 Karin Steinbach
Karin Steinbach Ilena Vincenti
Ilena Vincenti Doron Merkler
Doron Merkler- 1Department of Pathology and Immunology, University of Geneva, Geneva, Switzerland
- 2Division of Clinical Pathology, Geneva University Hospital, Geneva, Switzerland
Tissue-resident-memory CD8+ T cells (TRM) have been described as a non-circulating memory T cell subset that persists at sites of previous infection. While TRM in all non-lymphoid organs probably share a core signature differentiation pathway, certain aspects of their maintenance and effector functions may vary. It is well-established that TRM provide long-lived protective immunity through immediate effector function and accelerated recruitment of circulating immune cells. Besides immune defense against pathogens, other immunological roles of TRM are less well-studied. Likewise, evidence of a putative detrimental role of TRM for inflammatory diseases is only beginning to emerge. In this review, we discuss the protective and harmful role of TRM in organ-specific immunity and immunopathology as well as prospective implications for immunomodulatory therapy.
Introduction
During an infection, our immune defense operates in a sensitive balance in which the eradication of an invading pathogen should take place efficiently with the least possible damage to the body's own structures. For this, different subsets of immune cells have evolved, which form several lines of defense and are equipped with different functional specializations. Various leukocyte subsets—from broadly acting innate immune cells to antigen-specific and specialized lymphocytes—act together to constitute a joint defense reaction against infectious intruders. CD8+ (so-called cytotoxic) T lymphocytes are essential executors of the adaptive immune system and are particularly specialized in eliminating aberrant cells that are either infected with an intracellular pathogen or of tumorous nature. Regional and functional specialization can also be observed among CD8+ T cells, especially among memory T cells that provide long-term protection against reinfection with a previously encountered pathogen (1). While central memory (CM) T cells home to secondary lymphoid organs (SLO) where they provide a stem cell-like pool of highly-proliferative antigen-specific memory T cells, effector memory (EM) T cells lack homing receptors for SLOs and patrol the body, charged with effector molecules (2, 3). In the last decade, a third memory T cell subset, referred to as resident-memory (RM) T cells, has emerged as an important guardian providing potent local immune surveillance at sites of previous infection, especially at barrier sites in the body (4, 5). TRM procure superior protective immune memory in comparison to circulating memory T cells (6, 7) and presence of TRM in tumors is associated with enhanced tumor control and survival (8). The generation and maintenance of this non-circulating, “sessile” immune subset is therefore the focus of intensive research efforts, for example with the aim of developing more potent vaccines (9). Conversely, more and more reports start to emerge linking the presence of TRM with chronic inflammation and autoimmune diseases (10). Consequently, we need to deepen our understanding of TRM biology in order to consider protective and possible harmful functions of TRM into our strategies for new therapeutic approaches. There is currently a tendency to generalize the observed TRM functions across different organs, although some reports suggest that besides sharing a common differentiation program, TRM generation seems to be influenced by multiple factors and also adapt to the environment of their tissue of residence. In this review, we will focus on the presumed role of TRM in protective immunity, chronic inflammation and organ-specific autoimmune diseases. In particular, we will place special emphasis on CD8+ TRM, as they are the best studied TRM population so far. However, other resident lymphocyte populations have also been described. The latter include resident CD4+ memory T cells (11) and several resident invariant lymphocyte populations, such as liver NKT cells, gut-associated intraepithelial lymphocytes [including CD8αα T cells and (mucosal-associated invariant T) MAIT cells], and skin- and gut-resident memory γδ T cells (12–15). Moreover, resident innate lymphocyte (ILC) populations have been reported (16). Although we do not discuss these populations further in this review, some of our considerations might also apply to these cell subsets.
TRM Generation and Maintenance
The principal hallmark of bona fide TRM is their long-term persistence in non-lymphoid tissues (NLT) as a stable memory T cell pool independent of input from circulating T cells. TRM are often identified by a combination of surrogate markers (see Table 1), the most commonly used being CD69 and CD103, which are associated to their persisting and resident phenotype. Phenotypically, TRM resemble a mixture of TCM cells and effector T cells expressing markers associated with homeostatic proliferation and survival, such as Ki-67 and Bcl-2, and effector function, such as Granzyme B and co-inhibitory molecules (6, 32). Table 1 summarizes frequently used TRM markers in mice and humans. However, a mere phenotypical identification without functional analysis might include circulating T cell subsets that can transiently express e.g., CD69 and CD103 (33, 34). In order to unequivocally identify TRM, besides phenotypical analysis, functional experiments assessing TRM tissue egress, persistence, and their disequilibrium with peripheral TCM and TEM cells are usually performed (6, 35). TRM demonstrate a strong disequilibrium (>90%) in parabionts (36, 37) and remain stable in numbers even when recruitment of circulating T cells to NLT is inhibited (20, 27). In most NLT, with exception of the liver (25), TRM are anatomically separated from the blood and therefore not accessible to intravenously applied antibodies (32, 38, 39).
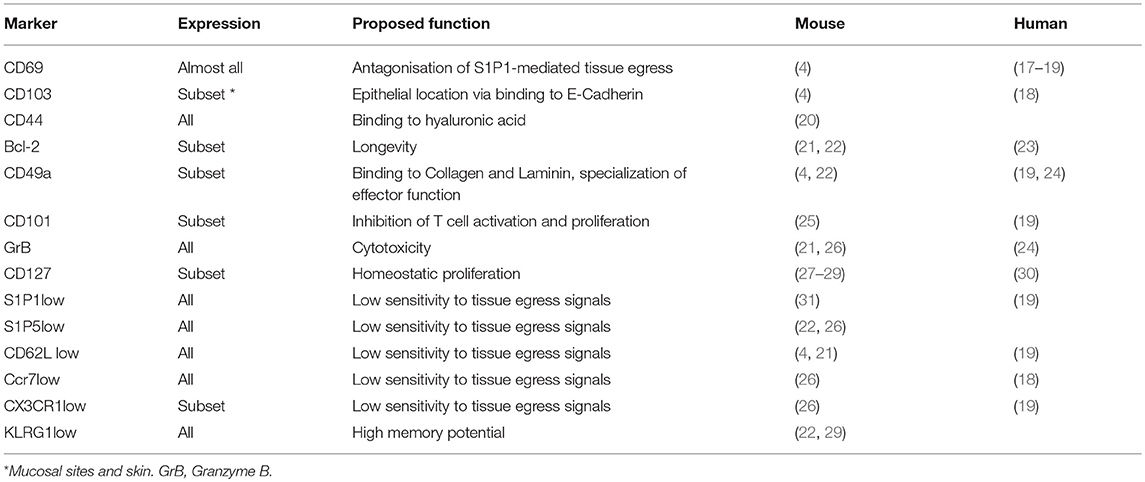
Table 1. Frequently used TRM markers in mice and humans.
In humans, TRM and TRM-like cells are mostly identified in a descriptive manner based on the homology with mouse TRM (17, 40) and by differential gene expression when compared to circulating memory T cell subsets (19, 24, 41). While functional analyses in humans are obviously more limited, studies in patients treated with immune-ablative regimens (42, 43), or transplantations of human tissue (44) indicate that human TRM -like populations identified on this basis likely constitute a similarly stable persisting T cell pool. TRM-like populations in human NLT vastly outnumber T cells in circulation (17, 30, 45, 46), something that cannot be found in mice housed in a specific-pathogen free (SPF) environment, but in pet shop mice (47). Human TRM will probably remain challenging to study, due to limited access to these cells and the lack of an in vitro culture system to this point. However, since not all aspects of human TRM biology can be reproduced in SPF mice, a combined approach of mouse and human research will be instrumental to extend our knowledge about the role of TRM in human health and disease.
TRM Differentiation and Maintenance Program
TRM mostly arise from CD127(IL7Rα)+KLRG1- memory precursor cells (22, 48, 49). Their differentiation into a long-term stably persisting and non-circulating cell population is based on two main requirements: the inhibition of tissue egress (residency) as well as longevity and/or homeostatic proliferation (maintenance). Once T cells have been recruited to the site of infection, TRM precursor cells probably receive local signals from their future tissue of residence that guide the timely activation and inhibition of specific transcriptional programs. The most common mechanism is the upregulation of CD69, which antagonizes sphingosin-1-phosphate-receptor-1 (S1P1)-mediated tissue egress, and thereby confers early tissue retention until TRM differentiation is complete (50–52). Most TRM express CD69 constitutively and in the absence of CD69, TRM generation in organs is strongly impaired (22). However, CD69 might be dispensable for long-term maintenance of fully-differentiated TRM, as has been described in the lung and the thymus (53, 54). Thus, temporary CD69 expression may be sufficient for TRM generation and may explain the absence of CD69 expression on a subset of long-term persisting TRM in the pancreas, salivary gland and female reproductive tract (37). Loss of S1P1, and potentially other tissue egress receptors, e.g., mediated by downregulation of the transcription factor KLF2 (31), together with expression of specific adhesion molecules, confers long-term tissue residency. Further, a combination of gene expression programs otherwise involved in the differentiation of both peripheral TCM and effector T cells ensure maintenance of a stable population of TRM by conserving proliferative capacity as well as acquisition of constitutive expression of effector molecules (49, 55). The transcription factors known to be involved in this process have been reviewed in detail recently (56, 57). TRM and TCM are probably generated from the same naive precursors (58), however, the gene expression profile of TRM is clearly distinct from peripheral memory T cells in mice (22, 59) and in humans (19, 24, 41). In mice, particularly the expression of transcription factors Blimp1, Hobit, and Runx3 in TRM precursors seems to be essential to acquire tissue residency (49, 59). For the maintenance of stable TRM population, a combination of signals stimulating longevity and homeostatic proliferation seems to be necessary. Most TRM express CD127 (IL7Rα), while expression of CD122 (IL2rβ), which can bind IL-2 as well as IL-15 when paired to CD132 (common γ chain, γc), seems to be more variable (22, 60). Previous studies have shown that IL-7 and IL-15-dependent longevity and homeostatic proliferation are maintaining TCM by Stat5 signaling (61–63). Likewise, both cytokines have been implied to contribute to TRM survival and maintenance (22, 64) and phosphorylation of Stat5 has been observed in a subset of brain TRM (32). However, the sources providing homeostatic signals assuring TRM long-term survival are so far still not completely known.
Tissue-specific Influences on TRM Differentiation and Maintenance
The gene expression program of TRM generated in different tissues is largely overlapping (19, 22, 59, 65), but some variations of this program as well as particular requirements for TRM differentiation seem to exist in different experimental settings, organs and even show inter-individual variability. A particular TRM phenotype and its functional characteristics are thus likely to be due to pathogen- and tissue-specific cues as well as the genetic background of the host (see Figure 1A). Moreover, most TRM markers are not homogeneously expressed in the whole resident population (18, 68), suggesting further specialization of a particular TRM population into functional subsets—even if they have been generated by one definite infection and harbor the same antigen-specificity. Differential gene expression programs and surface receptor expression on putative TRM subsets are likely to confer different tissue locations and functionality, as we will further discuss below. More detailed analysis, probably using single cell-based approaches will soon identify possible TRM subsets on a phenotype and functional basis.
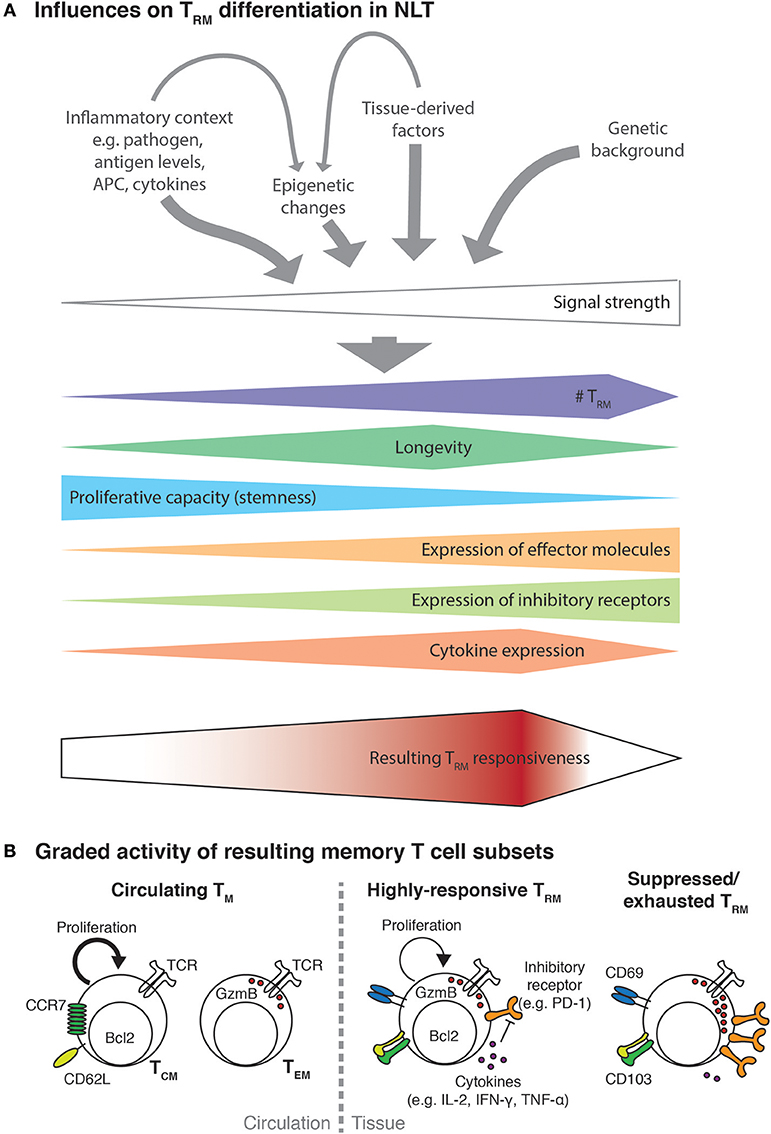
Figure 1. Multiple factors influence TRM functionality. (A) Activated T cells recruited to NLT will encounter an inflammatory environment shaped by the nature and extent of infection. The encountered signals will consist of different cytokines (e.g., Il-2, IL-12, IL-15, type I and type II interferons) potentially in concert with varying levels of cognate antigen presented on professional APC and infected cells as well as tissue-derived trophic factors, metabolites or the microbiome. Probably additionally influenced by the genetic background of the infected individual, the strength of the resulting signal to activated T cells will direct their expansion and differentiation into TRM. In analogy to the signal strength model of CD8+ T cell differentiation (66), higher signal strength will result in higher TRM numbers and be associated with more terminal differentiation, which manifests with progressive loss of proliferative capacity, acquisition of expression of effector molecules and increasing levels of inhibitory receptors. Encounter of very strong signals, such as during chronic infection, might lead to dysfunctional and exhausted TRM and even to their elimination. The combined effect of all these factors will then determine the responsiveness of the resulting TRM population to a secondary antigenic challenge or other inflammatory stimuli. (B) As a result of T cell activation and tissue-derived signal, circulating and resident memory T cells of different responsiveness will be generated. Circulating memory cells, namely central memory T cells (TCM) and effector memory T cells (TEM) show a delayed recruitment to the infected site. In addition, those cell subsets seem to specialize in either proliferative potential or immediate effector function. In contrast, a moderately strong TRM differentiation signal will result in high numbers of highly-responsive TRM that combine features of both TCM and TEM cells. Even though TRM may express inhibitory receptors such as PD-1 to some degree, they can overcome this regulation e.g., due to their high expression of inflammatory cytokines (67). Highly-responsive TRM can efficiently protect against re-infection but due to their low threshold for reactivation they could be prone to drive immunopathology or be involved in aberrant immune responses such as in allergies and autoimmune diseases. Alternatively, TRM can be subject to regulation by regulatory T cells and other mechanisms, which may impair their longevity and/or induce a suppressed phenotype.
One of the major incongruities of TRM differentiation in different organs is the dependency on local antigen expression. While TRM in the gut, skin and some mucosae can be generated and maintained independently of local antigen presentation (69–71), expression of local antigen seems to be required for the generation of TRM in the brain (29, 32). In theory, local antigen expression serves various purposes: In a very basic manner, local antigen expression will enhance recruitment and local expansion of TRM precursor cells and thereby increase the resulting TRM population (72). For some organs, local antigen expression might be strictly required for tissue entry of antigen-specific T cells, as suggested for the brain (73), and thereby be essential for TRM generation. In general, however, inflammatory cues, such as certain cytokines and chemokines, seem to be sufficient to promote TRM differentiation, such as evidenced by so-called “prime-and-pull” and “prime and trap” vaccination approaches, which efficiently generate TRM in skin, mucosae and the liver (25, 60, 70). Local antigen expression, and thus the local reactivation of TRM precursor cells by antigen-presenting cells (APC), might also serve the expression of cytokines and chemokines required to guide TRM differentiation and localization (55, 74), which could explain why in some experimental settings antigen is required (75), but not in others (22).
TRM heterogeneity is particularly evident with regard to their expression of adhesion molecules. TRM in different organs (and even further, different subsets of TRM) show sometimes combined, sometimes exclusive expression of adhesion molecules such as CD103 (IntegrinαE), CD49a (Integrinα1β1), LFA-1 (IntegrinαLβ2), and E-Cadherin (22, 24, 28, 37, 46, 76, 77). Depending on their interaction partner, adhesion molecule expression on a specific TRM subset probably serves its specific retention and positioning in their tissue of residence (68). CD103 mediates epithelial localization and TRM retention in the skin and gut by interacting with E-Cadherin (4, 22, 69, 78), while CD49a expression anchors TRM to the collagen matrix (79). Besides TRM localization, expression of adhesion molecules has also been linked to TRM functionality. CD103 expression has been associated with an enhanced cytotoxic capacity of CD8+ T cells toward E-Cadherin-expressing target cells (80). Likewise, CD49a expression by human skin TRM seems to discriminate between IFN-γ- and IL-17A-producing cells (24). We are however only beginning to understand how the exposure of TRM precursors to their specific inflammatory context affects TRM differentiation and functionality.
Cytokine redundancy (the common use of receptors and receptor subunits by different cytokines) and pleiotropy (multiple different functions exerted by one cytokine) are possible explanations for some of the observed variations in the dependency of TRM generation on cytokines in different experimental contexts. Interestingly, resting non-activated T cells share a common receptor (CD122/γc) for IL-2 and IL-15. It seems therefore likely that in conditions in which TRM precursors are exposed to e.g., high levels of IL-2 during the acute inflammatory response, IL-15 signaling becomes redundant for TRM generation. As mentioned above, both IL-7 and IL-15 can mediate pro-survival as well as homeostatic proliferation, and a certain functional redundancy might occur between these two cytokines, depending upon which receptors predominate on TRM or their precursors and which cytokine is available in the tissue niche occupied by TRM. Consistent with this idea, IL-15 dependency of TRM varies considerably between different organs and might be differentially required for TRM differentiation, survival and/ or homeostatic proliferation (81). This could also explain why expression levels of anti-apoptotic signaling molecules in TRM, such as Bcl-2, vary between organs, as do the rates of their spontaneous proliferation (22, 29, 32). Thus, it seems possible that for maintaining a stable TRM population, TRM longevity and potential for self-renewal can partly substitute for each other and the signals driving either process might therefore be functionally redundant to some extent. Similarly, transcriptional programming of TRM precursors might vary between one tissue to another. Hobit and Blimp1 have been described to play a partially redundant role during TRM differentiation, but depending on the tissue, TRM generation is more dependent on one of these transcription factors than the other (59). This indicates that transcriptional regulation of TRM differentiation could be incited in a different manner depending on the tissue niche and inflammatory context, possibly giving rise to TRM of different reactivity and functional potential (Figure 1A). In support of this concept, a recent study describes that the presence of pro-inflammatory cytokines like type I interferons and IL-12 drive differentiation of CD103– TRM (74), in contrast to the TGF-β-dependent differentiation of CD103+ TRM (22, 55, 78).
During their differentiation and long-term maintenance, TRM have to adapt to the metabolic environment of their tissue of residence. In most NLT, nutrients such as glucose and certain amino are more limited than in the circulation, and invading T cells need to adapt their metabolic processes to match their energy demands in this environment (82). While glucose plays a central role as energy source for all T subsets, activated T cells show especially high glycolysis rates and also fuel glucose-derived carbons into anabolic pathways such as fatty acid synthesis (83). Further, T cells are dependent on amino acid uptake and metabolism for full activation and differentiation (84–86). However, memory T cells critically rely on fatty acid oxidation (FAO) as an energy source (87–89), for which they synthesize long-chain fatty acids as substrates from glycolytic intermediates intracellularly (90). By contrast, TRM in the skin and adipose tissue rely on uptake of fatty acids from the extracellular space (91, 92), possibly due to the limited amount of glucose available for de novo fatty acid synthesis. However, it remains to be determined if TRM in more nutrient-rich organs such as the gut, liver and brain might show distinct tissue-specific metabolic adaptations.
Despite providing the energy for T cell expansion and survival, the metabolic environment also dictates T cell differentiation and effector function (93). Cytokine production, cytotoxicity, migration, and tissue invasiveness as well as the differentiation of memory T cells are instructed by metabolic changes (87, 94–97). One central regulator of this so-called metabolic reprogramming is mammalian target of rapamycin (mTOR) (98). mTOR is phosphorylated in response to TCR ligation, cytokine signaling as well as intracellular energy state. In turn, mTOR regulates CD8+ T cell differentiation via T-bet and Eomesodermin (99) as well as via the regulation of fatty acid metabolism (87). Inhibition of mTOR leads to a higher number of memory precursors and circulating memory T cells (94), by contrast, formation of long-lived TRM in mucosal tissues is impaired (100). Interestingly, activation of mTOR (together with phosphoinositol-3-kinase) induces downregulation of KLF2 and S1P1 in activated T cells (101), indicating that mTOR activation during TRM differentiation could contribute to establish tissue retention. In line with this, upregulation of CD69 on γδ T cells has been shown to enhance uptake of the amino acid tryptophan, which in turn enhanced mTOR- and arylhydrocarbon receptor (AhR)-dependent signaling pathways (102). AhR has been shown to be required for generation of TRM in the skin (103), further corroborating the idea of a mechanistic link between the metabolic, possibly tissue-specific, environment encountered by TRM precursors and the successful formation of a tissue-resident and long-lived T cell population.
Altogether, it seems likely that the combination of antigen load, inflammatory signals and nutrients in a tissue-specific niche creates a specific environmental context for TRM differentiation and maintenance (Figure 1). Given that some TRM niches, especially mucosal tissues and epithelial layers, undergo constant turnover and replacement of cells, it seems likely that the inflammation-induced TRM niche undergoes certain changes in cellular composition and expression of TRM-maintaining factors. To date, the exact sources of these determining factors still remain largely unknown. It might even be possible that the TRM-maintaining niche in some organs or under certain circumstances has only a limited lifespan, which could explain why TRM are not maintained long-term in some experimental settings (104). The environmental context probably determines not only the functional features of TRM residency and maintenance but also TRM responsiveness toward new inflammatory stimuli during a secondary infection (see Figure 1A). Future studies are needed to reveal more context-dependent variations in TRM generation and functionality, discovering new targets, potentially in a tissue-specific manner, for experimental and therapeutic manipulation of TRM.
TRM in Acute-Resolved Infection
TRM serve as a front-line defense against viral re-infection in various tissues. Due to their unique positioning, often directly at barrier surfaces, they can rapidly detect invading pathogens and provide immediate immune function. In comparison, immune surveillance by circulating memory T cells is slower and often allows virus spread for several days before sufficient recruitment, local expansion, and differentiation of peripheral memory T cells takes place to confine and successfully combat infection (27, 32). This notion is supported by a breadth of experimental models, that demonstrate accelerated pathogen control in the presence of TRM at the pathogen entry site in comparison to circulating memory T cells alone (9). Protective functions of TRM have been described for barrier tissues such as the skin (4, 27, 70, 105), the lung (106–109), the gut (48), and the reproductive tract (110). TRM localized to body surfaces may thus play an important role to prevent systemic infection by recurring pathogens invading via the skin and mucosae and to limit extensive tissue damage and scarring at the entry sites. As a consequence, TRM of a multitude of epitope specificities accumulate with age at these pathogen entry zones in free-living mice and humans (17, 47, 111). Interestingly, a protective role of TRM-mediated immune defense has been described also for internal organs such as the liver and the brain (25, 32), which display unique immune-regulatory functions (112, 113). As such, immune cell activation is impeded in these organs, e.g., due to low expression of MHC molecules, and often occurs with considerable delay, which increases the risk of persistent and widespread infection. The latter in turn can contribute to more severe immunopathology once an immune response is finally triggered. Similar to their positioning at epithelial surfaces in barrier tissues, TRM in the brain and liver are also preferentially located at potential pathogen entry sites, be it in meninges and close to brain blood vessels (32) or liver sinusoids (25). This enables TRM to quickly react and eliminate invading pathogens and thereby protect these vulnerable organs from potentially harmful inflammation.
Upon re-encounter of their cognate antigen, TRM employ two main paths to assure protection against the recurring pathogen. Firstly, they instantly provide highly potent cytotoxic effector functions that can eliminate the initially infected cells (barrier immunity) (27, 32). Indeed, a subset of TRM constitutively expresses Granzyme B, and perforin-mediated elimination of infected cells contributes to their protective effect in the brain (32). Secondly, TRM trigger a variety of local and recruited innate and adaptive immune mechanisms that can even provide bystander resistance to unrelated pathogens (39, 105, 110). TRM-derived interferon-γ (IFN-γ) plays an important role by stimulating the expression of adhesion molecules and chemokines that facilitate endothelial transgression of peripheral memory T and B cells (39). Further, the expression of IFN-γ-responsive genes—many of them with direct anti-viral functions—in uninfected bystander cells limits pathogen spread (105). Moreover, Granzyme B can deactivate a viral protein in neurons during latent HSV infection without inducing neuronal apoptosis (114) and IFN-γ can even purge viruses from infected cells in a non-cytolytic manner, a process that seems important to maintain tissue homeostasis in non-regenerative tissues such as the brain (115, 116). It is important to note that the protective capacity of TRM related to their cytotoxic activity and cytokine production requires the presentation of cognate antigen on MHC-I molecules, even though TRM can show signs of bystander activation in an inflammatory environment (32).
The protective capacity of TRM makes their generation a new objective for the development of vaccines. Indeed, skin vaccination and scarification during small pox vaccination that has now been associated with the generation of TRM has been shown to provide superior protective immunity than hypodermal injection (117). Alternatively, the above-mentioned “prime and pull” and “prime and trap” vaccination strategies, in which systemic administration of a vaccine is combined with local application of chemokines or antigen, improves immunological barrier functions through TRM generation (25, 60, 70). Interestingly, upon recruitment and activation in skin and mucosae, some T cells exit and give rise to SLO-associated TRM (118). Being positioned at entry sites for draining peripheral antigen, these SLO TRM provide a second line of defense and extend TRM-mediated immune memory to the draining lymphoid tissue (119). During antigenic re-challenge, TRM are the predominant population undergoing secondary expansion and together with recruited circulating T cells give rise to new generations of TRM (120, 121). This implies that protective immunity mediated by TRM can be boosted by repeated local immunizations. Further, infections with different pathogens can lead to a persisting TRM population that contains multiple specificities at once, which provide broader and more efficient protection (122). Future vaccination approaches implementing these new insights could thus improve T-cell-mediated protection at external and internal anatomical barrier sites.
TRM and Chronic Inflammation
Chronic inflammation results from repeated or continuous immune cell activation by recurrent or persisting antigens. Such responses are desirable to control latent-reactivating or persistent infections and to eliminate neoplastic cells. However, aberrant inflammation caused by environmental or self-antigens carries the risk of developing chronic inflammatory diseases, such as allergies and autoimmune diseases (AD). Indeed, TRM have been detected in several human inflammatory diseases (10, 123) (see Table 2). In principle, two main roles for TRM in chronic inflammatory settings can be envisaged. TRM can be drivers of chronic inflammation, thereby providing a compartmentalization of the immune response. And in a not necessarily exclusive scenario, TRM could trigger the bystander activation of allergen-reactive or self-reactive T cells and thereby serve as contributing triggers to chronic inflammatory diseases.

Table 2. TRM in human chronic inflammatory diseases.
TRM Functionality in the Context of Persisting Antigen
TRM in Chronic Infection
One of the earliest reports on resident T cell responses came from latently-infected sensory ganglia, in which HSV reactivation was controlled by a non-circulating T cell population (148, 149). Together with the above-mentioned observations during prime-and-boost vaccinations (122), this demonstrates that TRM may retain their inflammatory activity over repeated rounds of antigen stimulation. In the best scenario, this will prevent virus reactivation and ensure continuous virus latency and limitation of virus spread. Indeed, TRM can be detected in sanctuaries of persistent viruses such as human and mouse Cytomegalovirus (CMV) (150, 151), Hepatitis B virus (HBV) (67), Hepatitis C virus (HCV) (138), and Human Immunodeficiency Virus (HIV) (136, 152). Interestingly, high TRM numbers in HBV-infected liver and HIV-infected gut as well as clonal expansion of SLO TRM have been inversely correlated to virus loads and associated with spontaneous resolution of chronic infection (67, 136, 152), provoking interest in TRM-directed therapeutic approaches (137). Infection with most persisting viruses leads to chronic immune activation over time, including accumulation of a large virus-specific T cell population, a process referred to as “memory inflation” (153). Inflationary T cells can acquire a TRM-like phenotype and become resident, e.g., in the salivary gland, despite being probably an ontogenically-different T cell subset (154). Chronic inflammatory tissue damage is the common long-term consequence of persisting virus infection. Since HBV-specific TRM overcome immunosuppressive mechanisms in the liver and have high expression of pro-inflammatory cytokines like IL-2, IFN-γ, and TNF-α (67), it remains possible that TRM are also drivers of tissue damage in the context of chronic virus infections. Therefore, a potential harmful role of TRM in persisting infections merits further investigations, especially at the chronic stage of HBV and HCV infection.
So far, we understand very little about how and whether functional TRM can be generated in conditions in which their cognate antigen is continuously present. Chronic high levels of antigen in some persistent infections, such as Lymphocytic choriomeningitis virus (LCMV) clone 13 or latent CMV, seem to hamper de novo generation of TRM (69, 151). It is therefore likely that virus levels have to significantly contract after initial infection to allow for efficient TRM generation, even in the context of chronic infection. Interestingly, when TRM retention is impaired by lack of TGF-β signaling during chronic LCMV clone 13 infection in the gut, a stable population of anti-viral CD8+ T cells is maintained by continuous recruitment (78), indicating that impaired TRM formation can be compensated for. This suggests that depending on the cytokine milieu present during chronic infection, the local T cell pool might consist of variable proportions of TRM and recruited T cells.
TRM and Cancer
Tumors can be a source of neo-antigens stimulating anti-tumor CD8+ T cell responses (155, 156) and T cell infiltration is a prognostic marker for a beneficial outcome in some cancers (157, 158). Recent studies demonstrate, that a subset of tumor-infiltrating lymphocytes (TIL) in solid tumors resemble bona fide TRM and are associated with its epithelial layers (159–161). TRM-like TILs, in particular when they express CD103, have been associated with better prognosis (143–145, 162), a fact that could be explained by an enhanced cytotoxic efficiency upon interaction of CD103 on TRM with its ligand E-Cadherin on tumor cells (80). Accordingly, experimental strategies inducing tumor-specific TRM show superior tumor control in comparison to approaches that solely generate circulating tumor-specific effector T cells (163–165).
Tumor cells rely heavily on the uptake and metabolism of glucose and other nutrients, resulting in a metabolically-deprived tumor microenvironment (TME) (166, 167). Tumor-infiltrating T lymphocytes (TIL) are further subject of active immunosuppression by myeloid-derived suppressor cells (MDSC) and regulatory T cells (168). MDSC express ligands for immune checkpoint inhibitors (e.g., PD-L1 and PD-L2) and can also contribute to nutrient deprivation in the TME by uptake and metabolism of arginine. As a consequence of the increase of lactate in the TME, TIL lose cytotoxic effector functions and show impaired motility (169). TRM adapt to the metabolic environment of their tissue of residence by utilizing free fatty acids (92), and are under certain circumstances resistant to checkpoint blockade (67). This indicates that tumor-specific TRM might be better adapted to the immunosuppressive tumor microenvironment than their circulating counterparts (8). This opens new avenues for cancer immunotherapies. TRM already present in the tumor could be functionally enhanced by checkpoint inhibitors, potentially together with increasing their catabolic fatty acid metabolism. Indeed, administration of a PPAR-α agonist or free fatty acids increases the functionality of TIL in a melanoma model, especially in combination with anti-PD-1 treatment (170). Moreover, one could envisage to genetically engineer T cells for cell therapy with the aim to promote TRM generation. Recently, such an approach has been realized by modifying chimeric antigen receptor (CAR) T cells to express orthogonal IL-2 receptors allowing for a specific targeting of the transferred cell population (171). A better understanding of TRM differentiation and maintenance could inform a similar strategy aiming at increasing TRM differentiation, maintenance of functionality during CAR T cell therapy. In addition, reprogramming of tumor-infiltrating dendritic cells with β-glucan curdlan in a humanized mouse model of breast cancer enhances the differentiation of CD103+ TIL via DC-derived TGF-β production, resulting in rejection of an established tumor (172), highlighting how adoptive cell therapy can target TRM differentiation.
It remains unknown, how tumor-associated TRM are generated. Analogous to persistent infections (69), tumor-specific TRM are chronically exposed to their cognate antigen, which could impair their successful differentiation. Otherwise, one may speculate that tumor-associated TRM are already generated after development of the first tumor cells when there is still little cognate antigen present. This would imply that these TRM could also play a role in the control of tumor transformation, since they might co-reside with a primary tumor for years. Therapeutic induction of tumor-specific TRM, together with other resident populations, could enforce local anti-tumor immune response to the cancer (173) and may help to eradicate tumor cells from the body as well as reduce systemic side effects. To take advantage of this therapeutically, application of e.g., viral vectors that efficiently generate local TRM with only limited numbers of peripheral tumor-specific effector cells could be envisaged. The constitutive expression of checkpoint inhibitors by TRM (19) also harbors the hope that the anti-tumor activity of endogenous or therapeutically-induced TRM could be further enhanced by checkpoint inhibitor blockade (174). However, one has to keep in mind, that enhancing TRM activity and expansion might come with undesired side effects, and disinhibiting TRM might in turn give rise to tumors. Due to their localization and non-circulating behavior, TRM are refractory to most immune ablative therapies, as evidenced by mycosis fungoides, a human TRM-like skin lymphoma (42).
TRM-driven Chronic Inflammatory Diseases
T cells specific for self-antigens or environmental antigens are considered active drivers of diverse allergic reactions such as food and drug allergies, asthma, and diabetes, as well as autoimmune diseases such as psoriasis, inflammatory bowel disease, and multiple sclerosis (MS). In the past, these diseases were considered to be driven by effector or effector memory T cells that infiltrate the affected organ, however, several lines of evidence suggest that some of these chronic inflammatory diseases or some disease stages are instead predominantly driven by resident immune cells (10, 123). Therapeutic inefficacy of drugs inhibiting T cell recruitment indicates a putative TRM involvement in disease progression since TRM reside behind the blood-tissue barrier and are often refractory to systemic blockade or ablation. This applies, for example, to psoriasis (128) and progressive stages of MS (135). Interestingly, new exacerbations in fixed drug eruption (FDE) and psoriasis frequently occur at sites of previously-resolved skin lesions, indicative of an involvement of localized immune memory (175). Likewise, so-called smoldering lesions characterized by activated macrophages/microglia together with T cells at their fringes are almost exclusively observed in progressive MS (176, 177). Evidence of TRM persistence has been found in resolved psoriatic skin lesions (178) and in chronic MS lesions (135), in which they constitute the dominant T cell subset (our own unpublished observations). Indeed, psoriatic normal-appearing skin contains all immune components necessary to elicit lesion formation upon an environmental trigger (44) and compartmentalized inflammation has been correlated to cumulative brain damage during progressive MS. Beyond that, indications for TRM involvement were found in many other chronic inflammatory diseases (see Table 2), suggesting possible common mechanisms for their development and involvement in disease pathogenesis.
One hallmark of allergic exacerbations is the short time frame (24–48 h) after exposure to the environmental trigger, in which exacerbations occur as is observed in FDE, a T cell-driven allergic local cutaneous reaction to certain administered drugs (179). This short time interval between trigger and reaction cannot be explained by recruitment of circulating T cells, which typically takes longer (58). Evidently, analog time intervals are difficult to determine for flares of autoimmune diseases, where the trigger of an exacerbation is unknown. TRM generally have a low threshold for re-activation, and presence of their cognate antigen (e.g., by exogenous administration of peptide) is usually enough to elicit expression of cytolytic effector molecules and cytokines (39, 105, 110, 180). Likewise, induction of antigen expression in non-hematopoietic cells, without concomitant danger signals, is sufficient to elicit activation of resident T cells in the skin (180), probably via intermediary presentation of antigen on professional APC (180, 181). One may speculate that the reactivation threshold of TRM depends upon the residence organ and/or the TRM-generating stimulus, which could in turn result in organ-dependent susceptibility to allergic and self-reactive TRM responses (see Figure 1B).
Allergen- or self-reactive T cell responses are usually considered to be elicited by preceding sensitizing events, which are hypothesized to occur in two main ways. Sequence similarities between a pathogen and an allergen- or self-antigen (also called molecular mimicry) can elicit pathogen-specific T cells that may cross-react to allergens or self-antigen upon future exposure (182). In case these T cells differentiate into TRM, a compartmentalized allergic or autoreactive T cell response is the consequence. Alternatively, exposure of allergen- or self-reactive T cells to a pro-inflammatory environment, such as that generated during an infection, could elicit their bystander activation and recruitment (183). As mentioned above, non-specific inflammation can be sufficient to allow for TRM differentiation in the absence of their cognate antigen (25, 60, 69, 70), making this a plausible scenario for the generation of TRM specific for environmental or self-antigens. Moreover, bystander activation could also explain how TRM could serve as triggers for chronic inflammatory diseases. Antigenic challenge of TRM not only induces a new generation from the pre-existing TRM pool, but can also activate and recruit bystander T cells of unrelated antigen-specificity, which give rise to newly formed TRM (120, 121). Accumulation of TRM can further lead to the displacement of other pre-existing resident immune cells (47, 103). Although this might serve to replace the more resident innate immune system by a more specific and efficient resident adaptive immune system, it carries the risk of replacing mostly “naive” immune cells by more trained, and possibly more pro-inflammatory immune cell components. Indeed, TRM are often observed in clusters together with mature professional APCs and often CD4+ T helper cells as well as other immune cells, indicative of organ-associated lymphoid tissues (19, 29, 64, 184, 185). These specific niches are speculated to contribute to chronic inflammation, since they provide an optimal environment for T cell re-stimulation (186–188). Such structures have been shown to contribute to TRM maintenance by chemokine and cytokine production (64, 184), however, whether TRM actively sustain these immune cell clusters is not clear.
Altogether, this supports the idea that pathogen-specific TRM generated during an infection could trigger and/or drive chronic inflammatory diseases. A possible connection between TRM and chronic inflammation could also provide a mechanistic explanation for the observed epidemiological association of infections and the development or exacerbation of allergic and autoimmune diseases (189). In many chronic inflammatory diseases, not only CD8+ TRM, but also other immune cells such as T helper (Th) cell subsets, regulatory T cells, APCs and innate lymphocytes can probably become resident and thereby contribute to a compartmentalized immune response that is resistant to many systemic immunomodulatory therapies. Thus, more research efforts are needed to understand the requirements for the differentiation and maintenance of resident immune cells in order to be able to functionally impair or even deplete TRM in chronic inflammatory diseases. By identifying signaling pathways involved in TRM retention and maintenance, we are currently undertaking the first steps toward a specific targeting of TRM without global immunosuppression. One possibility could be to interfere with TRM metabolism. Pharmacological treatments with Rapamycin (100) or inhibitors of FAO such as Trimetazidine and Etomoxir (92) have already been shown to decrease TRM numbers in experimental models.
Immunoregulatory mechanisms are in place to prevent extensive TRM accumulation in some organs or their over-activation. TRM generation is intimately linked to TGF-β (55, 78), a cytokine associated to resolution of infection. This indicates that TRM differentiation might not occur in presence of chronic antigen exposure, thereby preventing extensive TRM generation in chronic inflammatory settings. Further, TRM can express inhibitory receptors, such as PD-1, Lag3, and Tim3 (19, 29, 49, 107), in principle making them susceptible to checkpoint inhibition. Although TRM have the possibility to overcome PD-1-associated inhibition (67), exhausted TRM have been detected in immune privileged sites such as the eye (190). TRM are also susceptible to regulatory T cell-mediated immunosuppression (191, 192). For the lung, liver and brain and other immune privileged sites, even mechanisms of natural suppression of TRM accumulation have been suggested (104) that could assure tissue homeostasis by prevention of TRM accumulation.
Conclusion
Vaccine strategies inducing TRM against recurring infections are promising approaches to improve immunological protection. Equally, tumor-specific TRM might help to eradicate aberrant tumor cells from the body and enforce a localization of this response, thereby minimizing systemic side effects. However, several challenges have to be overcome to realize these goals, which are firstly of a technical nature. TRM generation cannot be monitored in peripheral blood and therefore requires the taking of biopsies from target organs, which might not always be easily performed. Further, suitable vaccination vectors need to be designed that allow the efficient local induction of specific TRM and that do not result in unwanted side effects such as bystander-induced self-reactive TRM. Until now, most research studies have focused on the overwhelmingly positive role of TRM acting against infected or tumorous cells, however we still lack an appropriate understanding of the possible physiological consequences of TRM persistence. Further research efforts are warranted to better understand the role of TRM in chronic inflammatory diseases in order to identify the risks in amplifying TRM numbers or function. So far, we are lacking appropriate mouse models allowing specific genetic targeting of TRM and are not able to completely deplete already-established TRM. It is therefore instrumental to perform detailed preclinical and clinical studies to gain more insight into TRM biology and its adaptation during different experimental regimens and in different tissues to allow for a safe and efficient therapeutic tissue targeting of TRM.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. (2013) 31:137–61. doi: 10.1146/annurev-immunol-032712-095954
2. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
3. Graef P, Buchholz VR, Stemberger C, Flossdorf M, Henkel L, Schiemann M, et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity (2014) 41:116–26. doi: 10.1016/j.immuni.2014.05.018
4. Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. (2009) 10:524–30. doi: 10.1038/ni.1718
5. Mueller SN, Mackay LK. Tissue-resident memory T cells: local specialists in immune defence. Nat Rev Immunol. (2016) 16:79–89. doi: 10.1038/nri.2015.3
6. Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity (2014) 41:886–97. doi: 10.1016/j.immuni.2014.12.007
7. Carbone FR. Tissue-resident memory t cells and fixed immune surveillance in nonlymphoid organs. J Immunol. (2015) 195:17–22. doi: 10.4049/jimmunol.1500515
8. Amsen D, van Gisbergen K, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538–46. doi: 10.1038/s41590-018-0114-2
9. Rosato PC, Beura LK, Masopust D. Tissue resident memory T cells and viral immunity. Curr Opin Virol. (2017) 22:44–50. doi: 10.1016/j.coviro.2016.11.011
10. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
11. Iijima N, Iwasaki A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science (2014) 346:93–8. doi: 10.1126/science.1257530
12. Sheridan BS, Romagnoli PA, Pham QM, Fu HH, Alonzo F. III, Schubert WD, et al. gammadelta T cells exhibit multifunctional and protective memory in intestinal tissues. Immunity (2013) 39:184–195. doi: 10.1016/j.immuni.2013.06.015
13. Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science (2015) 350:981–5. doi: 10.1126/science.aac9593
14. Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell (2016) 164:1198–211. doi: 10.1016/j.cell.2016.02.048
15. Romagnoli PA, Sheridan BS, Pham QM, Lefrancois L, Khanna KM. IL-17A-producing resident memory gammadelta T cells orchestrate the innate immune response to secondary oral Listeria monocytogenes infection. Proc Natl Acad Sci USA. (2016) 113:8502–7. doi: 10.1073/pnas.1600713113
16. Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity (2013) 38:769–81. doi: 10.1016/j.immuni.2013.02.010
17. Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity (2013) 38:187–97. doi: 10.1016/j.immuni.2012.09.020
18. Thome JJ, Farber DL. Emerging concepts in tissue-resident T cells: lessons from humans. Trends Immunol. (2015) 36:428–35. doi: 10.1016/j.it.2015.05.003
19. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
20. Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, et al. Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med. (2010) 207:553–64. doi: 10.1084/jem.20090858
21. Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. (2006) 176:2079–83. doi: 10.4049/jimmunol.176.4.2079
22. Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. (2013) 14:1294–301. doi: 10.1038/ni.2744
23. Woon HG, Braun A, Li J, Smith C, Edwards J, Sierro F, et al. Compartmentalization of total and virus-specific tissue-resident memory CD8+ T cells in human lymphoid organs. PLoS Pathog. (2016) 12:e1005799. doi: 10.1371/journal.ppat.1005799
24. Cheuk S, Schlums H, Gallais Serezal I, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
25. Fernandez-Ruiz D, Ng WY, Holz LE, Ma JZ, Zaid A, Wong YC, et al. Liver-resident memory CD8(+) T cells form a front-line defense against malaria liver-stage infection. Immunity (2016) 45:889–902. doi: 10.1016/j.immuni.2016.08.011
26. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. (2012) 189:3462–71. doi: 10.4049/jimmunol.1201305
27. Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature (2012) 483:227–31. doi: 10.1038/nature10851
28. Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci USA. (2011) 108:16741–6. doi: 10.1073/pnas.1107200108
29. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci USA. (2010) 107:17872–9. doi: 10.1073/pnas.1010201107
30. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell (2014) 159:814–28. doi: 10.1016/j.cell.2014.10.026
31. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
32. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med. (2016) 213:1571–87. doi: 10.1084/jem.20151916
33. Lehmann J, Huehn J, de la Rosa M, Maszyna F, Kretschmer U, Krenn V, et al. Expression of the integrin alpha Ebeta 7 identifies unique subsets of CD25+ as well as CD25- regulatory T cells. Proc Natl Acad Sci USA. (2002) 99:13031–6. doi: 10.1073/pnas.192162899
34. Cibrian D, Sanchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol. (2017) 47:946–53. doi: 10.1002/eji.201646837
35. Gebhardt T, Palendira U, Tscharke DC, Bedoui S. Tissue-resident memory T cells in tissue homeostasis, persistent infection, and cancer surveillance. Immunol Rev. (2018) 283:54–76. doi: 10.1111/imr.12650
36. Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity (2004) 20:551–62. doi: 10.1016/S1074-7613(04)00103-7
37. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell (2015) 161:737–49. doi: 10.1016/j.cell.2015.03.031
38. Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature (2011) 477:216–9. doi: 10.1038/nature10339
39. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. (2013) 14:509–13. doi: 10.1038/ni.2568
40. Zhu J, Peng T, Johnston C, Phasouk K, Kask AS, Klock A, et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature (2013) 497:494–7. doi: 10.1038/nature12110
41. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol. (2016) 17:1467–78. doi: 10.1038/ni.3589
42. Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. (2012) 4:117ra117. doi: 10.1126/scitranslmed.3003008
43. Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. (2015) 7:279ra239. doi: 10.1126/scitranslmed.3010302
44. Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. (2004) 199:731–6. doi: 10.1084/jem.20031482
45. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. (2006) 176:4431–9. doi: 10.4049/jimmunol.176.7.4431
46. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS ONE (2011) 6:e16245. doi: 10.1371/journal.pone.0016245
47. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532:512–6. doi: 10.1038/nature17655
48. Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity (2014) 40:747–57. doi: 10.1016/j.immuni.2014.03.007
49. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature (2017) 552:253–7. doi: 10.1038/nature24993
50. Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature (2006) 440:540–4. doi: 10.1038/nature04606
51. Bankovich AJ, Shiow LR, Cyster JG. CD69 suppresses sphingosine 1-phosophate receptor-1 (S1P1) function through interaction with membrane helix 4. J Biol Chem. (2010) 285:22328–37. doi: 10.1074/jbc.M110.123299
52. Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, et al. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J Immunol. (2015) 194:2059–63. doi: 10.4049/jimmunol.1402256
53. Park SL, Mackay LK, Gebhardt T. Distinct recirculation potential of CD69(+)CD103(-) and CD103(+) thymic memory CD8(+) T cells. Immunol Cell Biol. (2016) 94:975–80. doi: 10.1038/icb.2016.60
54. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. (2016) 213:3057–73. doi: 10.1084/jem.20160938
55. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
56. Mackay LK, Kallies A. Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol. (2017) 38:94–103. doi: 10.1016/j.it.2016.11.004
57. Milner JJ, Goldrath AW. Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr Opin Immunol. (2018) 51:162–9. doi: 10.1016/j.coi.2018.03.017
58. Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med. (2015) 21:647–53. doi: 10.1038/nm.3860
59. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science (2016) 352:459–63. doi: 10.1126/science.aad2035
60. Shin H, Iwasaki A. A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature (2012) 491:463–7. doi: 10.1038/nature11522
61. Burchill MA, Goetz CA, Prlic M, O'Neil JJ, Harmon IR, Bensinger SJ, et al. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J Immunol. (2003) 171:5853–64. doi: 10.4049/jimmunol.171.11.5853
62. Willinger T, Freeman T, Hasegawa H, McMichael AJ, Callan MF. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J Immunol. (2005) 175:5895–903. doi: 10.4049/jimmunol.175.9.5895
63. Hand TW, Cui W, Jung YW, Sefik E, Joshi NS, Chandele A, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci USA. (2010) 107:16601–6. doi: 10.1073/pnas.1003457107
64. Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, et al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med. (2015) 21:1272–9. doi: 10.1038/nm.3962
65. Schenkel JM, Masopust D. Identification of a resident T-cell memory core transcriptional signature. Immunol Cell Biol. (2014) 92:8–9. doi: 10.1038/icb.2013.67
66. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
67. Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, et al. IL-2(high) tissue-resident T cells in the human liver: sentinels for hepatotropic infection. J Exp Med. (2017) 214:1567–80. doi: 10.1084/jem.20162115
68. Topham DJ, Reilly EC. Tissue-resident memory CD8(+) T cells: from phenotype to function. Front Immunol. (2018) 9:515. doi: 10.3389/fimmu.2018.00515
69. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
70. Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, et al. Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci USA. (2012) 109:7037–42. doi: 10.1073/pnas.1202288109
71. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. (2015) 16:406–14. doi: 10.1038/ni.3108
72. Khan TN, Mooster JL, Kilgore AM, Osborn JF, Nolz JC. Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med. (2016) 213:951–66. doi: 10.1084/jem.20151855
73. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An antigen-specific pathway for CD8 T cells across the blood-brain barrier. J Exp Med. (2007) 204:2023–30. doi: 10.1084/jem.20070064
74. Bergsbaken T, Bevan MJ, Fink PJ. Local inflammatory cues regulate differentiation and persistence of CD8(+) tissue-resident memory T cells. Cell Rep. (2017) 19:114–24. doi: 10.1016/j.celrep.2017.03.031
75. Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, et al. Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med. (2016) 213:3075–86. doi: 10.1084/jem.20160888
76. Haddadi S, Thanthrige-Don N, Afkhami S, Khera A, Jeyanathan M, Xing Z. Expression and role of VLA-1 in resident memory CD8 T cell responses to respiratory mucosal viral-vectored immunization against tuberculosis. Sci Rep. (2017) 7:9525. doi: 10.1038/s41598-017-09909-4
77. McNamara HA, Cai Y, Wagle MV, Sontani Y, Roots CM, Miosge LA, et al. Up-regulation of LFA-1 allows liver-resident memory T cells to patrol and remain in the hepatic sinusoids. Sci Immunol. (2017) 2:eaaj1996. doi: 10.1126/sciimmunol.aaj1996
78. Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity (2013) 39:687–96. doi: 10.1016/j.immuni.2013.08.019
79. Richter M, Ray SJ, Chapman TJ, Austin SJ, Rebhahn J, Mosmann TR, et al. Collagen distribution and expression of collagen-binding alpha1beta1 (VLA-1) and alpha2beta1 (VLA-2) integrins on CD4 and CD8 T cells during influenza infection. J Immunol. (2007) 178:4506–16. doi: 10.4049/jimmunol.178.7.4506
80. Le Floc'h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. (2007) 204:559–70. doi: 10.1084/jem.20061524
81. Schenkel JM, Fraser KA, Casey KA, Beura LK, Pauken KE, Vezys V, et al. IL-15-independent maintenance of tissue-resident and boosted effector memory CD8 T cells. J Immunol. (2016) 196:3920–6. doi: 10.4049/jimmunol.1502337
82. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science (2013) 342:1242454. doi: 10.1126/science.1242454
83. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
84. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
85. Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol. (2013) 191:4080–5. doi: 10.4049/jimmunol.1300923
86. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
87. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
88. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
89. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
90. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
91. Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, et al. White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity (2017) 47:1154–68.e6. doi: 10.1016/j.immuni.2017.11.009
92. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543:252–6. doi: 10.1038/nature21379
93. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
94. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi: 10.1038/nature08155
95. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
96. Chang CH, Curtis JD, Maggi LB Jr, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
97. Lee JH, Elly C, Park Y, Liu YC. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1alpha to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42:1062–74. doi: 10.1016/j.immuni.2015.05.016
98. Jones RG, Pearce EJ. MenTORing Immunity: mTOR signaling in the development and function of tissue-resident immune cells. Immunity (2017) 46:730–42. doi: 10.1016/j.immuni.2017.04.028
99. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32:67–78. doi: 10.1016/j.immuni.2009.10.010
100. Sowell RT, Rogozinska M, Nelson CE, Vezys V, Marzo AL. Cutting edge: generation of effector cells that localize to mucosal tissues and form resident memory CD8 T cells is controlled by mTOR. J Immunol. (2014) 193:2067–71. doi: 10.4049/jimmunol.1400074
101. Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. (2008) 9:513–21. doi: 10.1038/ni.1603
102. Cibrian D, Saiz ML, de la Fuente H, Sanchez-Diaz R, Moreno-Gonzalo O, Jorge I, et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat Immunol. (2016) 17:985–96. doi: 10.1038/ni.3504
103. Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, et al. Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA. (2014) 111:5307–12. doi: 10.1073/pnas.1322292111
104. Reagin KL, Klonowski KD. Incomplete memories: the natural suppression of tissue-resident memory CD8 T cells in the lung. Front Immunol. (2018) 9:17. doi: 10.3389/fimmu.2018.00017
105. Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, et al. T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science (2014) 346:101–5. doi: 10.1126/science.1254803
106. Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. (2011) 187:5510–4. doi: 10.4049/jimmunol.1102243
107. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. (2014) 95:215–24. doi: 10.1189/jlb.0313180
108. Gilchuk P, Hill TM, Guy C, McMaster SR, Boyd KL, Rabacal WA, et al. A distinct lung-interstitium-resident memory CD8(+) T cell subset confers enhanced protection to lower respiratory tract infection. Cell Rep. (2016) 16:1800–9. doi: 10.1016/j.celrep.2016.07.037
109. Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight (2016) 1:e85832. doi: 10.1172/jci.insight.85832
110. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science (2014) 346:98–101. doi: 10.1126/science.1254536
111. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest. (2018) 128:721–33. doi: 10.1172/JCI96957
112. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol. (2007) 28:12–8. doi: 10.1016/j.it.2006.11.004
113. Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. (2016) 13:88–110. doi: 10.1038/nrgastro.2015.200
114. Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science (2008) 322:268–71. doi: 10.1126/science.1164164
115. Griffin DE. Immune responses to RNA-virus infections of the CNS. Nat Rev Immunol. (2003) 3:493–502. doi: 10.1038/nri1105
116. Herz J, Johnson KR, McGavern DB. Therapeutic antiviral T cells noncytopathically clear persistently infected microglia after conversion into antigen-presenting cells. J Exp Med. (2015) 212:1153–69. doi: 10.1084/jem.20142047
117. Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat Med. (2010) 16:224–7. doi: 10.1038/nm.2078
118. Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity (2018) 48:327–38.e5. doi: 10.1016/j.immuni.2018.01.015
119. Schenkel JM, Fraser KA, Masopust D. Cutting edge: resident memory CD8 T cells occupy frontline niches in secondary lymphoid organs. J Immunol. (2014) 192:2961–4. doi: 10.4049/jimmunol.1400003
120. Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. (2018) 19:173–82. doi: 10.1038/s41590-017-0029-3
121. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. (2018) 19:183–91. doi: 10.1038/s41590-017-0027-5
122. Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK. Cutting edge: tissue-resident memory T cells generated by multiple immunizations or localized deposition provide enhanced immunity. J Immunol. (2017) 198:2233–7. doi: 10.4049/jimmunol.1601367
123. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. (2015) 7:269rv261. doi: 10.1126/scitranslmed.3010641
124. Bose T, Lee R, Hou A, Tong L, Chandy KG. Tissue resident memory T cells in the human conjunctiva and immune signatures in human dry eye disease. Sci Rep. (2017) 7:45312. doi: 10.1038/srep45312
125. Ickrath P, Kleinsasser N, Ding X, Ginzkey C, Beyersdorf N, Hagen R, et al. Accumulation of CD69+ tissue-resident memory T cells in the nasal polyps of patients with chronic rhinosinusitis. Int J Mol Med. (2018) 42:1116–24. doi: 10.3892/ijmm.2018.3653
126. Mizukawa Y, Yamazaki Y, Teraki Y, Hayakawa J, Hayakawa K, Nuriya H, et al. Direct evidence for interferon-gamma production by effector-memory-type intraepidermal T cells residing at an effector site of immunopathology in fixed drug eruption. Am J Pathol. (2002) 161:1337–47. doi: 10.1016/S0002-9440(10)64410-0
127. Teraki Y, Shiohara T. IFN-gamma-producing effector CD8+ T cells and IL-10-producing regulatory CD4+ T cells in fixed drug eruption. J Allergy Clin Immunol. (2003) 112:609–15. doi: 10.1016/S0091-6749(03)01624-5
128. Bhushan M, Bleiker TO, Ballsdon AE, Allen MH, Sopwith M, Robinson MK, et al. Anti-E-selectin is ineffective in the treatment of psoriasis: a randomized trial. Br J Dermatol. (2002) 146:824–31. doi: 10.1046/j.1365-2133.2002.04743.x
129. Teraki Y, Shiohara T. Preferential expression of alphaEbeta7 integrin (CD103) on CD8+ T cells in the psoriatic epidermis: regulation by interleukins 4 and 12 and transforming growth factor-beta. Br J Dermatol. (2002) 147:1118–26. doi: 10.1046/j.1365-2133.2002.05005.x
130. Ortega C, Fernández -A. S, Carrillo JM, Romero P, Molina IJ, Moreno JC, et al. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. (2009) 86:435–43. doi: 10.1189/JLB.0109046
131. Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, Ståhle M, et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol. (2014) 192:3111–20. doi: 10.4049/jimmunol.1302313
132. Li G, Larregina AT, Domsic RT, Stolz DB, Medsger TAJr, Lafyatis R, et al. Skin-resident effector memory CD8(+)CD28(-) T cells exhibit a profibrotic phenotype in patients with systemic sclerosis. J Invest Dermatol. (2017) 137:1042–50. doi: 10.1016/j.jid.2016.11.037
133. Kuric E, Seiron P, Krogvold L, Edwin B, Buanes T, Hanssen KF, et al. Demonstration of tissue resident memory CD8 T cells in insulitic lesions in adult patients with recent-onset type 1 diabetes. Am J Pathol. (2017) 187:581–8. doi: 10.1016/j.ajpath.2016.11.002
134. Radenkovic M, Uvebrant K, Skog O, Sarmiento L, Avartsson J, Storm P, et al. Characterization of resident lymphocytes in human pancreatic islets. Clin Exp Immunol. (2017) 187:418–27. doi: 10.1111/cei.12892
135. Machado-Santos J, Saji E, Troscher AR, Paunovic M, Liblau R, Gabriely G, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain (2018) 141:2066–82. doi: 10.1093/brain/awy151
136. Kiniry BE, Li S, Ganesh A, Hunt PW, Somsouk M, Skinner PJ, et al. Detection of HIV-1-specific gastrointestinal tissue resident CD8(+) T-cells in chronic infection. Mucosal Immunol. (2018) 11:909–20. doi: 10.1038/mi.2017.96
137. Bolte FJ, Rehermann B. Tissue-resident T cells in hepatitis B: a new target for cure? J Exp Med. (2017) 214:1564–6. doi: 10.1084/jem.20170842
138. Stelma F, de Niet A, Sinnige MJ, van Dort KA, van Gisbergen K, Verheij J, et al. Human intrahepatic CD69 + CD8+ T cells have a tissue resident memory T cell phenotype with reduced cytolytic capacity. Sci Rep. (2017) 7:6172. doi: 10.1038/s41598-017-06352-3
139. Ebert MP, Ademmer K, Muller-Ostermeyer F, Friess H, Buchler MW, Schubert W, et al. CD8+CD103+ T cells analogous to intestinal intraepithelial lymphocytes infiltrate the pancreas in chronic pancreatitis. Am J Gastroenterol. (1998) 93:2141–7. doi: 10.1111/j.1572-0241.1998.00610.x
140. Owens GC, Chang JW, Huynh MN, Chirwa T, Vinters HV, Mathern GW. Evidence for resident memory T cells in Rasmussen Encephalitis. Front Immunol. (2016) 7:64. doi: 10.3389/fimmu.2016.00064
141. Posavad CM, Zhao L, Dong L, Jin L, Stevens CE, Magaret AS, et al. Enrichment of herpes simplex virus type 2 (HSV-2) reactive mucosal T cells in the human female genital tract. Mucosal Immunol. (2017) 10:1259–69. doi: 10.1038/mi.2016.118
142. Woodberry T, Suscovich TJ, Henry LM, August M, Waring MT, Kaur A, et al. Alpha E beta 7 (CD103) expression identifies a highly active, tonsil-resident effector-memory CTL population. J Immunol. (2005) 175:4355–62. doi: 10.4049/jimmunol.175.7.4355
143. Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. (2018) 24:986–93. doi: 10.1038/s41591-018-0078-7
144. Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpreville V, et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol. (2015) 194:3475–86. doi: 10.4049/jimmunol.1402711
145. Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol. (2017) 18:940–50. doi: 10.1038/ni.3775
146. Webb JR, Milne K, Watson P, Deleeuw RJ, Nelson BH. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin Cancer Res. (2014) 20:434–44. doi: 10.1158/1078-0432.CCR-13-1877
147. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
148. Khanna KM, Bonneau RH, Kinchington PR, Hendricks RL. Herpes simplex virus-specific memory CD8+ T cells are selectively activated and retained in latently infected sensory ganglia. Immunity (2003) 18:593–603. doi: 10.1016/S1074-7613(03)00112-2
149. Verjans GM, Hintzen RQ, van Dun JM, Poot A, Milikan JC, Laman JD, et al. Selective retention of herpes simplex virus-specific T cells in latently infected human trigeminal ganglia. Proc Natl Acad Sci USA. (2007) 104:3496–501. doi: 10.1073/pnas.0610847104
150. Smith CJ, Caldeira-Dantas S, Turula H, Snyder CM. Murine CMV infection induces the continuous production of mucosal resident T cells. Cell Rep. (2015) 13:1137–48. doi: 10.1016/j.celrep.2015.09.076
151. Thom JT, Weber TC, Walton SM, Torti N, Oxenius A. The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep. (2015) 13:1125–36. doi: 10.1016/j.celrep.2015.09.082
152. Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, et al. Identification and characterization of HIV-specific resident memory CD8(+) T cells in human lymphoid tissue. Sci Immunol. (2018) 3:eaar4526. doi: 10.1126/sciimmunol.aar4526
153. O'Hara GA, Welten SP, Klenerman P, Arens R. Memory T cell inflation: understanding cause and effect. Trends Immunol. (2012) 33:84–90. doi: 10.1016/j.it.2011.11.005
154. Welten SPM, Sandu I, Baumann NS, Oxenius A. Memory CD8 T cell inflation vs tissue-resident memory T cells: same patrollers, same controllers? Immunol Rev. (2018) 283:161–75. doi: 10.1111/imr.12649
155. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
156. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
157. Fridman WH. The immune microenvironment as a guide for cancer therapies. Oncoimmunology (2012) 1:261–2. doi: 10.4161/onci.19651
158. Becht E, Giraldo NA, Dieu-Nosjean MC, Sautes-Fridman C, Fridman WH. Cancer immune contexture and immunotherapy. Curr Opin Immunol. (2016) 39:7–13. doi: 10.1016/j.coi.2015.11.009
159. Webb JR, Milne K, Nelson BH. PD-1 and CD103 are widely coexpressed on prognostically favorable intraepithelial CD8 T cells in human ovarian cancer. Cancer Immunol Res. (2015) 3:926–35. doi: 10.1158/2326-6066.CIR-14-0239
160. Boddupalli CS, Bar N, Kadaveru K, Krauthammer M, Pornputtapong N, Mai Z, et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight (2016) 1:e88955. doi: 10.1172/jci.insight.88955
161. Komdeur FL, Wouters MC, Workel HH, Tijans AM, Terwindt AL, Brunekreeft KL, et al. CD103+ intraepithelial T cells in high-grade serous ovarian cancer are phenotypically diverse TCRalphabeta+ CD8alphabeta+ T cells that can be targeted for cancer immunotherapy. Oncotarget (2016) 7:75130–44. doi: 10.18632/oncotarget.12077
162. Murray T, Fuertes Marraco SA, Baumgaertner P, Bordry N, Cagnon L, Donda A, et al. Very late antigen-1 marks functional tumor-resident CD8 T cells and correlates with survival of melanoma patients. Front Immunol. (2016) 7:573. doi: 10.3389/fimmu.2016.00573
163. Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martinez-Cano S, et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8(+) T cells. Nat Commun. (2017) 8:16073. doi: 10.1038/ncomms16073
164. Malik BT, Byrne KT, Vella JL, Zhang P, Shabaneh TB, Steinberg SM, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol. (2017) 2:eaam6346. doi: 10.1126/sciimmunol.aam6346
165. Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat Commun. (2017) 8:15221. doi: 10.1038/ncomms15221
166. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
167. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
168. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology (2013) 138:105–15. doi: 10.1111/imm.12036
169. Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, et al. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. (2015) 13:e1002202. doi: 10.1371/journal.pbio.1002202
170. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell (2017) 32:377–91.e9. doi: 10.1016/j.ccell.2017.08.004
171. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science (2018) 359:1037–42. doi: 10.1126/science.aar3246
172. Wu TC, Xu K, Banchereau R, Marches F, Yu CI, Martinek J, et al. Reprogramming tumor-infiltrating dendritic cells for CD103+ CD8+ mucosal T-cell differentiation and breast cancer rejection. Cancer Immunol Res. (2014) 2:487–500. doi: 10.1158/2326-6066.CIR-13-0217
173. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
174. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103(+) Tumor-resident CD8(+) T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res. (2018) 24:3036–45. doi: 10.1158/1078-0432.CCR-17-2257
175. Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol. (2010) 130:362–70. doi: 10.1038/jid.2009.247
176. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. (2015) 78:710–21. doi: 10.1002/ana.24497
177. Dal-Bianco A, Grabner G, Kronnerwetter C, Weber M, Hoftberger R, Berger T, et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. (2017) 133:25–42. doi: 10.1007/s00401-016-1636-z
178. Suarez-Farinas M, Fuentes-Duculan J, Lowes MA, Krueger JG. Resolved psoriasis lesions retain expression of a subset of disease-related genes. J Invest Dermatol. (2011) 131:391–400. doi: 10.1038/jid.2010.280
179. Shiohara T. Fixed drug eruption: pathogenesis and diagnostic tests. Curr Opin Allergy Clin Immunol. (2009) 9:316–21. doi: 10.1097/ACI.0b013e32832cda4c
180. Ali N, Zirak B, Truong HA, Maurano MM, Gratz IK, Abbas AK, et al. Skin-resident T cells drive dermal dendritic cell migration in response to tissue self-antigen. J Immunol. (2018) 200:3100–8. doi: 10.4049/jimmunol.1701206
181. Shin H, Kumamoto Y, Gopinath S, Iwasaki A. CD301b+ dendritic cells stimulate tissue-resident memory CD8+ T cells to protect against genital HSV-2. Nat Commun. (2016) 7:13346. doi: 10.1038/ncomms13346
182. Kohm AP, Fuller KG, Miller SD. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. Trends Microbiol. (2003) 11:101–5. doi: 10.1016/S0966-842X(03)00006-4
183. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. (2006) 19:80–94. doi: 10.1128/CMR.19.1.80-94.2006
184. Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun. (2016) 7:11514. doi: 10.1038/ncomms11514
185. Collins N, Hochheiser K, Carbone FR, Gebhardt T. Sustained accumulation of antigen-presenting cells after infection promotes local T-cell immunity. Immunol Cell Biol. (2017) 95:878–83. doi: 10.1038/icb.2017.60
186. Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T, et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat Immunol. (2014) 15:1064–9. doi: 10.1038/ni.2992
187. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
188. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. Ectopic lymphoid structures: powerhouse of autoimmunity. Front Immunol. (2016) 7:430. doi: 10.3389/fimmu.2016.00430
189. Munz C, Lunemann JD, Getts MT, Miller SD. Antiviral immune responses: triggers of or triggered by autoimmunity? Nat Rev Immunol. (2009) 9:246–58. doi: 10.1038/nri2527
190. Boldison J, Chu CJ, Copland DA, Lait PJ, Khera TK, Dick AD, et al. Tissue-resident exhausted effector memory CD8+ T cells accumulate in the retina during chronic experimental autoimmune uveoretinitis. J Immunol. (2014) 192:4541–50. doi: 10.4049/jimmunol.1301390
191. Graham JB, Da Costa A, Lund JM. Regulatory T cells shape the resident memory T cell response to virus infection in the tissues. J Immunol. (2014) 192:683–90. doi: 10.4049/jimmunol.1202153
Keywords: resident memory T cells, chronic, inflammation, infection, autoimmune
Citation: Steinbach K, Vincenti I and Merkler D (2018) Resident-Memory T Cells in Tissue-Restricted Immune Responses: For Better or Worse? Front. Immunol. 9:2827. doi: 10.3389/fimmu.2018.02827
Received: 31 July 2018; Accepted: 15 November 2018;
Published: 30 November 2018.
Edited by:
Weiguo Cui, Bloodcenter of Wisconsin, United StatesReviewed by:
Vandana Kalia, School of Medicine, University of Washington, United StatesBrian S. Sheridan, Stony Brook University, United States
Copyright © 2018 Steinbach, Vincenti and Merkler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Doron Merkler, ZG9yb24ubWVya2xlckB1bmlnZS5jaA==