 Mahin Abad Dar
Mahin Abad Dar Christoph Hölscher
Christoph Hölscher- Infection Immunology, Research Center Borstel, Borstel, Germany
Arginase-1 (Arg-1) is a marker for alternatively activated macrophages (AAM) and is mainly induced by the type 2 cytokines interleukin (IL)-4 and IL-13 through the common IL-4 receptor-alpha (Rα) subunit. Both, Arg-1 and AAM undermine macrophage effector functions against intracellular parasites and are therefore implicated in the susceptibility to infection with Trypanosoma cruzi, the causative agent of Chagas' disease. However, the involvement of Arg-1 in promoting intracellular replication of T. cruzi in AAM has not been proven so far in vivo. Because Arg-1 is only moderately expressed in T. cruzi-infected wildtype mice, we elucidated the role of Arg-1 and AAM during infection in IL-13-overexpressing (IL-13tg) mice, which are characterized by an inflammation-induced development of AAM and an accompanied elevated expression of Arg-1. In comparison to wildtype littermates, IL-13tg mice were highly susceptible to T. cruzi infection with enhanced parasitemia and impaired survival. Importantly, T. cruzi-infected IL-13tg mice developed an elevated alternative macrophage activation with increased arginase activity. To proof the hypothesis, that Arg-1 accounts for the increased susceptibility of IL-13tg mice, we blocked arginase activity in infected IL-13tg mice. Because this arginase inhibition resulted in a decreased susceptibility to experimental Chagas disease our study supports in summary the conclusion that IL-13/IL-4Rα-driven Arg-1 expression contributes to the permissiveness of the host to T. cruzi infection.
Introduction
Trypanosoma cruzi, a protozoan parasite that belongs to the family of Trypanosomatidae, is the causative agent of Chagas disease, a neglected tropical disease that is a major public health problem in Latin America (1). The complex lifecycle of T. cruzi involves several developmental stages in the insect vector and in vertebrates. In the vertebrate host, all nucleated cells can be infected by the parasite but macrophages are one of the most important cell types for T. cruzi replication during the acute phase of the infection. However, mechanisms that are involved in the permissiveness of host cells to T. cruzi infection are not completely understood so far.
A T helper type 1 (Th1) immune response characterized by the production of interferon-gamma (IFN-γ) is protective against experimental infection with T. cruzi (2–5) through the induction of classically activated macrophages (CAM). In these macrophages, the production of reactive nitrogen intermediates (RNI) by the activity of the inducible nitric oxide synthase 2 (NOS2) is central to the elimination of the intracellularly replicating amastigote developmental stage of T. cruzi. In contrast to a Th1 type immune response, the impact of a Th2-dominated immune response on the outcome of experimental Chagas disease is less clear. Th2 cells characterized by the secretion of interleukin (IL)-4 and IL-13 are only associated with an increased susceptibility to T. cruzi (6–10). However, downstream mechanisms that account for a Th2-mediated susceptibility after experimental infection with T. cruzi are not clearly established.
In general, the Th2-cytokines IL-4 and IL-13 mediate the development of alternatively activated macrophages (AAM) through binding to the IL-4 receptor-alpha (Rα) chain (11). Particularly, the induction of arginase (Arg)-1 in these cells is associated with an enhanced susceptibility to infection with parasites of the family Trypanosomatidae (12–15). In this context, there are two putative mechanisms by which IL-4Rα-induced Arg-1 may counteract effector functions against Trypanosomatidae in macrophages. At first, Arg-1 could antagonize the production of RNI by NOS2 either through depletion of L-arginine, the common substrate of these two enzymes, or by the inhibition of NOS2 through metabolites of the Arg-1 pathway (12–16). Furthermore, arginase activity results in the supply of polyamines capable to support the proliferation of intracellular parasites (15).
During experimental Leishmania major infection the IL-4Rα-mediated development of AAM and the associated strong Arg-1 induction account for the enhanced susceptibility of BALB/c mice (12, 13, 15, 17, 18). However, for infection with T. cruzi there is only proof based on in vitro experiments that the high arginase-activity in IL-4Rα-induced AAM supports the intracellular parasite replication by counteracting the antitrypanosomal effector mechanism mediated by NOS2 (19, 20). In experimental Chagas disease, however, it has been only shown that Arg-1 positive myeloid-derived suppressor cells (MDSC) infiltrate the heart of T. cruzi infected mice during the acute phase of the disease and that the degree of this infiltration correlates with the tissue parasite load (21, 22). Unfortunately, our knowledge about the function of AAM after infection with T. cruzi in vivo is rather limited but studies addressing the impact of IL-4Rα-induced Arg-1 on the permissiveness of host cells to this parasite are absolutely required because a human study points at a crucial role of the IL-4Rα for developing a cardiomyopathy, the major complication during the chronic phase of Chagas disease (23).
The poor expression of IL-4 and IL-13 after experimental infection of wildtype mice with T. cruzi nearly exclude a reasonable analysis of the contribution of the IL-4Rα-Arg-1 pathway in susceptibility to experimental Chagas disease. In the present study, we therefore took advantage of IL-13-overexpressing (tg) mice (24) and analyzed the outcome of T. cruzi infection under an increased Th2 immune response.
Results
Increased Susceptibility of IL-13-Overexpressing Mice to T. cruzi Infection
To investigate the role of IL-13 during experimental T. cruzi infection IL-13tg mice and wildtype littermates were infected i.p. with 50 blood trypomastigotes of the Tulahuen strain and the survival rate and parasite load during the acute phase of infection was determined (Figure 1). Compared to wildtype littermates, IL-13tg mice were highly susceptible to a normally sublethal dose of T. cruzi with a median survival of 18 days p.i. (Figure 1A). Moreover, IL-13tg mice developed a significantly higher parasitemia 13 and 15 days p.i. (Figure 1B). At the time point “moribund” at which the first infected animal had to be euthanized, the increased susceptibility of IL-13tg mice to T. cruzi was also reflected by the significantly increased tissue parasitism in the spleen when compared to the tissue parasite load in wildtype littermates (Figure 1C). Hence, increased levels of IL-13 result in an accelerated susceptibility to T. cruzi infection.
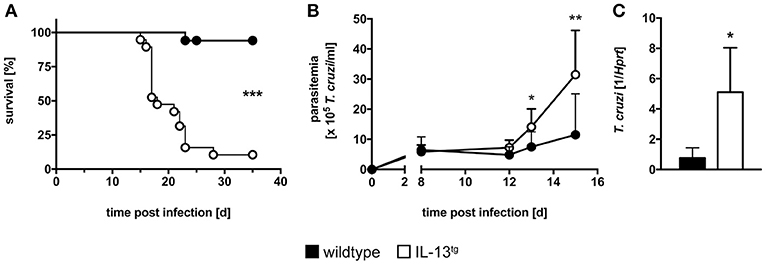
Figure 1. Increased susceptibility of infected IL-13tg mice. Transgene-negative littermate control (closed symbols) and IL-13tg (open symbols) mice were infected i.p. with 50 T. cruzi blood trypomastigotes for the analysis of parasitemia and survival and with 500 T. cruzi blood trypomastigotes for determination of the parasite tissue burden. (A) Survival and (B) parasitemia were assessed during infection. Results are expressed as the survival rate of 17–19 mice per group of two pooled experiments (A) or as mean parasitemia ±SD of 9–10 mice per group of one representative out of two experiments (B). (C) Tissue parasite burden in the spleen was analyzed by quantitative real-time PCR at the time point “moribund” at which the first animal had to be euthanized (16 days post infection in this particular experiment). Results are expressed as the means ±SD of 4–6 mice per group of one representative out of two experiments. Statistical analysis was performed using the Log Rank test (A) or Mann Whitney U test (B,C) defining differences between IL-13tg and wildtype mice as significant (*p ≤ 0.05; **p ≤ 0.01, ***p ≤ 0.001).
Histopathology in T. cruzi-Infected IL-13tg Mice
Mice often succumb to experimental Chagas disease after infection with reticulotropic strains due to a severe inflammation and subsequent immunopathology in infected organs (e.g., liver necrosis) (25, 26). To evaluate the degree of tissue damage in T. cruzi-infected wildtype and IL-13tg mice, sections of paraffin-embedded tissue from heart (Figures 2A,D), spleen (Figures 2B,E), and liver (Figures 2C,F) were prepared and stained with hematoxylin-eosin. Overall, no significant differences in the degree of pathology in organs form wildtype and IL-13tg mice were observed. However, quantification of heart inflammation revealed a tendentiously increased infiltration in cardiac tissue of mutant animals (Figure 2G). To quantify the extent of liver pathology after infection with T. cruzi, the content of the liver-derived enzymes ALT (Figure 2H) and AST (Figure 2I) were quantified in sera of infected mice. Both enzymes were found after infection in increasing quantities yet no differences between infected wildtype and IL-13tg mice could be observed. Importantly, at the time point “moribund” at which the first infected animal had to be euthanized, the amounts of ALT and AST in the serum of dying IL-13tg mice did not exceed those measured in surviving wildtype mice. Together, these data indicate that IL-13tg mice do not succumb to an increased inflammation and immunopathology. The elevated parasite load rather appears to be responsible for a premature death in the presence of high levels of IL-13.
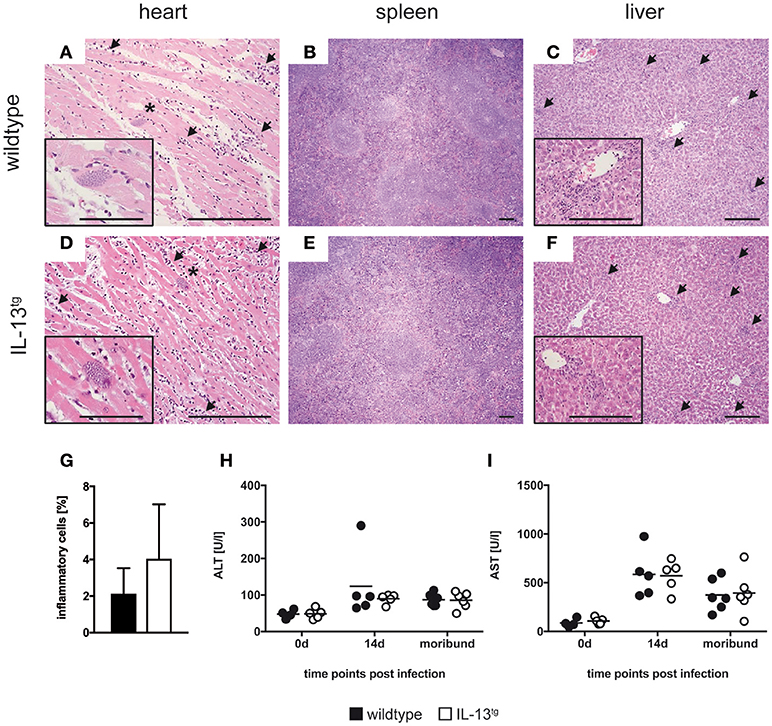
Figure 2. IL-13 has no influence on organ pathology during T. cruzi infection. Transgene-negative littermate control (closed symbols) and IL-13tg (open symbols) mice were infected i.p. with 500 T. cruzi blood trypomastigotes. (A–F) Organ sections of heart (A,D), spleen (B,E), and liver (C,F) from wildtype (A–C) and IL-13tg (D–F) mice were prepared at the time point “moribund” at which the first animal had to be euthanized (16 days post infection in this particular experiment) and stained with hematoxylin and eosin. Representative photomicrographs from 4 to 6 mice per group of one representative out of two experiments are shown (bar, 200 μm; arrow, infiltration; *, parasite nest). (G) Quantification of heart inflammation as depicted in (A) and (D). (H,I) Liver-derived enzymes ALT (H) and AST (I) were quantified in sera at the indicated time points after infection. Results are expressed as the means of 4–6 mice per group of one representative out of two experiments.
Cytokine Expression in T. cruzi-Infected Il-13tg Mice
During experimental Chagas disease, cytokines are important for inducing a protective Th1 immune response and subsequent effector mechanisms in macrophages (2, 4). However, the release of these inflammatory mediators has to be tightly regulated to prevent an immunopathological sequelae (27). To examine the influence of IL-13 overexpression on the cytokine response during experimental Chagas disease, different pro-inflammatory and anti-inflammatory/regulatory cytokines were analyzed by quantitative real time RT-PCR in spleen homogenates of uninfected and T. cruzi-infected mice (Figure 3).
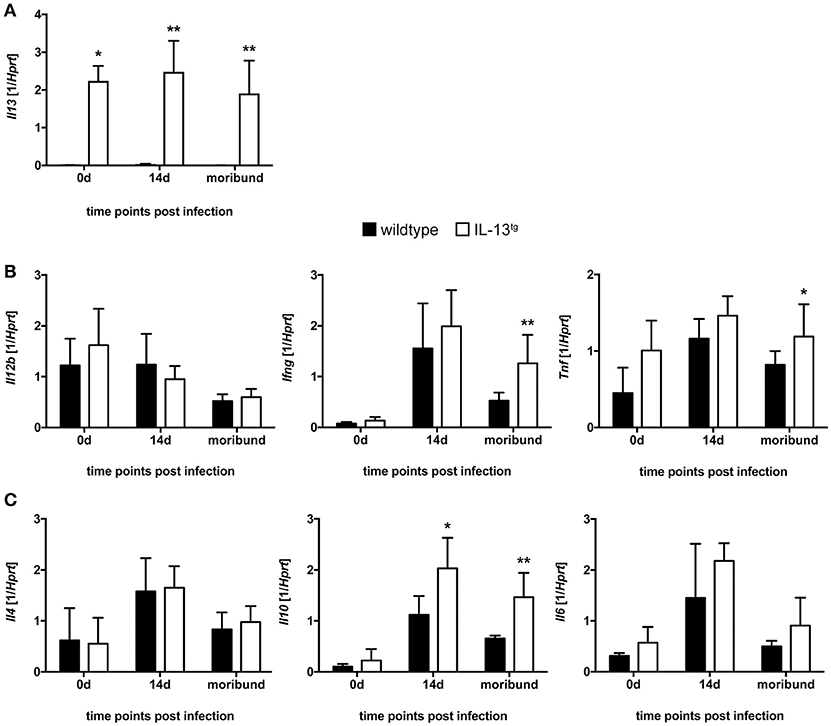
Figure 3. Cytokine expression in T. cruzi-infected wildtype and IL-13tg mice. Transgene-negative littermate control (closed bars) and IL-13tg (open bars) mice were infected i.p. with 500 T. cruzi blood trypomastigotes. mRNA expression of (A) Il13, (B) Il12b, Ifng, Tnf, (C) Il4, Il10, and Il6 in spleens was quantified by real-time PCR at the indicated time points post infection (“moribund” indicates the time point at which the first animal had to be euthanized; 16 days post infection in this particular experiment). Results are expressed as the means ±SD of 4 - 6 mice per group of one representative out of two experiments. Statistical analysis was performed using the Mann Whitney U test defining differences between IL-13tg and wildtype mice as significant (*p ≤ 0.05; **p ≤ 0.01).
Although Il13 gene expression was highly induced in spleens of IL-13tg mice (Figure 3A), gene expression of the pro-inflammatory cytokines Il12b, Ifng, Tnf (Figure 3B) and the anti-inflammatory/regulatory cytokines and Il4 and Il6 (Figure 3C) were not significantly different between wildtype and IL-13tg mice 14 days after T. cruzi infection. Only the anti-inflammatory cytokine Il10 was significantly increased in spleen homogenates of IL-13tg mice at this time point (Figure 3C). At the time point “moribund” at which the first infected animal had to be euthanized however, transcript levels of Ifng and Tnf were found to be significantly elevated in IL-13tg (Figure 3B) and the significantly increased gene expression of Il10 was still apparent in these mice (Figure 3C). Together, these results reveal that high amounts of IL-13 during T. cruzi infection are not accompanied by a biased cytokine response.
NOS2 Expression and RNI Production in Spleens of T. cruzi-Infected IL-13tg Mice
During experimental Chagas disease, LRG-47- and NOS2-dependent effector mechanisms in CAM are crucial to eliminate T. cruzi (2, 28). Because IL-13 is known to modulate the activation status of macrophages, LRG-47 and NOS2 expression as well as RNI production were analyzed in the spleen of T. cruzi infected wildtype and IL-13tg mice (Figure 4).
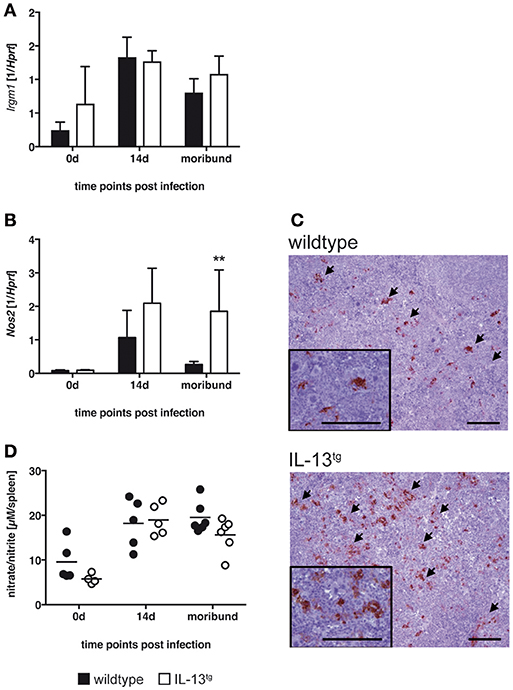
Figure 4. Classical macrophage activation is not altered in IL-13tg mice after infection with T. cruzi. Transgene-negative littermate control (closed bars) and IL-13tg (closed bars) mice were infected i.p. with 500 T. cruzi blood trypomastigotes. (A,B) mRNA expression of Irgm (A) and Nos2 (B) in spleens were quantified by real-time PCR at the indicated time points post infection (“moribund” indicates the time point at which the first animal had to be euthanized; 16 days post infection in this particular experiment). Results are expressed as the means ±SD of 4–6 mice per group of one representative out of two experiments. Statistical analysis was performed using the Mann Whitney U test defining differences between IL-13tg and wildtype mice as significant (**p ≤ 0.01). (C) For immunohistochemical analysis, spleens were isolated at the time point “moribund” at which the first animal had to be euthanized (16 days post infection in this particular experiment). Histological sections were stained for NOS2 and counterstained with hematoxylin. Representative photomicrographs from 4 to 6 mice per group of one representative out of two experiments are shown (bar, 200 μm; arrow, NOS2-positive cells). (D) Nitrate/nitrite levels in spleen homogenates were determined by the Griess reaction after reduction of nitrate to nitrite. Results are expressed as means of 4–6 mice per group of one experiment.
Gene expression of Irgm was comparable in spleens of both, wildtype and IL-13tg mice during the course of experimental Chagas disease (Figure 4A). Nos2 gene expression was strongly induced in both mouse strains (Figure 4B). Thereafter, the quantity of Nos2 transcripts declined in wildtype mice whereas in moribund IL-13tg mice Nos2 expression was significantly enhanced. Immunohistochemical staining of NOS2 revealed an enhanced appearance of this enzyme in spleens of moribund IL-13tg mice when compared to corresponding wildtype mice (Figure 4C). Because NOS2 metabolizes L-arginine eventually to RNI which are effective against the intracellular parasite, we quantified the content of these reactive intermediates in homogenates of spleens isolated from uninfected and T. cruzi-infected wildtype and IL-13tg mice (Figure 4D). After infection, the production of RNI was induced in spleens of both, wildtype and IL-13tg mice. Thereafter, the amount of intermediates appeared to decline in spleen homogenates isolated from moribund IL-13tg mice and were found to be slightly reduced compared to corresponding wildtype mice. Together, after infection with T. cruzi the expression of NOS2 was not impaired in the presence of high IL-13 concentrations. In contrast, NOS2 levels increased during the course of disease. However, this was not reflected by an also elevated production of RNI.
IL-13 Mediates Alternatively Activated Macrophages in IL-13tg Mice
IL-13 induces an alternative activation of macrophages thereby favoring the growth of intracellular parasites (17, 29). AAM are characterized by enhanced expression of unique markers such as Retnla, Chi3l3, and Arg1. To evaluate whether these markers are induced in IL-13tg mice after infection with T. cruzi, we performed quantitative real time RT-PCR of spleen homogenates (Figures 5A–C). Before infection, Retnla (Figure 5A) and Chi3l3 (Figure 5B) gene expression was hardly detectable. After infection of wildtype mice with T. cruzi, Retnla was not expressed in wildtype mice whereas Chi3l3 was induced to quantifiable levels. However, transcript levels of both Retnla (Figure 5A) and Chi3l3 (Figure 5B) were significantly enhanced in spleens of infected IL-13tg mice. Arg1 was barely expressed in spleens of T. cruzi-infected wildtype mice (Figure 5C). Compared to this, Arg1 gene expression was significantly increased approximately 12–18 fold in spleen homogenates prepared from infected IL-13tg mice (Figure 5C). We next analyzed the expression of Arg-1 in spleens of moribund IL-13tg mice and corresponding wildtype mice by immunohistochemistry (Figure 5D). Staining of Arg-1 disclosed a very strong appearance of the enzyme in IL-13tg mice that was strikingly different from corresponding wildtype mice in which Arg-1 was hardly detectable (Figure 5D). To eventually examine arginase enzyme activity in wildtype and IL-13tg mice during experimental Chagas disease, the urea concentration after L-arginine hydrolysis in spleen homogenates was quantified (Figure 5E). Before infection, no enzyme activity was detectable in spleens of both wildtype and IL-13tg mice. After infection with T. cruzi, a rather low arginase activity was induced in wildtype mice but, in contrast, this activity was significantly increased approximately 4 fold in spleen homogenates of IL-13tg mice.
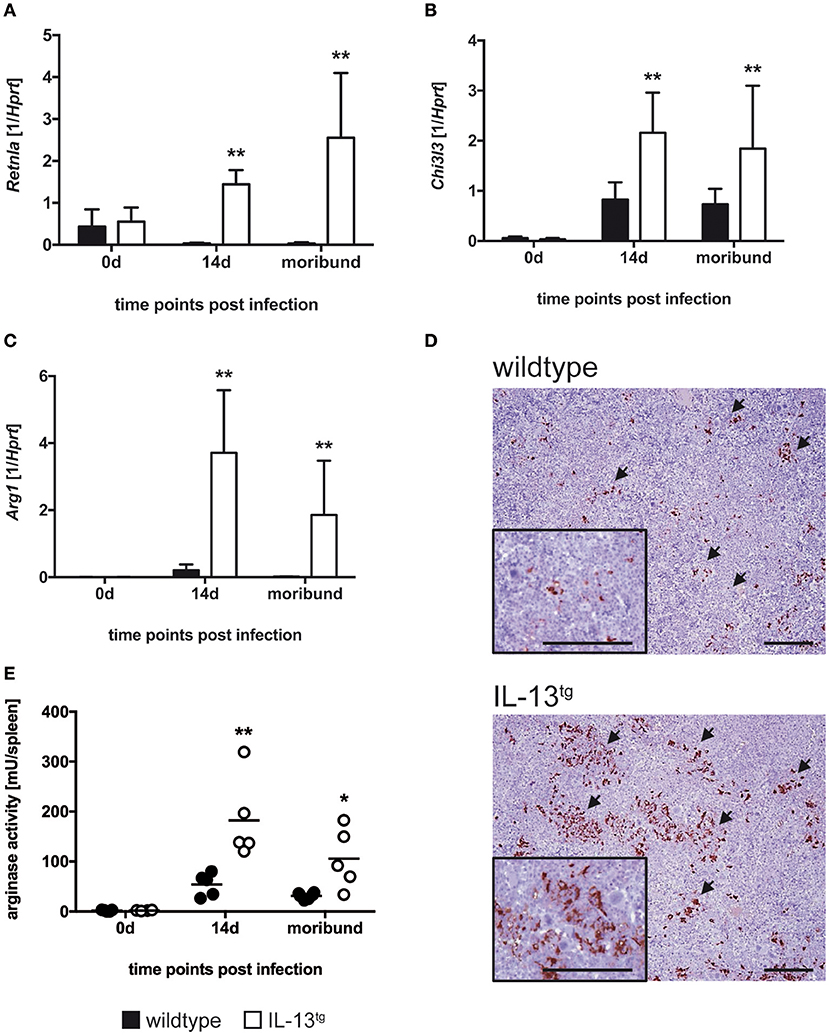
Figure 5. Increased alternative macrophage activation in T. cruzi-infected IL-13tg mice. Transgene-negative littermate control (closed bars) and IL-13tg (open bars) mice were infected i.p. with 500 T. cruzi blood trypomastigotes. (A–C) mRNA expression of Retnla (A), Chi3l3 (B), and Arg1 (C) in spleens were quantified by real-time PCR at the indicated time points post infection (“moribund” indicates the time point at which the first animal had to be euthanized; 16 days post infection in this particular experiment). Results are expressed as the means SD± of 4–6 mice per group of one representative out of two experiments. Statistical analysis was performed using the Mann Whitney U test defining differences between IL-13tg and wildtype mice as significant (**p ≤ 0.010). (D) For immunohistochemical analysis, spleens were isolated at the time point “moribund” at which the first animal had to be euthanized (16 days post infection in this particular experiment). Histological sections were stained for Arg-1 and counterstained with hematoxylin. Representative photomicrographs from 4 to 6 mice per group of one representative out of two experiments are shown (bar, 200 μm; arrow, Arg-1-positive cells). (E) Arginase activity in spleen homogenates were determined by arginase assay at the indicated time points post infection (“moribund” indicates the time point at which the first animal had to be euthanized; 16 days post infection in this particular experiment). Results are expressed as means ±SD of 4–6 mice per group of one experiment. Statistical analysis was performed using the Mann Whitney U test defining differences between IL-13tg and wildtype mice as significant (*p ≤ 0.05; **p ≤ 0.01).
Arg-1 Accounts for the Increased Susceptibility of Il-13tg Mice to Experimental Chagas Disease
So far, we have shown that during experimental Chagas disease overexpression of IL-13 resulted in high Arg-1 expression and arginase activity that was associated with a decreased survival rate and enhanced parasitemia. Because Arg-1 is implicated in the impairment of effector functions in T. cruzi-infected macrophages in vitro (20), we next sought to demonstrate that Arg-1 in fact accounted for the increased susceptibility of IL-13tg mice to T. cruzi infection. We therefore infected wildtype and IL-13tg mice with T. cruzi, treated transgenic mice either with PBS or with the arginase inhibitor Nω-hydroxy-nor-arginine (nor-NOHA) and monitored survival and determined parasitemia (Figure 6). T. cruzi-infected IL-13tg mice reached a median survival time of 19 days (Figure 6A). In contrast, the median survival time of infected nor-NOHA-treated IL-13tg mice was extended to 28 days. In addition to the survival rate, the parasitemia was determined at the time point “moribund” at which the first infected animal had to be euthanized (Figure 6B). The concentration of T. cruzi in the blood of infected wildtype mice was approximately 5 × 105 trypomastigotes/ml and was with 7.5 × 105 trypomastigotes/ml increased in IL-13tg mice. Although not statistically significant different, Arg-1 inhibition resulted in a decrease in parasitemia of IL-13tg to 3 × 105 trypomastigotes/ml (Figure 6B). Hence, Arg-1 appears to be a key factor induced by IL-13 that mediates susceptibility to T. cruzi in IL-13tg mice.
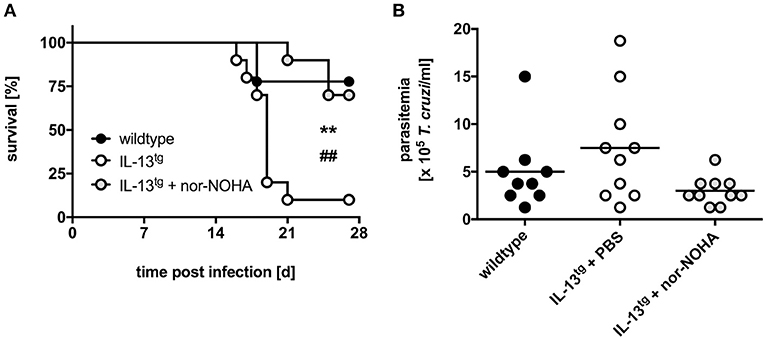
Figure 6. Inhibition of Arg-1 results in a decreased susceptibility of infected IL-13tg mice to T. cruzi infection. Transgene-negative littermate control and IL-13tg mice were infected i.p. with 50 T. cruzi blood trypomastigotes for the analysis of parasitemia and survival. Arginase activity was inhibited in IL-13tg mice by daily i.p. administration with 600 μg nor-NOHA. PBS-treated IL-13tg mice and wildtype mice served as control groups. (A) Survival during the course of infection and (B) parasitemia at 16 days of infection. Results are expressed as percent survival (A) and mean parasitemia (B) of 9–10 mice per group of two pooled experiments. Statistical analysis was performed using the Log Rank test defining differences between IL-13tg and wildtype mice (*,** p ≤ 0.01) and between IL-13tg and IL-13tg mice treated with nor-NOHA (#,##p ≤ 0.01) as significant.
Discussion
A Th2 immune response accompanied by AAM and Arg-1 is assumed to undermine the protective Th1 immune response against infections with trypanosmatids and thus contributes to susceptibility. In contrast to the well characterized model system for cutaneous leishmaniasis not much is known about the impact of the IL-4Rα-Arg-1 axis on the course of experimental Chagas disease. However, a human study points toward a crucial role of the IL-4Rα for developing of cardiomyopathy during Chagas disease, the major complication during the chronic phase of the infection (23). So far, most studies addressing the function of IL-4Rα-mediated signaling in experimental Chagas disease employed IL-4-deficient(−/−) and STAT6−/− mice or neutralization of IL-4 by administration of monoclonal antibodies (7, 10, 30, 31). However, these reports revealed conflicting results with respect to IL-4Rα as a susceptibility factor during infection with T. cruzi. Because the induction of the IL-4Rα ligands IL-4 and IL-13 is rather low the analysis of the outcome of T. cruzi infection in IL-4−/− and STAT6−/− or under anti-IL-4 treatment is limited. Hence, to determine the hypothetical effect of the IL-4Ra on the outcome of Chagas disease (23) we infected mice with T. cruzi which overexpressed IL-13 specifically in T cells (24). Our study is the first that clearly shows a contribution of the IL-4Rα ligand IL-13 to the susceptibility to experimental Chagas disease in vivo. As it has been shown for cutaneous leishmaniasis (29), T. cruzi-infected IL-13tg mice presented an increased parasite replication accompanied by an enhanced mortality compared to infected wildtype mice. Moreover, the support of the parasite growth by IL-13 appeared to be a crucial mechanism for the increased susceptibility.
The effective immune response against T. cruzi is initiated by a pro-inflammatory Th1 immune response leading to high amounts of IFNγ which in turn induces effector responses in CAM such as LRG-47 and NOS2 (2, 28). Hence, the common underlying mechanism of an increased susceptibility to T. cruzi infection is thought to be an impaired release of pro-inflammatory mediators such as IL-12 (4) and a reduced production of IFNγ (2). Therefore, in our study IL-13 might have suppressed this pro-inflammatory immune response leading to an insufficient IFNγ production and subsequent impaired CAM induction. However, we did not observe this kind of IL-13-mediated suppression of protective Th1 cytokines. To the contrary, the release of pro-inflammatory cytokines in the spleen of T. cruzi-infected IL-13tg mice was rather increased. Accordingly, the IFNγ/TNF-mediated expression of LRG-47 was similar and the expression of NOS2 was even enhanced in IL-13-overexpressing mice. However, this increased enzyme expression did not result in an also elevated release of RNI by CAM in IL-13tg mice during experimental Chagas disease.
An exacerbated inflammatory immune response also contributes to immunopathology during experimental Chagas disease which is controlled by IL-10 (27, 32). Because in the present study overexpression of IL-13 also resulted in a significantly augmented expression of IL-10, an inflammation-induced pathology mediated by the increased release of the Th1 cytokines IFNγ and TNF appears to be greatly counterregulated even though the cellular infiltration in cardiac tissue was tendentiously increased. Hence, the amplified levels of IL-10 may account for the largely checked inflammatory tissue inflammation in T. cruzi-infected IL-13tg mice. Taken together, IL-13 overexpression did not negatively affect the generation of NOS2-dependent effector responses in CAM, the pro-inflammatory cytokine release and inflammation during experimental Chagas disease.
Whereas classical macrophage activation was rather similar in T. cruzi-infected IL-13tg and wildtype mice, overexpression of IL-13 lead to an elevated induction of markers for alternative macrophage activation with a strikingly increased Arg-1 production and enhanced arginase activity. Hence, our study confirms, that during infection IL-13 promotes a strong alternative macrophage activation (29, 33, 34). Additionally, the increased levels of IL-10 in T. cruzi-infected IL-13tg mice may add to alternative macrophage activation in these animals as this cytokine is also able to induce Arg-1 directly or through the upregulation of the IL-4Rα on macrophages (35, 36). Because the induction of AAM could create optimal conditions for an uncontrolled parasite proliferation by the effects of Arg-1 (15, 18), we examined the overall impact of arginase activity on the susceptibility of IL-13tg mice to T. cruzi infection and inhibited the enzyme by administering nor-NOHA. This treatment reduced arginase activity in infected IL-13tg mice to levels observed in corresponding wildtype animals (data not shown) and significantly ameliorated the outcome of experimental Chagas disease when compared to untreated IL-13tg mice. Hence, Arg-1 appears to be a key molecule that mediates the IL-13-dependent susceptibility to T. cruzi infection.
There are several mechanisms by which Arg-1 may undermine protective immune responses against intracellular parasites. IL-13 induces via the IL-13/IL-4 type II receptor and STAT6 Arg-1 in AAM but also in MDSC which has also been described to promote susceptibility to T. cruzi infection (21, 37). The fact that Arg-1 and NOS2 coexist in the here examined T. cruzi-infected IL-13tg mice may favor a role of MDSC in the outcome of infection. The suppressive mechanisms of MDSC e.g. by local L-arginine consumption appear to affect T cells (38, 39). Because both enzymes Arg-1 and NOS2 metabolizes L-arginine, the suppressive capacity of MDSCs depends on IL-4Rα-mediated Arg-1 and/or IFNγ-induced NOS2 activities (38, 40, 41). Although we have not directly addressed the cellular source of IFNγ, the phenotype of MDSC or the T cell response in T. cruzi-infected IL-13tg mice, the simultaneous occurrence of increased NOS2 and Arg-1 indicates that MDSCs contribute to the susceptibility of IL-13tg mice to experimental Chagas disease.
In addition to a suppressive effect of L-arginin depletion on T cells by MDSCs, IL-13-induced Arg-1 may also have a direct effect on parasite replication in host cells like macrophages. Accordingly, macrophages with elevated arginase activity, induced through the IL-4Rα or the T. cruzi component cruzipain, promote parasite growth which could be inhibited by blockade of enzyme activity (19, 20).
In host cells, Arg-1 induction can support parasite replication by competitive depletion of the common substrate L-arginine which is also used by NOS2 to generate RNI (42). Hence, although NOS2 is elevated in T. cruzi-infected IL-13tg mice the simultaneously enhanced IL-4Rα-mediated arginase activity may explain the rather reduced levels of RNI by an overall increased consumption of L-arginine. Along this line, IL-4Rα ligation has recently been shown to induce asymmetric dimethylarginine (ADMA) (43), which represents an endogenous inhibitor of NOS2 activity (44). Because experimental T. cruzi infection of mice results in increased levels of ADMA concomitant with a reduced NOS2 activity (45) the IL-13-mediated induction of this methylated derivate of L-arginine may - although independent of Arg-1 - also inhibit effector responses in macrophages.
Additionally, arginase activity results in the production of polyamines, which appear to be essential for intracellular parasite replication (15). Parasites are able to generate polyamines endogenously but can also utilize to a great extent host polyamines. They are synthesized by metabolic processes that are similar in parasites and the host including arginase, which catalyzes the enzymatic hydrolysis of L-arginine to L-ornithine and urea, and ornithine decarboxylase, which catalyzes the enzymatic decarboxylation of L-ornithine in putrescine (46, 47). In turn, putrescine is a substrate for spermidine synthase to synthetize spermidine which is essential for growth and survival of trypanosomatid parasites. Hence, within AAM the microenvironment with an increased activity of host Arg-1 and subsequent elevated levels of host polyamines represent a “land of plenty” for intracellular parasites. Additionally, trypanosomatid parasites have a unique metabolism, in which spermidine is further metabolized by trypanothione reductase which provides an intracellular antioxidant system essential to survive the hostile intracellular environment (46–48). Together, during infection with T. cruzi IL-13 induces an increased arginase activity and subsequently elevated amounts of polyamines. This excess supply of host polyamines may also account for the arginase-dependent uncontrolled parasite replication in T. cruzi-infected IL-13tg mice.
In summary, we here give evidence that IL-4Rα-induced Arg-1 mediates susceptibility to acute experimental Chagas disease by several - mutually not exclusive - mechanisms downstream of Arg-1 including L-arginine depletion in MDSCs and AAM and polyamine synthesis. A correlation between Arg-1, arginase activity and susceptibility was already shown in patients with visceral leishmaniasis (49, 50). However, although there is a clear link of IL4RA polymorphism and cardiomyopathy in human Chagas disease (23), the impact of Arg-1 and the subsequent functional effects have not been investigated so far. Nevertheless, such studies will possibly unravel novel therapeutic targets.
Methods
Mice and Parasites
IL-13tg mice on a BABL/c background were kindly provided by Andrew McKenzie (MRC Laboratory of Molecular Biology, Cambridge, UK) and bred under specific-pathogen-free conditions in the animal facility of the Research Center Borstel. For experiments, female transgenic mice and transgene-negative littermates aged 6 to 8 weeks were infected intraperitoneally (i.p.) with the T. cruzi strain Tulahuen (WHO reference strain M/HOM/CH/00/Tulahuen C2). To obtain high numbers of T. cruzi blood form trypomastigotes for infection experiments and to prevent the transfer of lymphocytes and antibodies, SCID mice mice (purchased from Charles River, Sulzfeld, Germany) were i.p. infected with 1 × 107 parasites in cryopreserved T. cruzi stabilates. At day 12 post infection, blood was collected from infected SCID, mixed with heparin and parasites were enriched in the plasma by differential centrifugation. Parasites were resuspended in PBS/0.5% glucose and used for infection. An infection dose of 500 blood trypomastigotes was used to induce detectable inflammatory cytokine responses and pathology. For determining parasitemia and mortality, a sublethal dose of 50 parasites was used (26, 51). For arginase-1 inhibition, mice were treated daily with 600 μg/mouse Nor-NOHA (N(omega)-hydroxy-l-arginine; Alexis Biochemicals) in 200 μl PBS starting 2 days before infection. Control animals were treated with the same amount of PBS. During infection experiments, mice were kept under barrier conditions in the BSL 3 facility at the Research Center Borstel in individually ventilated cages. All experiments were conducted according to the German animal protection laws and were approved by the Animal Research Ethics Board of the Ministry of Environment, Kiel, Germany.
Determination of Parasitemia and Tissue Parasite Load
Blood parasitemia was determined in 3 μl of tail vein blood that was lyzed in 27 μl NH4Cl (0.87% [wt/vol]). Viable parasites were counted in a Neubauer chamber.
Tissue parasite burdens were analyzed by quantitative real-time PCR of genomic DNA isolated from the spleen. A 70 bp sequence of the 140/116-kDa antigen gene of T. cruzi (accession no. U15616) was amplified with the forward primer 5-ACT CAT CGG GTT TGA AGC AT-3, the reverse primer 5-GCC AGG GTC TAG TAC TCT TTG CT-3, and the internal probe 5-CAG CAG GC-3_ A 107 bp stretch of the murine hypoxanthine–guanine phosphoribosyltransferase (hprt) gene, used for quantification of host DNA, was amplified with the forward primer 5-GTG GCC CTC TGT GTG CTC-3, the reverse primer 5-TCT ACA GTC ATA GGA ATG GAT CTA TCA-3, and the internal probe 5-ACC TGC TG-3. Quantitative PCR was performed on a Light Cycler 480 (Roche Diagnostics Corporation, Indianapolis, IN). Data were analyzed employing the “Second Derivative Maximum” and “Standard Curve Method.”
Histopathology and Immunohistochemistry
For histolopathological and immunohistochemical analysis, heart, spleen and liver were isolated at the time point “moribund” at which the first animal had to be euthanized and fixed in 4% formalin/PBS, set in paraffin blocks and sectioned (2 μm). Histopathological analyses were performed using standard protocols for hematoxylin/eosin staining. For quantification of heart inflammation, serial sections of heart tissue were analyzed for the degree of cellular infiltaraion using the ImageJ software (NIH, Bethesda, Maryland) as described (52). For NOS2 staining, tissue sections were stained with a polyclonal rabbit anti-mouse NOS2 antiserum (Upstate) as previously described (53). For Arg-1 staining, sections were stained using a mouse anti-mouse arginase-1 antibody (BD Transduction) and the Vector M.O.M. immunodetection kit (Vector Laboratories).
AST and ALT Assay
Serum was prepared using the BD Microtainer SST Tubes (BD Pharmingen) and the aspartate transaminase (AST), and alanine transaminase (ALT) activity was determined by using the Reflotron System of Diagnosis (Roche Diagnostics).
Quantitative Real-Time PCR
Organs were homogenized in 4M guanidinium-isothiocyanate buffer and total RNA was extracted by acid phenol extraction. cDNA was obtained using murine moloney leukemia virus reverse transcriptase (RvertAid, Invitrogen) and oligo-dT (12-18mer; Sigma) as a primer. Quantitative PCR was performed on a Light Cycler 480 Instrument (Roche Diagnostics). Data were analyzed employing the “Second Derivative Maximum” and “Standard Curve Method” using hypoxanthine-guanine phosphoribosyltransferase (hprt) as a housekeeping gene to calculate the level of gene expression in relation to hprt. The following primer and probe sets were employed: Arg1: sense 5′-CCT GGA ACT GAA AGG AAA G-3′, antisense 5′-TTG GCA GAT ATG CAG GGA GT-3′, probe 2; Chi3l3: 5′-GAA CAC TGA GCT AAA AAC TCT CCT G-3′, antisense 5′- GAG ACC ATG GCA CTG AAC G-3′, probe 88; Hprt: sense 5′-TCC TCC TCA GAC CGC TTT T-3′, antisense 5′-CCT GGT TCA TCA TCG CTA ATC-3′, probe 95; Ifng: sense 5′-ATC TGG AGG AAC TGG CAA AA-3′, antisense 5′-TTC AAG ACT TCA AAG AGT CTG AGG TA-3′, probe 21; Il4: sense 5′-CAT CGG CAT TTT GAA CGA G-3′, antisense 5′-CGA GCT CAC TCT CTG TGG TG-3′, probe 2; Il6: sense 5′- CT ACC AAA CTG GAT ATA ATC AGG A-3′, antisense 5′-CCA GGT AGC TAT GGT ACT CCA-′, probe 6; Il12b: sense 5′-CCA TCA GCA GAT CAT TCT AGA CAA-3′, antisense 5′-CGC CAT TAT GAT TCA GAG ACT G-3′, probe 78; Il13: sense 5′-CCT CTG ACC CTT AAG GAG CTT AT-3′, antisense 5′-CGT TGC ACA GGG GAG TCT-3′, probe 17; Irgm: sens 5′-AAG GCC ACT AAC ATC GAA TCA-3′, antisense 5′-TGC CTT ATC TCA CTT AAT ACT CCT-3′, probe 82; Nos2: sense 5′-CTT TGC CAC GGA CGA GAC-3′, antisense 5′-TCA TTG TAC TCT GAG GGC TGA C-3′, probe 13; Retnla: sense 5′-TAT GAA CAG ATG GGC CTC CT-3′, antisense 5′-GGC AGT TGC AAG TAT CTC CT-3′, probe 3; Tnf : sense 5′-TGC CTA TGT CTC AGC CTC TTC-3′, antisense 5′-GAG GCC ATT TGG GAA CTT CT-3′, probe 49 (all from Roche).
Arginase Activity and Nitrate Production in Spleen Homogenates
To detect reactive nitrogen intermediates (RNI) in uninfected and infected mice, spleen homogenates were collected at different time points. After deproteination of homogenates using Micron YM-30 centrifugal filters (Millipore, Schwalbach, Germany), NO3 was converted into NO2 utilizing a commercial nitrate reductase kit (Cayman; Axxora, Lörrach, Germany). After adding Griess reagents, the content of NO2 was determined by photometric measurement reading the absorbance at 540 nm on a microplate reader (Sunrise; Tecan, Männedorf, Switzerland) as previously described (54). To determine arginase activity in murine tissue, weighed pieces of organs were homogenized in 100 μl of 0.1% Triton X-100 (Sigma) containing a protease inhibitor cocktail (Roche). Fifty microliter of 10 mM MnCl2 (Merck, Darmstadt, Germany) and 50 mM Tris-HCl (Merck) were added to all samples and the enzyme was activated by heating for 10 min at 55°C. Arginine hydrolysis was conducted by incubating 25 μl of the activated lysate with 25 μl of 0.5 M L-arginine (Merck) at 37°C for 60 min. The reaction was stopped with 400 μl of H2SO4 (96%)/H3PO4 (85%)/H2O (1/3/7, v/v/v). As a degree of arginase activity, the urea concentration was measured at 540 nm after addition of 25 μl α-isonitrosopropiophenone (Sigma; dissolved in 100% ethanol) followed by heating at 95°C for 45 min. One unit of arginase activity is defined as the amount of enzyme that catalyzes the formation of 1 μmol urea/min.
Statistical Analysis
Quantifiable data are expressed as means of individual determinations and standard deviation. Statistical analysis was performed using the unpaired Student's t-test, the Mann-Whitney U test, the Kruskal-Wallis test or the Log-Rank test defining different error probabilities (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001).
Author Contributions
MA performed the experiments, analyzed the data, and wrote the manuscript; CH conceived and designed the experiments and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was funded by the German Bundesministerium für Bildung und Forschung (KMU-innovativ 5, Förderkennzeichen 0315814) and the Open-Access-Publicationsfonds of the Leibniz society. We are thankful to Andrew McKenzie for providing breeding pairs of IL-13tg mice. The expert technical support of Alexandra Hölscher is highly appreciated. We are also grateful to Kerstin Traxel and Marion Schuldt for supplying and cleaning the lab and to Christine Keller and Ilka Monath for organizing the animal facility and taking care of the mice.
References
1. World-Health-Organization. Chagas disease (American trypanosomiasis). In: Fact Sheets. (Geneva: WHO) (2018).
2. Holscher C, Kohler G, Muller U, Mossmann H, Schaub GA, Brombacher F. Defective nitric oxide effector functions lead to extreme susceptibility of Trypanosoma cruzi-infected mice deficient in gamma interferon receptor or inducible nitric oxide synthase. Infect Immun. (1998) 66:1208–15.
3. Antunez MI, Cardoni RL. Early IFN-gamma production is related to the presence of interleukin (IL)-18 and the absence of IL-13 in experimental Trypanosoma cruzi infections. Immunol Lett. (2001) 79:189–96. doi: 10.1016/S0165-2478(01)00283-8
4. Muller U, Kohler G, Mossmann H, Schaub GA, Alber G, Di Santo JP, et al. IL-12-independent IFN-gamma production by T cells in experimental Chagas' disease is mediated by IL-18. J Immunol. (2001) 167:3346–53. doi: 10.4049/jimmunol.167.6.3346
5. Galvao Da Silva AP, Jacysyn JF, De Almeida Abrahamsohn I. Resistant mice lacking interleukin-12 become susceptible to Trypanosoma cruzi infection but fail to mount a T helper type 2 response. Immunology (2003) 108:230–7. doi: 10.1046/j.1365-2567.2003.01571.x
6. Hoft DF, Lynch RG, Kirchhoff LV. Kinetic analysis of antigen-specific immune responses in resistant and susceptible mice during infection with Trypanosoma cruzi. J Immunol. (1993) 151:7038–47.
7. Petray PB, Rottenberg ME, Bertot G, Corral RS, Diaz A, Orn A, et al. Effect of anti-gamma-interferon and anti-interleukin-4 administration on the resistance of mice against infection with reticulotropic and myotropic strains of Trypanosoma cruzi. Immunol Lett. (1993) 35:77–80. doi: 10.1016/0165-2478(93)90151-Q
8. Barbosa De Oliveira LC, Curotto De Lafaille MA, Collet De Araujo Lima GM, De Almeida Abrahamsohn I. Antigen-specific Il-4- and IL-10-secreting CD4+ lymphocytes increase in vivo susceptibility to Trypanosoma cruzi infection. Cell Immunol. (1996) 170:41–53. doi: 10.1006/cimm.1996.0132
9. Hiyama K, Hamano S, Nakamura T, Nomoto K, Tada I. IL-4 reduces resistance of mice to Trypanosoma cruzi infection. Parasitol Res. (2001) 87:269–74. doi: 10.1007/PL00008577
10. Soares MB, Silva-Mota KN, Lima RS, Bellintani MC, Pontes-De-Carvalho L, Ribeiro-Dos-Santos R. Modulation of chagasic cardiomyopathy by interleukin-4: dissociation between inflammation and tissue parasitism. Am J Pathol. (2001) 159:703–9. doi: 10.1016/S0002-9440(10)61741-5
11. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
12. Iniesta V, Gomez-Nieto LC, Corraliza I. The inhibition of arginase by N(omega)-hydroxy-l-arginine controls the growth of Leishmania inside macrophages. J Exp Med. (2001) 193:777–84. doi: 10.1084/jem.193.6.777
13. Iniesta V, Gomez-Nieto LC, Molano I, Mohedano A, Carcelen J, Miron C, et al. Arginase I induction in macrophages, triggered by Th2-type cytokines, supports the growth of intracellular Leishmania parasites. Parasite Immunol. (2002) 24:113–8. doi: 10.1046/j.1365-3024.2002.00444.x
14. Duleu S, Vincendeau P, Courtois P, Semballa S, Lagroye I, Daulouede S, et al. Mouse strain susceptibility to trypanosome infection: an arginase-dependent effect. J Immunol. (2004) 172:6298–303. doi: 10.4049/jimmunol.172.10.6298
15. Kropf P, Fuentes JM, Fahnrich E, Arpa L, Herath S, Weber V, et al. Arginase and polyamine synthesis are key factors in the regulation of experimental leishmaniasis in vivo. FASEB J. (2005) 19:1000–2. doi: 10.1096/fj.04-3416fje
16. Gobert AP, Daulouede S, Lepoivre M, Boucher JL, Bouteille B, Buguet A, et al. L-Arginine availability modulates local nitric oxide production and parasite killing in experimental trypanosomiasis. Infect Immun. (2000) 68:4653–7. doi: 10.1128/IAI.68.8.4653-4657.2000
17. Holscher C, Arendse B, Schwegmann A, Myburgh E, Brombacher F. Impairment of alternative macrophage activation delays cutaneous leishmaniasis in nonhealing BALB/c mice. J Immunol. (2006) 176:1115–21. doi: 10.4049/jimmunol.176.2.1115
18. Schleicher U, Paduch K, Debus A, Obermeyer S, Konig T, Kling JC, et al. TNF-mediated restriction of arginase 1 expression in myeloid cells triggers type 2 NO synthase activity at the site of infection. Cell Rep. (2016) 15:1062–75. doi: 10.1016/j.celrep.2016.04.001
19. Stempin C, Giordanengo L, Gea S, Cerban F. Alternative activation and increase of Trypanosoma cruzi survival in murine macrophages stimulated by cruzipain, a parasite antigen. J Leukoc Biol. (2002) 72:727–34. doi: 10.1189/jlb.72.4.727
20. Stempin CC, Tanos TB, Coso OA, Cerban FM. Arginase induction promotes Trypanosoma cruzi intracellular replication in Cruzipain-treated J774 cells through the activation of multiple signaling pathways. Eur J Immunol. (2004) 34:200–9. doi: 10.1002/eji.200324313
21. Cuervo H, Pineda MA, Aoki MP, Gea S, Fresno M, Girones N. Inducible nitric oxide synthase and arginase expression in heart tissue during acute Trypanosoma cruzi infection in mice: arginase I is expressed in infiltrating CD68+ macrophages. J Infect Dis. (2008) 197:1772–82. doi: 10.1086/529527
22. Cuervo H, Guerrero NA, Carbajosa S, Beschin A, De Baetselier P, Girones N, et al. Myeloid-derived suppressor cells infiltrate the heart in acute Trypanosoma cruzi infection. J Immunol. (2011) 187:2656–65. doi: 10.4049/jimmunol.1002928
23. Florez O, Martin J, Gonzalez CI. Interleukin 4, interleukin 4 receptor-alpha and interleukin 10 gene polymorphisms in Chagas disease. Parasite Immunol. (2011) 33:506–11. doi: 10.1111/j.1365-3024.2011.01314.x
24. Emson CL, Bell SE, Jones A, Wisden W, Mckenzie AN. Interleukin (IL)-4-independent induction of immunoglobulin (Ig)E, and perturbation of T cell development in transgenic mice expressing IL-13. J Exp Med. (1998) 188:399–404. doi: 10.1084/jem.188.2.399
25. Hamano S, Himeno K, Miyazaki Y, Ishii K, Yamanaka A, Takeda A, et al. WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity (2003) 19:657–67. doi: 10.1016/S1074-7613(03)00298-X
26. Bohme J, Rossnagel C, Jacobs T, Behrends J, Holscher C, Erdmann H. Epstein-Barr virus-induced gene 3 suppresses T helper type 1, type 17 and type 2 immune responses after Trypanosoma cruzi infection and inhibits parasite replication by interfering with alternative macrophage activation. Immunology (2016) 147:338–48. doi: 10.1111/imm.12565
27. Holscher C, Mohrs M, Dai WJ, Kohler G, Ryffel B, Schaub GA, et al. Tumor necrosis factor alpha-mediated toxic shock in Trypanosoma cruzi-infected interleukin 10-deficient mice. Infect Immun. (2000) 68:4075–83. doi: 10.1128/IAI.68.7.4075-4083.2000
28. Santiago HC, Feng CG, Bafica A, Roffe E, Arantes RM, Cheever A, et al. Mice deficient in LRG-47 display enhanced susceptibility to Trypanosoma cruzi infection associated with defective hemopoiesis and intracellular control of parasite growth. J Immunol. (2005) 175:8165–72. doi: 10.4049/jimmunol.175.12.8165
29. Matthews DJ, Emson CL, Mckenzie GJ, Jolin HE, Blackwell JM, Mckenzie AN. IL-13 is a susceptibility factor for Leishmania major infection. J Immunol. (2000) 164:1458–62. doi: 10.4049/jimmunol.164.3.1458
30. Abrahamsohn IA, Da Silva AP, Coffman RL. Effects of interleukin-4 deprivation and treatment on resistance to Trypanosoma cruzi. Infect Immun. (2000) 68:1975–9. doi: 10.1128/IAI.68.4.1975-1979.2000
31. Tarleton RL, Grusby MJ, Zhang L. Increased susceptibility of Stat4-deficient and enhanced resistance in Stat6-deficient mice to infection with Trypanosoma cruzi. J Immunol. (2000) 165:1520–5. doi: 10.4049/jimmunol.165.3.1520
32. Hunter CA, Ellis-Neyes LA, Slifer T, Kanaly S, Grunig G, Fort M, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. (1997) 158:3311–6.
33. Muller U, Stenzel W, Kohler G, Werner C, Polte T, Hansen G, et al. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol. (2007) 179:5367–77. doi: 10.4049/jimmunol.179.8.5367
34. Heitmann L, Abad Dar M, Schreiber T, Erdmann H, Behrends J, Mckenzie AN, et al. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J Pathol. (2014) 234:338–50. doi: 10.1002/path.4399
35. Schreiber T, Ehlers S, Heitmann L, Rausch A, Mages J, Murray PJ, et al. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J Immunol. (2009) 183:1301–12. doi: 10.4049/jimmunol.0803567
36. Qualls JE, Neale G, Smith AM, Koo MS, Defreitas AA, Zhang H, et al. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci Signal. (2010) 3:ra62. doi: 10.1126/scisignal.2000955
37. Goni O, Alcaide P, Fresno M. Immunosuppression during acute Trypanosoma cruzi infection: involvement of Ly6G (Gr1(+))CD11b(+)immature myeloid suppressor cells. Int Immunol. (2002) 14:1125–34. doi: 10.1093/intimm/dxf076
38. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. (2008) 222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x
39. Rodriguez PC, Ochoa AC, Al-Khami AA. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front Immunol. (2017) 8:93. doi: 10.3389/fimmu.2017.00093
40. Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol. (2005) 5:641–54. doi: 10.1038/nri1668
41. Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111:4233–44. doi: 10.1182/blood-2007-07-099226
42. Rutschman R, Lang R, Hesse M, Ihle JN, Wynn TA, Murray PJ. Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J Immunol. (2001) 166:2173–7. doi: 10.4049/jimmunol.166.4.2173
43. Pattnaik B, Bodas M, Bhatraju NK, Ahmad T, Pant R, Guleria R, et al. IL-4 promotes asymmetric dimethylarginine accumulation, oxo-nitrative stress, and hypoxic response-induced mitochondrial loss in airway epithelial cells. J Allergy Clin Immunol. (2016) 138:130–41.e9. doi: 10.1016/j.jaci.2015.11.036
44. Blackwell S. The biochemistry, measurement and current clinical significance of asymmetric dimethylarginine. Ann Clin Biochem. (2010) 47:17–28. doi: 10.1258/acb.2009.009196
45. Carbajosa S, Rodriguez-Angulo HO, Gea S, Chillon-Marinas C, Poveda C, Maza MC, et al. L-arginine supplementation reduces mortality and improves disease outcome in mice infected with Trypanosoma cruzi. PLoS Negl Trop Dis. (2018) 12:e0006179. doi: 10.1371/journal.pntd.0006179
46. Colotti G, Ilari A. Polyamine metabolism in Leishmania: from arginine to trypanothione. Amino Acids (2011) 40:269–85. doi: 10.1007/s00726-010-0630-3
47. Ilari A, Fiorillo A, Genovese I, Colotti G. Polyamine-trypanothione pathway: an update. Future Med Chem. (2017) 9:61–77. doi: 10.4155/fmc-2016-0180
48. Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Trypanothione: a novel bis(glutathionyl)spermidine cofactor for glutathione reductase in trypanosomatids. Science (1985) 227:1485–7. doi: 10.1126/science.3883489
49. Abebe T, Takele Y, Weldegebreal T, Cloke T, Closs E, Corset C, et al. Arginase activity - a marker of disease status in patients with visceral leishmaniasis in ethiopia. PLoS Negl Trop Dis. (2013) 7:e2134. doi: 10.1371/journal.pntd.0002134
50. Takele Y, Abebe T, Weldegebreal T, Hailu A, Hailu W, Hurissa Z, et al. Arginase activity in the blood of patients with visceral leishmaniasis and HIV infection. PLoS Negl Trop Dis. (2013) 7:e1977. doi: 10.1371/annotation/9e89dee8-e2b7-4559-bd4e-933692403410
51. Erdmann H, Behrends J, Holscher C. During acute experimental infection with the reticulotropic Trypanosoma cruzi strain Tulahuen IL-22 is induced IL-23-dependently but is dispensable for protection. Sci Rep. (2016) 6:32927. doi: 10.1038/srep32927
52. Jensen EC. Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec. (2013) 296:378–81. doi: 10.1002/ar.22641
53. Reiling N, Holscher C, Fehrenbach A, Kroger S, Kirschning CJ, Goyert S, et al. Cutting edge: toll-like receptor (TLR)2- and TLR4-mediated pathogen recognition in resistance to airborne infection with Mycobacterium tuberculosis. J Immunol. (2002) 169:3480–4. doi: 10.4049/jimmunol.169.7.3480
Keywords: Chagas disease, interleukin-13, arginase-1, Trypanosoma cruzi, macrophage-cell
Citation: Abad Dar M and Hölscher C (2018) Arginase-1 Is Responsible for IL-13-Mediated Susceptibility to Trypanosoma cruzi Infection. Front. Immunol. 9:2790. doi: 10.3389/fimmu.2018.02790
Received: 30 June 2018; Accepted: 13 November 2018;
Published: 29 November 2018.
Edited by:
Debora Decote-Ricardo, Universidade Federal Rural do Rio de Janeiro, BrazilReviewed by:
Phileno Pinge-Filho, Universidade Estadual de Londrina, BrazilNúria Gironès, Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Spain
Copyright © 2018 Abad Dar and Hölscher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Hölscher, Y2hvZWxzY2hlckBmei1ib3JzdGVsLmRl