Abstract
Resident memory (TRM) cells are a distinct tissue-localized T cell lineage that is crucial for protective immunity in peripheral tissues. While a great deal of effort has focused on defining their role in immunity to infections, studies now reveal TRM cells as a vital component of the host immune response to cancer. Characterized by cell-surface molecules including CD103, CD69, and CD49a, TRM-like tumor-infiltrating lymphocytes (TILs) can be found in a wide range of human cancers, where they portend improved prognosis. Recent studies in mouse tumor models have shown that TRM cells are induced by cancer vaccines delivered in peripheral tissue sites, or by the depletion of regulatory T cells. Such tumor-specific TRM cells are recognized as both necessary and sufficient for long-lived protection against tumors in peripheral tissue locations. TRM responses against tumor/self-antigens can concurrently result in the development of pathogenic TRM responses to self, with a growing number of autoimmune diseases and inflammatory pathologies being attributed to TRM responses. This review will recount the path to discovering the importance of resident memory CD8 T cells as they pertain to cancer immunity. In addition to highlighting key studies that directly implicate TRM cells in anti-tumor immunity, we will highlight earlier work that implicitly suggested their importance. Informed by studies in infectious disease models, and instructed by a clear role for TRM cells in autoimmunity, we will discuss strategies for therapeutically promoting TRM responses in settings where they don't naturally occur.
Introduction
Cancer can be considered a disease of immune dysfunction, with a failure of immune recognition leading to the outgrowth of malignant cells as tumors (1). Tumor development has been said to occur in three distinct steps: inefficient elimination of early transformed cells, development of a state of equilibrium between tumor cells and immune cells, and tumor escape from immune pressure (2). While innate immune cells are important for early tumor immune surveillance, T cells are fundamentally recognized for their crucial role in the antigen-specific recognition and elimination of malignantly transformed cells (2). Indeed a wealth of studies from humans and mouse models establishes a particularly potent role for CD8 T cells in controlling the outgrowth of malignancies (3).
The recent success of T cell immune checkpoint inhibitor (ICI) therapies for cancer has revealed CD8 T cells as potent mediators of immunity against advanced cancers (4, 5). Following effective priming in lymph nodes, T cells traffic to tumors and other peripheral tissues. In a growing number of cases, CD8 T cells have been shown to mediate the regression of large bulky tumors, resulting in durable long-term disease remissions (5). The persistence of such responses is fundamentally thought to be based on the ability of T cells to act as potent effectors and, subsequently, generate long-lived memory (6). T cell memory is antigen-specific, and can provide durable host-wide protection. As the field of cancer immunotherapy advances rapidly, it is now crucial to understand how the dissemination and maintenance of tumor-specific T cells can be optimally achieved.
Studies in infectious disease models have provided a wealth of information regarding memory CD8 T cell generation and localization. Classical definitions of memory T cells derive from the belief that T cells localize and recirculate predominantly throughout the blood and secondary lymphoid organs (7, 8). Such memory T cells were traditionally defined as being comprised of both central memory (TCM) and effector memory (TEM) subsets (9). TCM cells were shown to persist and recirculate through the blood, bone marrow, lymph nodes, and spleen; whereas TEM cells were shown to recirculate predominantly through blood, and peripheral tissues (10). In the early 2000's, large and persistent populations of antigen (Ag)-specific CD8 T cells in peripheral tissues were initially classified as TEM cells in recirculation from the blood (8, 11).
These early classifications of T cell memory were quickly brought to bear on the question of what T cell subset provides the best immunity against cancer. A series of mouse adoptive T cell therapy studies published a decade ago showed that in vitro activated melanoma Ag (gp100)-specific TCM-like CD8 cells have a greater ability to control established melanomas in comparison with clonally-identical Teff/TEM-like cells (12, 13). Subsequent work in humans identified a third major subset of memory T cells known as stem cell-like memory (TSCM) cells (14). This less-differentiated T cell subset was capable of generating both TCM and TEM cells, and was shown in adoptive immunotherapy studies to have even greater anti-melanoma potency as compared with TCM cells (15, 16). However, these early studies relied on definitions of memory that had been generated from a myopic focus on blood and lymphoid tissues. The concept that tumor-specific T cells could persist in peripheral tissues and tumors, without recirculation from the blood, was not yet being seriously considered.
Studies in viral models have now revealed a distinct lineage of memory T cells that resides in peripheral tissues and can provide orders of magnitude stronger protection than their TCM cell counterparts (17). It is now recognized that peripheral host cells are surveyed overwhelmingly by TRM cells that vastly outnumber their recirculating counterparts in peripheral tissues (18). The role of these tissue-resident memory (TRM) cells in immune responses against cancer is only beginning to be explored. However, early studies have revealed that TRM cells are induced by vaccination, present in human tumors, and sustained by the same molecular mechanisms that were defined by infectious disease models. As the concepts of tumor immunity and autoimmunity remain closely linked, a better understanding of TRM responses to cancer has also provided new insights regarding a role for TRM cells in autoimmune disease. In turn, lessons regarding TRM responses in autoimmune disease have begun to inform the field of tumor immunotherapy.
The goal of this review is to discuss new advances in our understanding of resident-memory T cells as they pertain to cancer immunity and associated autoimmunity. In addition to discussing recent studies that have directly implicated TRM cells in anti-tumor immunity, we will highlight key early studies that implicitly suggested a contribution from TRM cells before their existence was known. As the field has grown out of studies in infectious diseases, we will draw heavily on such models in forming the groundwork for studies in cancer. The focus of this article will be on CD8 TRM cells as key mediators of the anti-tumor response, but not to imply an unimportant role for CD4 T cells. While CD4 TRM cells have been described in multiple infectious disease settings (19), their role in immunity to cancer remains as yet undefined.
Features of TRM cells in infectious disease models
CD8 TRM cells are defined based on their long-term persistence in peripheral tissues without recirculation from the blood. Since the earliest discovery of extra-lymphoid memory T cells in peripheral tissues of mice infected with vesicular stomatitus virus (VSV), and listeria monocytogenes (LM) infections (11), TRM responses have been documented in response to a myriad of infections including lymphocytic choriomeningitis virus (LCMV) (20, 21), herpes simplex virus (HSV) (20, 22, 23), chlamydia (24), influenza (23, 25), vaccina virus (VACV) (17), human immunodeficiency virus (HIV) (26), tuberculosis (TB) (27), mouse cytolomegalovirus (MCMV) (28), and human papilloma virus (HPV) (29, 30). Thus, the formation of TRM responses upon productive host infection can be viewed as a rule rather than an exception.
Phenotypic features of TRM cells
As a unique memory T cell lineage, CD8 TRM cells can be distinguished from other T cell subsets based on their cell surface phenotype. Like all memory T cells, TRM cells are differentiated from naïve T cells based on their expression of CD44; a marker of antigen experience (31). TRM cells also lack expression of CD62L (L-selectin); which differentiates them from naïve T cells and TCM cells that require CD62L for entry into secondary lymphoid organs (10). To distinguish TRM cells from effector and TEM cells, more detailed phenotypic considerations are necessary, and tissue retention markers; most notably CD103 and CD49a (VLA-1) are typically used. CD103 is a TGF-β induced molecule that promotes TRM cell tissue retention by binding to e-cadherin, which is expressed on normal host epithelial cells (32). CD49a promotes tissue retention and survival through binding to collagenase type IV (33). While studies have largely focused on CD103 and CD49a as markers of tissue residency, it is important to note that their expression is not absolute, nor required. CD103 expression has been shown to be dispensable for TRM formation in the liver (34) and gut (35), and CD49a is dispensable for TRM formation in the lung (27). Additional retention markers have been used to identify TRM cells, such as cutaneous leukocyte antigen (CLA) in the skin (36, 37) and LFA-1 in the liver (38). Thus, while TRM cells are exclusively CD44hi and CD62Llow they can express an array of tissue-specific retention markers, with no single marker being sufficient to definitively identify a TRM population.
Another key phenotypic feature of TRM cells is the expression of CD69, a marker of T cell activation, which blocks T cell expression S1PR1 (20). CD69 thus promotes tissue retention and residency by interfering with the ability to sense the S1P gradient that is essential for tissue egress (39, 40). Similarly, TRM cells typically lack the expression of CCR7, which cooperates with S1PR1 for tissue egress through lymphatic vessels. However, as with CD103 and CD49a, CD69 can be dispensable for TRM formation (18) and has been shown to be non-definitive in distinguishing recirculating T cells from TRM cells in the steady state (41). In humans, a role for CD69 may be more pronounced than in mice, as peripheral tissue TRM cells in healthy individuals overwhelmingly expressed (42).
TRM cells occupy a unique niche in their tissue of residence, and take on a dendritic morphology that is uncharacteristic of circulating memory T cells (43). TRM cells continually scan the peripheral tissue where an initial insult occurred (44), exhibiting limited migratory ability, and tending to accumulate at sites of antigen persistence (45). TRM cells adapt well to their surroundings by exploiting the features of tissues in which they reside. In skin, TRM cells have been shown to cluster around niches formed by keratinocytes, near hair follicles, which secrete IL-15, IL-7 (46), and TGF-β (47). In anatomical regions with a high tissue turnover rate, such as the lamina propria of the gut, immune cells such as macrophages support the formation of TRM aggregates (48). In other barrier tissues, TRM cells occupy de novo niches, such as repair-associated memory depots (RAMD) in the lung, and mucosa-associated lymphoid tissue (MALT) in the female reproductive tract (FRT) (49, 50). Localization to barrier sites of mucosal tissues exerts a metabolic burden that typically limits the persistence of T cells. However, TRM cells utilize fatty acid beta-oxidative phosphorylation to support their longevity (51–53). In contrast to conventional memory T cells which conduct their own fatty acid synthesis, TRM cells in the skin have been shown to express high levels of fatty acid binding proteins FABP4 and 5, to facilitate the necessary uptake of fatty acids (51). These properties of TRM cells enable them to function in diverse peripheral tissue niches.
Transcriptional profiles of TRM cells
Transcriptional profiling has demonstrated that TRM cells are distinct from their TCM and TEM counterparts, and thus represent a unique T cell lineage (20, 54). Although unique genes define TRM subpopulations in different tissues (20), a core transcriptional signature has been proposed for TRM cells (54, 55). This signature highlights the distinctive nature of TRM cells as a hybrid between effector and memory cells, which are armed in an effector like-state even during quiescence (54, 55).
Master transcriptional regulators of TRM cell differentiation across multiple tissue types include Hobit, Blimp1 (42), and Runx3 (54). In contrast, Tbx21 (T-bet) and Eomes—the master regulators of effector and lymphoid memory T cell lineages—have been shown to impede the development of TRM cells (56). Hobit and its homolog Blimp1 act in synergy as negative regulators of tissue egress, by directly binding to S1pr1, Ccr7, and Tcf7 motifs in mice (55). Additionally, Blimp-1 has been shown to initiate cytotoxic function while Hobit maintains deployment-ready cytotoxicity in TRM cells (57). On the other hand, Runx3 acts to promote the expression of TRM tissue retention markers such as CD103 and CD69 (54). In addition to these canonical TRM transcription factors, the NR4A family has also been shown to be highly upregulated in TRM cells, with the absence of Nr4a1 resulting in a reduced capacity to generate TRM (58).
While most in-depth transcriptional analyses have, to date, been conducted in murine infectious disease models, transcriptional characteristics of TRM cells from humans are also beginning to be reported. CD8 TRM cells from human lungs showed high CD69 expression, and variable CD103 expression (59). These lung TRM cells could be distinguished from their circulating counterparts by high levels of GZMB, IFNG, TNF, and NOTCH1 transcripts, with NOTCH signaling shown to promote IFNG gene expression (59). Separate studies showed that CD69+ memory T cells across multiple tissues of human cadavers exhibit a conserved transcriptional profile including ITGA1 (CD49a), ITGAE (CD103), and PDCD1 (PD-1) expression (42). In contrast to mouse TRM cells however, human cells lacked expression of ZNF683 (HOBIT) and PRDM1 (BLIMP-1) (42). It is important to note that TRM cell transcriptional signatures have been largely generated from pooled T cell samples, thus lacking single cell resolution and missing the complexity and heterogeneity that potentially exists within a TRM cell pool. Single-cell RNA-sequencing of TRM cells from mice and humans may, in the future, reveal heterogeneous CD8 TRM cell subsets.
Protective function of TRM cells
TRM cells have been shown to play a dominant role in protection against peripheral infections, in some cases mediating orders of magnitude stronger protection than lymphoid memory T cells (17). However, because infections generate both resident and circulating memory T cell compartments, specialized techniques have been needed to isolate the contribution of TRM cells from that of their lymphoid memory counterparts (60). FTY720 is a small molecule S1PR antagonist that inhibits T cell egress from lymph nodes, and thereby prevents circulating memory T cell subsets from accessing peripheral tissues (61). In mice infected by VACV-OVA through skin scarification (s.s.), it was shown that treatment with FTY720 had no effect on protection against cutaneous viral re-challenge, indicating that skin TRM cells are sufficient for long-lived protection (17). In the setting of influenza viral infection, a protective role for lung TRM cells was also first established by studies involving FTY720 treatment (62). Low dose monoclonal antibody (mAb) depletion strategies can also be employed based on their ability to efficiently deplete circulating and lymphoid T cells, while sparing tissue-resident T cell niches (49). Using this technique, HSV skin infection was shown to generate long-lived protective immunity that was unperturbed by the depletion of circulating memory T cells (20). Lastly, the surgical joining procedure known as parabiosis, has been used to isolate the contribution of TCM from that of TRM. Parabiosis allows the equilibration of circulating immune compartments between immune and naïve mice, thus transferring circulating memory to a naive recipient (8). Through elegant parabiosis studies in conjunction with the use of FTY720, skin CD8+ TRM cells were shown to be superior to TCM in protecting against cutaneous VACV re-infection (17). These fundamental studies established a crucial role for TRM cells in mediating long-lived protection against peripheral infections.
Resident memory T cell responses to cancer
The above characteristics of TRM cells have, more recently, been recognized for their relevance to cancer immunity. Indeed, as infections occur in peripheral tissues, so do cancers arise in the same tissues. As such, it stands to reason that populations of tumor-specific TRM cells can occupy tumors themselves, and the tissues from which they arise (Figure 1). Despite this, the role of TRM cells in mediating immunity to cancer has only recently been described.
Figure 1
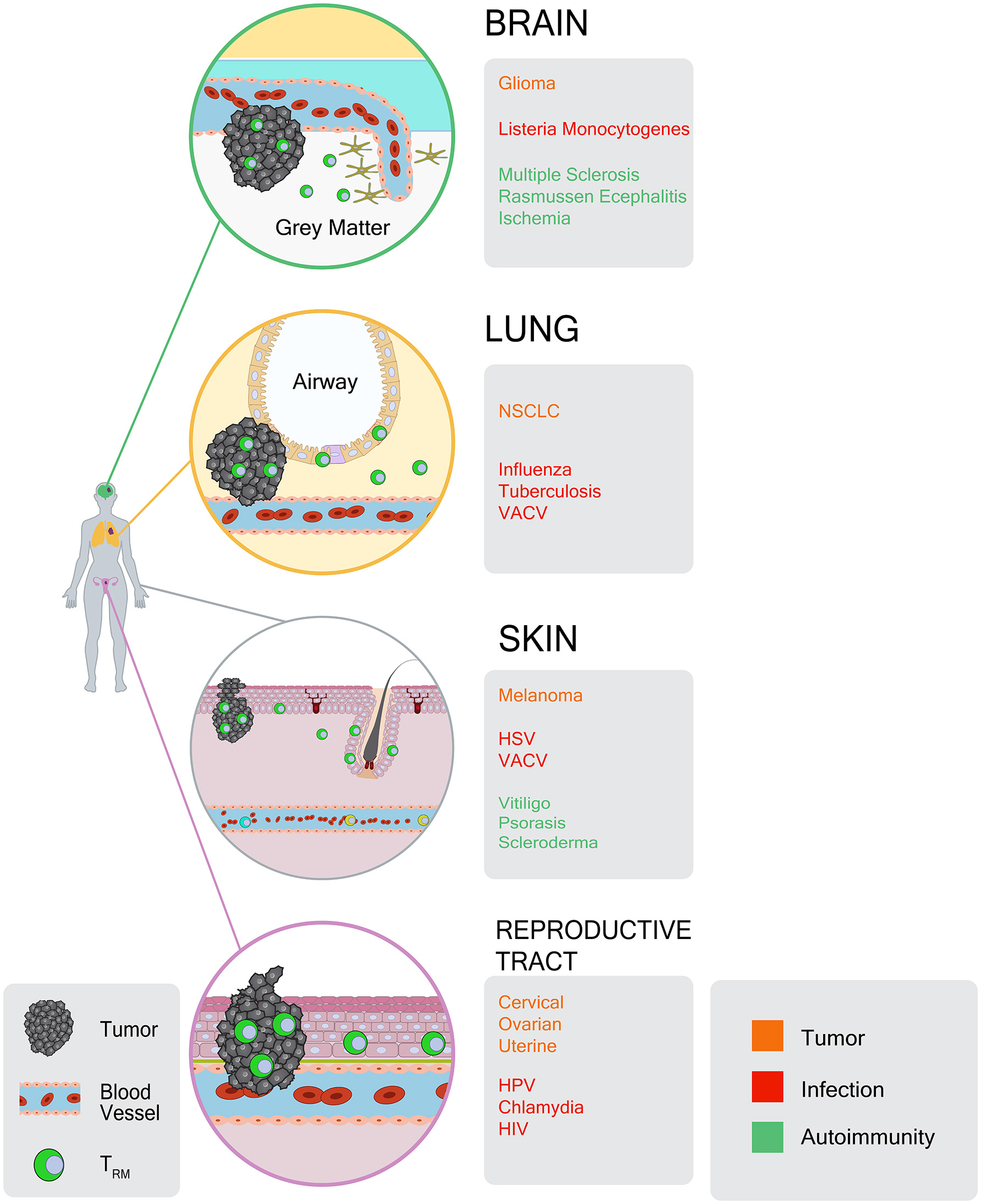
Atlas of TRM responses in peripheral tissues and tumors. CD8+ TRM cells occupy a diverse array of tissues and tumors. For each tissue location, gray boxes indicate: (1) tumor types in which TRM-like TILs have been identified (orange), (2) infections that give rise to TRM responses (red), and (3) autoimmune or inflammatory conditions in which TRM cells have been identified (green).
Prior to the term “resident memory,” a growing body of literature had already identified CD103 expressing CD8 T cells within human tumors, and linked these cells to improved prognosis. With CD103 now recognized as a common TRM cell marker, these studies can be viewed as the earliest evidence of tumor infiltrating lymphocytes (TILs) having TRM-like properties. Importantly, these findings formed the ground work for the extensive characterization of TRM cell transcriptional signatures in a variety of human tumors, and mechanistic mouse work to establish a crucial role for this memory T cell subset in immunity to cancer.
Identification of CD103+CD8+ TILs
Studies examining CD103 expression on tumor infiltrating lymphocytes date back 30 years, with an initial focus on the identification of cellular localization patterns within tumors. A 1988 analysis was the first to identify CD103-positive tumor infiltrating lymphocytes (TILs) (63). These studies found that a majority of TILs in gastrointestinal tumors were positive for CD103 (at the time referred to as HML1), and that these cells were localized throughout the tumor mass (63). A decade later, studies involving pancreatic cancer patients revealed that approximately 20% of all CD8 T cells in tumors expressed CD103 as well as CD45RO, a human memory T cell marker (64). Unlike the prior study (65) these studies showed CD103+ T cell aggregation in fibrous stromal tissue, and exclusion from tumor cell nests (64). Tumor-excluded T cell distribution was further supported by a 2001 analysis of bladder cancer samples in which the majority of CD103+CD8+ TILs were found on the periphery of the tumor, potentially suggesting immune failure (66). However, a 2003 study reported high levels of CD103+CD8+ T cells infiltrating intratumoral regions of microsatellite-unstable colorectal cancers (65). Later work revealed that CD103+ TILs were not limited to tumor masses, and could also be found in the ascites fluid of high-grade serous ovarian cancer (67). This early collection of studies established CD103 expression on CD8+ TILs in various solid cancers, but indicated their variable distribution throughout the tumor microenvironment (TME).
With CD103 substantiated as marker of tumor-infiltrating CD8 T cells, several early studies also sought to define the function of CD103 in the interaction between T cells and tumor cells. Attempts at defining these interactions focused predominantly on two molecules: TGF-β, as an inducer of CD103 expression (68, 69), and e-cadherin, as the only known binding partner for CD103 (70). An early study showed that TGF-β-induced CD103 expression significantly enhanced the lysis of e-cadherin-transduced pancreatic cancer cells by CD8 T cells in vitro (71). This was found to occur by increasing the adhesion of CTL's to tumor cells, a process which depended on the expression of both CD103 and e-cadherin (71). However, it remained unknown whether such interactions occur in response to tumor cell encounter, and whether Ag recognition plays a role in CD103 induction. Subsequent studies involving human HER2/NEU-specific T cell clones revealed that cognate antigen and TGF-β were both required for the in vitro upregulation of CD103 on T cells (72). Furthermore, a human colon cancer cell line was capable of secreting enough TGF-β to promote T cell expression of CD103 upon Ag-recognition in vitro, suggesting that tumor-derived TGF-β could induce CD103 expression on TILs (72). This study further showed that T cells exposed to TGF-β during priming, will re-express CD103 more readily upon recall (72). Follow-up studies in a human lung cancer xenograft mouse model showed that CD103 is induced upon T cell trafficking to tumors, and that in vivo neutralization of TGF-β impairs the recruitment of CD103+ CD8 T cells into the TME (73). These studies together substantiated the role of tumor-derived TGF-β in the induction of CD103 on intratumoral T cells.
While CD103 upregulation on TILs was shown to be TGF-β dependent, the mechanism governing this process in cancer still remained unclear. In 2014, it was shown that TGF-β induces Smad2/3 phosphorylation which allows nuclear translocation and binding to the proximal regulatory elements of CD103 (74). Indeed pSmad2/3 nuclearization was recently used as a marker of CD103+ TIL's in human cervical cancers, in which TGF-β signaling is abundant (75). The key role that TGF-β plays in CD103+ TIL biology was further validated using a tumor-specific human T cell clone, by showing that binding of TGF-β to its receptor promoted the recruitment and phosphorylation of integrin-linked kinase (ILK) to the CD103 intracellular domain, inducing integrin inside-out signaling that may further promote TRM cell migration and function (76, 77).
Subsequent studies have also shed light on the putative role of CD103/e-cadherin interactions in promoting tumor cell killing. In vitro studies used CD103 expressing tumor-antigen specific T cell lines from lung cancer patients to demonstrate that e-cadherin expression was required by tumor cells for effective killing via granule polarization and exocytosis (78). It was separately shown that CD103 ligand engagement together with TCR binding, enhances the strength of human TIL/tumor cell interactions (79). CD103 engagement with e-cadherin was found to shape immunological synapse morphology, which was essential for the polarization of cytokine and lytic granules containing granzyme B and IFN-γ (79). A co-stimulatory role for CD103 was subsequently established by showing that CD103 triggering promotes phosphorylation of ERK1/2 kinases and phospholipase Cγ1 (PLCγ1)—a process that was required for cytotoxicity by tumor-specific human T cells (80). CD103-downstream signaling in TILs was separately shown to require paxillin phosphorylation and binding to the CD103 cytoplasmic tail, potentiating effector function against cognate lung tumor cells (81). Thus, ligation of CD103 has been proposed to enhance tumoricidal T cell functions in vivo.
While the above studies employed e-cadherin-expressing tumor cell lines and/or transfectants, it should be noted that the expression of e-cadherin is often lost on epithelial cells during malignant transformation and tumorigenesis (82). This may suggest that CD103-ecadherin interactions are unlikely to play a major role in the TME. This is underscored by analyses of bladder (66), ovarian (83), lung (84), and pancreatic cancer (85) specimens showing either a lack of co-localization between CD103-positivie TILs and e-cadherin expressing tumor cells, or a lack of correlation between e-cadherin expression levels and CD103+ TIL infiltration. However, some studies do report the contrary (86). Interestingly, CD103 has been shown to have unknown binding partners in peripheral tissue (87, 88), and one could speculate that such partners function in engaging CD103 on CD8 T cells in e-cadherin negative tumors, or negative regions within tumors. Alternately, following upregulation of CD103 by tumor-derived TGF-β, this integrin might serve as a marker of the TRM transcriptional program, rather than a functional player that engages its ligand in the TME.
Prognostic significance of CD103+CD8+ TILs
While early studies confirmed the existence of CD103+CD8+ TlLs in solid tumors, evidence that these cells had prognostic value for patients did not appear in the literature until 2014, when it was shown that this subset is strongly associated with survival in high-grade serous ovarian carcinoma (HGSOC) (83). In this study, CD103+ TILs were clearly localized to intratumoral regions, as opposed to associated tumor stroma. Interestingly, the presence of CD8+CD103− cells conferred no benefit when compared to tumors devoid of all CD8 T cells, suggesting that the CD103+CD8+ subset dominates the protective response in HGSOC (83).
Studies followed in several tumor types, all demonstrating correlation between CD103+CD8+ cells localized in tumor nests (also referred to as “intraepithelial” regions), and improved patient prognosis. In urothelial (bladder) cancer, high intratumoral CD103+CD8+ TIL density was inversely correlated with tumor size and could be used to predict improved overall survival (86). Similarly, a large breast cancer study demonstrated that CD103+ TIL infiltration to intratumoral (but not stromal) regions of tumor masses was prognostic of survival in a basal-like subtype (89). Investigation of endometrial adenocarcinoma showed that CD103 expression clearly delineated CD8 T cells localized to intratumoral regions (as opposed to stromal regions), and was an independent predictor of improved survival, particularly for high risk disease (90). Intraepithelial CD103-positive TILs were separately showed to be a valuable biomarker for therapeutic response in cervical cancer patients undergoing chemotherapy and radiation therapy (91). In contrast to earlier studies, these studies all supported CD103 as a marker of T cells infiltrating tumor nests as opposed to stroma. Importantly, these studies highlighted the superior prognostic strength of CD103+CD8+ TILs as compared with total CD8+ TILs.
Several studies have also supported this association for lung cancer. Early work showed that total CD103+CD8+ TILs have prognostic value in non-small cell lung cancer (NSCLC) patients with regards to early disease-free survival (92). Further subsetting of NSCLC patients showed that high numbers of CD103+ TILs in tumor nests were an independent predictor of disease-free survival for patients with pulmonary squamous cell carcinoma (84). Interestingly, this study also reported a strong correlation between smoking status and increased density of CD103+ TILs (84). A separate study of human NSCLC tumors showed that CD8 T cells in intratumoral regions were more likely to express CD103 than those in stromal regions, which was again highly predictive of survival (93). Further validation of CD103 as a biomarker was provided through stratification of NSCLC patients in The Cancer Genome Atlas (TCGA) database, which showed that patients with high tumor ITGAE (CD103) expression have improved overall survival (93).
For pancreatic cancer, the association between CD103 expression and prognosis is less straight-forward. Whereas high numbers of total CD8 T cells predicted improved prognosis in pancreatic ductal cell adenocarcinoma (PDAC), CD103+CD8+ TIL numbers did not predict survival, nor did CD103+CD8+ TILs in intratumoral regions (85). Interestingly though, a high ratio of CD103+ TILs in intratumoral vs. stromal locations was predictive of prognosis, potentially indicating the importance of spatial relationships between TRM-like cell subsets in PDAC (85). Indeed TRM cell localization to intratumoral regions would appear to situate them for optimal tumor control, although CD103+ TIL subsets may not be prognostic in all tumor types.
Phenotypic characteristics of TRM-like TILs
More detailed phenotypic analyses of CD8+CD103+ TILs have revealed additional TRM-like characteristics of these cells. Investigation of HGSCOC, endometrial adenocarcinoma, and ovarian cancer, all showed that CD8+CD103+ TILs express high levels of the exhaustion marker PD-1 (75, 83, 90). CD8 T cells in pediatric glial tumors exhibited a CD45RO+ CD69+ CCR7− TRM-like phenotype, in addition to multiple inhibitory checkpoints including PD-1, PD-L1, and TIGIT (94). Detailed flow cytometric and qPCR analyses of CD103+CD8+ TILs from lung cancer patients also revealed high levels of PDCD1 (PD-1) and HAVCR2 (TIM3), and low levels of the tissue egress marker S1PR1 (92). Using an immunofluorescence technique to visualize TRM in NSCLC, co-expression of CD49a (VLA-1) was identified on CD103+CD8+ T cells (93). This study also showed elevated levels of PD-1 and TIM-3 on CD103+CD8+ TILs as compared to CD103-negative TILs. Accordingly Cy-TOF analysis of melanoma-infiltrating T cells showed that a CD69+ subset (among which ~50% expressed CD103), co-expressed high levels of inhibitory checkpoint molecules CTLA-4 and PD-1 (95). Many of these studies have thus hypothesized that TRM-like TILs are key targets of immune checkpoint inhibitor therapy.
It is important to note that inhibitory checkpoint molecules, while overwhelmingly expressed on TRM-like TILs, have demonstrated somewhat more variable expression on TRM cells in peripheral infection models. PD-1 is expressed on CD103+CD8+ brain TRM cells in response to listeria monocytogenes, MCMV, and VSV infection (96, 97). Interestingly, upregulation of PD-1 on brain TRM cells is independent of chronic antigen stimulation or inflammation (98). However, a study of VSV infection found PD-1 expression absent on TRM cells in the brain (99). In the skin, HSV infection induces TRM cells that express PD-1 (20, 100), and CTLA-4 (20). However, VACV infection produced PD-1-negative TRM cells in the skin (17), and LCMV infection induced TRM cells in the small intestine that lacked PD-1 (101). Thus, inhibitory checkpoint molecule expression does not appear to be a defining feature of TRM cells in peripheral infection models. Moreover, it remains unclear if PD-1 expression levels differ between CD103+ cells in normal peripheral tissues and those in tumors.
Considering the widespread expression of inhibitory checkpoint molecules on CD103+ TILs, it has remained unclear if these cells represent true TRM cells or, rather, exhausted TILs that simply express CD103 in the TGF-β rich TME. Indeed no studies as-yet have demonstrated the long-term persistence of CD103+ CD8 T cells in tumors, without ongoing input from circulation—the true hallmark of a TRM response. In HGSCOC tumors it was further shown that CD103+ TILs have phenotypic characteristics of varying T cell differentiation states including TCM, TEM, and TRM cells (75), which may suggest that these cells are replenished from circulation. In the next section we will highlight several studies that transcriptionally profiled CD8+ TILs to generate a more comprehensive profile of their gene expression signature. These results, although varied across different tumor types, collectively support the conclusion that subsets of TILs are regulated by TRM transcriptional programs.
Transcriptional profiles of TRM-like TILs
Transcriptional profiling of TILs from NSCLC tumors has provided new insights into the characteristics of TRM-like TILS that extend beyond lung cancer. NSCLC tumors with a high TIL infiltration score were shown to have more pronounced gene expression characteristics of TRM cells including higher transcript levels for ITGAE (CD103), CD69, ITGA1 (CD49a), CXCR6, PDCD1 (PD-1), HAVCR2 (TIM3), LAG3, and TIGIT, but lower expression of KLRG1, CCR7, SELL (CD62L), and S1PR1 (102). Even among tumors with high CD8 T cell density, high expression of CD103 conferred patient survival advantage (102). Focusing on CD103hi CD8 TILs, this study also identified elevated expression of components of the NOTCH signaling pathway, as well as CD39, the cell surface ectonucleotidase that dephosphorylates ATP (102). This CD103+CD39+ TIL subset was the focus of a subsequent study that identified this populations in head and neck squamous cell carcinoma (HNSCC), melanoma, HNSCC, ovarian, lung, and rectal cancer tumors (103). In HNSCC patients, the CD103+CD39+ TIL population was found to be a better predictor of survival as compared with CD39 and/or CD103-negative CD8 T cell subsets (103). Transcriptional analysis of this sorted CD103+CD39+ subset from five tumors (HNSCC and ovarian) revealed enrichment of gene transcripts associated with exhaustion and reduced expression of T cell recirculation associated genes, suggesting that CD103+CD39+ TILs may have the most pronounced TRM-like character in tumors (103).
TIL characterization through bulk RNA-sequencing provides a wealth of transcriptional data, but obstructs the detection of small heterogenous populations within the TME. On the other hand, single-cell RNA sequencing (scRNA-seq) allows for fine resolution of TIL sub-populations at a cost of failing to detect poorly expressed transcripts. Indeed scRNA-seq of over six thousand TILs isolated from two patients with triple negative breast cancer (TNBC) showed at least four differential clusters of CD8 T cells, all expressing CD69 (104). One TIL cluster, identified based on high CD103 expression, was shown to express low levels of transcripts for KLRG1 and SELL (CD62L), as well as tissue egress related genes, KLF2 and S1PR1 (104). This TIL subset also expressed high transcript for inhibitory checkpoint genes HAVCR2, PDCD1, CTLA4, TIGIT, and LAG3, and cytotoxicity-related genes GZMB (granzyme) and PRF1 (perforin). Coupled with bulk RNA-seq data on sorted TIL populations, this study showed that CD8+CD103+ TILs in breast cancer exhibit multiple features of TRM differentiation. Importantly, this TRM-like TIL gene signature was predictive of survival in TNBC patients from the METABRIC consortium, and could be used to distinguish melanoma responders to ICI therapy (104).
Further highlighting the heterogenicity of the TIL population, another study sequenced >12,000 TILs from twelve NSCLC tumors and identified seven clusters with one expressing the TRM specific transcription factor, ZNF683 (HOBIT) (105). In contrast to the above study in TNBC, HOBIT-expressing lung TRM-like TILs expressed low levels of CD103 and showed reduced expression of PD-1 as compared to other CD8 clusters, suggesting a unique transcriptional program in NSCLC TRM-like TILs (105). As in TNBC, the scRNA-seq derived gene signature was used to stratify patients in a TCGA lung adenocarcinoma (LUAD) dataset, to show that enrichment of a CD8-ZNF683 gene-signature is predictive of survival when compared to other TIL derived signatures (105).
Taken together, the above studies highlight variability in TRM-like TIL gene signatures in cancer. Discrepancies likely arise due to unique tumor microenvironmental factors associated with individual patients, potentially relating to tumor tissue of origin, stage, and mutational status and/or burden. In comparing TRM-like TILs to bona fide TRM cells in normal peripheral tissues, it is also important to note that tumors, by their nature, lack many of the structural and molecular features of normal tissues that provide a hospitable niche for TRM cells. Thus, one might expect that TRM-like TILs would never perfectly match TRM cells that reside in the normal tissue counterpart of a tumor. Despite this, the discovery that TILs with features of TRM cells portend improved patient survival across multiple tumor types, underscores the importance of these cells, and represents a key recent advance in the field of cancer immunology.
Role of TRM cells in mediating immunity to cancer; lessons from mouse models
While the above studies provide strong correlative associations between TRM cells and improved patient survival, until very recently, formal evidence that TRM cells can mediate immunity to cancer was lacking. In retrospect, preclinical cancer vaccine studies published 15 years ago, inferred the crucial contribution of a long-lived, tissue-localized T cell population, however the importance of resident memory had not yet been recognized. This section will illustrate the path to our discovery of a role for TRM cells in mediating durable anti-tumor immunity.
Early indications of TRM cells from studies of cancer vaccination route
One of the earliest studies to infer TRM cell responses to cancer, published in 2003, revealed that melanoma-specific memory CD8 T cells distribute to distinct tissue locations, depending on the route of vaccination (106). By administering a peptide-pulsed dendritic cell (DC) vaccine via various routes, it was shown that only subcutaneous vaccination could reliably protect against a subcutaneous melanoma tumor rechallenge (106). This concept of regionally localized tumor-specific CD8 T cell memory was revolutionary at the time, although lymphoid tissues remained the focus of this early work. As such, tumor protection in these mice was attributed to memory populations in local lymph nodes, rather than in peripheral tissues (106).
Subsequent work extended the concept of tissue-localized tumor immunity, while further demonstrating that tumor-specific CD8 T cells could localize to peripheral tissue. In a 2010 study, mice infected through various routes with recombinant vaccinia virus (rVACV) expressing OVA257−264, were challenged with an OVA-expressing B16 melanoma cell line 6 weeks later (107). Reminiscent of the 2003 study, optimal protection against dermal melanoma rechallenge was only afforded by prior infection in the skin (107). This study further identified OVA-specific T cells in the skin, and referred to them as “skin-resident TEM cells” (107). While it was inferred that such TEM cells could mediate tumor protection in the skin, no formal experiments were performed to isolate the effects of this tissue-resident population from those of the lymphoid memory T cell compartment, both of which were present in tumor-bearing mice.
The field again approached this concept in 2013, with studies showing that growth of orthotopic head and neck TC1 tumors (implanted in the tongue) could only be inhibited when prior vaccination had been delivered via the intranasal (i.n.) route (108). In this study, i.n. vaccination with Shiga toxin B subunit fused to the HPV16-E7 tumor/viral peptide (STxB-E7) gave rise to E739−47-specific CD8+ T cells in mucosa-draining lymph nodes. These tumor-specific T cells expressed CD103 and CD49a however, in contrast to the 2010 study, T cell responses were not analyzed in peripheral (mucosal) tissue itself. Interestingly, mAb-mediated blockade of CD49a was shown to block CD8 T cell infiltration into TC1 tumors during acute STxB-E7 treatment, indicating CD49a as an important determinant of T cell infiltration into tumors (108). One other report in 2016 similarly showed that in vivo mAb-mediated blockade of CD49a or CD103 significantly impaired the control of subcutaneous B16-OVA tumors, supporting important roles for these molecules in T cell mediated anti-tumor immunity (109). While these studies revealed important new concepts regarding vaccine route and markers of tissue (or tumor) residence, the question of whether TRM cells directly mediate tumor protection remained open.
A key role for TRM cells in mediating anti-tumor immunity
Several notable studies in the past 2 years have now definitively demonstrated a role for TRM cells in mediating immunity to cancer. Importantly, each of these studies employed techniques originally used in infectious disease models, to isolate the contribution of TRM cells from that of the lymphoid memory compartment. In doing so, these studies illustrate a definitive role for TRM cells in providing long-lived protection against multiple tumor types, and in various tissue locations.
In follow-up work to the 2014 study involving STxB-E7 vaccination, it was shown that intranasal vaccination indeed generates a large pool of Ag-specific memory T cells in lung mucosal tissue (93). This was in clear contrast to intramuscular vaccination, which generated effector-like T cells in the spleen. E7-specific CD8 T cells in the lung expressed TRM markers including CD103 and CD49a, that were absent on T cells in the spleen. Further transcriptomic analysis of E7 Ag-specific CD8 T cells from the spleen showed that they expressed higher levels of lymphoid homing and tissue exit markers (i.e., Sell and S1pr1) compared with lung, while lacking adhesion and retention markers. Three key experiments were conducted to implicate TRM cells in the recall response against E7-expressing TC1 head and neck tumors. First, FTY720 was used to illustrate a minimal contribution of circulating T cells to tumor protection (93). Second, in vivo mAb-mediated TGF-β blockade was used to demonstrate a reduction in the generation of TRM populations, in conjunction with significantly decreased tumor protection. Finally, parabiosis was used to demonstrate that vaccinated mice were protected against tumor rechallenge, while no protection was afforded to parabiosed naive mice. These studies thus convincingly showed a dominant role for E7-specific TRM cells in protection against orthotopic head and neck cancer.
A thorough investigation of VACV-OVA vaccination route separately implicated TRM cells as important players in anti-tumor immunity (110). Mice infected with VACV-OVA by the dermal, nasal, or peritoneal routes showed distinct patterns of antigen-specific T cell memory formation in circulation and in peripheral tissues (110). Using i.p. vaccination to generate circulating memory without resident memory, or FTY720 as a means for blocking T cell access to the skin, it was shown that either circulating or resident memory are sufficient for protection against B16-OVA re-challenge in the skin (110). Moreover, parabiotic transfer of circulating memory to naïve recipient mice conferred reduced tumor protection compared with vaccinated parabiotic donor mice, demonstrating that TRM cells are significant contributors to tumor immunity induced by viral vaccination (110).
This same year, our own work illustrated that TRM cells in the skin are both necessary and sufficient for long-lived protection against B16 melanoma (111). Employing therapeutic depletion of regulatory T cells to break tolerance to melanoma differentiation antigens, followed by surgery to curatively excise residual B16 primary tumors, we identified the formation of tumor/self (gp100) Ag-specific CD8 T cells in the skin with a CD44hi CD62Llow CD103+ CD69+ CLA+ TRM phenotype (Figure 2) (111). These tumor-specific T cells persisted for several months even following extended FTY720 treatment or upon skin grafting onto T cell deficient mice, indicating their true TRM nature. Importantly, generation of this TRM population in the skin depended on CD8 T cell expression of CD103 and Fut7 (the enzymatic determinant of cellular CLA production) (111). As Treg-depleted mice also generated memory T cell responses in lymphoid tissues, a requirement for TRM cells in protection against B16 dermal re-challenge was shown by two methods. First, long-lived tumor immunity was shown to be unperturbed by the continual administration of FTY720. Second, genetic knockout of CD103 in the mouse CD8 T cell compartment, while having no effect on lymphoid memory generation, was shown to completely abrogate skin tumor protection. Thus, CD103-dependent TRM cells in the skin were the sole mediators of long-lived immunity against the dermal B16 melanoma (111).
Figure 2

Skin resident-memory T cell responses to melanoma in the context of autoimmune vitiligo. Tumor-specific TRM precursor populations are recruited from circulation into the skin by chemokine signals. Following entrance into the skin, these T cells kill melanocytes, develop into TRM cells, and produce IFN-γ. Tumor-specific TRM cells populate the dermis, epidermis, and hair follicles, where IL-15 is produced. CD103-dependent TRM cells mediate the durable recall response to melanoma in the dermis.
This study also extended a link between tumor immunity and autoimmunity formed by our earlier work that autoimmunity against normal melanocytes (i.e., vitiligo) maintains lymphoid memory T cell responses against melanoma/melanocyte shared antigens (112, 113). Indeed we found that TRM cells only developed in the skin of mice with treatment-related vitiligo (111). Vitiligo was shown to be required for the seeding of gp100-specific TRM precursors throughout pigmented and depigment skin, but preferentially in melanocyte-depleted hair follicles (111) (Figure 2). The concept of generating TRM responses against tumor/self-antigens was also illustrated in studies involving intradermal DNA vaccination against gp100 (114). This study showed that vaccination induced the development of gp100-specific TRM cells in the skin, in association with autoimmune vitiligo. Importantly TRM cells (but not circulating memory T cells) were refractory to low dose anti-CD8 mAb depletion, a technique used to demonstrate that TRM cells mediate long-lived protection against B16 tumor rechallenge (114). Thus, in the generation of protective TRM responses to tumor antigens that are shared by normal tissues, autoimmunity clearly plays an important role.
These mechanistic studies in mouse tumor models, coupled with data from patient tumor TILs, now clearly affirm the relevance of TRM cells to tumor immunity. Preclinical immunotherapy studies further illustrate that established methods for vaccinating against tumor antigens can be highly effective at generating TRM responses to cancer (93, 110, 114). Reaffirming studies of 15 years ago, vaccination route is crucial for generating the proper TRM responses to tumors in various tissue locations (106). Treg depletion also generates TRM responses against shared tumor/self-antigens, likely relating to the role of Tregs in controlling peripheral tissue autoimmunity (111). While correlative data support the notion that immune checkpoint inhibitor therapies act on phenotypically exhausted TRM-like TILs in solid tumors, such therapies have not yet been shown to induce de novo TRM responses in cancer patients. Regardless, the knowledge that ~20% of anti-PD-1 treated melanoma patients develop vitiligo (115) may imply that melanoma-specific TRM responses are generated or awakened in such patients. Indeed, skin immune-related adverse events of ICI therapy have excellent prognostic value for melanoma patients (116), further underscoring the idea that autoimmunity supports TRM responses to cancer. The next section will discuss a role for TRM cells in mediating autoimmunity; a class of diseases that has long instructed the field of tumor immunology.
Role of TRM cells in autoimmune disease
Skin autoimmunity
Skin autoimmune diseases provide the most compelling evidence of TRM-mediated pathogenic responses against self. In accordance with the above studies in melanoma, three recent studies have demonstrated melanocyte Ag-specific CD8 TRM cells in vitiligo-affected patient skin (non-melanoma associated) (117–119). The first of these showed that vitiligo-associated TRM cells display a CD8+CD103+CD49a+ phenotype, and become localized to both the epidermis and dermis of lesional patient skin (117). These cells are highly functional based on their production of perforin, granzyme B, and IFN-γ upon in vitro restimulation (117). TRM cells in vitiligo-affected skin also expressed the tissue homing receptor CXCR3 (118), consistent with prior reports that vitiligo is mediated by IFN-γ induced ligands for this receptor; CXCL9 and 10 (120). Additional phenotypic analysis of vitiligo-associated TRM cells revealed the expression of CD122, the alpha chain of IL-15R (119); a classic memory T cell marker that supports TRM populations in viral infection models (20, 46). Importantly, mechanistic studies in a T cell receptor transgenic (TCR Tg) CD8 T cell-induced mouse model of vitiligo, revealed that anti-CD122 mAb treatment repigmented vitiligo-affected skin in a highly durable fashion (119). This study was the first to show that factors supporting TRM cell maintenance can serve as targets to impair autoimmunity. Interestingly no differences were observed between TRM responses in patients with active vs. stable disease (118, 119), further underscoring the concept that vitiligo is a disease of immune memory.
Corollary studies of CD8 T cell responses in psoriasis patients show that not all TRM responses are created equal. Psoriasis, like vitiligo, occurs in lesional patches of skin, but is recognized as an IL-17-driven disease. Accordingly CD8+ TRM cells in psoriatic plaque skin preferentially produced IL-17 upon restimulation, exhibit a CD103+ CD49a− CLA+ CCR6+ IL-23R+ phenotype (117, 121), and lack expression of CXCR3 (118). Interestingly, cytokine production was maintained by psoriasis-associated TRM cells even in patients that had undergone long term treatment and had resolved disease (117, 118), again supporting the highly durable nature of TRM responses in autoimmune disease. Contrary to the long-held belief that psoriasis is mediated by γδ T cells, active and clinically resolved psoriasis lesions were shown to contain oligoclonal T cell populations that were overwhelmingly of the αβ T cell lineage, and that produced both IL-17 and IL-22 (122). These studies collectively indicate a role for Tc17-like TRM cells in the pathogenesis of psoriasis.
Although less well-studied, TRM responses have recently been reported in conjunction with scleroderma—a disease of unclear etiology, characterized primarily by fibrosis of the skin. Analysis of skin from patients with early stage scleroderma revealed higher proportions of CD8+CD28− T cells that expressed the TRM marker CD69, and also overwhelmingly expressed CCR10, although largely lacked CD103 (123). Functional analyses indicated a proportion of restimulated cells that were capable of making granzyme B, IFN-γ and IL-13, a pro-fibrotic cytokine (123). This study, together with the above studies in vitiligo and psoriasis patients, highlights the diverse functional programs adapted by TRM cells in the context of aberrant skin pathologies.
Other autoimmune and inflammatory conditions
Several reports have provided evidence of TRM responses in other autoimmune diseases and inflammatory conditions, particularly in diseases of the brain and CNS. With growing evidence that CD8 T cells may play a role in the pathogenesis of multiple sclerosis (124), work in mouse MS models has suggested that Ag-specific CD8 TRM cells in the CNS could contribute to pathogenesis (125). The creation of TCR Tg mice expressing a TCR specific for an MHC-I restricted epitope of an astrocyte-expressed protein resulted in MS-like disease, with the brains of mice becoming infiltrated by CD8 T cells with a CD44+CD69+CD103+ TRM -like phenotype (125). Evidence of MS-associated TRM cells also derives from lesional brain specimens of MS patients. In a small subset of patients with high inflammatory infiltrates, CD103-expressing CD8 T cells were identified, although these cells did not express CD69 (126). In addition to MS, a study of Rasmussen's Encephalitis, a rare pediatric neuroinflammatory disease of unknown etiology, also reported CD8 αβ T cells in seven out of seven RE brain surgery specimens, >50% of which expressed CD103 (127). Mouse studies show that TRM cells may accumulate naturally with age in the CNS, evidenced by the appearance of a CD8 +CD44hi CD62Llow CD69hi PD1+ subset (128). In an ischemic stroke model in aged mice, restimulation of brain CD8 T cells induced the production of TNF-α, IFN-γ, and CCL2 (128). Despite this, the question remains of whether brain CD8 TRM cells are pathogenic. CD8+CD103+ TRM cells from brains of mice with MS-like disease did not produce cytokines (125). In lupus-prone mice, CD8+CD44hiCD62lowCD69+ TRM-like cells accumulated in the brain, but their ablation exacerbated neuropsychiatric lupus, suggesting that these cells might instead serve a regulatory role (129).
As of yet, few studies have convincingly identified TRM cells in other autoimmune disease types. An investigation of lesional biopsies from recent-onset type-1 diabetes (T1D) patients reported CD8+CD69+CD103+ TRM-like cells in diseased islets (130), although a separate study showed a preponderance of CD8 T cells with similar phenotypes in normal, healthy human islets (131). Transcriptomes of T1D patient islets were more skewed to the production of inflammatory cytokines including IFN-γ IL-18, and IL-22 (130), although it is as yet unclear if these pathogenic cytokines are derived from TRM cells. It has been speculated that TRM cells mediate, or contribute to, a host of human autoimmune and inflammatory conditions (132). Advancements in our understanding of TRM biology should guide further investigations of TRM-mediated pathology.
TRM responses against normal self-tissues can be instructive with regards to cancer immunity. First, they reveal that self Ag-specific T cells can be maintained as TRM responses, in the peripheral tissues where carcinomas arise (Figure 1). Such T cells would seem ideally situated to provide surveillance against tumorigenesis, progression, and metastasis, and may naturally do so. Second, they illustrate that TRM cells can adopt a number of stable immune effector states, producing IFN-γ, IL-17, IL-22, IL-13, and/or granzyme B, all of which have separately been shown to oppose tumor growth. In particular, vitiligo and psoriasis-associated TRM cells maintain stable phenotypes and provide durable recall responses, with more apparent functional diversity than was originally recognized based on viral infection models. Our knowledge of mechanisms driving pathogenic self Ag-specific TRM cells, coupled with our understanding of TRM responses against foreign infections, can greatly inform our understanding of TRM responses to cancer.
Optimizing TRM cell responses for cancer immunotherapy
The discovery that standard cancer vaccine and immunotherapy approaches provide long-lived protection through TRM responses—rather than TCM responses, as originally speculated—indicates that promoting TRM responses is a worthwhile goal for the field. The challenge remains in generating TRM responses when (and where) they don't naturally develop. The final section of this review will highlight current knowledge regarding TRM cell behavior that can aid in promoting such responses in the setting of cancer immunotherapy.
Optimal TRM cell precursor populations
The ultimate goal of adoptive T cell therapy is to provide long-lived cancer cures. In patients with metastatic solid tumors, T cells must seek and destroy tumor cells in multiple peripheral tissue locations. TRM cells seem ideal for this job, although successfully delivering these populations presents clear challenges. Because TRM cells are, by definition, resident in peripheral tissue, their in vitro generation for intravenous delivery is a complex proposition. In considering the systemic administration of a TRM response, one must consider the precursor populations that can optimally seed tissues in a receptive host.
TRM precursor seeding
Studies in mouse models largely support the conclusion that timing is crucial for TRM precursor seeding, and that this event must occur early during the course of T cell priming and differentiation. In LCMV infection, early effector cells migrated into intestinal epithelium, seeding a TRM population within 7 days of the initial infection (41). Similarly, in HSV infection, a delay of 2 weeks did not alter TCM formation, but it dramatically reduced TRM cell seeding in the skin, resulting in a loss of protection against viral rechallenge (32). This window of opportunity is affirmed by studies of mucosal chlamydia vaccination, wherein TRM precursors seeded uterine mucosa within 7 days of vaccination (24). Our own studies indicate that tumor-specific TRM precursors are primed early in response to immunotherapy, as they could be isolated from melanoma-draining lymph nodes just 8 days following therapeutic depletion of regulatory T cells (111). These studies support the idea that T cells at an early differentiated state have the greatest propensity to form TRM.
Despite this, some evidence supports the idea that TCM cell plasticity can give rise to TRM. In studies employing VACC-OVA as a dermal vaccine, sorted OVA-specific TCM cells that were re-transferred in vivo, generated skin TRM populations in response to VACC-OVA recall infection (110). On a per-cell basis, TCM cells were shown to be more effective at generating TRM cells compared with naïve T cells (110). Similarly, when administered to tumor-bearing mice, Ag-specific TCM cells were better at accessing tumors and acquiring a TRM phenotype, compared with their naïve counterparts. As TRM and TCM cells represent distinct lineages, it remains unclear how such memory T cell reprogramming occurs. While it has been shown that TRM and TCM cells have a common clonal origin (133), TCM cells do not naturally convert to TRM cells in the post-infection setting (17). Thus, factors relating to secondary infection with VACC-OVA, or tumor growth itself, may alter the plasticity of TCM cells (110). Indeed prior preclinical approaches to melanoma adoptive immunotherapy with in vitro generated gp100-specific TCM, TSCM, and Tc17 subsets (12, 15, 134) may naturally give rise to TRM cells in a tumor-bearing host. This is consistent with the observation that these treatments all induce overt vitiligo (12, 15, 134)—a disease of skin-resident memory.
TRM precursor phenotypes
With regards to phenotype, epithelium-infiltrating TRM precursors in infectious disease models have been shown to express CXCR3 and lack expression of the terminal effector T cell marker KLRG1 (20, 111). Deficiency in CXCR3 reduces the overall number of TRM cells in the skin (20, 135, 136), and mAb-mediated CXCR3 blockade can prevent TRM formation (34). Accordingly, we identified CXCR3+KLRG1− T cells in tumor-draining lymph nodes, that were capable of seeding tumor-specific Trm responses in the skin following i.v. adoptive transfer (Figure 2) (111). CXCR3 is also reported as a mediator of T cell access to solid tumors (137), thus its role in the seeding of TRM precursors further underscores the importance of CXCR3 expression on T cells in an immunotherapy setting.
In our melanoma model, we also observed CD103 expression on TRM precursors in tumor-draining lymph nodes, and found that genetic loss of CD103 impaired early T cell lodgement in the skin (111). This is in contrast to viral models in which CD103 is only expressed upon T cell entry into the epidermis (20), although we also observed a further increase in CD103 expression following T cell entry into the skin. As our TRM cells were primed in response to dermal melanoma growth, early CD103 expression in our model could result from tumor-derived TGF-β entering tumor-draining lymph nodes. Numerous infectious disease studies have shown that CD103 expression is required for the long-lived, antigen-independent maintenance and retention of TRM cells (20, 47, 101, 138, 139). However, this unexpected role for CD103 in promoting skin lodgment indicates that CD103 may also be a useful feature of tumor-specific TRM precursors.
TRM precursor transcriptional programs
In considering transcription factors that program TRM development, whereas Hobit expression is restricted to late events in peripheral tissues (55), Runx3 expression occurs earlier, and appears to program TRM precursor behavior. Identified by an siRNA screen of factors that promote tissue residence in vivo, Runx3 was found to suppress Tbet expression, enforce Itgae expression, and suppress multiple genes associated with tissue egress (54). Runx3 expressing T cells were transcriptionally distinct from TCM and TEM precursor populations as early as 7 days post infection. Of therapeutic importance, Runx3 overexpression in LCMV-specific CD8 T cells promoted T cell access to viral antigen-expressing B16 tumors, enforced the acquisition of a TRM phenotype in tumors, and enhanced T cell anti-tumor activity (54). These studies establish that Runx3 promotes TRM characteristics and anti-tumor efficacy of transferred T cells (54), making it an attractive target for expression in the adoptive immunotherapy setting.
Optimal host tissue microenvironment
Tumor cell dissemination from primary tumors to peripheral tissue locations is predicated on Paget's seed and soil hypothesis of 1898, which states that tumor cells (the seed) can only take up residence in suitable tissues (the soil) (140). This principle can also be applied to TRM cell seeding in peripheral tissues. Indeed among T cells, TRM cells are unique in their propensity for tissue. Providing the proper “soil” for precursor T cell residence will be a critical step toward supporting TRM function, both in tumors and in cancer-prone tissues. Moreover, the properties of TRM cells that enable them to function in diverse peripheral tissue niches, might imbue them with the unique ability to persist and function in solid tumors.
Metabolic factors
Metabolic characteristics of TRM cells that support their function in peripheral tissues may also support their function in tumors. In a VACV skin infection model, fatty acid binding protein 4 and 5 (FABP4/5) were shown to be among the highest expressed genes in TRM cells, enabling the metabolism of exogenous free fatty acids in the skin (51). TRM cells in the tumor microenvironment must compete for nutrients with tumor cells, which use high levels of glucose and glutamine (141). Indeed Tregs have been shown to function well in the TME due to their ability to utilize both glycolysis and fatty acid metabolism (142). Reliance on fatty acid catabolism has recently been shown to be essential for CD8+ TIL function (143). The PPAR agonists fenofibrate, and bezafibrate, which both promote fatty acid oxidation, have each been shown to improve T cell anti-tumor activity (143, 144), although it remains to be seen if these effects are due to improved TRM responses. Regardless, the metabolic requirements of TRM cells may make them ideally suited to persist and function in a metabolically hostile TME.
Chemokines and cytokines
Chemokine cues are crucial for the seeding of TRM precursors, and can be used therapeutically to pull TRM cells into peripheral tissues. Our finding that melanoma-specific CXCR3+ precursors cells only induce TRM responses in vitiligo-affected hosts (111), together with studies showing that the CXCR3/CXCL9/10 axis is crucial for vitiligo development (120, 145), underscores how an autoimmune tissue microenvironment can provide a hospitable niche for tumor-specific TRM cell seeding (Figure 2). CXCL9 and 10 have also been used therapeutically to seed TRM responses based on a “prime and pull” approach (146). In one study, subcutaneous vaccination elicited a systemic T-cell response against HSV, followed by topical CXCL9/10 application to the vaginal mucosa as a means to pull activated T cells into tissue (146). The resulting TRM responses and long-term protection against HSV were comparable to that of mice that that had been immunized intravaginally (146). Similarly, when immunizing against mTB in the lung, parental vaccination followed by intranasal administration of CXCL16, pulled CXCR6 cognate receptor-expressing TRM cells into the lung where they provided long-lived protection (147). CXCR10 has separately been shown to promote TRM formation in the skin (135, 148), suggesting the possibility of a similar approach involving CCL27. These studies show that, even when precursors are generated in a systemic manner, chemokine signals in a specific tissue location can induce functional TRM.
Inflammatory sensitizing agents can also modify peripheral tissue in a way that promotes robust TRM responses. Using the chemical sensitizing agent dinitrofluorobenzene (DNFB), local inflammation in skin recruited effector T cells and converted them to a TRM phenotype (32). Similarly, topical diphenyl cyclopropinone (DPCP) applied to human skin induced contact dermatitis and the subsequent formation of TRM (133). Accordingly, DPCP application to human skin has been shown to induce high levels of cutaneous CXCL9 and 10 (149). Interestingly, DPCP recently received orphan drug approval for the topical treatment of cutaneous melanoma metastases (150), having demonstrating dramatic local efficacy against cutaneous melanoma metastases in one patient that received concurrent immune checkpoint inhibitor therapy (151). It is tempting to speculate that TRM cells participated in this anti-tumor response, which was also accompanied by vitiligo (151).
Once T cells access peripheral tissues, both TGF-β and IL-15 are recognized as fundamentally important TRM survival cues (20, 56). While TGF-β has come to be known for its roles in promoting tumor growth (152), suppressing T cell function (153), and enforcing Treg stability (154), its key role in TRM cell generation suggests its value in certain immunotherapy contexts. On the other hand, IL-15 has long been recognized for its role in supporting anti-tumor T cell responses (155, 156). Studies show that TRM cells preferentially accumulate at sites of high IL-15 production, such as hair follicles in the skin (46, 111). Accordingly, we found that melanoma/melanocyte (gp100)-specific TRM cells cluster in hair follicles of vitiligo-affected skin (111). In conjunction with the finding that gp100-specific T cells express CD122 (IL-15Ra) and require IL-15 for their pathogenic role in melanocyte destruction (119), IL-15 may also enhance melanoma-specific TRM maintenance. Although it is important to note exceptions to the requirement for IL-15, which are surprising considering its canonical role as a homeostatic memory cytokine (157). In fact, certain TRM responses (i.e., in the female reproductive tract) exhibit no dependence on IL-15 for proliferation or survival (157). TRM cells that re-engage antigen in peripheral tissues appear to decrease their reliance on IL-15 (157), indicating that antigen also serves an important role in peripheral tissue.
Antigen and costimulatory requirements
As TRM precursors seed tissue, their differentiation and maintenance has been shown to be shaped by the local engagement of Ag. Indeed following VACV infection, TRM responses were increased ten to 50-fold if their cognate antigen had been engaged in the skin (158, 159). We also observed an important role for peripheral antigen in generating tumor-specific TRM responses (111). Following priming in response to a B16-OVA tumor in the dermis, TRM cells that re-engaged their target antigen on melanocytes in the skin (gp100-specific) were present in far greater numbers than those that could not (OVA-specific) (111). In vaccinating against a tumor-specific neoantigen, TRM responses in peripheral (non-tumor) tissues might best be generated by a proposed “prime and trap” approach (34). This was demonstrated in the context of malaria vaccination, wherein the expression of cognate antigen on hepatocytes served to trap circulating CD8 effector T cells in the liver, where they underwent conversion to TRM cells (34).
While memory T cells are defined by their ability to persist in the absence of Ag, in some settings chronic Ag exposure might also support TRM cell persistence. Following HPV vaccination of patients with cervical intraepithelial neoplasia, CD8+ T cells expressed CD69, and localized to tertiary lymphoid structures in neoplastic cervical tissue, where they expressed Ki67 as evidence of cognate antigen engagement (160). In considering TRM responses against a tumor/self Ag, we showed that functional TRM could be generated against a melanoma antigen (gp100), but only in conjunction with autoimmunity against normal host melanocytes (111). While this may reflect the chemokine/cytokine environment of the autoimmune tissue niche, it may alternately reflect a role for persistent antigen exposure. Interestingly, we identified melanoma-specific TRM cells throughout the skin, although they were preferentially localized to hair follicles containing white hairs, suggesting an absence of local antigen (111). It remains possible that, in the context of autoimmune disease, TRM cells develop into both Ag-dependent and independent subsets.
Although less well-studied, peripheral costimulation may also play a crucial role in programming the TRM response. Recent studies of intranasal flu vaccination showed that boosting with 4-1BBL in a replication defective adenovirus vector generates a robust lung parenchymal CD69+ CD103+/− TRM population (161). Boosting required local (intranasal) 4-1BBL installation, and worked by recruiting additional Ag-specific T cells from circulation into the lungs (161). Lung TRM responses generated in this fashion were highly durable and provided protection for at least 1 year after boost (161). Earlier work also suggests that agonistic OX-40 mAb treatment can promote lung TRM responses, although these studies involved parenteral vaccination (162). Future work is needed to determine how and where to provide the most potent costimulatory signals for optimal TRM formation.
Conclusions
The above findings represent a fundamental advance in our understanding CD8 T cell responses to cancer. While the success of ICI therapy has reaffirmed the long-held belief that CD8 T cells are crucial for tumor immunity, an evolving knowledge of memory T cell function in peripheral tissues has informed our understanding of the type of T cell that may be most needed. Innovations in single cell cytometry and RNA sequencing have rapidly been brought to bear on the characterization of tumor-infiltrating lymphocytes and, together with mechanistic studies in mice, provide compelling evidence that TRM cells are players in the immune response to human cancer.
Several key questions remain. While TRM-like TILs portend improved prognosis for a growing number of cancers, future studies should address how to fully revive such cells in the TME, and how to generate greater numbers of TRM precursors through immunotherapeutic means. A role for TRM cells in tumor immunosurveillance has not yet been established, and it will be interesting to learn if CD8 TRM cells can also limit tumorigenesis. Finally, the field has focused heavily on CD8+ T cells, although a role for CD4 TRM populations is not yet understood. Local TRM helper subsets might be greatly beneficial to tumor immunity, whereas TRM regulatory cells might be particularly detrimental in the TME.
The knowledge that TRM cells can be generated by cancer vaccine and immunotherapy regimens represents a paradigm-shift for a field that has long monitored tumor-specific T cells in the blood. Thus, going forward one must recognize a need to monitor TRM responses in peripheral tissues and tumors of cancer patients receiving immunotherapy. Such peripheral T cell responses might provide the best indication of responsiveness to therapy, and long-term survival. Informed by studies in infectious disease models, and instructed by the involvement of TRM cells in autoimmunity, future research efforts will hopefully overcome the barriers to promoting effective TRM responses to cancer.
Statements
Author contributions
AM and MT both conceptualized, wrote, revised, and approved of the final submitted manuscript.
Funding
AM was supported by NIH F31CA232554-01. MT was supported by NIH R21CA209375-01, R01CA225028, R01CA214062, R01CA205965, P30CA023108-38, and the generous philanthropy of the Knights of the York Cross of Honour.
Acknowledgments
The authors would like to thank Lana M. Ugurtas for her expertise and generous assistance in the design of figure illustrations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
BurnetM. Cancer; a biological approach. I. The processes of control. Br Med J. (1957) 1:779–86. 10.1136/bmj.1.5022.779
2.
DunnGPOldLJSchreiberRD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity (2004) 21:137–48. 10.1016/j.immuni.2004.07.017
3.
DurgeauAVirkYCorgnacSMami-ChouaibF. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. (2018) 9:14. 10.3389/fimmu.2018.00014
4.
WolchokJDSaengerY. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist (2008) 13(Suppl. 4):2–9. 10.1634/theoncologist.13-S4-2
5.
WeiSCDuffyCRAllisonJP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. 10.1158/2159-8290.CD-18-0367
6.
ReadingJLGálvez-CancinoFSwantonCLladserAPeggsKSQuezadaSA. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol Rev. (2018) 283:194–212. 10.1111/imr.12657
7.
UsherwoodEJHoganRJCrowtherGSurmanSLHoggTLAltmanJDet al. Functionally heterogeneous CD8(+) T-cell memory is induced by Sendai virus infection of mice. J Virol. (1999) 73:7278–86.
8.
KlonowskiKDWilliamsKJMarzoALBlairDALingenheldEGLefrançoisL. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20:551–62. 10.1016/S1074-7613(04)00103-7
9.
SallustoFLenigDFörsterRLippMLanzavecchiaA. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. 10.1038/44385
10.
SallustoFGeginatJLanzavecchiaA. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. (2004) 22:745–63. 10.1146/annurev.immunol.22.012703.104702
11.
MasopustDVezysVMarzoALLefrançoisL. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7. 10.1126/science.1058867
12.
KlebanoffCAGattinoniLTorabi-PariziPKerstannKCardonesARFinkelsteinSEet al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci USA. (2005) 102:9571–6. 10.1073/pnas.0503726102
13.
GattinoniLKlebanoffCAPalmerDCWrzesinskiCKerstannKYuZet al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. (2005) 115:1616–26. 10.1172/JCI24480
14.
GattinoniLLugliEJiYPosZPaulosCMQuigleyMFet al. A human memory T cell subset with stem cell-like properties. Nat Med. (2011) 17:1290–7. 10.1038/nm.2446
15.
GattinoniLZhongXSPalmerDCJiYHinrichsCSYuZet al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. (2009) 15:808–13. 10.1038/nm.1982
16.
FarberDLYudaninNARestifoNP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. (2014) 14:24–35. 10.1038/nri3567
17.
JiangXClarkRALiuLWagersAJFuhlbriggeRCKupperTS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483:227–31. 10.1038/nature10851
18.
SteinertEMSchenkelJMFraserKABeuraLKManloveLSIgyártóBZet al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161:737–49. 10.1016/j.cell.2015.03.031
19.
MuruganandahVSathkumaraHDNavarroSKupzA. A systematic review: the role of resident memory T cells in infectious diseases and their relevance for vaccine development. Front Immunol. (2018) 9:1574. 10.3389/fimmu.2018.01574
20.
MackayLKRahimpourAMaJZCollinsNStockATHafonMLet al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. 10.1038/ni.2744
21.
SchenkelJMFraserKAVezysVMasopustD. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. (2013) 14:509–13. 10.1038/ni.2568
22.
WakimLMWaithmanJvan RooijenNHeathWRCarboneFR. Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science (2008) 319:198–202. 10.1126/science.1151869
23.
ZammitDJTurnerDLKlonowskiKDLefrançoisLCauleyLS. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity (2006) 24:439–49. 10.1016/j.immuni.2006.01.015
24.
StaryGOliveARadovic-MorenoAFGondekDAlvarezDBastoPAet al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science (2015) 348:aaa8205. 10.1126/science.aaa8205
25.
HoganRJUsherwoodEJZhongWRobertsAADuttonRWHarmsenAGet al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol. (2001) 166:1813–22. 10.4049/jimmunol.166.3.1813
26.
BuggertMNguyenSSalgado-Montes de OcaGBengschBDarkoSRansierAet al. Identification and characterization of HIV-specific resident memory CD8(+) T cells in human lymphoid tissue. Sci Immunol. (2018) 3:eaar4526. 10.1126/sciimmunol.aar4526
27.
HaddadiSThanthrige-DonNAfkhamiSKheraAJeyanathanMXingZ. Expression and role of VLA-1 in resident memory CD8 T cell responses to respiratory mucosal viral-vectored immunization against tuberculosis. Sci Rep. (2017) 7:9525. 10.1038/s41598-017-09909-4
28.
HofmannMPircherH. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci USA. (2011) 108:16741–6. 10.1073/pnas.1107200108
29.
TrimbleCLClarkRAThoburnCHansonNCTasselloJFrosinaDet al. Human papillomavirus 16-associated cervical intraepithelial neoplasia in humans excludes CD8 T cells from dysplastic epithelium. J Immunol. (2010) 185:7107–14. 10.4049/jimmunol.1002756
30.
ÇuburuNGrahamBSBuckCBKinesRCPangYYDayPMet al. Intravaginal immunization with HPV vectors induces tissue-resident CD8+ T cell responses. J Clin Invest. (2012) 122:4606–20. 10.1172/JCI63287
31.
BuddRCCerottiniJCMacDonaldHR. Phenotypic identification of memory cytolytic T lymphocytes in a subset of Lyt-2+ cells. J Immunol. (1987) 138:1009–13.
32.
MackayLKStockATMaJZJonesCMKentSJMuellerSNet al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA. (2012) 109:7037–42. 10.1073/pnas.1202288109
33.
RichterMVTophamDJ. The alpha1beta1 integrin and TNF receptor II protect airway CD8+ effector T cells from apoptosis during influenza infection. J Immunol. (2007) 179:5054–63. 10.4049/jimmunol.179.8.5054
34.
Fernandez-RuizDNgWYHolzLEMaJZZaidAWongYCet al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity (2016) 45:889–902. 10.1016/j.immuni.2016.08.011
35.
BergsbakenTBevanMJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. (2015) 16:406–14. 10.1038/ni.3108
36.
SheridanBSLefrancoisL. Regional and mucosal memory T cells. Nat Immunol. (2011) 12:485–91. 10.1038/ni.2029
37.
ClarkRAChongBMirchandaniNBrinsterNKYamanakaKDowgiertRKet al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. (2006) 176:4431–9. 10.4049/jimmunol.176.7.4431
38.
McNamaraHACaiYWagleMVSontaniYRootsCMMiosgeLAet al. Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol. (2017) 2:eaaj1996. 10.1126/sciimmunol.aaj1996
39.
MackayLKBraunAMacleodBLCollinsNTebartzCBedouiSet al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. (2015) 194:2059–63. 10.4049/jimmunol.1402256
40.
BankovichAJShiowLRCysterJG. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem. (2010) 285:22328–37. 10.1074/jbc.M110.123299
41.
MasopustDChooDVezysVWherryEJDuraiswamyJAkondyRet al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. 10.1084/jem.20090858
42.
KumarBVMaWMironMGranotTGuyerRSCarpenterDJet al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. 10.1016/j.celrep.2017.08.078
43.
AriottiSBeltmanJBChodaczekGHoekstraMEvan BeekAEGomez-EerlandRet al. Tissue-resident memory CD8+ T cells continuously patrol skin epithelia to quickly recognize local antigen. Proc Natl Acad Sci USA. (2012) 109:19739–44. 10.1073/pnas.1208927109
44.
ZaidAMackayLKRahimpourABraunAVeldhoenMCarboneFRet al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA. (2014) 111:5307–12. 10.1073/pnas.1322292111
45.
MackayLKWakimLvan VlietCJJonesCMMuellerSNBannardOet al. Maintenance of T cell function in the face of chronic antigen stimulation and repeated reactivation for a latent virus infection. J Immunol. (2012) 188:2173–8. 10.4049/jimmunol.1102719
46.
AdachiTKobayashiTSugiharaEYamadaTIkutaKPittalugaSet al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med. (2015) 21:1272–9. 10.1038/nm.3962
47.
MohammedJBeuraLKBobrAAstryBChicoineBKashemSWet al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol. (2016) 17:414–21. 10.1038/ni.3396
48.
BergsbakenTBevanMJFinkPJ. Local inflammatory cues regulate differentiation and persistence of CD8(+) tissue-resident memory T cells. Cell Rep. (2017) 19:114–24. 10.1016/j.celrep.2017.03.031
49.
TakamuraSYagiHHakataYMotozonoCMcMasterSRMasumotoTet al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. (2016) 213:3057–73. 10.1084/jem.20160938
50.
TakamuraS. Niches for the long-term maintenance of tissue-resident memory T cells. Front Immunol. (2018) 9:1214. 10.3389/fimmu.2018.01214
51.
PanYTianTParkCOLofftusSYMeiSLiuXet al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543:252–6. 10.1038/nature21379
52.
HanSJGlatman ZaretskyAAndrade-OliveiraVCollinsNDzutsevAShaikJet al. White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity (2017) 47:1154–68.e6. 10.1016/j.immuni.2017.11.009
53.
PanYKupperTS. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol. (2018) 9:1347. 10.3389/fimmu.2018.01347
54.
MilnerJJTomaCYuBZhangKOmilusikKPhanATet al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552:253–7. 10.1038/nature24993
55.
MackayLKMinnichMKragtenNALiaoYNotaBSeilletCet al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352:459–63. 10.1126/science.aad2035
56.
MackayLKWynne-JonesEFreestoneDPellicciDGMielkeLANewmanDMet al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43:1101–11. 10.1016/j.immuni.2015.11.008
57.
KragtenNAMBehrFMVieira BragaFARemmerswaalEBMWesselinkTHOjaAEet al. Blimp-1 induces and Hobit maintains the cytotoxic mediator granzyme B in CD8 T cells. Eur J Immunol. (2018) 48:1644–6210.1002/eji.201847771
58.
BoddupalliCSNairSGraySMNowyhedHNVermaRGibsonJAet al. ABC transporters and NR4A1 identify a quiescent subset of tissue-resident memory T cells. J Clin Invest. (2016) 126:3905–16. 10.1172/JCI85329
59.
HombrinkPHelbigCBackerRAPietBOjaAEStarkRet al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol. (2016) 17:1467–78. 10.1038/ni.3589
60.
GebhardtTPalendiraUTscharkeDCBedouiS. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. (2018) 283:54–76. 10.1111/imr.12650
61.
MandalaSHajduRBergstromJQuackenbushEXieJMilliganJet al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science (2002) 296:346–9. 10.1126/science.1070238
62.
WuTHuYLeeYTBouchardKRBenechetAKhannaKet al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. (2014) 95:215–24. 10.1189/jlb.0313180
63.
JarryACerf-BensussanNBrousseNGuy-GrandDMuzeauFPotetF. Same peculiar subset of HML1 + lymphocytes present within normal intestinal epithelium is associated with tumoral epithelium of gastrointestinal carcinomas. Gut (1988) 29:1632–8. 10.1136/gut.29.12.1632
64.
AdemmerKEbertMMüller-OstermeyerFFriessHBüchlerMWSchubertWet al. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. (1998) 112:21–6. 10.1046/j.1365-2249.1998.00546.x
65.
QuinnEHawkinsNYipYLSuterCWardR. CD103+ intraepithelial lymphocytes–a unique population in microsatellite unstable sporadic colorectal cancer. Eur J Cancer (2003) 39:469–75. 10.1016/S0959-8049(02)00633-0
66.
CresswellJRobertsonHNealDEGriffithsTRKirbyJA. Distribution of lymphocytes of the alpha(E)beta(7) phenotype and E-cadherin in normal human urothelium and bladder carcinomas. Clin Exp Immunol. (2001) 126:397–402. 10.1046/j.1365-2249.2001.01652.x
67.
WebbJRWickDANielsenJSTranEMilneKMcMurtrieEet al. Profound elevation of CD8+ T cells expressing the intraepithelial lymphocyte marker CD103 (alphaE/beta7 Integrin) in high-grade serous ovarian cancer. Gynecol Oncol. (2010) 118:228–36. 10.1016/j.ygyno.2010.05.016
68.
AustrupFRebstockSKilshawPJHamannA. Transforming growth factor-beta 1-induced expression of the mucosa-related integrin alpha E on lymphocytes is not associated with mucosa-specific homing. Eur J Immunol. (1995) 25:1487–91. 10.1002/eji.1830250602
69.
RihsSWalkerCVirchowJCBoerCKroegelCGiriSNet al. Differential expression of alpha E beta 7 integrins on bronchoalveolar lavage T lymphocyte subsets: regulation by alpha 4 beta 1-integrin crosslinking and TGF-beta. Am J Respir Cell Mol Biol. (1996) 15:600–10. 10.1165/ajrcmb.15.5.8918367
70.
AgaceWWHigginsJMSadasivanBBrennerMBParkerCM. T-lymphocyte-epithelial-cell interactions: integrin alpha(E)(CD103)beta(7), LEEP-CAM and chemokines. Curr Opin Cell Biol. (2000) 12:563–8. 10.1016/S0955-0674(00)00132-0
71.
FrenchJJCresswellJWongWKSeymourKCharnleyRMKirbyJA. T cell adhesion and cytolysis of pancreatic cancer cells: a role for E-cadherin in immunotherapy?Br J Cancer (2002) 87:1034–41. 10.1038/sj.bjc.6600597
72.
LingKLDulphyNBahlPSalioMMaskellKPirisJet al. Modulation of CD103 expression on human colon carcinoma-specific CTL. J Immunol. (2007) 178:2908–15. 10.4049/jimmunol.178.5.2908
73.
FranciszkiewiczKLeFloc'h AJalilAVigantFRobertTVergnonIet al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. (2009) 69:6249–55. 10.1158/0008-5472.CAN-08-3571
74.
MokraniMKlibiJBluteauDBismuthGMami-ChouaibF. Smad and NFAT pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J Immunol. (2014) 192:2471–9. 10.4049/jimmunol.1302192
75.
KomdeurFLWoutersMCWorkelHHTijansAMTerwindtALBrunekreeftKLet al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRalphabeta+ CD8alphabeta+ T cells that can be targeted for cancer immunotherapy. Oncotarget (2016) 7:75130–44. 10.18632/oncotarget.12077
76.
BoutetMGauthierLLeclercMGrosGde MontprevilleVThéretNet al. TGFbeta signaling intersects with CD103 integrin signaling to promote T-lymphocyte accumulation and antitumor activity in the lung tumor microenvironment. Cancer Res. (2016) 76:1757–69. 10.1158/0008-5472.CAN-15-1545
77.
Mami-ChouaibFBlancCCorgnacSHansSMalenicaIGranierCet al. Resident memory T cells, critical components in tumor immunology. J Immunother Cancer (2018) 6:87. 10.1186/s40425-018-0399-6
78.
LeFloc'h AJalilAVergnonILeMaux Chansac BLazarVBismuthGet al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. (2007) 204:559–70. 10.1084/jem.20061524
79.
FranciszkiewiczKLeFloc'h ABoutetMVergnonISchmittAMami-ChouaibF. CD103 or LFA-1 engagement at the immune synapse between cytotoxic T cells and tumor cells promotes maturation and regulates T-cell effector functions. Cancer Res. (2013) 73:617–28. 10.1158/0008-5472.CAN-12-2569
80.
LeFloc'h AJalilAFranciszkiewiczKValidirePVergnonIMami-ChouaibF. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cgamma-dependent pathway. Cancer Res. (2011) 71:328–38. 10.1158/0008-5472.CAN-10-2457
81.
GauthierLCorgnacSBoutetMGrosGValidirePBismuthGet al. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8(+) resident memory T cells in tumors. Cancer Res. (2017) 77:7072–82. 10.1158/0008-5472.CAN-17-1487
82.
HigginsJMMandlebrotDAShawSKRussellGJMurphyEAChenYTet al. Direct and regulated interaction of integrin alphaEbeta7 with E-cadherin. J Cell Biol. (1998) 140:197–210. 10.1083/jcb.140.1.197
83.
WebbJRMilneKWatsonPDeleeuwRJNelsonBH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. 10.1158/1078-0432.CCR-13-1877
84.
KohJKimSKimMYGoHJeonYKChungDH. Prognostic implications of intratumoral CD103+ tumor-infiltrating lymphocytes in pulmonary squamous cell carcinoma. Oncotarget (2017) 8:13762–9. 10.18632/oncotarget.14632
85.
LohneisPSinnMBischoffSJühlingAPelzerUWislockaLet al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer (2017) 83:290–301. 10.1016/j.ejca.2017.06.016
86.
WangBWuSZengHLiuZDongWHeWet al. CD103+ tumor infiltrating lymphocytes predict a favorable prognosis in urothelial cell carcinoma of the bladder. J Urol. (2015) 194:556–62. 10.1016/j.juro.2015.02.2941
87.
JenkinsonSEWhawellSASwalesBMCorpsEMKilshawPJFarthingPM. The α E(CD103)β 7 integrin interacts with oral and skin keratinocytes in an E-cadherin-independent manner. Immunology (2011) 132:188–96. 10.1111/j.1365-2567.2010.03352.x
88.
BrownDWFurnessJSpeightPMThomasGJLiJThornhillMHet al. Mechanisms of binding of cutaneous lymphocyte-associated antigen-positive and alphaebeta7-positive lymphocytes to oral and skin keratinocytes. Immunology (1999) 98:9–15. 10.1046/j.1365-2567.1999.00855.x
89.
WangZQMilneKDerocherHWebbJRNelsonBHWatsonPH. CD103 and intratumoral immune response in breast cancer. Clin Cancer Res. (2016) 22:6290–7. 10.1158/1078-0432.CCR-16-0732
90.
WorkelHHKomdeurFLWoutersMCPlatAKlipHGEgginkFAet al. CD103 defines intraepithelial CD8+ PD1+ tumour-infiltrating lymphocytes of prognostic significance in endometrial adenocarcinoma. Eur J Cancer (2016) 60:1–11. 10.1016/j.ejca.2016.02.026
91.
KomdeurFLPrinsTMvan de WallSPlatAWismanGBAHollemaHet al. CD103+ tumor-infiltrating lymphocytes are tumor-reactive intraepithelial CD8+ T cells associated with prognostic benefit and therapy response in cervical cancer. Oncoimmunology (2017) 6:e1338230. 10.1080/2162402X.2017.1338230
92.
DjenidiFAdamJGoubarADurgeauAMeuriceGde MontprévilleVet al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. (2015) 194:3475–86. 10.4049/jimmunol.1402711
93.
NizardMRousselHDinizMOKarakiSTranTVoronTet al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. (2017) 8:15221. 10.1038/ncomms15221
94.
VasquezJCHuttnerAZhangLMarksAChanABaehringJMet al. SOX2 immunity and tissue resident memory in children and young adults with glioma. J Neurooncol. (2017) 134:41–53. 10.1007/s11060-017-2515-8
95.
BoddupalliCSBarNKadaveruKKrauthammerMPornputtapongNMaiZet al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight (2016) 1:e88955. 10.1172/jci.insight.88955
96.
WakimLMWoodward-DavisALiuRHuYVilladangosJSmythGet al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. (2012) 189:3462–71. 10.4049/jimmunol.1201305
97.
PrasadSHuSShengWSChauhanPSinghALokensgardJR. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J Neuroinflammation (2017) 14:82. 10.1186/s12974-017-0860-3
98.
Shwetank AbdelsamedHAFrostELSchmitzHMMockusTEYoungbloodBAet al. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol Cell Biol. (2017) 95:953–9. 10.1038/icb.2017.62
99.
WakimLMWoodward-DavisABevanMJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. (2010) 107:17872–9. 10.1073/pnas.1010201107
100.
ParkSLZaidAHorJLChristoSNPrierJEDaviesBet al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. (2018) 19:183–91. 10.1038/s41590-017-0027-5
101.
CaseyKAFraserKASchenkelJMMoranAAbtMCBeuraLKet al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. (2012) 188:4866–75. 10.4049/jimmunol.1200402
102.
GanesanAPClarkeJWoodOGarrido-MartinEMCheeSJMellowsTet al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. (2017) 18:940–50. 10.1038/ni.3775
103.
DuhenTDuhenRMontlerRMosesJMoudgilTde MirandaNFet al. Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun. (2018) 9:2724. 10.1038/s41467-018-05072-0
104.
SavasPVirassamyBYeCSalimAMintoffCPCaramiaFet al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. (2018) 24:986–93. 10.1038/s41591-018-0078-7
105.
GuoXZhangYZhengLZhengCSongJZhangQet al. Publisher Correction: global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. (2018) 24:1628. 10.1038/s41591-018-0167-7
106.
MullinsDWSheasleySLReamRMBullockTNFuYXEngelhardVH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med. (2003) 198:1023–34. 10.1084/jem.20021348
107.
LiuLZhongQTianTDubinKAthaleSKKupperTS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med. (2010) 16:224–7. 10.1038/nm.2078
108.
SandovalFTermeMNizardMBadoualCBureauMFFreyburgerLet al. Mucosal imprinting of vaccine-induced CD8(+) T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. (2013) 5:172ra20. 10.1126/scitranslmed.3004888
109.
MurrayTFuertes MarracoSABaumgaertnerPBordryNCagnonLDondaAet al. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. (2016) 7:573. 10.3389/fimmu.2016.00573
110.
EnamoradoMIborraSPriegoECuetoFJQuintanaJAMartínez-CanoSet al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. (2017) 8:16073. 10.1038/ncomms16073
111.
MalikBTByrneKTVellaJLZhangPShabanehTBSteinbergSMet al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol. (2017) 2:eaam6346. 10.1126/sciimmunol.aam6346
112.
ByrneKTCôtéALZhangPSteinbergSMGuoYAllieRet al. Autoimmune melanocyte destruction is required for robust CD8+ memory T cell responses to mouse melanoma. J Clin Invest. (2011) 121:1797–809. 10.1172/JCI44849
113.
ByrneKTZhangPSteinbergSMTurkMJ. Autoimmune vitiligo does not require the ongoing priming of naive CD8 T cells for disease progression or associated protection against melanoma. J Immunol. (2014) 192:1433–9. 10.4049/jimmunol.1302139
114.
Gálvez-CancinoFLópezEMenaresEDíazXFloresCCáceresPet al. Vaccination-induced skin-resident memory CD8(+) T cells mediate strong protection against cutaneous melanoma. Oncoimmunology (2018) 7:e1442163. 10.1080/2162402X.2018.1442163
115.
HuaCBoussemartLMateusCRoutierEBoutrosCCazenaveHet al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. (2016) 152:45–51. 10.1001/jamadermatol.2015.2707
116.
Freeman-KellerMKimYCroninHRichardsAGibneyGWeberJS. Nivolumab in resected and unresectable metastatic melanoma: characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res. (2016) 22:886–94. 10.1158/1078-0432.CCR-15-1136
117.
CheukSSchlumsHGallaisSérézal IMartiniEChiangSCMarquardtNet al. CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity (2017) 46:287–300. 10.1016/j.immuni.2017.01.009
118.
BonifaceKJacqueminCDarrigadeASDessartheBMartinsCBoukhedouniNet al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol. (2018) 138:355–64. 10.1016/j.jid.2017.08.038
119.
RichmondJMStrassnerJPZapataLGargMRidingRLRefatMAet al. Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. (2018) 10:eaam7710. 10.1126/scitranslmed.aam7710
120.
RashighiMAgarwalPRichmondJMHarrisTHDresserKSuMWet al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. (2014) 6:223ra23. 10.1126/scitranslmed.3007811
121.
CheukSWikénMBlomqvistLNylénSTalmeTStåhleMet al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. (2014) 192:3111–20. 10.4049/jimmunol.1302313
122.
MatosTRO'MalleyJTLowryELHammDKirschIRRobinsHSet al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. (2017) 127:4031–41. 10.1172/JCI93396
123.
LiGLarreginaATDomsicRTStolzDBMedsgerTALafyatisRet al. Skin-resident effector memory CD8(+)CD28(-) T cells exhibit a profibrotic phenotype in patients with systemic sclerosis. J Invest Dermatol. (2017) 137:1042–50. 10.1016/j.jid.2016.11.037
124.
HusebyESHusebyPGShahSSmithRStadinskiBD. Pathogenic CD8 T cells in multiple sclerosis and its experimental models. Front Immunol. (2012) 3:64. 10.3389/fimmu.2012.00064
125.
SasakiKBeanAShahSSchuttenEHusebyPGPetersBet al. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol. (2014) 192:3029–42. 10.4049/jimmunol.1302911
126.
Machado-SantosJSajiETröscherARPaunovicMLiblauRGabrielyGet al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain (2018) 141:2066–82. 10.1093/brain/awy151
127.
OwensGCChangJWHuynhMNChirwaTVintersHVMathernGW. Evidence for resident memory T cells in rasmussen encephalitis. Front Immunol. (2016) 7:64. 10.3389/fimmu.2016.00064
128.
RitzelRMCrapserJPatelARVermaRGrenierJMChauhanAet al. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol. (2016) 196:3318–30. 10.4049/jimmunol.1502021
129.
MorawskiPAQiCFBollandS. Non-pathogenic tissue-resident CD8(+) T cells uniquely accumulate in the brains of lupus-prone mice. Sci Rep. (2017) 7:40838. 10.1038/srep40838
130.
KuricESeironPKrogvoldLEdwinBBuanesTHanssenKFet al. Demonstration of tissue resident memory CD8 T cells in insulitic lesions in adult patients with recent-onset type 1 diabetes. Am J Pathol. (2017) 187:581–8. 10.1016/j.ajpath.2016.11.002
131.
RadenkovicMUvebrantKSkogOSarmientoLAvartssonJStormPet al. Characterization of resident lymphocytes in human pancreatic islets. Clin Exp Immunol. (2017) 187:418–27. 10.1111/cei.12892
132.
ParkCOKupperTS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. 10.1038/nm.3883
133.
GaideOEmersonROJiangXGulatiNNizzaSDesmaraisCet al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med. (2015) 21:647–53. 10.1038/nm.3860
134.
BowersJSNelsonMHKundimiSBaileySRHuffLWSchwartzKMet al. Dendritic cells in irradiated mice trigger the functional plasticity and antitumor activity of adoptively transferred Tc17 cells via IL12 signaling. Clin Cancer Res. (2015) 21:2546–57. 10.1158/1078-0432.CCR-14-2294
135.
ZaidAHorJLChristoSNGroomJRHeathWRMackayLKet al. Chemokine receptor-dependent control of skin tissue-resident memory T cell formation. J Immunol. (2017) 199:2451–9. 10.4049/jimmunol.1700571
136.
SrivastavaRKhanAAChilukuriSSyedSATranTTFurnessJet al. CXCL10/CXCR3-dependent mobilization of herpes simplex virus-specific CD8(+) TEM and CD8(+) TRM cells within infected tissues allows efficient protection against recurrent herpesvirus infection and disease. J Virol. (2017) 91:e00278–17. 10.1128/JVI.00278-17
137.
MullinsIMSlingluffCLLeeJKGarbeeCFShuJAndersonSGet al. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. (2004) 64:7697–701. 10.1158/0008-5472.CAN-04-2059
138.
ZhangNBevanMJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. 10.1016/j.immuni.2013.08.019
139.
WuTCXuKBanchereauRMarchesFYuCIMartinekJet al. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol Res. (2014) 2:487–500. 10.1158/2326-6066.CIR-13-0217
140.
PagetS. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. (1989) 8:98–101.
141.
AnastasiouD. Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer (2017) 116:277–86. 10.1038/bjc.2016.412
142.
PacellaIProcacciniCFocaccettiCMiacciSTimperiEFaicchiaDet al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA. (2018) 115:E6546–55. 10.1073/pnas.1720113115
143.
ZhangYKurupatiRLiuLZhouXYZhangGHudaihedAet al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32:377–91 e9. 10.1016/j.ccell.2017.08.004
144.
ChowdhuryPSChamotoKKumarAHonjoT. PPAR-induced fatty acid oxidation in T cells increases the number of tumor-reactive CD8+ T cells and facilitates anti-PD-1 therapy. Cancer Immunol Res. (2018) 6:1375–87. 10.1158/2326-6066.CIR-18-0095
145.
RichmondJMMasterjohnEChuRTedstoneJYoudMEHarrisJE. CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol. (2017) 137:982–5. 10.1016/j.jid.2016.10.048
146.
ShinHIwasakiA. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491:463–7. 10.1038/nature11522
147.
LeeLNRonanEOde LaraCFrankenKLOttenhoffTHTchilianEZet al. CXCR6 is a marker for protective antigen-specific cells in the lungs after intranasal immunization against Mycobacterium tuberculosis. Infect Immun. (2011) 79:3328–37. 10.1128/IAI.01133-10
148.
XiaMHuSFuYJinWYiQMatsuiYet al. CCR10 regulates balanced maintenance and function of resident regulatory and effector T cells to promote immune homeostasis in the skin. J Allergy Clin Immunol. (2014) 134:634–44 e10. 10.1016/j.jaci.2014.03.010
149.
GulatiNSuárez-FariñasMFuentes-DuculanJGilleaudeauPSullivan-WhalenMCorrea da RosaJet al. Molecular characterization of human skin response to diphencyprone at peak and resolution phases: therapeutic insights. J Invest Dermatol. (2014) 134:2531–40. 10.1038/jid.2014.196
150.
DamianDLSawRPThompsonJF. Topical immunotherapy with diphencyprone for in transit and cutaneously metastatic melanoma. J Surg Oncol. (2014) 109:308–13. 10.1002/jso.23506
151.
GulatiNCarvajalRDPostowMAWolchokJDKruegerJG. Definite regression of cutaneous melanoma metastases upon addition of topical contact sensitizer diphencyprone to immune checkpoint inhibitor treatment. Exp Dermatol. (2016) 25:553–4. 10.1111/exd.13030
152.
SeoaneJGomisRR. TGF-beta family signaling in tumor suppression and cancer progression. Cold Spring Harb Perspect Biol. (2017) 9:a022277. 10.1101/cshperspect.a022277
153.
LiuMLiSLiMO. TGF-beta control of adaptive immune tolerance: a break from treg cells. Bioessays (2018) 40:e1800063. 10.1002/bies.201800063
154.
WanYYFlavellRA. 'Yin-Yang' functions of transforming growth factor-beta and T regulatory cells in immune regulation. Immunol Rev. (2007) 220:199–213. 10.1111/j.1600-065X.2007.00565.x
155.
KlebanoffCAGattinoniLPalmerDCMuranskiPJiYHinrichsCSet al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res. (2011) 17:5343–52. 10.1158/1078-0432.CCR-11-0503
156.
NgSSMNagyBAJensenSMHuXAliceaCFoxBAet al. Heterodimeric IL15 treatment enhances tumor infiltration, persistence, and effector functions of adoptively transferred tumor-specific t cellsin the absence of lymphodepletion. Clin Cancer Res. (2017) 23:2817–30. 10.1158/1078-0432.CCR-16-1808
157.
SchenkelJMFraserKACaseyKABeuraLKPaukenKEVezysVet al. IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J Immunol. (2016) 196:3920–6. 10.4049/jimmunol.1502337
158.
KhanTNMoosterJLKilgoreAMOsbornJFNolzJC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med. (2016) 213:951–66. 10.1084/jem.20151855
159.
MuschaweckhABuchholzVRFellenzerAHesselCKönigPATaoSet al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med. (2016) 213:3075–86. 10.1084/jem.20160888
160.
MaldonadoLTeagueJEMorrowMPJotovaIWuTCWangCet al. Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med. (2014) 6:221ra13. 10.1126/scitranslmed.3007323
161.
ZhouACWagarLEWortzmanMEWattsTH. Intrinsic 4-1BB signals are indispensable for the establishment of an influenza-specific tissue-resident memory CD8 T-cell population in the lung. Mucosal Immunol. (2017) 10:1294–309. 10.1038/mi.2016.124
162.
Salek-ArdakaniSMoutaftsiMSetteACroftM. Targeting OX40 promotes lung-resident memory CD8 T cell populations that protect against respiratory poxvirus infection. J Virol. (2011) 85:9051–9. 10.1128/JVI.00619-11
Summary
Keywords
TRM, TIL, CD103, TCM, melanoma, vitiligo, immunotherapy, biomarker
Citation
Molodtsov A and Turk MJ (2018) Tissue Resident CD8 Memory T Cell Responses in Cancer and Autoimmunity. Front. Immunol. 9:2810. doi: 10.3389/fimmu.2018.02810
Received
17 September 2018
Accepted
14 November 2018
Published
29 November 2018
Volume
9 - 2018
Edited by
Weiguo Cui, Bloodcenter of Wisconsin, United States
Reviewed by
Kim Klonowski, University of Georgia, United States; Tania H. Watts, University of Toronto, Canada
Updates
Copyright
© 2018 Molodtsov and Turk.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mary Jo Turk mary.jo.turk@dartmouth.edu
This article was submitted to Immunological Memory, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.