (ILT3 and 4) are also abundantly expressed in the solid tumor microenvironment [reviewed in 156)]. Furthermore, by providing a persistent source of antigen while avoiding clearance, tumors potentially promote T cell exhaustion. As discussed above, checkpoint blockade has been a successful approach to sustain T cell function, and blockade of inhibitory receptors such as T-cell membrane protein-3 (TIM-3), lymphocyte-activation protein-3 (LAG-3), T cell Ig and ITIM domain (TIGIT), cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), and programmed death-1 (PD-1) or their cognate ligands are being tested in clinical trials to reverse or prevent exhaustion [reviewed in (47)]. The upregulation of these receptors has been previously reported to abrogate the persistence and activity of the anti-tumor response of CAR T cells (155). Accordingly, John et al. reported that combining anti-Her2 CAR T cells and PD-1 blocking antibodies enhances tumor growth inhibition in association with decreased frequencies of GR1+ CD11b+ MDSCs (157). Strategies in which CAR T cells are engineered to secrete immune checkpoint inhibitors such as anti-PD-L1 (110), and -PD-1 (158) antibodies or PD-1-blocking single-chain variable fragments (112) possess the advantage of increasing the local delivery of these agents to the tumor microenvironment, while avoiding toxicities associated with systemic checkpoint blockade. Co-expression of a dominant-negative PD-1 receptor with mesothelin-targeted CAR T cells has also been shown to render these cells resistant to PD-1-induced inhibition and to significantly improve their in vivo anti-tumor efficacy following a single administration (155). The Clustered Regularly Interspaced Short Palindromic (CRISPR)/CRISPR associated protein 9 (Cas9) provides a robust and multiplexable genome editing tool that permits knock-out of inhibitory receptors (Figure 2). This system can be used to knock-out PD-1 and CTLA-4 on allogeneic universal CAR T cells (159). Finally, it is intriguing to consider the possibility of directing CAR transgenes to specific genomic loci encoding inhibitory receptors using recently developed viral and non-viral technologies (160, 161).
Concluding Remarks
Many pre-clinical studies indicate that adoptive cell transfer therapy with autologous T cells is a powerful approach for the treatment of cancer. In contrast to the recent FDA approvals of CAR T cells in hematologic malignancies, the effectiveness of this approach for a variety of more common non-hematopoietic cancers is much lower. As was underscored in this review, CAR T cells may hold great promise for the treatment of solid tumors; these malignancies have a high-unmet medical need and are generally considered incurable with present therapies. However, the achievement of complete and durable remissions for patients with non-hematopoietic cancers will require optimization of CAR T cells in the areas of improving antigen targeting, enhancing T cell trafficking, bolstering intrinsic T cell potency and arming these lymphocytes to do battle in the face of multiple immunosuppressive barriers imposed by the solid tumor microenvironment. Both current and future advances in cellular engineering, site-specific genome editing and synthetic biology will undoubtedly bolster the safety, reliability and efficacy of CAR T cell therapy for a variety of diseases. Thus, while there are currently some detours on the road to clinical success, CAR T cells are on the fast track to becoming a potentially curative modality for many different cancers.
Author Contributions
KL, RY, and JF conceptualized, wrote, and edited the manuscript. AB, MD, JM, SL, DD, and BL provided feedback and edited the manuscript.
Funding
Publication of this open-access article was supported through internal funding from the Center for Cellular Immunotherapies at the University of Pennsylvania.
Conflict of Interest Statement
RY, MD, JM, SL, BL, and JF hold patents related to CD19-directed CAR T cell therapy. RY, MD, JM, SL and JF receive research funding from Novartis Pharmaceutical Company. RY, MD, SL and JF also receive research funding from Tmunity Therapeutics Incorporated. BL is a co-founder and equity holder in Tmunity Therapeutics, is a consultant for Novartis and CRC Oncology, and serves on the Scientific Advisory Boards of Avectas, Brammer Bio, Cure Genetics, and Incysus.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DD and handling Editor declared their shared affiliation at the time of review.
References
1. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
2. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
3. Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
4. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
5. Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. (2015) 7:303ra139. doi: 10.1126/scitranslmed.aac5415
6. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
7. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54. doi: 10.1056/NEJMoa1708566
8. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
9. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, prospective evaluation comparing intensity of lymphodepletion before adoptive transfer of tumor-infiltrating lymphocytes for patients with metastatic melanoma. J Clin Oncol. (2016) 34:2389–97. doi: 10.1200/JCO.2016.66.7220
10. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
11. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
12. Lu YC, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. (2014) 20:3401–10. doi: 10.1158/1078-0432.CCR-14-0433
13. Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. (2015) 125:3981–91. doi: 10.1172/JCI82416
14. Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. (2016) 22:433–8. doi: 10.1038/nm.4051
15. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
16. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science (2015) 350:1387–90. doi: 10.1126/science.aad1253
17. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
18. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science (2017) 356:200–5. doi: 10.1126/science.aak9510
19. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
20. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
21. Brocker T, Karjalainen K. Adoptive tumor immunity mediated by lymphocytes bearing modified antigen-specific receptors. Adv Immunol. (1998) 68:257–69. doi: 10.1016/S0065-2776(08)60561-1
22. Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer (2003) 3:35–45. doi: 10.1038/nrc971
23. Maus MV, Fraietta JA, Levine BL, Kalos M, Zhao Y, June CH. Adoptive immunotherapy for cancer or viruses. Annu Rev Immunol. (2014) 32:189–225. doi: 10.1146/annurev-immunol-032713-120136
24. June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. (2018) 379:64–73. doi: 10.1056/NEJMra1706169
25. Mullaney BP, Pallavicini MG. Protein-protein interactions in hematology and phage display. Exp Hematol. (2001) 29:1136–46. doi: 10.1016/S0301-472X(01)00693-2
26. Gacerez AT, Arellano B, Sentman CL. How chimeric antigen receptor design affects adoptive T cell therapy. J Cell Physiol. (2016) 231:2590–8. doi: 10.1002/jcp.25419
27. Brocker T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood (2000) 96:1999–2001.
28. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
29. Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. (2006) 24:e20–2. doi: 10.1200/JCO.2006.05.9964
30. Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, et al. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. (1997) 159:5921–30.
31. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
32. Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. (2009) 17:1453–64. doi: 10.1038/mt.2009.83
33. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
34. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
35. Garfall AL, Maus MV, Hwang WT, Lacey SF, Mahnke YD, Melenhorst JJ, et al. Chimeric Antigen receptor T cells against CD19 for multiple myeloma. N Engl J Med. (2015) 373:1040–7. doi: 10.1056/NEJMoa1504542
36. Ramos CA, Savoldo B, Torrano V, Ballard B, Zhang H, Dakhova O, et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest. (2016) 126:2588–96. doi: 10.1172/JCI86000
37. Ormhoj M, Bedoya F, Frigault MJ, Maus MV. CARs in the lead against multiple myeloma. Curr Hematol Malig Rep. (2017) 12:119–25. doi: 10.1007/s11899-017-0373-2
38. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. (2007) 15:825–33. doi: 10.1038/sj.mt.6300104
39. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
40. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
41. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
42. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
43. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
44. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. (2015) 7:275ra222. doi: 10.1126/scitranslmed.aaa4963
45. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
46. Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. (2013) 31:999–1008. doi: 10.1038/nbt.2725
47. Mirzaei HR, Rodriguez A, Shepphird J, Brown CE, Badie B. Chimeric antigen receptors T cell therapy in solid tumor: challenges and clinical applications. Front Immunol. (2017) 8:1850. doi: 10.3389/fimmu.2017.01850
48. Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patane M, et al. Constitutive and TNFalpha-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR-T cell therapy. Sci Transl Med. (2018) 10:eaao2731. doi: 10.1126/scitranslmed.aao2731
49. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. (2008) 180:4901–9. doi: 10.4049/jimmunol.180.7.4901
50. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
51. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. (2013) 31:71–5. doi: 10.1038/nbt.2459
52. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16:566–81. doi: 10.1038/nrc.2016.97
53. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
54. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164:770–9. doi: 10.1016/j.cell.2016.01.011
55. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:99573. doi: 10.1172/jci.insight.99573
56. Wing A, Fajardo CA, Posey AD Jr, Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res. (2018) 6:605–16. doi: 10.1158/2326-6066.CIR-17-0314
57. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
58. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
59. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
60. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. (2009) 15:808–13. doi: 10.1038/nm.1982
61. Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer (2012) 12:671–84. doi: 10.1038/nrc3322
62. Klebanoff CA, Scott CD, Leonardi AJ, Yamamoto TN, Cruz AC, Ouyang C, et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J Clin Invest. (2016) 126:318–34. doi: 10.1172/JCI81217
63. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. doi: 10.1038/nm.2446
64. Roth A, Yssel H, Pene J, Chavez EA, Schertzer M, Lansdorp PM, et al. Telomerase levels control the lifespan of human T lymphocytes. Blood (2003) 102:849–57. doi: 10.1182/blood-2002-07-2015
65. Topp MS, Riddell SR, Akatsuka Y, Jensen MC, Blattman JN, Greenberg PD. Restoration of CD28 expression in CD28- CD8+ memory effector T cells reconstitutes antigen-induced IL-2 production. J Exp Med. (2003) 198:947–55. doi: 10.1084/jem.20021288
66. Ghassemi S, Nunez-Cruz S, O'Connor RS, Fraietta JA, Patel PR, Scholler J, et al. Reducing ex vivo culture improves the anti-leukemic activity of Chimeric Antigen Receptor (CAR)-T cells. Cancer Immunol Res. (2018) 6:1100–9. doi: 10.1158/2326-6066.CIR-17-0405
67. Klebanoff CA, Crompton JG, Leonardi AJ, Yamamoto TN, Chandran SS, Eil RL, et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight (2017) 2:95103. doi: 10.1172/jci.insight.95103
68. Bowers JS, Majchrzak K, Nelson MH, Aksoy BA, Wyatt MM, Smith AS, et al. PI3Kdelta inhibition enhances the antitumor fitness of adoptively transferred CD8(+) T cells. Front Immunol. (2017) 8:1221. doi: 10.3389/fimmu.2017.01221
69. Lin J-X, Migone T-S, Tseng M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity (1995) 2:331–9. doi: 10.1016/1074-7613(95)90141-8
70. Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med. (2002) 195:1523–32. doi: 10.1084/jem.20020066
71. Berard M, Brandt K, Paus SB, Tough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. (2003) 170:5018–26. doi: 10.4049/jimmunol.170.10.5018
72. Wallace DL, Berard M, Soares MV, Oldham J, Cook JE, Akbar AN, et al. Prolonged exposure of naive CD8+ T cells to interleukin-7 or interleukin-15 stimulates proliferation without differentiation or loss of telomere length. Immunology (2006) 119:243–53. doi: 10.1111/j.1365-2567.2006.02429.x
73. Kaneko S, Mastaglio S, Bondanza A, Ponzoni M, Sanvito F, Aldrighetti L, et al. IL-7 and IL-15 allow the generation of suicide gene–modified alloreactive self-renewing central memory human T lymphocytes. Blood (2009) 113:1006–15. doi: 10.1182/blood-2008-05-156059
74. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
75. Loh YH, Hartung O, Li H, Guo C, Sahalie JM, Manos PD, et al. Reprogramming of T cells from human peripheral blood. Cell Stem Cell (2010) 7:15–9. doi: 10.1016/j.stem.2010.06.004
76. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
77. Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. (2016) 126:3479–94. doi: 10.1172/JCI86437
78. Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature (2017) 552:362–7. doi: 10.1038/nature24633
79. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545:452–6. doi: 10.1038/nature22367
80. Youngblood B, Hale JS, Kissick HT, Ahn E, Xu X, Wieland A, et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature (2017) 552:404–9. doi: 10.1038/nature25144
81. Pace L, Goudot C, Zueva E, Gueguen P, Burgdorf N, Waterfall JJ, et al. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science (2018) 359:177–86. doi: 10.1126/science.aah6499
82. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
83. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
84. Li J, Wientjes MG, Au JL. Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J. (2010) 12:223–32. doi: 10.1208/s12248-010-9181-5
85. Cho A, Howell VM, Colvin EK. The extracellular matrix in epithelial ovarian cancer-a piece of a puzzle. Front Oncol. (2015) 5:245. doi: 10.3389/fonc.2015.00245
86. Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. (2016) 35:655–67. doi: 10.1007/s10555-016-9650-0
87. Enblad G, Karlsson H, Loskog AS. CAR T-cell therapy: the role of physical barriers and immunosuppression in lymphoma. Hum Gene Ther. (2015) 26:498–505. doi: 10.1089/hum.2015.054
88. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics (2016) 3:16006. doi: 10.1038/mto.2016.6
89. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA. (2012) 109:15101–8. doi: 10.1073/pnas.1213353109
90. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
91. Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. (2000) 60:2497–503.
92. Eikenes L, Tari M, Tufto I, Bruland OS, de Lange Davies C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br J Cancer (2005) 93:81–8. doi: 10.1038/sj.bjc.6602626
93. Goodman TT, Olive PL, Pun SH. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int J Nanomed. (2007) 2:265–74.
94. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
95. Long KB, Gladney WL, Tooker GM, Graham K, Fraietta JA, Beatty GL. IFNgamma and CCL2 cooperate to redirect tumor-infiltrating monocytes to degrade fibrosis and enhance chemotherapy efficacy in pancreatic carcinoma. Cancer Discov. (2016) 6:400–13. doi: 10.1158/2159-8290.CD-15-1032
96. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
97. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
98. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
99. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2015) 28:831–3. doi: 10.1016/j.ccell.2015.11.002
100. Klampatsa A, Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, et al. Intracavitary 'T4 immunotherapy' of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. (2017) 393:52–9. doi: 10.1016/j.canlet.2017.02.015
101. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
102. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol. (2017) 95:356–63. doi: 10.1038/icb.2016.128
103. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. (2012) 18:1672–83. doi: 10.1158/1078-0432.CCR-11-3050
104. Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves antiglioma activity of IL13Ralpha2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. (2017) 5:571–81. doi: 10.1158/2326-6066.CIR-16-0376
105. Tang L, Zheng Y, Melo MB, Mabardi L, Castano AP, Xie YQ, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. (2018) 36:707–16. doi: 10.1038/nbt.4181
106. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. (2017) 20:3025–33. doi: 10.1016/j.celrep.2017.09.002
107. Legler DF, Johnson-Leger C, Wiedle G, Bron C, Imhof BA. The alpha v beta 3 integrin as a tumor homing ligand for lymphocytes. Eur J Immunol. (2004) 34:1608–16. doi: 10.1002/eji.200424938
108. Fu X, Rivera A, Tao L, Zhang X. Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int J Cancer (2013) 133:2483–92. doi: 10.1002/ijc.28269
109. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
110. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
111. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood (2017) 129:1039–41. doi: 10.1182/blood-2016-09-738245
112. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
113. Long KB, Collier AI, Beatty GL. Macrophages: key orchestrators of a tumor microenvironment defined by therapeutic resistance. Mol Immunol. (2017). doi: 10.1016/j.molimm.2017.12.003. [Epub ahead of print].
114. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
115. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the immunosuppressive tumor microenvironment of hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. (2017) 7:1154–67. doi: 10.1158/2159-8290.CD-16-0850
116. Beatty GL, O'Hara M. Chimeric antigen receptor-modified T cells for the treatment of solid tumors: defining the challenges and next steps. Pharmacol Ther. (2016) 166:30–9. doi: 10.1016/j.pharmthera.2016.06.010
117. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA, et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. (2015) 64:817–29. doi: 10.1007/s00262-015-1692-6
118. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
119. Ligtenberg MA, Mougiakakos D, Mukhopadhyay M, Witt K, Lladser A, Chmielewski M, et al. Coexpressed catalase protects chimeric antigen receptor-redirected t cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J Immunol. (2016) 196:759–66. doi: 10.4049/jimmunol.1401710
120. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. (2010) 10:490–500. doi: 10.1038/nri2785
121. Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. (2011) 71:6915–20. doi: 10.1158/0008-5472.CAN-11-1156
122. Bollard CM, Rossig C, Calonge MJ, Huls MH, Wagner HJ, Massague J, et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood (2002) 99:3179–87. doi: 10.1182/blood.V99.9.3179
123. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J Clin Oncol. (2018) 36:1128–39. doi: 10.1200/JCO.2017.74.3179
124. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
125. Kobayashi K, Omori K, Murata T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. (2018) 37:347–54. doi: 10.1007/s10555-018-9740-2
126. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. (2010) 285:27571–80. doi: 10.1074/jbc.M110.127100
127. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. (2016) 4:541–51. doi: 10.1158/2326-6066.CIR-15-0263
128. Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, et al. Metabolic heterogeneity in human lung tumors. Cell (2016) 164:681–94. doi: 10.1016/j.cell.2015.12.034
129. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
130. Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. (2009) 183:6095–101. doi: 10.4049/jimmunol.0803510
131. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
132. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
133. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. (2013) 14:1173–82. doi: 10.1038/ni.2714
134. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature (2016) 540:236–41. doi: 10.1038/nature20165
135. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32:377–91 e379. doi: 10.1016/j.ccell.2017.08.004
136. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight (2017) 2:93411. doi: 10.1172/jci.insight.93411
137. Schodel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, et al. Hypoxia, Hypoxia-inducible transcription factors, and renal cancer. Eur Urol. (2016) 69:646–57. doi: 10.1016/j.eururo.2015.08.007
138. Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer (2002) 2:38–47. doi: 10.1038/nrc704
139. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell (2009) 15:501–13. doi: 10.1016/j.ccr.2009.03.018
140. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene (2010) 29:625–34. doi: 10.1038/onc.2009.441
141. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi: 10.1182/blood-2006-06-031856
142. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–42 e813. doi: 10.1016/j.cell.2016.09.031
143. Yusuf I, Fruman DA. Regulation of quiescence in lymphocytes. Trends Immunol. (2003) 24:380–6. doi: 10.1016/S1471-4906(03)00141-8
144. O'Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends Immunol. (2015) 36:71–80. doi: 10.1016/j.it.2014.12.004
145. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
146. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. (2013) 110:14336–41. doi: 10.1073/pnas.1221740110
147. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
148. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
149. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eil RL, et al. Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res. (2015) 75:296–305. doi: 10.1158/0008-5472.CAN-14-2277
150. Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBioMedicine (2016) 4:50–61. doi: 10.1016/j.ebiom.2016.01.019
151. Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. (2016) 23:63–76. doi: 10.1016/j.cmet.2015.11.002
152. Sukumar M, Kishton RJ, Restifo NP. Metabolic reprograming of anti-tumor immunity. Curr Opin Immunol. (2017) 46:14–22. doi: 10.1016/j.coi.2017.03.011
153. Kawalekar OU, RS OC, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T Cells. Immunity (2016) 44:712. doi: 10.1016/j.immuni.2016.01.021
154. Irving M, Vuillefroy de Silly R, Scholten K, Dilek N, Coukos G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: don't forget the fuel. Front Immunol. (2017) 8:267. doi: 10.3389/fimmu.2017.00267
155. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
156. Vasaturo A, Di Blasio S, Peeters DG, de Koning CC, de Vries JM, Figdor CG, et al. Clinical implications of co-inhibitory molecule expression in the tumor microenvironment for DC vaccination: a game of stop and go. Front Immunol. (2013) 4:417. doi: 10.3389/fimmu.2013.00417
157. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. (2013) 19:5636–46. doi: 10.1158/1078-0432.CCR-13-0458
158. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. (2017) 23:6982–92. doi: 10.1158/1078-0432.CCR-17-0867
159. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget (2017) 8:17002–11. doi: 10.18632/oncotarget.15218
160. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543:113–7. doi: 10.1038/nature21405
Keywords: CAR T cell, immunotherapy, cancer, solid tumor, microenvironment, adoptive cell therapy, non-hematopoietic malignancy
Citation: Long KB, Young RM, Boesteanu AC, Davis MM, Melenhorst JJ, Lacey SF, DeGaramo DA, Levine BL and Fraietta JA (2018) CAR T Cell Therapy of Non-hematopoietic Malignancies: Detours on the Road to Clinical Success. Front. Immunol. 9:2740. doi: 10.3389/fimmu.2018.02740
Received: 16 August 2018; Accepted: 07 November 2018;
Published: 03 December 2018.
Edited by:
John-Maher, King's College London, United KingdomReviewed by:
Hinrich Abken, University of Cologne, GermanyDavid Marc Davies, King's College London, United Kingdom
Copyright © 2018 Long, Young, Boesteanu, Davis, Melenhorst, Lacey, DeGaramo, Levine and Fraietta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph A. Fraietta, amZyYWlAdXBlbm4uZWR1