 Federica Barzaghi1,2,3*†
Federica Barzaghi1,2,3*† Federica Minniti4†
Federica Minniti4† Margherita Mauro4
Margherita Mauro4 Massimiliano De Bortoli4Rita Balter4Elisa Bonetti4Ada Zaccaron4
Massimiliano De Bortoli4Rita Balter4Elisa Bonetti4Ada Zaccaron4 Virginia Vitale4Maryam Omrani2
Virginia Vitale4Maryam Omrani2 Matteo Zoccolillo2,3
Matteo Zoccolillo2,3 Immacolata Brigida2
Immacolata Brigida2 Maria Pia Cicalese1,2Massimo Degano5Michael S. Hershfield6
Maria Pia Cicalese1,2Massimo Degano5Michael S. Hershfield6 Alessandro Aiuti1,2,7Anastasiia V. Bondarenko8
Alessandro Aiuti1,2,7Anastasiia V. Bondarenko8 Matteo Chinello4Simone Cesaro4
Matteo Chinello4Simone Cesaro4- 1Pediatric Immunohematology and Bone Marrow Transplantation Unit, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 2San Raffaele Telethon Institute for Gene Therapy, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 3Department of Systems Medicine, Tor Vergata University, Rome, Italy
- 4Paediatric Hematology-Oncology, Ospedale della Donna e del Bambino, Verona, Italy
- 5Biocrystallography Unit, Division of Immunology, Transplantation and Infectious Diseases, IRCCS San Raffaele Scientific Institute, Milan, Italy
- 6Department of Medicine and Biochemistry, Duke University School of Medicine, Durham, NC, United States
- 7Vita-Salute San Raffaele University, Milan, Italy
- 8Department of Pediatric Infectious Diseases and Immunology, Medical Academy of Postgraduate Education, Kiev, Ukraine
Adenosine deaminase 2 (ADA2) deficiency is an auto-inflammatory disease due to mutations in cat eye syndrome chromosome region candidate 1 (CECR1) gene, currently named ADA2. The disease has a wide clinical spectrum encompassing early-onset vasculopathy (targeting skin, gut and central nervous system), recurrent fever, immunodeficiency and bone marrow dysfunction. Different therapeutic options have been proposed in literature, but only steroids and anti-cytokine monoclonal antibodies (such as tumor necrosis factor inhibitor) proved to be effective. If a suitable donor is available, hematopoietic stem cell transplantation (HSCT) could be curative. Here we describe a case of ADA2 deficiency in a 4-year-old Caucasian girl. The patient was initially classified as autoimmune neutropenia and then she evolved toward an autoimmune lymphoproliferative syndrome (ALPS)-like phenotype. The diagnosis of ALPS became uncertain due to atypical clinical features and normal FAS-induced apoptosis test. She was treated with G-CSF first and subsequently with immunosuppressive drugs without improvement. Only HSCT from a 9/10 HLA-matched unrelated donor, following myeloablative conditioning, completely solved the clinical signs related to ADA2 deficiency. Early diagnosis in cases presenting with hematological manifestations, rather than classical vasculopathy, allows the patients to promptly undergo HSCT and avoid more severe evolution. Finally, in similar cases highly suspicious for genetic disease, it is desirable to obtain molecular diagnosis before performing HSCT, since it can influence the transplant procedure. However, if HSCT has to be performed without delay for clinical indication, related donors should be excluded to avoid the risk of relapse or partial benefit due to a hereditary genetic defect.
Introduction
Deficiency of adenosine deaminase 2 (DADA2) is a genetic auto-inflammatory disease caused by autosomal recessive mutations in adenosine deaminase 2 (ADA2) gene, mapped on chromosome 22q11 (1). Adenosine deaminase 2 (ADA2) is an enzyme secreted in the plasma by myeloid cells that catalyzes the conversion of adenosine to inosine (2). The pathomechanism of DADA2 remains unclear, but immunomodulatory function of ADA2 is crucial. Indeed, in vitro studies suggest that ADA2 works as an autocrine factor activating cells of the immune system (3). Thus, deficiency of ADA2 results in multiple immune abnormalities, such as increased pro-inflammatory M1 macrophages (as opposed to anti-inflammatory M2 macrophages), upregulation of genes associated with neutrophil activation, increased secretion of pro-inflammatory cytokines including TNF and interleukin-1b (IL-1b) and endothelial damage (4, 5).
DADA2 was described for the first time in 2014 by two independent studies (4, 6). The predominant clinical presentation includes recurrent fever, early-onset vasculopathy involving skin, gut and central nervous system (1, 7, 8). Blood tests usually show elevation of acute phase reactants (erythrocyte sedimentation rate and C-reactive protein) (1). Recent evidences suggest a broader clinical spectrum encompassing also features of autoimmunity, immunodeficiency and bone marrow dysfunction (1, 8–14). The diagnosis of DADA2 is based on the measurement of plasma ADA2 enzymatic activity and on the identification of biallelic mutations in ADA2.
Splenomegaly or hepatosplenomegaly are frequently reported in patients with DADA2 deficiency (1, 4, 11, 15, 16) as well as cytopenia (1, 4, 11, 17). Nevertheless, their presence in addition to clinical and biological features defining an Autoimmune Lymphoproliferative Syndrome (ALPS)-like phenotype is less common. Only two cases of DADA2 presenting with clinical features resembling ALPS have been recently described (17, 18). Another patient experienced hemophagocytic lymphohistocytosis on a background of ALPS-like manifestations (19).
We describe a case of ADA2 deficiency presenting as ALPS-like disease, unresponsive to immunosuppressive therapy, successfully treated with allogeneic hematopoietic stem cell transplantation (HSCT).
Case Presentation
A 4-year old Caucasian female child was referred to our Center for diagnostic assessment of severe neutropenia lasting for 6 months that was not responsive to the administration of granulocyte-colony stimulating factor (G-CSF) up to a maximum dose of 20 mcg/kg daily.
The patient was born after a full-term gestation, from non-consanguineous parents. The birth weight was 3,600 g. Familial medical history was unremarkable. Parents reported recurrent infections of upper respiratory tract and stomatitis during the first years of life, before the occurrence of severe neutropenia.
Physical examination was normal, except for the presence of splenomegaly (long axis 11.5 cm at ultrasound scan), whereas blood count revealed profound neutropenia (absolute neutrophil count, ANC, 0.02 × 109/L) without anemia and thrombocytopenia. Bone marrow examination showed the presence of rare myeloid precursors with normal representation of the erythroid and megakaryocyte lineages. The clonogenic study of bone marrow progenitors in vitro showed a normal growth of erythroid cells whereas the growth of myeloid cells was below the normal range: CFU-E/BFU-E 37 (normal range 27–81/2 × 104); CFU-GEMM 1 (normal range 0–10/2 × 104); CFU-GM 30 (normal range 33–100/2 × 104). Interestingly the patient bone marrow plasma markedly inhibited the growth of progenitor cells obtained from healthy voluntary bone marrow donors: CFU-E/BFU-E/(2 × 105) from 88 (controls) to 4 (controls + patient plasma), CFU-GEMM/(2 × 105) from 17 to 9. An extensive diagnostic work-up excluded other potential causes of neutropenia such as Kostmann disease, Shwachman-Diamond syndrome, Fanconi anemia and paroxysmal nocturnal hemoglobinuria. Finally, a misdiagnosed form of autoimmune neutropenia was hypothesized. Indeed, anti-neutrophil IgG were 171.1 mg/dL (normal value < 33.5 mg/dL), IgM were 24.9 mg/dL (normal value < 32.2 mg/dL). Hence, the patient underwent a 4-week trial with prednisone at 2.5 mg/kg/day that led to complete remission of neutropenia (ANC 3.360 × 109/L). Prednisone was slowly tapered. Nevertheless, fever occurred at every attempt of reducing the dose of prednisone below 1 mg/kg/day. Given the significant side effects after 3 months of steroid therapy, introduction of steroid-sparing agents (cyclosporine first and mycophenolate mofetil later) was attempted. However, neutropenia did not improve and the patient remained steroid dependent.
After 4 months, she experienced a single episode of livedo reticularis at the lower limbs. The histological examination showed a livedoid thrombotic vasculopathy. Moreover, the patient was hospitalized eight times due to episodes of fever, abdominal pain and neutropenia with elevated inflammation markers. Abdominal CT scan, esophagogastroduodenoscopy (EGDS) and colonoscopy showed no abnormalities, as well as gastrointestinal biopsies.
The patient was assessed also for ALPS (Table 1) (20) because of autoimmune neutropenia and splenomegaly: she displayed increased double negative T-lymphocytes in the peripheral blood (2.5%, normal value <1.7% of total lymphocytes), IL-18 (1.125 pg/ml, normal value 36–258 pg/ml), IL-10 (7.2 pg/ml, normal value <1 pg/ml). However, vitamin B12 levels where only mildly elevated as compared to patients with classical ALPS (775 pg/mL, normal value 191–663) (21); similarly, FAS-induced apoptosis test was normal (survivor T-lymphocytes after FAS antigenic stimulation 42%) (22). Sirolimus was started (23, 24) and withdrawn after 2 months, due to the occurrence of fever and abdominal pain. Prednisone was continued alone. No mutations were found in ALPS-disease causative genes (FAS, FASL, CAS10). The control of clonogenic tests 7 months after starting immunosuppression showed a good growth of CFU-E/BFU-E (120/2 × 104) and CFU-GEMM (52/2 × 104); the growth of bone marrow progenitor cells derived from healthy donors was no more inhibited by the patient's bone marrow plasma.
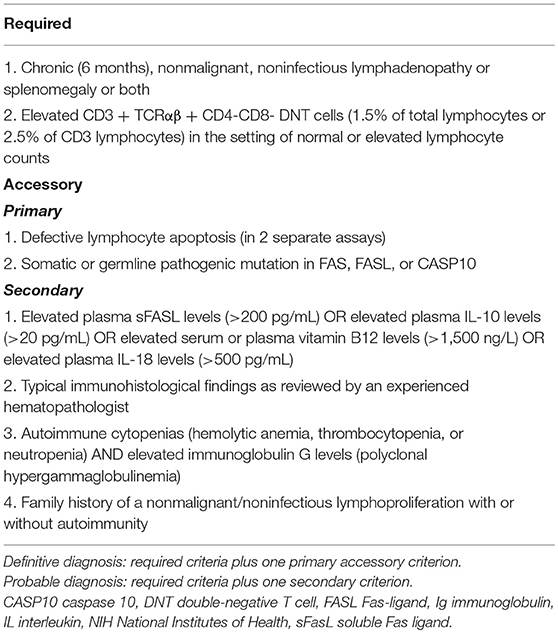
Table 1. Diagnostic criteria for ALPS defined by 2009 NIH consensus (20).
Considering the persistence of neutropenia despite the administration of G-CSF and immunosuppressive therapy, the patient was candidate to allogeneic HSCT. During pre-transplant workup, brain magnetic resonance (MRI) revealed a 5-mm signal alteration in the right cortical-subcortical frontal basal region consistent with a petechial spot.
HSCT was performed at 5 years and 4 months of age. The patient underwent a myeloablative conditioning with busulfan, thiotepa, fludarabine and rituximab (Table 2). Graft-vs.-host disease (GVHD) prophylaxis consisted of anti-thymocite globulin, short methotrexate, cyclosporine (from day 0 to day +28) than replaced by tacrolimus (from day +29). Neutrophil engraftment occurred on day +26, and platelet engraftment occurred on day +34. Complete donor chimerism was detected on day +33. Main complications after HSCT were gut and skin grade II acute GVHD requiring additional therapy with prednisone at 2 mg/kg/day, tapered slowly due to frequent relapses. Further complications were: CMV reactivation (on day + 31) treated successfully with valganciclovir and osteonecrosis of the knees (on day + 138), treated with vitamin D, calcium, bisphosphonates, physio-kinesiotherapy and hyperbaric oxygen therapy.

Table 2. Conditioning regimen.
Considering the atypical clinical evolution, the inadequate response to therapy and the absence of mutations, the diagnosis of ALPS was questioned. We performed molecular analysis by Haloplex target system platform (Illumina) on the patient's buccal swab obtained after HSCT. The patient resulted compound heterozygous for two novel mutations in ADA2 gene. The variant located at exon 9 determines an amino acidic substitution (c.1367 A>G, p.Y456C) and the variant located in exon 8 introduces a premature stop codon causing the synthesis of a truncated protein (c.1196. G>A, p.W399*). Both parents were healthy heterozygous carriers. These new mutations are reported as detrimental for the protein according to different prediction tools (e.g., PROVEAN, PolyPhen).
The high-resolution crystal structure of the homodimeric wild type protein was previously determined (PDB ID 3LGD, corresponding to UniProt Q9NZK5 amino acid residues 29-510, Zavialov et al.,http://dx.doi.org/10.1074/jbc.M109.083527). To address the effects of the two mutations, we performed a structural and energetic analysis of the enzyme. The p.Y456C mutation affects a hydrophobic pocket of ADA2 (Figure 1A). Of note, this is a novel mutation but the p.Y453C mutation, which affects the same hydrophobic pocket, is one of the most common found in patients with DADA2 (1). The p.Y456C mutation carried by our patient heavily destabilizes the ADA2 structure (ΔΔ G = 4.53 ± 0.12 kcal/mol from n = 5 independent simulations) as calculated using the program FoldX (25). The p.W399* mutation instead leads to a truncation of the protein C-terminus, removing key residues required for the correct folding of the active site, and resulting in a solvent-exposed catalytic Zn2+ ion (Figure 1B). Thus, both mutations are predicted to dramatically reduce the enzymatic activity of ADA2 through structural destabilization of the protein.
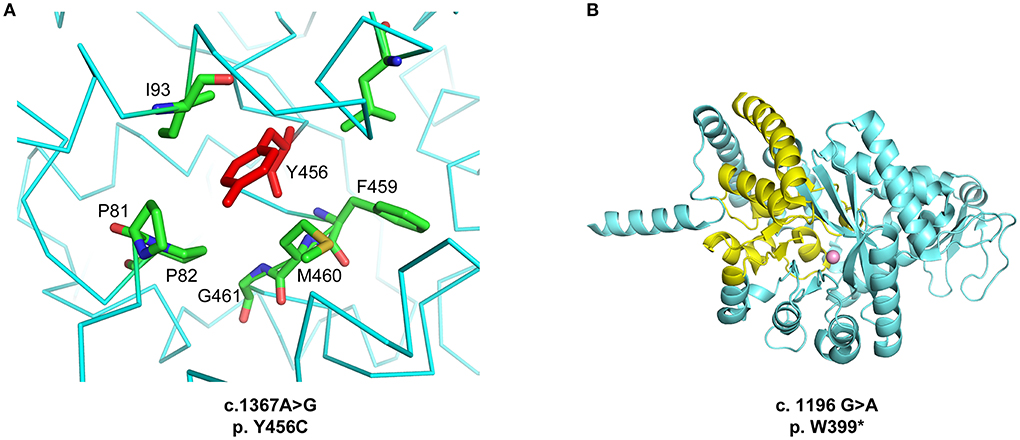
Figure 1. (A) Location of the p.Y456C mutation. The Y456 residue is located in an α-helical segment and inserted between hydrophobic residues. (B) Structural abnormality of the ADA2 protein generated by the p.W399* mutation. In yellow, the portion of the protein that is lacking due to the stop codon introduced by the mutation. Figures generated with Pymol (http://www.pymol.org).
Pre-HSCT plasma of the patient was not available but we could assess ADA2 activity in the parents, which resulted lower than healthy controls as expected in carriers, supporting the deleterious effect of the mutations on the protein function. Moreover, patient's ADA2 plasma activity after HSCT was normal, in the absence of clinical manifestations of the disease and in the presence of full donor chimerism (Table 3). These results strongly suggest DADA2 as cause of the hematological manifestations of the patient.
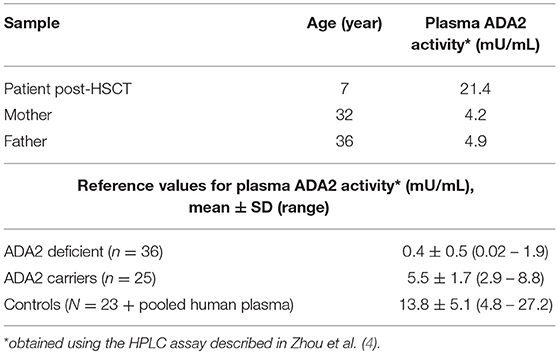
Table 3. Plasma ADA2 activity in the index family.
The patient gradually recovered from HSCT complications and immunosuppressive therapy was withdrawn. She is now alive and well 28 months after HSCT enjoying a normal life.
Discussion
Clinical phenotype and age at onset of DADA2 vary widely without clear correlation to ADA2 genotype (8).
Although not described initially, hematological disorders appear to be an emergent clinical manifestation of DADA2. A various degree of hematological dysfunction affects approximately 43% of patients, in the absence of evident vasculitis (14). Anemia, including pure red cell aplasia, and lymphopenia are the most frequent findings, while neutropenia is a rare clinical manifestation (1, 13, 14).
Initially, our patient was classified as an ALPS-like based on autoimmune neutropenia, splenomegaly and positive biomarkers. Subsequently, the diagnosis of ALPS became less certain because of the lack of response to immunosuppressive drugs, atypical clinical features (livedo reticularis and recurrent severe abdominal pain), normal FAS-induced apoptosis test, and lack of mutations of ALPS-related genes. Only widening the genetic evaluation after HSCT in the index case and in the parents, DADA2 was diagnosed. Of note, clinical presentation of DADA2 as ALPS-like disease have been reported in two patients (17, 18) who were successfully treated with HSCT and anti-TNF medications respectively.
Of note, during the active phase of the disease, the patient's bone marrow plasma markedly inhibited the growth of progenitor cells obtained from healthy bone marrow donors. This finding and the reversion of inhibition after administration of steroids, suggests the presence of pro-inflammatory mediators inhibiting the growth of bone marrow progenitor cells, as observed in the setting of acquired aplastic anemia, where plasma pro-inflammatory cytokines block the differentiation of bone marrow progenitor cells (26). This further supports the possible benefit derived by the administration of TNF-inhibitors in ADA2 patients before transplant, in order to reduce the inflammatory burden also in the bone marrow niches (14). Current literature suggests different therapeutic options for DADA2: the choice between medical therapy and HSCT depends on predominant symptoms and severity of the clinical phenotype. Being an inflammatory condition, high doses of steroids are usually useful to control the disease, although a steroid-dependence often occurs (5), as in the case herein described. If inflammatory symptoms and vasculitis are predominant, anti–tumor necrosis factor agents are usually beneficial (6, 7, 11, 14). HSCT is a potential definitive treatment, since it provides ADA2 producing monocytes, which are able to restore ADA2 plasma activity and control the disease manifestations (15, 17, 19, 27). Moreover, as recently reported by Hashem et al. (28), bone marrow dysfunction and immunodeficiency are the most frequent indications leading to transplant.
Our experience suggests that ADA2 deficiency should be considered as differential diagnosis in cases of severe autoimmune neutropenia evolving to ALPS-like disease not responsive to standard immunosuppressive approaches. Early diagnosis based on these manifestations could allow prompt therapeutic intervention anticipating the onset of vasculopathy and related severe complications.
Importantly, clinicians should aim at obtaining the molecular diagnosis of putative genetic diseases before transplant, since it could influence the management of patients candidate to HSCT. This particularly applies in ADA2 deficiency, considering the high risk of complications due to the persisting inflammation and the impaired endothelial integrity (17, 19, 29). In this light, anti-TNF therapy administered before the conditioning can reduce vascular inflammation (14). With the same aim, conditioning should be tailored, for example replacing Busulfan with Treosulfan or other agents displaying lower endothelial toxicity. Another strategy can foresee prophylactic Defibrotide to reduce the risk of sinusoidal obstruction syndrome. In addition, the selection of the donor can favor unrelated donor avoiding carrier familial donors who might display partial reduction of ADA2 activity. This partial defect can be amplified in the presence of mixed chimerism, leaving a residual and potentially clinically significant enzymatic defect (28). Finally, evaluating ADA2 activity after transplant, especially in case of mixed chimerism, allows monitoring the efficacy and promptly detect the disease recurrence (27). Further studies are needed to define the appropriate timing of HSCT and as well as intensity of conditioning regimen.
Concluding Remarks
Early diagnosis of DADA2 in cases presenting with hematological manifestations, rather than classical vasculopathy, is crucial, allowing the patients to undergo HSCT, if indicated, before the arousal of more severe manifestations of the disease. Moreover, physicians can adjust the transplant procedure based on the features of the disease, in order to minimize complications and create the most favorable conditions to allow an efficacious control of the disease. Finally, this case highlights the importance of reaching a genetic diagnosis before HSCT. If transplant cannot be delayed, a related donor should be excluded in all cases of genetic disease suspicion without precise molecular diagnosis: an unrelated donor would give the possibility to avoid relapse or partial benefit due to a common genetic defect.
Ethics Statement
The study was performed in accordance with the Declaration of Helsinki. The patient was enrolled in a study approved by the San Raffaele Ethical Committee (TIGET06). Written informed consent was obtained from parents for biological sample collection and data publication.
Author Contributions
FM, MM, MC, MDB, RB, EB, AZ, VV, and SC participated to clinical care of the patient and related clinical data. IB, MZ, and MO performed the molecular analysis by Haloplex. IB, MZ, MO, FB, and MPC contributed to the selection of variants. MD analyzed the effect of mutations on the protein structure. MH performed the dosage of ADA2 enzymatic activity. FB, FM, MM, AA, and SC designed the study, analyzed results and wrote the paper. AVB referred the patient and participated in the clinical management.
Funding
The study was supported by grants of the Italian Ministero della Salute (Programma di rete, NET-2011-02350069), the European Commission (ERARE-3-JTC 2015 EUROCID) and Fondazione Telethon (TIGET Core grant C6). FB and MZ conducted this study as part of their Ph.D. in Immunologia Medicina Molecolare e Biotecnologie Applicate, Tor Vergata University, Rome, Italy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient and her family for their trust and for allowing publication of clinical information.
References
1. Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. (2018) 38:569–78. doi: 10.1007/s10875-018-0525-8
2. Zavialov AV, Gracia E, Glaichenhaus N, Franco R, Zavialov AV, Lauvau G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J Leukoc Biol. (2010) 88:279–90. doi: 10.1189/jlb.1109764
3. Kaljas Y, Liu C, Skaldin M, Wu C, Zhou Q, Lu Y, et al. Human adenosine deaminases ADA1 and ADA2 bind to different subsets of immune cells. Cell Mol Life Sci. (2017) 74:555–70. doi: 10.1007/s00018-016-2357-0
4. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
5. Caorsi R, Penco F, Schena F, Gattorno M. Monogenic polyarteritis: the lesson of ADA2 deficiency. Pediatr Rheumatol Online J. (2016) 14:51. doi: 10.1186/s12969-016-0111-7
6. Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. (2014) 370:921–31. doi: 10.1056/NEJMoa1307362
7. Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. (2017) 76:1648–56. doi: 10.1136/annrheumdis-2016-210802
8. Hashem H, Kelly SJ, Ganson NJ, Hershfield MS. Deficiency of adenosine deaminase 2 (DADA2), an inherited cause of polyarteritis nodosa and a mimic of other systemic rheumatologic disorders. Curr Rheumatol Rep. (2017) 19:70. doi: 10.1007/s11926-017-0699-8
9. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. (2016) 177:316–20. doi: 10.1016/j.jpeds.2016.06.058
10. Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. (2016) 36:179–86. doi: 10.1007/s10875-016-0245-x
11. Schepp J, Proietti M, Frede N, Buchta M, Hubscher K, Rojas Restrepo J, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheumatol. (2017) 69:1689–700. doi: 10.1002/art.40147
12. Skrabl-Baumgartner A, Plecko B, Schmidt WM, Konig N, Hershfield M, Gruber-Sedlmayr U, et al. Autoimmune phenotype with type I interferon signature in two brothers with ADA2 deficiency carrying a novel CECR1 mutation. Pediatr Rheumatol Online J. (2017) 15:67. doi: 10.1186/s12969-017-0193-x
13. Cipe FE, Aydogmus C, Serwas NK, Keskindemirci G, Boztug K. Novel Mutation in CECR1 Leads to Deficiency of ADA2 with Associated Neutropenia. J Clin Immunol. (2018) 38:273–7. doi: 10.1007/s10875-018-0487-x
14. Michniacki TF, Hannibal M, Ross CW, Frame DG, DuVall AS, Khoriaty R, et al. Hematologic Manifestations of Deficiency of Adenosine Deaminase 2 (DADA2) and Response to Tumor Necrosis Factor Inhibition in DADA2-Associated Bone Marrow Failure. J Clin Immunol. (2018) 38:166–73. doi: 10.1007/s10875-018-0480-4
15. Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of Adenosine Deaminase Type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheumatol. (2016) 68:2314–22. doi: 10.1002/art.39699
16. Sahin S, Adrovic A, Barut K, Ugurlu S, Turanli ET, Ozdogan H, et al. Clinical, imaging and genotypical features of three deceased and five surviving cases with ADA2 deficiency. Rheumatol Int. (2018) 38:129–36. doi: 10.1007/s00296-017-3740-3
17. Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (Oxford) (2016) 55:902–10. doi: 10.1093/rheumatology/kev439
18. Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 mimicking autoimmune lymphoproliferative syndrome in the absence of livedo reticularis and vasculitis. Pediatr Blood Cancer (2018) 65. doi: 10.1002/pbc.26912
19. Van Eyck LJr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. (2015) 135:283–7.e285. doi: 10.1016/j.jaci.2014.10.010
20. Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood (2010) 116:e35–40. doi: 10.1182/blood-2010-04-280347
21. Bowen RA, Dowdell KC, Dale JK, Drake SK, Fleisher TA, Hortin GL, et al. Elevated vitamin B(1)(2) levels in autoimmune lymphoproliferative syndrome attributable to elevated haptocorrin in lymphocytes. Clin Biochem. (2012) 45:490–2. doi: 10.1016/j.clinbiochem.2012.01.016
22. Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, et al. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. (2010) 125:946–9 e946. doi: 10.1016/j.jaci.2009.12.983
23. Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood (2011) 118:5741–51. doi: 10.1182/blood-2011-07-325217
24. Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, et al. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood (2016) 127:17–28. doi: 10.1182/blood-2015-07-657981
25. Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. (2005) 33:W382–388. doi: 10.1093/nar/gki387
26. Zeng Y, Katsanis E. The complex pathophysiology of acquired aplastic anaemia. Clin Exp Immunol. (2015) 180:361–70. doi: 10.1111/cei.12605
27. Bucciol G, Delafontaine S, Segers H, Bossuyt X, Hershfield MS, Moens L, et al. Hematopoietic Stem Cell Transplantation in ADA2 Deficiency: Early Restoration of ADA2 Enzyme Activity and Disease Relapse upon Drop of Donor Chimerism. J Clin Immunol. (2017) 37:746–50. doi: 10.1007/s10875-017-0449-8
28. Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood (2017) 130:2682–8. doi: 10.1182/blood-2017-07-798660
Keywords: ADA2 deficiency, HSCT, ALPS, ADA2, CECR1, autoimmunity, immunodeficiency, neutropenia
Citation: Barzaghi F, Minniti F, Mauro M, De Bortoli M, Balter R, Bonetti E, Zaccaron A, Vitale V, Omrani M, Zoccolillo M, Brigida I, Cicalese MP, Degano M, Hershfield MS, Aiuti A, Bondarenko AV, Chinello M and Cesaro S (2019) ALPS-Like Phenotype Caused by ADA2 Deficiency Rescued by Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 9:2767. doi: 10.3389/fimmu.2018.02767
Received: 10 August 2018; Accepted: 12 November 2018;
Published: 14 January 2019.
Edited by:
Sergio Rosenzweig, National Institutes of Health (NIH), United StatesReviewed by:
V. Koneti Rao, National Institutes of Health (NIH), United StatesIvona Aksentijevich, National Human Genome Research Institute (NHGRI), United States
Copyright © 2019 Barzaghi, Minniti, Mauro, De Bortoli, Balter, Bonetti, Zaccaron, Vitale, Omrani, Zoccolillo, Brigida, Cicalese, Degano, Hershfield, Aiuti, Bondarenko, Chinello and Cesaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Federica Barzaghi, YmFyemFnaGkuZmVkZXJpY2FAaHNyLml0
†These authors have contributed equally to this work