 Alessio Mylonas
Alessio Mylonas Curdin Conrad
Curdin Conrad- Department of Dermatology, University Hospital CHUV, Lausanne, Switzerland
Chronic plaque psoriasis is a common debilitating skin disease. The identification of the pathogenic role of the TNF/IL-23/TH17 pathway has enabled the development of targeted therapies used in the clinic today. Particularly, TNF inhibitors have become a benchmark for the treatment of numerous chronic inflammatory diseases such as psoriasis. Although being highly effective in psoriasis treatment, anti-TNFs can themselves induce psoriasis-like skin lesions, a side effect called paradoxical psoriasis. In this review, we provide a comprehensive look at the different cellular and molecular players involved in classical plaque psoriasis and contrast its pathogenesis to paradoxical psoriasis, which is clinically similar but immunologically distinct. Classical psoriasis is a T-cell mediated autoimmune disease driven by TNF, characterised by T-cells memory, and a relapsing disease course. In contrast, paradoxical psoriasis is caused by the absence of TNF and represents an ongoing type-I interferon-driven innate inflammation that fails to elicit T-cell autoimmunity and lacks memory T cell-mediated relapses.
Introduction
Psoriasis is a distinctly human, chronic, inflammatory skin disease, affecting 2–3% of the population worldwide, with prevalence varying considerably according to race and geographic location (1). Clinically, plaque type psoriasis, the most common form of psoriasis, is characterised by well-demarcated erythematous lesions covered with silvery-white scales. These lesions are histologically reflected by keratinocyte hyperproliferation leading to epidermal hyperplasia (acanthosis), characteristic elongation of the rete ridges (papillomatosis), thickening of the cornified layer (hyperkeratosis), and incomplete keratinocyte differentiation resulting in retention of nuclei in the stratum corneum (parakeratosis). Leukocytes, including T-cells, dendritic cells, neutrophils, and macrophages make up a considerable dermal and epidermal immune cell infiltrate. Psoriasis is caused by the interaction of predisposing genetic factors and environmental triggers leading to dysregulated innate and adaptive immune responses. Today, psoriasis is widely regarded as a T-cell-mediated autoimmune disease and skin infiltrating T lymphocytes play key effector roles by driving disease development and maintenance. Dendritic cells producing TNF and IL-23 stimulate activation of both CD4+ and CD8+ T-cells, which in turn migrate into the epidermis. Upon recognition of autoantigens, T-cells produce TH17-cytokines such as IL-17A, IL-17F, and IL-22, which drive the psoriatic phenotype by inducing keratinocyte hyperproliferation. In support of this, several single nucleotide polymorphisms cluster throughout this pathway including genes in the TNF/dendritic cell activation pathway (TNFAIP3, REL, TN1P1, NFKBIA) as well as in the T-cell activation (HLA-Cw6, ERAP1/ZAP70, ETS1, SOCS1, TNFRSF9), and TH17/TC17-differentiation pathways (IL23A, IL23R) (2, 3). Consequently, antibodies targeting the pathogenic TNF/IL-23/IL-17 pathway have revolutionised psoriasis treatment over the past 15 years and are widely used in the clinic today.
In particular, TNF blockade has become the benchmark in management of numerous chronic inflammatory diseases, such as rheumatoid arthritis, Crohn's disease, and psoriasis (4–7). As such, more than two million patients have already been treated with anti-TNFs and, with the advent of biosimilars, these figures are expected to grow further over the coming years. Yet, targeting TNF is not without consequence, as TNF is a potent pro-inflammatory cytokine known to coordinate immune responses and play an important role in limiting the spread of infectious pathogens. Thus, TNF blockade leads to an increased risk of infections and slightly increased risk for certain malignancies. However, more surprisingly, anti-TNF treatment can also induce new psoriasis-like skin lesions in about 2–5% of treated patients (8).
As anti-TNFs are amongst the most potent anti-inflammatory drugs used in the treatment of psoriasis, developing psoriasis-like skin lesions due to TNF blockade was somewhat paradoxical—hence the designation “paradoxical psoriasis.”
This review aims to provide a focussed overview of the latest developments in the T-cell and cytokine networks in classical psoriasis, and contrast them to paradoxical psoriasis induced by anti-TNFs, which is clinically similar to psoriasis but immunologically distinct. Finally, these findings will be put into perspective with future avenues of research and possible clinical interventions.
Classical Psoriasis
The Established Role of T-Helper and the Revisited Cytotoxic T-Cells
The pathogenic role for T-cells in psoriasis is well-established and stems from the following clinical observations and experimental findings: Immunosuppressive agents, such as cyclosporine, or therapies specifically targeting T-cells are efficacious in psoriasis treatment (9–12). HLA-Cw6 represents the strongest genetic risk variant associated with psoriasis (13). Molecular analysis of psoriasis tissue showed that lesional T-cells are oligoclonal (14) and recognise epidermal autoantigens (15–18). Finally, clinically relevant xenotransplant models of psoriasis have demonstrated an essential functional role for T-cells (19–21).
T-cells migrate into inflamed skin through expression of the skin-homing Cutaneous Lymphocyte-associated Antigen (CLA) (22), LFA-1 and α4β1 (23), and the chemokine receptors CCR8 and CCR10 (24). More specifically TH1 cells use CXCR3 and CCR4 (25), whereas TH17 cells use CCR4 and CCR6 (26). Among the most well-described chemokines involved in T-cell migration to the skin are CCL27 (27, 28), and CCL20 (29) produced by keratinocytes upon an inflammatory trigger. While circulating T-cells certainly play an important role in skin immunopathology, there are twice as many T-cells residing in normal healthy skin than are present in the circulation (22). Moreover, pathogenic oligoclonal T-cells remain resident in resolved psoriatic skin lesions suggesting that disease recurrence might be initiated through reactivation of skin-resident T-cells (30). Indeed, these skin-resident memory T-cells were found to be sufficient to drive psoriasis development without further recruitment of circulating cells (19, 20). Activation within the skin led to proliferation of T-cells in the dermal compartment, which preceded keratinocyte hyperproliferation. In fact, the psoriatic phenotype was only induced by migration of T-cells into the epidermis and blockade of the epidermal infiltration by T-cells prevented the development of a psoriatic lesion (20). These findings suggest that intraepidermal T-cells reflect key effector cells in psoriasis.
Traditionally, much attention has been given to differentiated CD4+ T-cell subsets across chronic inflammatory diseases (31–34), including psoriasis (35). However, CD8+ T-cells, which are present in healthy skin as tissue resident memory T-cells (36), have been shown to produce a similar cytokine profile (37). In psoriasis, dermal T-cell infiltrates are mostly comprised of CD4+ cells, whereas the majority of T-cells in the epidermis—which represent key effector cells—are CD8+ (19). Indeed, we could recently show that intraepidermal CD8+ T-cells are functionally essential for psoriasis (38).
Psoriasis has been studied extensively from a genetics perspective, with HLA class I alleles known for more than 40 years to be heavily implicated (39). The HLA-Cw6 variant is the strongest psoriasis susceptibility allele and has 10-fold higher association with early-onset severe psoriasis. As to how exactly class I HLA molecules might contribute to the pathogenesis of psoriasis is not entirely clear. But in light of the fundamental role of epidermal CD8+ T-cells in psoriasis, the fact that lesional T-cells are of oligoclonal origin and CD8+ T-cells recognise peptide antigens presented on MHC class I molecules suggest a role for epidermal (auto-)antigens in psoriasis. As mentioned above, epidermal CD8+ T-cells in psoriasis are key effectors in psoriasis (20), and they are of oligoclonal origin (14, 30)—thus potentially recognising common antigens. Taken together with HLA-Cw6 representing the strongest genetic risk variant associated with psoriasis, this suggests that recognition of epidermal (auto-)antigens by CD8+ T-cells is pathogenic in psoriasis.
Indeed, the streptococcal M protein from Streptococcus pyogenes has been identified as an antigen target of primarily CD8+ T-cells (40). T-cells directed against the streptococcal M-protein had the ability to react to keratin 14, which is overexpressed in psoriatic skin, due to sequence homology and antigenic similarity (molecular mimicry). Thus, the immune response to a streptococcal infection could divert T-cells toward skin antigens and cause skin pathology. Intriguingly, streptococcal throat infections are a well-known trigger factor for onset and exacerbation of psoriasis.
Other recently identified epidermal autoantigens include keratin 7 (41) and the antimicrobial peptide LL37 expressed by keratinocytes (17) as well as the melanocyte antigen ADAMTSL5 (18). Finally, CD1a-restricted lipids were also found to elicit T-cell responses in psoriatic patients (42). Interestingly, CD1a-autoreactive T-cells isolated from skin were identified as TH22 cells producing IL-22 (43), a cytokine overexpressed in psoriasis and known to drive keratinocyte hyperproliferation.
Antigen-recognition by T-cells is thought to play a pivotal role in psoriasis, but an all-encompassing consensus on the nature of autoreactivity has yet to be reached. Despite this, all of the identified auto-antigens to date are significantly upregulated in psoriatic skin as compared to uninvolved or healthy skin. Because the majority can be induced locally upon injury, the prevailing model postulates that skin trauma could lead to upregulation of putative auto-antigens and their recognition by tissue-resident antigen-experienced T-cells in psoriasis patients.
Cytokine Networks: The TNF/IL-23/IL-17 Axis
Nowadays, the pathogenic role of the TNF/IL-23/TH17 axis in psoriasis is well-known and numerous biologics targeting the different cytokines of this pro-inflammatory pathway are widely used in the clinic. Yet, the arrival of TNF blockers in the early 2000s completely revolutionised the management of psoriasis and other chronic inflammatory diseases. Despite underwhelming results of anti-TNF in sepsis (44), the successful use in rheumatoid arthritis (RA) spurred trials in other chronic inflammatory diseases such as Crohn's disease, psoriasis and psoriatic arthritis (45).
TNF is known to be potently produced by immune and non-immune cells including macrophages, T-cells, dendritic cells (DC), neutrophils, and fibroblasts. One of its major roles is to mount appropriate adaptive immune responses to tumors and pathogens. This is achieved through several mechanisms. Induction of DC-maturation leads to upregulation of CD40, CD80, CD83, and CD86 thereby potentiating T-cell receptor (TCR)-mediated responses and amplifying weak antigen affinity interactions (46). It also serves to limit the immune-suppressive effects of regulatory T-cells (47) and to enhance proliferation and survival of committed effector memory T-cells. In line with these findings, TNF is critically required to mount effective CD8+ T-cell responses against tumors and for the recruitment of T-cells into tumor sites (48). These pro-inflammatory effects of TNF are corroborated in psoriasis, where TNF is found to dictate the inflammatory environment in several ways (49–52). In detailed histological and molecular investigations, it was found to be mostly produced by mature conventional DCs. Blockade of TNF leads to an initial reduction of the chemokine CCL20, which preferentially recruits TH17 cells into inflamed tissue, coinciding with loss of IL-17 and diminution of dermal and epidermal T-cells. In addition, it leads to normalisation of DC numbers and reduction of IL-23 cytokine expression, followed by normalised keratinocyte differentiation, and eventually to histological improvement and clinical response. Taken together, TNF maintains a pro-inflammatory environment that primes pathogenic TH17 T-cells through induction of IL-23, maintaining them at the site of inflammation, and sustaining TH17 cytokine production (53, 54).
Though TNF might contribute to increased IL-17 production by TH17 cells (55), IL-23 directly governs TH17 cytokine production both by critically participating in TH17 cell polarisation as well as by stimulating production of IL-17 by differentiated TH17 cells (56, 57). Initial supportive evidence for a functional role of IL-23 in psoriasis included the clinical efficacy of an anti-p40 monoclonal antibody (blocking both IL-12 and IL-23) in psoriasis (58) and the association of a single nucleotide polymorphism in the IL23R gene in psoriasis patients (55, 59). Confirmation soon followed by the successful use of IL-23-specific antibodies in clinically relevant mouse models and then in patients (60, 61). In addition, IL-4 abrogated TH17 cell-mediated inflammation by selectively silencing IL-23 in antigen-presenting cells while sparing IL-12/TH1 immunity (62) and resulting in therapeutic outcome. In psoriasis, IL-23 is mainly produced by activated DCs but keratinocytes and other non-immune cells probably contribute to its production. In fact, it has been shown recently that TNF-dependent epigenetic control of IL-23 expression in keratinocytes plays a role in chronic skin inflammation (63).
TH17 cytokines, such as IL-17A, IL-17F, and IL-22, represent the key effector cytokines in psoriasis pathogenesis as they directly drive the development of a psoriatic phenotype. They induce epidermal hyperproliferation, attract neutrophils to the skin, and activate keratinocytes to produce chemokines and antimicrobial peptides, which sustain the inflammatory process (64). There are six homologous IL-17 cytokines (A through F) which are produced by either haematopoietic or non-haematopoietic cells, can signal through different combinations of receptors, and mediate distinct biological activities. The individual members are reviewed in this issue by Brembilla et al. (65). Besides the aforementioned effects in the pathogenesis of psoriasis, IL-17 can act in synergy with TNF to further potentiate expression of multiple pro-inflammatory mediators known to play a role in psoriasis, such as IL-8, beta-defensins, S100A proteins, IL-19, and CCL20 (29, 66–68). As such, concurrent inhibition of TNF and IL17 might result in more effective therapy. Though, a bi-specific dual variable domain immunoglobulin targeting both cytokines did not demonstrate increased efficacy compared to anti-TNF in RA (69), this remains to be tested in psoriasis.
While antibodies targeting IL-17 have shown great efficacy in psoriasis (53, 70), blockade of IL-22 failed to meet primary end points in clinical trials indicating distinct role for IL-17 and IL-22 in psoriasis. IL-22, which is produced by TH17 cells (71) and exclusively by a distinct TH22 subpopulation (72, 73), had previously been regarded as a very promising target. In psoriasis, IL-22 is found to be produced by dermal CD4+ but also by epidermal CD8+ TC17 and CD4+ TH22 cells (38, 74, 75), as well as Innate Lymphoid Cells (76, 77), and mast cells (78). Interestingly, skin-resident T-cells mediating disease memory in clinically resolved psoriasis plaques were found to be epidermal TC17 and TH22 cells producing mainly IL-22 (75). Upon binding to its heterodimeric receptor consisting of IL-22RA1 and IL10Rβ (79), which is expressed on keratinocytes (80), IL-22 induces proliferation of keratinocytes and inhibits terminal maturation (81, 82). While transient expression of IL-22 upon skin injury promotes epidermal remodelling with re-epithelisation of skin wounds (74), its chronic expression by psoriatic T-cells drives keratinocyte hyperproliferation and epidermal hyperplasia. In line with this, in transgenic mice, IL-22 was sufficient to induce a skin phenotype that resembles psoriasis (82), and the psoriatic phenotype induced by skin injection of IL-23 was abrogated in IL-22-deficient mice (83). However, the pathogenic function of IL-22 shows redundancy with other members of the IL-20 subfamily of cytokines such as IL-19 and IL-20, potentially rendering its blockade clinically ineffective in humans.
Detailed knowledge about the pathogenesis of chronic plaque psoriasis and the central role for the TNF/IL-23/TH17 pathway has led to the development of therapies targeting the pathogenic cytokines, including anti-TNFs, anti-p40 (IL-12/IL-23), anti-p19 (IL-23 specific), anti-IL-17A, and anti-IL-17 receptor antibodies. This pathway and its pathogenic mechanisms in classical plaque psoriasis are illustrated in Figure 1. However, less is known about instigators of psoriasis and pathogenic upstream triggers of acute cutaneous inflammation.
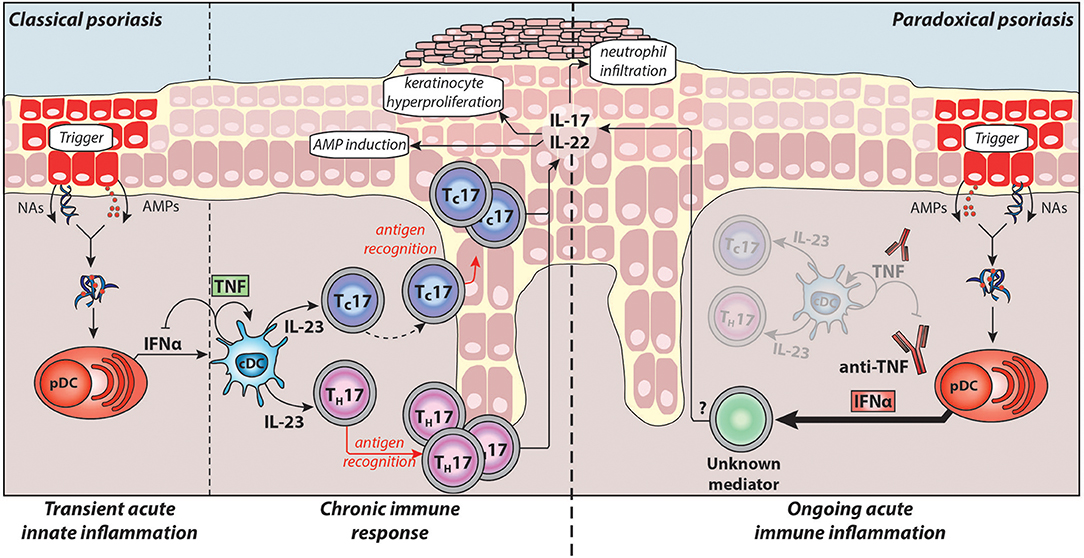
Figure 1. Pathogenesis of classical plaque psoriasis and paradoxical psoriasis. Antimicrobial peptides (AMPs), which are produced by keratinocytes upon skin injury or released by neutrophils, form complexes with nucleic acids (NAs) released by dying cells. These complexes activate plasmacytoid dendritic cells (pDC) to produce large amounts IFNα during the acute/early phase of psoriasis pathogenesis. IFNα activates conventional dendritic cells (cDCs), which in turn produce TNF and IL-23. TNF induces the maturation of cDCs and pDCs, which lose their ability to produce IFNα. Thus, in classical psoriasis, early IFNα production gets relayed by TNF that controls and limits the IFNα production by pDCs via a negative feedback loop (through induction of pDC maturation). Subsequently, IL-23 and other pro-inflammatory cytokines produced by cDCs drive the activation of potentially autoreactive T-cells, which proliferate and, particularly CD8+ TC cells, migrate into the epidermis. Upon antigen recognition they produce the TH17 cytokines IL-17 and IL-22 that induce keratinocyte hyperproliferation, attract neutrophils to the skin, and upregulate AMP production providing a positive feedback loop eventually resulting in the psoriatic phenotype (chronic/late phase). Normally, during anti-TNF therapy, the absence of TNF and consequently of downstream cytokines suppresses pathogenic T-cells thereby alleviating classical psoriasis. However, in patients developing paradoxical psoriasis, TNF blockade inhibits pDC maturation and leads to sustained IFNα production. In addition, as cDC cannot mature in absence of TNF, paradoxical psoriasis fails to elicit a T cell mediated autoimmune response. Thus, paradoxical psoriasis remains in an ongoing IFNα-driven acute immune inflammation independent of T-cells. The exact pathogenic downstream mechanism of IFNα-driven paradoxical psoriasis skin lesions remains to be fully elucidated.
Type I Interferons, Setting the Tone for Autoimmunity?
Type I IFNs are key cytokines in antiviral host defence due to their ability to limit viral replication and to induce an effective antiviral immune response (84, 85). They promote maturation of myeloid DCs and priming of CD8+ T-cells (86), induce T-cell proliferation (87), and sustain their survival (88). In addition, type-I IFNs stimulate differentiation of B-cells into antibody-secreting plasma cells (89). Thus, type-I IFNs are essential for the induction of an effective immune response against viruses. Although produced by all nucleated cells, they are preferentially expressed by a rare type of circulating cells called plasmacytoid dendritic cells (pDCs) (90). Upon viral recognition through endosomal Toll-like receptors (TLR) 7 and 9, pDCs produce extraordinary amounts of type-I IFN, and, therefore, have also been called professional IFN producing cells (90, 91). Under normal conditions, pDCs are not present in peripheral tissues, but they get recruited to the skin in case of infection, injury, autoimmunity, and cancer (64). Skin wounding induces rapid skin infiltration of pDCs and transient expression of type-I IFNs, which accelerate re-epithelialisation (92).
As self-nucleic acids are abundantly released into the extracellular environment during apoptotic and necrotic cell death, it is essential that pDCs avoid inappropriate activation by host-derived nucleic acid, but retain the ability to quickly respond to viral DNA and RNA. To achieve this, TLRs that sense nucleic acids are located intracellularly within endosomes, which prevents activation by extracellular self-DNA/RNA but allows immune response to viruses that actively invade the cells. Moreover, extracellular nucleases rapidly degrade nucleic acid released by dying cells without affecting DNA or RNA contained within viruses (93). Thus, under normal circumstances host-derived self-DNA/RNA released by apoptotic or damaged cells cannot activate TLR9 and 7. However, they can become potent triggers of pDC activation and type-I IFN production in the presence of endogenous antimicrobial peptides (AMP) such as LL37 and beta-defensins (94–96). AMPs are typically not expressed in healthy skin under steady-state conditions, but are transiently produced by keratinocytes or released by infiltrating neutrophils in response to skin wounding or infections (97–99). Their cationic and amphipathic structure allows AMPs to interact with and disrupt microbial membranes, which typically contain a high degree of negative charges (100). Besides their role as direct effector molecules against microorganisms, AMPs are also involved in the initiation of inflammation by breaking innate tolerance to otherwise inert extracellular self-DNA and self-RNA. Cationic AMPs bind to negatively charged fragments of nucleic acid to form aggregated and condensed structures that are resistant to extracellular degradation. Translocation of these complexes into endosomes and activation of TLR7 and 8 (RNA), or TLR9 (DNA) lead to sustained production of IFNα and IFNβ by pDCs (94–96).
Under physiological conditions, AMP-expression with activation of pDCs by AMP-nucleic acid complexes is transient, controlled by the damaging or infectious stimulus. By contrast, in psoriasis, the expression of AMPs is persistent and leads to sustained production of type-I IFNs by pDCs, which accumulate in the dermis of early developing psoriatic lesions (101). Subsequently, these trigger activation of myeloid DCs and autoreactive T-cells. Recent work has demonstrated that IFNα particularly drives the activation and skin infiltration of pathogenic CD8+ T-cells in psoriasis (102). Moreover, IFNα conditioned DCs produce large amounts of IL-23 (103, 104) indicating an important role for type-I IFNs in driving TH/TC17-mediated (auto)immunity in psoriasis. Indeed, depletion of pDCs or blocking type-I IFN signalling both inhibited psoriasis development confirming that its overexpression by pDCs reflects a critical early/acute event in the pathogenesis of psoriasis (101). This is further supported by the observations that de novo psoriasis or pre-existing psoriasis can be triggered and/or aggravated by IFNα therapy (105–108) and the TLR7 agonist imiquimod (109, 110). Interestingly, epidermal trauma, which induces AMP expression by keratinocytes and attracts pDCs into the skin, is also a typical trigger of psoriasis known as Koebner phenomenon.
Taken together, type-I IFNs play a critical role in the acute/early phase of psoriasis pathogenesis by (1) activating dermal myeloid DC, (2) inducing their maturation by upregulating co-stimulatory molecules and HLA molecules, and (3) participating in TH/TC17 polarisation of autoimmune T-cells through induction of IL-23 production by myeloid DCs (Figure 1).
Paradoxical Psoriasis
Almost two decades of clinical experience with anti-TNFs have provided considerable advances in our understanding of the biology of TNF. More than 2 million patients have been treated with anti-TNFs so far.
Expected side effects such as increased susceptibility to infection and a slightly increased risk for malignancies have been confirmed (111–113), though the cancer risk still remains a matter of debate (114, 115). However, the observation that anti-TNFs, which are normally extremely effective in the treatment of chronic inflammatory diseases, could lead to aggravation of pre-existing autoimmune diseases and onset of new inflammatory diseases, was unexpected and a paradox. In fact, lupus-like syndrome can be observed in 0.5–1% of anti-TNF treated patients and 2–5% of patients develop psoriasis-like skin lesions, called paradoxical psoriasis (8, 116, 117). They represent important side effects in the treatment of major chronic autoimmune diseases as they potentially necessitate treatment cessation. Since the first description of paradoxical psoriasis (117, 118), numerous cases have been reported (119–121). Paradoxical psoriasis appears independently of the underlying disease or the type of anti-TNF agent used and regresses upon discontinuation of therapy, which suggests that paradoxical psoriasis does represent a side effect of TNF blockade and not de novo psoriasis. Though the side effect is a well-established phenomenon, its pathogenesis had remained elusive and only recently, the dysbalance of TNF and type-I IFN (yin-yang of TNF and IFNα) has been confirmed as a pathogenic mechanism underlying paradoxical psoriasis (8).
The first clues for a link between anti-TNF therapy and increased type-I IFN expression came from the observation that anti-TNF therapy induces an IFN signature in blood of juvenile arthritis patients (122). Likewise, anti-TNF treatments promote formation of anti-nuclear antibodies (123), which are associated with increased type-I IFN levels in SLE patients (124). Furthermore, anti-TNFs can induce or aggravate lupus, a well-known type-I IFN-driven autoimmune disease (125, 126). Indeed, patients with anti-TNF induced paradoxical psoriasis showed an increased IFN signature in lesional skin (127). Recently, we could confirm that TNF controls the production of type I-IFN by pDCs and that anti-TNF induces its unabated overexpression driving paradoxical psoriasis (8).
Upon activation, pDCs produce type-I IFNs first, which is relayed by their production of TNF. TNF induces maturation of pDCs, which upregulate costimulatory molecules and lose their ability to produce interferons (8, 128). Thereby, TNF limits the duration of type-I IFN production by pDCs, while conversely, TNF blockade decreases pDC maturation and extends their ability to produce type-I IFN. This supports a yin-yang model of TNF and type-I IFN. In classical plaque psoriasis, early transient overexpression of type-I IFN is replaced by a dominant TNF-driven chronic inflammation. In contrast, TNF blockade leads to an ongoing type-I IFN mediated acute inflammation in paradoxical psoriasis (Figure 1).
Another important distinction between the two entities is that in paradoxical psoriasis, unlike classical psoriasis, T-cells play a redundant role (8). Hence, both classical and paradoxical psoriasis are induced by pDC-derived type-I IFN. But while classical psoriasis develops into a T-cell mediated autoimmune disease, paradoxical psoriasis represents an ongoing type-I IFN-driven innate immune response that fails to elicit T cell autoimmunity. In line with this, there are no relapses of paradoxical psoriasis upon discontinuation of anti-TNF therapy, which supports lack of T-cell mediated disease memory in paradoxical psoriasis.
It remains unclear what triggers the activation of pDCs and eventually drives paradoxical psoriasis. Potentially certain environmental factors such as microbes could trigger development of paradoxical psoriasis, as a considerable number of patients have been found to have concurrent superinfections (129). Furthermore, the yin-yang of TNF and type-I IFN, the pathogenic mechanism underlying paradoxical psoriasis, is inherently true for healthy individuals as much as patients. But only 2–5% of anti-TNF treated patients develop paradoxical psoriasis indicating that there is another key determining factor such as genetic predisposition for paradoxical psoriasis. Among polymorphisms associated with psoriasis, five have been identified to also be associated to paradoxical psoriasis, and these include IL23R, FBXL19, CTLA4, SLC12A8, and TAP1 (130) though it remains to be determined exactly how they would fit in the pathological mechanism.
Though classical and paradoxical psoriasis have similarities in their clinical presentation, many distinctions have been identified in recent years as to the pathogenic mechanism. Table 1 summarises key similarities and differences between the two entities, and highlights potential treatment strategies.
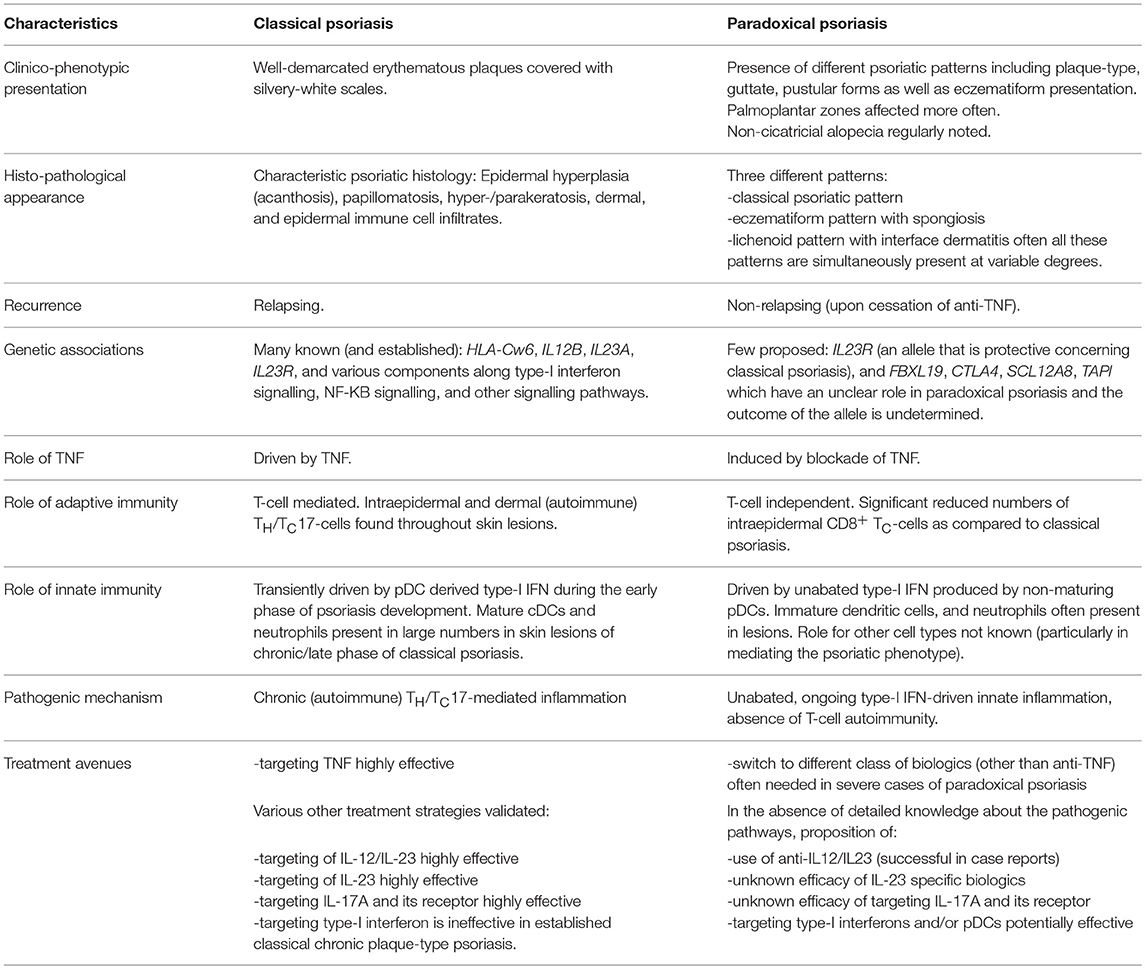
Table 1. Classical vs. Paradoxical psoriasis- differences, similarities, and treatment strategies.
Outlook
Detailed knowledge on classical plaque psoriasis, particularly by identifying the relevant role of the TNF/IL-23/TH17 axis in its pathogenesis, has allowed for novel, more targeted therapies.
Though newer treatments targeting IL-23 and IL-17 show better efficacy, today, anti-TNFs remain a gold-standard in psoriasis management. Yet important immunological side effects of TNF blockade, such as paradoxical psoriasis and lupus-like syndrome, may require premature discontinuation of an otherwise effective treatment option for patients. These side effects are caused by an unabated type-I IFN-driven immune response making it an intriguing target in these patients. While, anti-IFNs have not shown efficacy in chronic plaque psoriasis confirming the distinct inflammatory pathways in chronic and acute forms of classical psoriasis (131), they provided promising results in SLE. Therefore, type-I IFN blockade might be a valuable treatment option in acute forms of psoriasis such as erythrodermic or guttate psoriasis as well as in paradoxical psoriasis. However, simultaneous inhibition of the interferon pathway together with the ongoing TNF blockade might increase the infectious risk too considerably. Therefore, targeting IFN-producing pDCs (i.e., via anti-ILT-7, anti-BDCA2) or inhibiting TLR 7 and 9, thereby blocking pDC activation, could provide more suitable therapeutic options in patients that need continuation of their anti-TNF treatment. In this way, production of type-I IFNs by monocytes and stromal cells would remain intact and might allow sufficient immune responses toward infectious agents.
Despite considerable advances in the understanding of paradoxical psoriasis and its pathogenesis, several questions are still unanswered. Downstream mechanisms that mediate the interferon-driven psoriatic phenotype of paradoxical psoriasis remain unknown as IFNα does not directly induce keratinocyte hyperproliferation. The identification of cytokines involved and their cellular source might provide additional novel targets for therapeutic intervention. In addition, biomarkers to predict side effects such as paradoxical psoriasis and lupus-like syndrome could help optimising the management of patients with chronic inflammatory diseases.
Author Contributions
AM and CC wrote and edited the manuscript and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge funding from the Gottfried & Julia Bangerter-Rhyner Foundation, the University Hospital of Lausanne (CHUV), and the Faculty of Biology and Medicine of the University of Lausanne (FBM-UNIL).
References
1. Parisi R, Symmons DP, Griffiths CE, Ashcroft DM, et al. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. (2013) 133:377–85. doi: 10.1038/jid.2012.339
2. Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. (2012) 44:1341–8. doi: 10.1038/ng.2467
3. Tsoi LC, Spain SL, Ellinghaus E, Stuart PE, Capon F, Knight J, et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat Commun. (2015) 6:7001. doi: 10.1038/ncomms8001
4. Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, et al. Treatment of rheumatoid arthritis with a recombinant human Tumour Necrosis Factor Receptor (p75)-Fc fusion protein. N Engl J Med. (1997) 337:141–8.
5. Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. (2003) 349:2014–2023. doi: 10.1056/NEJMoa030409
6. Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. (2005) 353:2462–76. doi: 10.1056/NEJMoa050516
7. Taylor PC, Feldmann M. Anti-TNF biologic agents: still the therapy of choice for rheumatoid arthritis. Nat Rev Rheumatol. (2009) 5:578–82. doi: 10.1038/nrrheum.2009.181
8. Conrad C, Di Domizio J, Mylonas A, Belkhodja C, Demaria O, Navarini AA, et al. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat Commun. (2018) 9:25. doi: 10.1038/s41467-017-02466-4
9. Weinshenker BG, Bass BH, Ebers GC, Rice GP. Remission of psoriatic lesions with muromonab-CD3 (Orthoclone OKT3) treatment. J Am Acad Dermatol. (1989) 20:1132–3.
10. Bachelez H, Flageul B, Dubertret L, Fraitag S, Grossman R, Brousse N, et al. Treatment of recalcitrant plaque psoriasis with a humanized non-depleting antibody to CD4. J Autoimmun (1998) 11:53–62.
11. Gottlieb SL, Gilleaudeau P, Johnson R, Estes L, Woodworth TG, Gottlieb AB, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests primary immune, but not keratinocyte, pathogenic basis. Nat Med. (1995) 1:442–8.
12. Krueger GG, Ellis CN. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol. (2003) 148:784–8. doi: 10.1046/j.1365-2133.2003.05239.x
13. Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, Jenisch S, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. (2006) 78:827–51. doi: 10.1086/503821
14. Prinz JC, Vollmer S, Boehncke WH, Menssen A, Laisney I, Trommler P. Selection of conserved TCR VDJ rearrangements in chronic psoriatic plaques indicates a common antigen in psoriasis vulgaris. Eur J Dermatol. (1999) 29:3360–8.
15. Vollmer S, Menssen A, Prinz JC. Dominant lesional T cell receptor rearrangements persist in relapsing psoriasis but are absent from nonlesional skin: evidence for a stable antigen-specific pathogenic T cell response in psoriasis vulgaris. J Invest Dermatol. (2001) 117:1296–301. doi: 10.1046/j.0022-202x.2001.01494.x
16. Sigmundsdottir H, Sigurgeirsson B, Troye-Blomberg M, Good MF, Valdimarsson H, Jonsdottir I. Circulating T cells of patients with active psoriasis respond to streptococcal M-peptides sharing sequences with human epidermal keratins. Scand J Immunol. (1997) 45:688–97.
17. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. (2014) 5:5621. doi: 10.1038/ncomms6621
18. Arakawa A, Siewert K, Stöhr J, Besgen P, Kim SM, Rühl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. (2015) 212:2203–12. doi: 10.1084/jem.20151093
19. Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. (2004) 199:731–6. doi: 10.1084/jem.20031482
20. Conrad C, Boyman O, Tonel G, Tun-Kyi A, Laggner U, de Fougerolles A, et al. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat Med. (2007) 13:836–42. doi: 10.1038/nm1605
21. Wrone-Smith T, Nickoloff BJ. Dermal injection of immunocytes induces psoriasis. J Clin Invest. (1996) 98:1878–87. doi: 10.1172/JCI118989
22. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. (2006) 176:4431–9. doi: 10.4049/jimmunol.176.7.4431
23. Teraki Y, Miyake A, Takebayashi R, Shiohara T. Homing receptor and chemokine receptor on intraepidermal T cells in psoriasis vulgaris. Exp Dermatol. (2004) 29: 658–63. doi: 10.1111/j.1365-2230.2004.01638.x
24. Schaerli P, Ebert L, Willimann K, Blaser A, Roos RS, Loetscher P, et al. A skin-selective homing mechanism for human immune surveillance T cells. J Exp Med. (2004) 199:1265–75. doi: 10.1084/jem.20032177
25. Al-Banna NA, Vaci M, Slauenwhite D, Johnston B, Issekutz TB. CCR4 and CXCR3 play different roles in the migration of T cells to inflammation in skin, arthritic joints, and lymph nodes. Eur J Immunol. (2014) 44:1633–43. doi: 10.1002/eji.201343995
26. Pène J, Chevalier S, Preisser L, Vénéreau E, Guilleux MH, Ghannam S, et al. Chronically inflamed human tissues are infiltrated by highly differentiated Th17 Lymphocytes. J Immunol. (2008) 180:7423–30. doi: 10.4049/jimmunol.180.11.7423
27. Homey B, Alenius H, Müller A, Soto H, Bowman EP, Yuan W, et al. CCL27–CCR10 interactions regulate T cell–mediated skin inflammation. Nat Med. (2002) 8:157–66. doi: 10.1038/nm0202-157
28. Sigmundsdottir H, Pan J, Debes GF, Alt C, Habtezion A, Soler D, et al. DCs metabolize sunlight-induced vitamin D3 to 'program' T cell attraction to the epidermal chemokine CCL27. Nat Immunol. (2007) 8:285–93. doi: 10.1038/ni1433
29. Homey B, Dieu-Nosjean MC, Wiesenborn A, Massacrier C, Pin JJ, Oldham E, et al. Up-regulation of macrophage inflammatory protein-3 /CCL20 and CC chemokine receptor 6 in psoriasis. J Immunol. (2000) 164:6621–32. doi: 10.4049/jimmunol.164.12.6621
30. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. (2017) 127:4031–41. doi: 10.1172/JCI93396
31. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. (2006) 6:329–33. doi: 10.1038/nri1807
32. Raphael I, Nalawade S, Eagar TN, Forsthuber TG T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74:5–17. doi: 10.1016/j.cyto.2014.09.011
33. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol. (2018) 18:121–33. doi: 10.1038/nri.2017.118
34. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
35. Diani M, Altomare G, Reali E. T helper cell subsets in clinical manifestations of psoriasis. J Immunol Res. (2016) 2016:7692024. doi: 10.1155/2016/7692024
36. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. (2015) 7:279ra39. doi: 10.1126/scitranslmed.3010302
37. Hijnen D, Knol EF, Gent YY, Giovannone B, Beijn SJ, Kupper TS, et al. CD8(+) T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-gamma, IL-13, IL-17, and IL-22. J Invest Dermatol. (2013) 133:973–9. doi: 10.1038/jid.2012.456
38. Di Meglio P, Villanova F, Navarini AA, Mylonas A, Tosi I, Nestle FO, et al. Targeting CD8(+) T cells prevents psoriasis development. J Allergy Clin Immunol. (2016) 138:274–276.e276. doi: 10.1016/j.jaci.2015.10.046
39. Tiilikainen A, Lassus A, Karvonen J, Vartiainen P, Julin M. Psoriasis and HLA-Cw6. Br J Dermatol. (1980) 102:179–84.
40. Johnston A, Gudjonsson JE, Sigmundsdottir H, Love TJ, Valdimarsson H. Peripheral blood T cell responses to keratin peptides that share sequences with streptococcal M proteins are largely restricted to skin-homing CD8+ T-cells. Clin Exp Dermatol. (2004) 138:83–93. doi: 10.1111/j.1365-2249.2004.02600.x
41. Yunusbaeva M, Valiev R, Bilalov F, Sultanova Z, Sharipova L, Yunusbayev B. Psoriasis patients demonstrate HLA-Cw*06:02 allele dosage-dependent T cell proliferation when treated with hair follicle-derived keratin 17 protein. Sci Rep. (2018) 8:6098. doi: 10.1038/s41598-018-24491-z
42. Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, et al. Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med. (2016) 213:2399–412. doi: 10.1084/jem.20160258
43. de Jong A, Peña-Cruz V, Cheng TY, Clark RA, Van Rhijn I, Moody DB. CD1a-autoreactive T cells are a normal component of the human alphabeta T cell repertoire. Nat Immunol. (2010) 11:1102–9. doi: 10.1038/ni.1956
44. Fisher CJ, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. New Engl J Med. (1996) 334:1697–702. doi: 10.1056/Nejm199606273342603
45. Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF therapy: past, present and future. Int Immunol. (2015) 27:55–62. doi: 10.1093/intimm/dxu102
46. Maney NJ, Reynolds G, Krippner-Heidenreich A, Hilkens CMU. Dendritic cell maturation and survival are differentially regulated by TNFR1 and TNFR2. J Immunol. (2014) 193:4914–23. doi: 10.4049/jimmunol.1302929
47. Chen X, Hamano R, Subleski JJ, Hurwitz AA, Howard OM, Oppenheim JJ. Expression of costimulatory TNFR2 induces resistance of CD4+FoxP3- conventional T cells to suppression by CD4+FoxP3+ regulatory T cells. J Immunol. (2010) 185:174–82. doi: 10.4049/jimmunol.0903548
48. Calzascia T, Pellegrini M, Hall H, Sabbagh L, Ono N, Elford AR, et al. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J Clin Invest. (2007) 117:3833–45. doi: 10.1172/JCI32567
49. Gottlieb AB. Tumor necrosis factor blockade: mechanism of action. J Investig Dermatol Symp Proc. (2007) 12:1–4. doi: 10.1038/sj.jidsymp.5650029
50. Tan JK, Aphale A, Malaviya R, Sun Y, Gottlieb AB. Mechanisms of action of etanercept in psoriasis. J Investig Dermatol Symp Proc. (2007) 12:38–45. doi: 10.1038/sj.jidsymp.5650037
51. Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. (2007) 204:3183–94. doi: 10.1084/jem.20071094
52. Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. (2009) 124:1022–10.e1-395. doi: 10.1016/j.jaci.2009.08.046
53. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
54. Gordon KB, Colombel JF, Hardin DS. Phase 3 Trials of Ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. (2016) 375:2102. doi: 10.1056/NEJMc1610828
55. Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. (2007) 80:273–90. doi: 10.1086/511051
56. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
57. Gaffen SL, Jain R, Garg AV, Cua DJ, The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
58. Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature (2007) 445:866–73. doi: 10.1038/nature05663
59. Capon F, Di Meglio P, Szaub J, Prescott NJ, Dunster C, Baumber L, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. (2007) 122:201–6. doi: 10.1007/s00439-007-0397-0
60. Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, McClanahan TK, et al. Cutting edge: a critical functional role for IL-23 in psoriasis. J Immunol. (2010) 185:5688–91. doi: 10.4049/jimmunol.1001538
61. Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature (2015) 521:222–6. doi: 10.1038/nature14175
62. Guenova E, Skabytska Y, Hoetzenecker W, Weindl G, Sauer K, Tham M, et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proc Natl Acad Sci USA. (2015) 112:2163–8. doi: 10.1073/pnas.1416922112
63. Li H, Yao Q, Mariscal AG, Wu X, Hülse J, Pedersen E, et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun. (2018) 9:1420. doi: 10.1038/s41467-018-03704-z
64. Conrad C, Meller S, Gilliet M. Plasmacytoid dendritic cells in the skin: to sense or not to sense nucleic acids. Semin Immunol. (2009) 21:101–9. doi: 10.1016/j.smim.2009.01.004
65. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
66. Wang CQF, Akalu YT, Suarez-Farinas M, Gonzalez J, Mitsui H, Lowes MA, et al. IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: potential relevance to psoriasis. J Invest Dermatol. (2013) 133:2741–52. doi: 10.1038/jid.2013.237
67. Albanesi C, Cavani A, Girolomoni G. IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-g and TNF-a. J Immunol. (1998) 162:494–502.
68. Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. (2011) 131:677–87. doi: 10.1038/jid.2010.340
69. Genovese MC, Weinblatt ME, Aelion JA, Mansikka HT, Peloso PM, Chen K, et al. ABT-122, a bispecific DVD-immunoglobulin targeting TNF- and IL-17A, in RA with inadequate response to methotrexate: a randomized, double-blind study. Arthritis Rheumatol. (2018) 70:1710–20. doi: 10.1002/art.40580
70. Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti–interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. (2012) 366:1190–9. doi: 10.1056/NEJMoa1109997
71. Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
72. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63. doi: 10.1038/ni.1767
73. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. (2009) 10:864–71. doi: 10.1038/ni.1770
74. Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest. (2009) 119:3573–85. doi: 10.1172/JCI40202
75. Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. (2014) 192:3111–20. doi: 10.4049/jimmunol.1302313
76. Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol. (2014) 134:2351–60. doi: 10.1038/jid.2014.146
77. Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, et al. Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol. (2014) 134:984–91. doi: 10.1038/jid.2013.477
78. Mashiko S, Bouguermouh S, Rubio M, Baba N, Bissonnette R, Sarfati M. Human mast cells are major IL-22 producers in patients with psoriasis and atopic dermatitis. J Allergy Clin Immunol. (2015) 136:351–359.e351. doi: 10.1016/j.jaci.2015.01.033
79. Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem. (2000) 275:31335–9. doi: 10.1074/jbc.M005304200
80. Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity (2004) 21:241–54. doi: 10.1016/j.immuni.2004.07.007
81. Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. (2005) 174:3695–702. doi: 10.4049/jimmunol.174.6.3695
82. Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med. (2009) 87:523–36. doi: 10.1007/s00109-009-0457-0
83. Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature (2007) 445:648–51. doi: 10.1038/nature05505
84. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. (2005) 23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843
85. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
86. Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. (2003) 4:1009–15. doi: 10.1038/ni978
87. Tough DF, Borrow P, Sprent J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science (1996) 272:1947–50.
88. Marrack P, Kappler J, Mitchell T. Type I interferons keep activated T cells alive. J Exp Med. (1999) 189:521–30.
89. Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity (2003) 19:225–34. doi: 10.1016/s1074-7613(03)00208-5
90. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science (1999) 284:1835–7.
91. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. (1999) 5:919–23. doi: 10.1038/11360
92. Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J Exp Med. (2010) 207:2921–30. doi: 10.1084/jem.20101102
93. Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr Opin Immunol. (2008) 20:401–7. doi: 10.1016/j.coi.2008.06.008
94. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature (2007) 449:564–9. doi: 10.1038/nature06116
95. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. (2009) 206:1983–94. doi: 10.1084/jem.20090480
96. Lande R, Chamilos G, Ganguly D, Demaria O, Frasca L, Durr S, et al. Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur J Immunol. (2015) 45:203–13. doi: 10.1002/eji.201344277
97. Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, et al. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. (2001) 117:91–7. doi: 10.1046/j.1523-1747.2001.01340.x
98. Schauber J, Dorschner RA, Coda AB, Büchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. (2007) 117:803–11. doi: 10.1172/JCI30142
99. Sorensen O, Arnljots K, Cowland JB, Bainton DF, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood (1997) 90:2796–803.
100. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature (2002) 415:389–95. doi: 10.1038/415389a
101. Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. (2005) 202:135–43. doi: 10.1084/jem.20050500
102. Stockenhuber K, Hegazy AN, West NR, Ilott NE, Stockenhuber A, Bullers SJ, et al. Foxp3(+) T reg cells control psoriasiform inflammation by restraining an IFN-I-driven CD8(+) T cell response. J Exp Med. (2018) 215:1987–98. doi: 10.1084/jem.20172094
103. Lapenta C, Santini SM, Spada M, Donati S, Urbani F, Accapezzato D, et al. IFN-alpha-conditioned dendritic cells are highly efficient in inducing cross-priming CD8(+) T cells against exogenous viral antigens. Eur J Immunol. (2006) 36:2046–60. doi: 10.1002/eji.200535579
104. Santini SM, Lapenta C, Donati S, Spadaro F, Belardelli F, Ferrantini M. Interferon-alpha-conditioned human monocytes combine a Th1-orienting attitude with the induction of autologous Th17 responses: role of IL-23 and IL-12. PLoS ONE (2011) 6:e17364. doi: 10.1371/journal.pone.0017364
105. Funk J, Langeland T, Schrumpf E, Hanssen LE. Psoriasis induced by interferon-a. Br J Dermatol. (1991) 125:463–5.
106. Lemmenmeier E, Gaus B, Schmid P, Hoffmann M. A case of erythrodermia from exacerbated psoriasis vulgaris due to treatment of acute hepatitis C. BMC Dermatol. (2016) 16:5. doi: 10.1186/s12895-016-0042-5
107. Damiani G, Franchi C, Pigatto P, Altomare A, Pacifico A, Petrou S, et al. Outcomes assessment of hepatitis C virus-positive psoriatic patients treated using pegylated interferon in combination with ribavirin compared to new Direct-Acting Antiviral agents. World J Hepatol. (2018) 10:329–36. doi: 10.4254/wjh.v10.i2.329
108. Pauluzzi P, Kokelj F, Perkan V, Pozzato G, Moretti M. Psoriasis exacerbation induced by interferon-alpha. Report of two cases. Acta Derm Venereol. (1993) 73:395.
109. Gilliet M, Conrad C, Geiges M, Cozzio A, Thürlimann W, Burg G, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. (2004) 140:1490–5. doi: 10.1001/archderm.140.12.1490
110. Eyerich K, Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. (2018) 32:692–703. doi: 10.1111/jdv.14673
111. Burmester GR, Panaccione R, Gordon KB, McIlraith MJ, Lacerda AP. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Ann Rheum Dis. (2013) 72:517–24. doi: 10.1136/annrheumdis-2011-201244
112. Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS. Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc Patient Saf. (2013) 5:79–99. doi: 10.2147/DHPS.S28801
113. Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA (2006) 295:2275–85. doi: 10.1001/jama.295.19.2275
114. Wu CY, Chen DY, Shen JL, Ho HJ, Chen CC, Kuo KN, et al. The risk of cancer in patients with rheumatoid arthritis taking tumor necrosis factor antagonists: a nationwide cohort study. Arthritis Res Ther. (2014) 16:449. doi: 10.1186/s13075-014-0449-5
115. Costenbader KH, Glass R, Cui J, Shadick N. Risk of serious infections and malignancies with anti-TNF antibody therapy in rheumatoid arthritis. JAMA (2006) 296:2201; author reply: 2203–4. doi: 10.1001/jama.296.18.2201-a
116. Aringer M, Smolen JS. The role of tumor necrosis factor-alpha in systemic lupus erythematosus. Arthritis Res Ther. (2008) 10:202. doi: 10.1186/ar2341
117. Sfikakis PP, Iliopoulos A, Elezoglou A, Kittas C, Stratigos A. Psoriasis induced by anti-tumor necrosis factor therapy: a paradoxical adverse reaction. Arthritis Rheum. (2005) 52:2513–8. doi: 10.1002/art.21233
118. Baeten D, Kruithof E, Van den Bosch F, Van den Bossche N, Herssens A, Mielants H, et al. Systematic safety follow up in a cohort of 107 patients with spondyloarthropathy treated with infliximab: a new perspective on the role of host defence in the pathogenesis of the disease? Ann Rheum Dis. (2003) 62:829–34. doi: 10.1136/ard.62.9.829
119. Cohen JD, Bournerias I, Buffard V, Paufler A, Chevalier X, Bagot M, et al. Psoriasis induced by tumor necrosis factor-alpha antagonist therapy: a case series. J Rheumatol. (2007) 34:380–5.
120. de Gannes GC, Ghoreishi M, Pope J, Russell A, Bell D, Adams S, et al. Psoriasis and pustular dermatitis triggered by TNF-{alpha} inhibitors in patients with rheumatologic conditions. Arch Dermatol. (2007) 143:223–31. doi: 10.1001/archderm.143.2.223
121. Brown G, Wang E, Leon A, Huynh M, Wehner M, Matro R, et al. Tumor necrosis factor-alpha inhibitor-induced psoriasis: Systematic review of clinical features, histopathological findings, and management experience. J Am Acad Dermatol. (2017) 76:334–41. doi: 10.1016/j.jaad.2016.08.012
122. Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFNa in autoimmune diseases. Proc Natl Acad Sci USA. (2005) 102:3372–7. doi: 10.1073/pnas.0408506102
123. Ivashkiv LB. Type I interferon modulation of cellular responses to cytokines and infectious pathogens: potential role in SLE pathogenesis. Autoimmunity (2003) 36:473–9. doi: 10.1080/08916930310001605882
124. Ytterberg SR, Schnitzer TJ. Serum interferon levels in patients with systemic lupus erythematosous. Arthritis Rheum. (1982) 25:401–7.
125. Bengtsson AA, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, et al. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus (2000) 9:664–71. doi: 10.1191/096120300674499064
126. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science (2001) 294:1540–3. doi: 10.1126/science.1064890
127. Seneschal J, Milpied B, Vergier B, Lepreux S, Schaeverbeke T, Taïeb A. Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol. (2009) 161:1081–8. doi: 10.1111/j.1365-2133.2009.09329.x
128. Soumelis V, Liu YJ. From plasmacytoid to dendritic cell: morphological and functional switches during plasmacytoid pre-dendritic cell differentiation. Eur J Immunol. (2006) 36:2286–92. doi: 10.1002/eji.200636026
129. Cleynen I, Vermeire S. Paradoxical inflammation induced by anti-TNF agents in patients with IBD. Nat Rev Gastroenterol Hepatol. (2012) 9:496–503. doi: 10.1038/nrgastro.2012.125
130. Cabaleiro T, Prieto-Pérez R, Navarro R, Solano G, Román M, Ochoa D, et al. Paradoxical psoriasiform reactions to anti-TNFalpha drugs are associated with genetic polymorphisms in patients with psoriasis. Pharmacogenomics J (2016) 16:336–40. doi: 10.1038/tpj.2015.53
Keywords: plaque psoriasis, paradoxical psoriasis, TNF, IL-23, TH17, type I-interferon
Citation: Mylonas A and Conrad C (2018) Psoriasis: Classical vs. Paradoxical. The Yin-Yang of TNF and Type I Interferon. Front. Immunol. 9:2746. doi: 10.3389/fimmu.2018.02746
Received: 17 August 2018; Accepted: 07 November 2018;
Published: 28 November 2018.
Edited by:
Eva Reali, Istituto Ortopedico Galeazzi (IRCCS), ItalyReviewed by:
Silvia Vidal, Sant Pau Institute for Biomedical Research, SpainYumin Xia, Second Affiliated Hospital of Xi'an Jiaotong University, China
Copyright © 2018 Mylonas and Conrad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Curdin Conrad, Y3VyZGluLmNvbnJhZEBjaHV2LmNo