 Inga Kavazović1
Inga Kavazović1 Felix M. Wensveen
Felix M. Wensveen- 1Department of Histology & Embryology, University of Rijeka, Rijeka, Croatia
- 2Department of Experimental Immunology, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
Effector and memory CD8 T cells have an intrinsic difference in the way they must approach antigen; effector cells need to address the pathogen at hand and therefore favor outgrowth of only high-affinity clones. In contrast, the memory pool benefits from greater clonal diversity to recognize and eliminate pathogens with mutations in their immunogenic epitopes. Effector and memory fates are ultimately the result of the same three signals that control T cell activation; T cell receptor (TCR) engagement together with co-stimulation and cytokines. Great progress has been made in our understanding of the transcriptional programs that drive effector or memory differentiation. However, how these two different programs result from the same initial cues is still a matter of debate. An emerging image is that not only the classical three signals determine T cell differentiation, but also the ability of cells to access these signals relative to that of other activated clones. Inter-clonal competition is therefore not only a selective force, but also a mediator of CD8 T cell fate. How this is regulated on a transcriptional level, especially in the context of a selective “hunger game” based on antigen-affinity in which only cells of high-affinity are supposed to survive, is still poorly defined. In this review, we discuss recent literature that illustrates how antigen-affinity dependent inter-clonal competition shapes effector and memory populations in an environment of antigen affinity-driven selection. We argue that fine-tuning of TCR signal intensity presents an attractive target for regulating the scope of CD8 T cell vaccines.
Introduction
CD8 T cells play a critical role in the protection of our body from the occurrence and recurrence of intracellular pathogens and tumors. To recognize the large number of potential threats, the naïve CD8 T cell pool consists of millions of clones, each unique based on its antigen receptor. To prevent an excessive use of resources for the maintenance of these cells, each clone is present at low frequency. Only upon activation do antigen-specific clones expand to form the effector and memory pools (1–4). Naïve CD8 T cells need three separate signals for optimal effector and memory generation: (1) antigen recognition by the T cell receptor (TCR), (2) co-stimulation, and (3) cytokines (5). These three signals are not hierarchically equal. Generally, only in case of TCR engagement do co-stimulation and cytokines contribute to T cell activation. Moreover, the affinity of the TCR determines the capacity of an activated cell to access vital co-stimulatory molecules, cytokines and nutrients (6). Considering the vast diversity of the naïve CD8 T cell pool, statistical probability dictates that for any given antigen, many more low- than high-affinity clones exist. To mount an efficient CD8 T cell response, selection of activated clones based on antigen-specificity must take place (6, 7). We have therefore proposed a fourth factor that controls effector and memory T cell formation: “competitive fitness”—the ability to compete for extracellular signals with other activated T cell clones based on antigen affinity (8).
The parameters that determine competitive fitness differ between effector and memory cells, because of the difference in the way that these pools must approach antigen. Upon infection, the effector pool is faced with an actively replicating pathogen and therefore only the most efficient, high-affinity clones are selected into its ranks (6, 9). Immunological memory must protect the host against re-infection with a previously encountered pathogen. Due to selective pressure on the original pathogen as it moves through its host population, re-infections are more likely to occur with a variant carrying mutations in its immunogenic epitopes (10–12). Hence, selection of memory clones is a trade-off between specificity and diversity. Too much specificity restricts antigen-recognition, which precludes responsiveness against mutated pathogens. Too much diversity impairs efficiency of recall responses. In both mice and humans increasing the diversity of the memory pool enables recognition of a larger fraction of the potential pathogen-carried sequence space resulting in a higher probability of recognizing mutated pathogens (13, 14). How clonal selection within the effector and memory cell populations is regulated is only partially understood. Here, we propose a crucial role of TCR signaling in an affinity-based inter-clonal competition which shapes clonal diversity and regulates effector and memory differentiation.
The Impact of Signal Intensity on CD8 T Cell Differentiation
The initiating event for CD8 T cell activation is recognition of an antigen embedded in the major histocompatibility complex (pMHC) on an antigen-presenting cell (APC) by the TCR. This results in the activation of a network of signaling cascades that mediate differentiation, proliferation, and survival (15, 16). Upon activation, a single naïve CD8 T cell has the potential to give rise to various effector and memory CD8 T cell subsets (17, 18). Divergent cell fates depend on the intensity of the cumulative signal activating an individual CD8 T cell (19). This signal strength represents the sum of different factors such as the affinity and avidity of TCR binding to antigen-pMHC complexes, co-stimulation, and cytokines (8, 20–22).
Initially, it was proposed that only a cumulative signal of high overall strength allows T cell activation and formation of effector and memory cells (23). This was based on the observation that only cells of high-affinity vigorously expand upon activation (24). The model was challenged by the finding that even very weak TCR-pMHC interactions promote proliferation and generation of functional memory (25, 26). In addition, even a brief 2 h priming phase was shown to be sufficient to induce the complete diversity of effector and memory CD8 T cell subsets (27–29). To analyze these processes more directly, SIINFEKL (Ova)-specific OT-1 cells were transferred to naïve recipient mice, which were subsequently infected with L. monocytogenes (LM) expressing Ova or altered peptide ligands (APL) that bind the OT-1 TCR with lower affinity. This revealed that even weak ligands are sufficient to activate naïve cells and mediate formation of both effector and memory T cells (30). This raised the question how the immune system prevents that clones of low specificity and efficiency expand and exhaust the limited amount of available resources. The answer came from the observation that the potency to induce effector cell proliferation positively correlates with the intensity of the TCR signal (24, 30–32). Decreasing the cumulative signal strength by pretreating mice with antibiotics before L. monocytogenes infection and thus lowering antigenic load resulted in reduced expansion of antigen specific effector T cells (33, 34). In addition to a proliferative advantage of high-affinity cells, activated effector CD8 T cells were shown to undergo negative selection of low-affinity clones based on a reduced capacity of these cells to access and thus outcompete other clones for limited resources (8). Upon activation T cells induce expression of the IL-2 receptor in an antigen-affinity dependent manner (6, 30). IL-2 mediates survival by triggering the PI3K signaling cascade and sustaining the pro-survival protein Mcl-1 (Figure 1). High-affinity effector cells therefore have a competitive survival advantage over low-affinity cells in their ability to access IL-2. This selection process narrows clonal diversity, since only highly specific clones are allowed to generate progeny and create an almost monoclonal effector CD8 T cell pool (6, 8). Animals lacking Noxa, a pro-apoptotic antagonist of Mcl-1, have a reduced survival threshold for effector cells and therefore showed reduced dependency on IL-2. As a result, these mice had an increased number of low-affinity clones contributing to the effector pool, which was of reduced anti-viral potential (6).
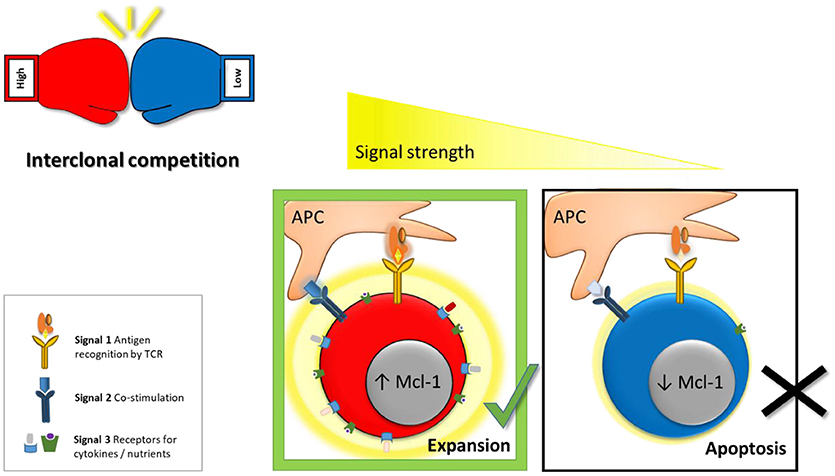
Figure 1. Model for inter-clonal competition between effector cells based on antigen-affinity. For efficient activation and optimal effector CD8 T cell formation 3 signals are required (1) antigen recognition by the TCR, (2) co-stimulation, and (3) cytokines. We proposed as a fourth factor “competitive fitness”—the ability to compete for these signals with other activated T cell clones. Cumulative signal strenght (visualized by a graded yellow halo) is the main factor controling the capacity of activated lymphocytes to access vital co-stimulatory molecules, cytokines and nutrients (e.g., glucose, amino acids). Thus, high-affinity effector cells have a competitive advantage over low-affinity cells in their ability to access these signals. In addition, high-affinity cells take-up more IL-2 which in turn mediates survival of high-affinity clones by triggering the PI3K signaling cascade and sustaining pro-survival proteins such as Mcl-1. Hence, low-affinity clones undergo negative selection through apoptosis to ensure that only the fittest, high-affinity clones contribute to the antiviral response.
Co-stimulation and cytokines greatly contribute to the cumulative activating signal intensity and therefore have a major impact on TCR-affinity mediated selection of CD8 T cell clones. CD28-driven co-stimulation is essential for proper CD8 T cell responses after weak TCR-pMHC interactions. Conversely, high antigen doses and prolonged antigen stimulation can compensate for a lack of CD28 co-stimulation in vivo (35, 36). CD27-driven co-stimulation promotes production of IL-2 in activated T cells (37). Animals deficient for CD27 therefore have reduced access to IL-2, resulting in a less clonally diverse effector response of increased overall affinity (13). Notably, expression of CD70, the ligand of CD27, is regulated by antigen avidity (13, 38–40), but whether this contributes to the diversity of the effector response is unknown. Similarly, cytokines impact cell fate decisions and clonal selection mechanisms. CD8 T cells activated in the presence of high levels of IL-2 or IL-12 exhibit increased proliferation rates and superior effector functions (23, 30, 33, 41–43). Exogenous addition of IL-2 rescued survival of low-affinity cells (6), indicating that stronger inflammatory responses will allow for more clones to contribute to the effector response, though this does not necessarily promote their dominance.
In summary, TCR signaling is not an on/off switch. Rather, it enables integration of signals with different intensities, which are further amplified by the right cytokines and co-stimulatory molecules. Fine-tuning of TCR signal intensity shapes T cell differentiation and clonal selection.
Inter-Clonal Competition in the Context of Effector and Memory Formation
Even though a naïve cell can generate both effector and memory cells (44), memory potential is associated with weaker activating signals. Very low affinity antigens are still able to induce memory formation but have a strongly reduced capacity to induce effector differentiation (30, 45). Exogenous factors such as IL-2, IL-12, or CD28 co-stimulation add to the cumulative activating signal and help activated cells to obtain an effector phenotype (43, 46). Very high levels of stimulation, in contrast, push T cells “beyond” an effector stage into exhaustion (47, 48). Various models have been proposed how activating signal strength regulates CD8 memory formation. The “decreasing potential” model suggests that memory formation is the “default” state of activated T cells and that effector memory or effector cell differentiation is only possible if a certain level of activation is reached(49, 50). Whether this level represents a binary threshold, or whether effector potential is gradually increased in response to increasing signal strength is a matter debate and appears to depend on the molecules that are used to determine threshold values(43, 51–55).
Mostly, the impact of affinity on effector and memory potential has been interrogated by presenting a single (TCR-transgenic) T cell clone, with high- or low-affinity ligands (30, 45). However, a biologically more relevant question is how signal strength is linked to memory formation, not at the level of a single clone but in the context of the entire antigen-responsive population. Statistical probability dictates that for a given antigen, many more high-affinity than low-affinity cells exist within the naïve T cell pool. Hence, molecular mechanisms are in place to ensure that preferentially cells of high-efficiency are selected into both the effector and memory cell pools (6–8, 13). The impact of cumulative signal intensity is therefore not only a checkpoint controlling effector vs. memory fate decisions, but also controls the competitive fitness of cells in a selective environment that regulates the diversity of antigen-experienced T cell populations. To shed more light on this concept, experiments were performed in which a pool of individually labeled OT-1 cells was transferred to a host which was subsequently infected with LM-Ova. Analysis of donor cells revealed that even within a monoclonal high-affinity population, a relatively small fraction of clones dominates the effector response (17, 18). This would suggest that only a small number of cells reaches the cumulative signaling threshold required for CD8 T cell expansion. When a sufficiently high number of monoclonal cells is transferred, stochastic effects are negated, which ensures that in experimental settings donor cells usually make a significant contribution to the effector response (18). However, in a physiological setting, each clone is present at very low frequency (3). This indicates that inter-clonal competition becomes an important factor that controls shaping of the antigen-specific cell pool. Indeed, when mice were transferred with only a single OT-1 cell, in less than one third of animals these cells could be recovered after infection with LM-Ova (18). The recruitment of antigen-specific cells into the immune response is highly efficient and nearly complete (56), excluding limited antigen-exposure as a determining factor. Thus, considering the fact that effector cells are derived from a small number of precursors that is able to generate exponential expansion (17, 18), small differences in competitive fitness will ensure highly selective outgrowth of clones.
In the cell-tracing experiment, cells that did not undergo massive expansion generally adopted a memory-like phenotype (18). Together with the observation that low-affinity cells preferentially form memory, the question arises whether only the effector pool is selected for high-affinity clones and that the memory pool allows contribution of all activated cells. Studies in which the clonal diversity of effector and memory cells was directly compared showed that the effector cell pool is much more restricted in its clonal diversity than the memory pool directed against the same antigen (7, 13, 57). However, clones that dominate the effector pool are also dominant in the memory population, albeit to a lower degree (7, 13, 57). Low cumulative signal strength favors memory formation and is associated with reduced proliferation (30, 31). Why then, is the memory response not completely dominated by low-affinity cells? One possibility is that high-affinity cells have a selective advantage also during memory formation. Another option is that they preferentially use a different mechanism to form memory than low-affinity cells. These models are not mutually exclusive and experimental evidence for both exist (Figure 2). Mice deficient for the co-stimulatory molecule CD27 generate a memory pool of comparable size as wild type controls yet is almost devoid of low-affinity clones (13). Similarly, low-affinity cells have a higher dependence on TNF receptor signaling during recall (58). This indicates that low-affinity memory precursors have increased dependence on factors that contribute to the cumulative activating signal and thus have a survival disadvantage when competing for these factors.
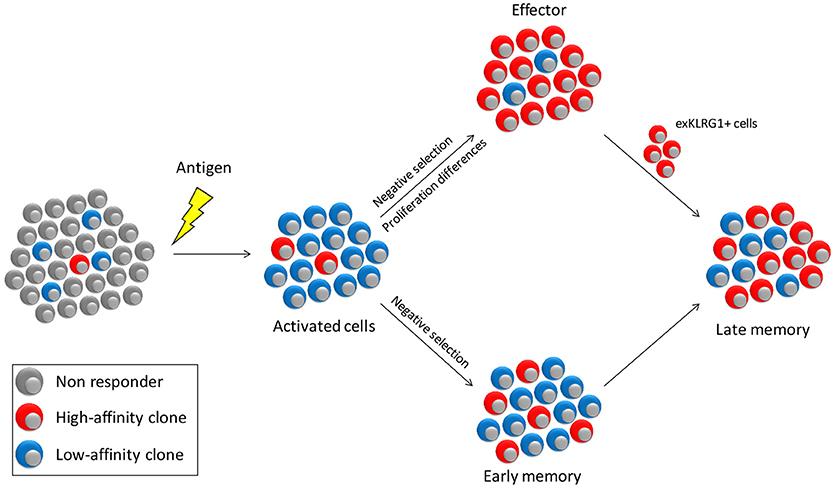
Figure 2. Model for affinity selection within the effector and memory pools. Antigen encounter will activate a small number of high affinity cells and a much larger number of cells with lower affinity. The effector pool is stringently selected for cells of the highest affinity both through negative selection of low-affinity cells and through a proliferative advantage of high- over low-affinity cells. In contrast, affinity-based selection of early memory is less strong, since differences in proliferation between high- and low-affinity cells is less pronounced. Selection of high-affinity cells will therefore primarily occur due to competition for survival factors such as CD27 co-stimulation and cytokines. Late memory is supplemented by exKLRG1 cells, which are predominantly of high-affinity, thus increasing the overall affinity of the memory pool.
Antigen-experienced cells can be subdivided based on different parameters, but a common segregation uses IL-7Rα (CD127) and KLRG1. Memory precursors (MPECs) are defined as CD127+KLRG1− whereas short lived effectors (SLECs) have the converse phenotype. A recent study indicates, however, that with the CD127+KLRG1+ cell pool and even among SLECs, cells exist that form “exKLRG1+” memory after clearance of a pathogen (59). Even though the frequency of cells with memory potential in these pools is much lower than amongst MPECs, the high number of KLRG1+ cells formed during an immune response ensures that in absolute numbers exKLRG1+ cells make a significant contribution to the memory pool (59). High-affinity cells preferentially form cells with a SLEC phenotype, whereas low-affinity cells more rapidly become MPECs (30, 45). Even though direct experimental evidence is still lacking, these findings indicate that low-affinity memory cells are formed directly, whereas high-affinity memory is also derived from exKLRG1+ effector type cells (Figure 2).
Maintenance of CD8 memory cells is independent of antigen and predominantly depends on cytokines such as IL-15 and IL-7 (60, 61). Whereas expression of cytokine receptors differs between cells of high- and low-affinity early after activation, at later time-points these differences are lost (6, 30). In the first weeks after clearance of a pathogen, the avidity of the antigen-specific pool therefore still changes as long-lived effector cells undergo apoptosis (62). However, once the clonal composition of the memory pool is established it remains stable for months to years after initial infection, both in humans and mice (63–66). Thus, clonal diversity of the memory CD8 T cell pool appears to be a long-term investment of the immune system to counter viral mutants.
An open question is how memory cell formation is influenced by inter-clonal competition on a molecular level. Various factors important for effector cell formation are induced in a way that directly correlates with antigen affinity, such as T-bet, IRF4, and Blimp-1 and these suppress expression of memory-associated molecules, such as Eomes and Tcf7 (54, 67, 68). The transcription factor IRF4 was found to regulate expansion of effector cells by promoting the metabolic switch to aerobic glycolysis in a TCR affinity–dependent manner. IRF4 expression was higher in high-affinity clones, ensuring their preferential expansion and effector differentiation over low-affinity clones (69, 70). Surprisingly, both Eomes and Tcf7 are induced upon activation of T cells (71, 72) and expression of Eomes can even be higher in high- than in low-affinity cells, dependent on the level of stimulation (45, 70). Notably, both T-bet and Eomes are essential for CD8 T cells to obtain a normal effector cell phenotype (72). The ratio between these molecules, rather than their expression level therefore appears to determine whether a cell obtains a memory or effector cell phenotype (46). How this dynamic regulation of transcription factors is regulated in the context of affinity-based selection in effector and memory cell pools remains to be elucidated.
In summary, the impact of cumulative signal intensity on effector vs. memory cell differentiation should be viewed in the context of clonal selection strategies that shape the antigen-specific cell pools. The impact of affinity on cell fate decisions appears to have evolved in order to ensure selection of only highly specific cells in the effector cell pool, whilst allowing sufficient diversity of CD8 T cell memory in a pool that is still dominated by high-affinity cells.
Therapeutic Potential of Clonal Diversity
Enhanced diversity within the memory CD8 T cell pool is of particular benefit against re-infections with rapidly mutating viruses (73). For example, HIV patients appear to benefit from greater clonal diversity of their virus-specific T cell response (13, 14, 73–77). As an effective vaccine against HIV remains elusive, future strategies may involve manipulation of IL-2 levels and/or co-stimulatory molecules during priming to broaden the scope of the immune response. Evolutionary, increased clonal diversity of the memory pool compared to the effector pool is an acceptable strategy, as it does not appear to greatly reduce recall capacity against the original antigen. Cd27−/− mice, which generate a memory CD8 T cell pool that almost exclusively consists of high-affinity cells do not show an increased recall response following re-infection with a pathogen carrying high-affinity ligands (13). Similarly, co-transfer of high- and low-affinity memory cells directed against the same antigen does not result in a reduced ability of high-affinity cells to expand upon antigen re-encounter (45). In fact, re-encounter of the same antigen further skews the secondary effector pool in favor of high-affinity clones. In addition, re-infection with pathogens carrying a mutated immuno-dominant epitope promotes selective outgrowth of previously low-frequency clones that have now become of high-affinity (13, 65). Thus, clonal selection plays an important role both during primary and secondary responses, but does not affect functionality of subdominant clones. Increasing memory diversity of a vaccine against pathogens is therefore unlikely to reduce the overall effectiveness of protection.
Vaccination against tumors should target only transformed cells while avoiding unnecessary damage of healthy tissue. Reducing the number of targeted epitopes included in a vaccine lowers chances of off-target effects, but also limits the effectiveness of a vaccine and allows for more rapid outgrowth of cells with mutations in their immunogenic epitopes. Rather, anti-tumor vaccination in combination with a strategy that narrows the scope of the immune response per epitope holds promise for a more efficient and specific treatment. A better understanding of the molecular mechanisms that control the diversity of the T cell response are therefore of crucial importance (8, 78, 79).
The degree of heterogeneity within the CD8 T cell response depends on the ability of activated clones to integrate signals from the TCR, co-stimulatory molecules and cytokines, but also their relative fitness in an environment of rapidly expanding cells competing for the same resources. Recent studies demonstrate the importance TCR signal strength in regulating T cell differentiation, but much remains unknown about the molecular mechanisms that control the clonal selection strategies that shape the diversity of the effector and memory pools. Deeper insight in the transcriptional network underlying affinity-based clonal selection therefore holds great promise for the development of novel, more efficient CD8 T cell vaccines with an altered scope.
Author Contributions
IK and FMW wrote the manuscript. IK, FMW, and BP participated in drafting and editing the text and figures. All authors gave final approval to the version submitted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Netherlands Organization for Scientific Research (Veni grant 91614029 to FMW), the European Commission (PCIG14-GA-2013-630827 to FMW), a University of Rijeka Support grant (865.10.2101 to FMW) and the Croatian Science Foundation (IP-2016-06-8027 to FMW and IP-2016-06-9306 to BP).
References
1. Casrouge A, Beaudoing E, Dalle S, Pannetier C, Kanellopoulos J, Kourilsky P. Size estimate of the alpha beta TCR repertoire of naive mouse splenocytes. J Immunol. (2000) 164:5782–7. doi: 10.4049/jimmunol.164.11.5782
2. Bousso P, Casrouge A, Altman JD, Haury M, Kanellopoulos J, Abastado JP, et al. Individual variations in the murine T cell response to a specific peptide reflect variability in naive repertoires. Immunity (1998) 9:169–78. doi: 10.1016/S1074-7613(00)80599-3
3. Blattman JN, Antia R, Sourdive DJ, Wang X, Kaech SM, Murali-Krishna K, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. J Exp Med. (2002) 195:657–64. doi: 10.1084/jem.20001021
4. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science (1999) 286:958–61. doi: 10.1126/science.286.5441.958
5. Curtsinger JM, Mescher MF. Inflammatory cytokines as a third signal for T cell activation. Curr Opin Immunol. (2010) 22:333–40. doi: 10.1016/j.coi.2010.02.013
6. Wensveen FM, van Gisbergen KP, Derks IA, Gerlach C, Schumacher TN, van Lier RA, et al. Apoptosis threshold set by Noxa and Mcl-1 after T cell activation regulates competitive selection of high-affinity clones. Immunity (2010) 32:754–65. doi: 10.1016/j.immuni.2010.06.005
7. Wensveen FM, Klarenbeek PL, van Gisbergen KP, Pascutti MF, Derks IA, van Schaik BD, et al. Pro-apoptotic protein Noxa regulates memory T cell population size and protects against lethal immunopathology. J Immunol. (2013) 190:1180–91. doi: 10.4049/jimmunol.1202304
8. Wensveen FM, van Gisbergen KP, Eldering E. The fourth dimension in immunological space: how the struggle for nutrients selects high-affinity lymphocytes. Immunol Rev. (2012) 249:84–103. doi: 10.1111/j.1600-065X.2012.01156.x
9. Butz EA, Bevan MJ. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity (1998) 8:167–75. doi: 10.1016/S1074-7613(00)80469-0
10. Koup RA. Virus escape from CTL recognition. J Exp Med. (1994) 180:779–82. doi: 10.1084/jem.180.3.779
11. Ince WL, Gueye-Mbaye A, Bennink JR, Yewdell JW. Reassortment complements spontaneous mutation in influenza A virus NP and M1 genes to accelerate adaptation to a new host. J Virol. (2013) 87:4330–8. doi: 10.1128/JVI.02749-12
12. Brehm MA, Pinto AK, Daniels KA, Schneck JP, Welsh RM, Selin LK. T cell immunodominance and maintenance of memory regulated by unexpectedly cross-reactive pathogens. Nat Immunol. (2002) 3:627–34. doi: 10.1038/ni806
13. van Gisbergen KP, Klarenbeek PL, Kragten NA, Unger PP, Nieuwenhuis MB, Wensveen FM, et al. The costimulatory molecule CD27 maintains clonally diverse CD8(+) T cell responses of low antigen affinity to protect against viral variants. Immunity (2011) 35:97–108. doi: 10.1016/j.immuni.2011.04.020
14. Pantaleo G, Demarest JF, Schacker T, Vaccarezza M, Cohen OJ, Daucher M, et al. The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia. Proc Natl Acad Sci USA. (1997) 94:254–8. doi: 10.1073/pnas.94.1.254
15. Cantrell D. Signaling in lymphocyte activation. Cold Spring Harb Perspect Biol. (2015) 2015:7. doi: 10.1101/cshperspect.a018788
16. Brownlie RJ, Zamoyska R. T cell receptor signalling networks: branched, diversified and bounded. Nat Rev Immunol. (2013) 13:257–69. doi: 10.1038/nri3403
17. Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science (2013) 340:635–9. doi: 10.1126/science.1235487
18. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Graf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science (2013) 340:630–5. doi: 10.1126/science.1235454
19. Lanzavecchia A, Sallusto F. Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol. (2002) 2:982–7. doi: 10.1038/nri959
20. Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science (1999) 283:680–2. doi: 10.1126/science.283.5402.680
21. Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature (1995) 375:148–51. doi: 10.1038/375148a0
22. Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol. (2011) 186:5039–45. doi: 10.4049/jimmunol.1003650
23. Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol. (2004) 16:217–25. doi: 10.1016/j.coi.2004.02.005
24. Busch DH, Pamer EG. T cell affinity maturation by selective expansion during infection. J Exp Med. (1999) 189:701–10. doi: 10.1084/jem.189.4.701
25. Hamilton SE, Wolkers MC, Schoenberger SP, Jameson SC. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat Immunol. (2006) 7:475–81. doi: 10.1038/ni1326
26. Goldrath AW, Bevan MJ. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity (1999) 11:183–90. doi: 10.1016/S1074-7613(00)80093-X
27. van Stipdonk MJ, Lemmens EE, Schoenberger SP. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nat Immunol. (2001) 2:423–9. doi: 10.1038/87730
28. Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. (2000) 165:6833–9. doi: 10.4049/jimmunol.165.12.6833
29. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. (2001) 2:415–22. doi: 10.1038/87720
30. Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature (2009) 458:211–4. doi: 10.1038/nature07657
31. Hommel M, Hodgkin PD. TCR affinity promotes CD8+ T cell expansion by regulating survival. J Immunol. (2007) 179:2250–60. doi: 10.4049/jimmunol.179.4.2250
32. Wherry EJ, Puorro KA, Porgador A, Eisenlohr LC. The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J Immunol. (1999) 163:3735–45.
33. Badovinac VP, Porter BB, Harty JT. CD8+ T cell contraction is controlled by early inflammation. Nat Immunol. (2004) 5:809–17. doi: 10.1038/ni1098
34. Badovinac VP, Harty JT. Manipulating the rate of memory CD8+ T cell generation after acute infection. J Immunol. (2007) 179:53–63. doi: 10.4049/jimmunol.179.1.53
35. Kundig TM, Shahinian A, Kawai K, Mittrucker HW, Sebzda E, Bachmann MF, et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity (1996) 5:41–52. doi: 10.1016/S1074-7613(00)80308-8
36. Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. (2003) 3:939–51. doi: 10.1038/nri1248
37. Peperzak V, Xiao Y, Veraar EA, Borst J. CD27 sustains survival of CTLs in virus-infected nonlymphoid tissue in mice by inducing autocrine IL-2 production. J Clin Invest. (2010) 120:168–78. doi: 10.1172/JCI40178
38. Wensveen FM, Unger PP, Kragten NA, Derks IA, ten Brinke A, Arens R, et al. CD70-driven costimulation induces survival or Fas-mediated apoptosis of T cells depending on antigenic load. J Immunol. (2012) 188:4256–67. doi: 10.4049/jimmunol.1102889
39. Penaloza-MacMaster P, Ur Rasheed A, Iyer SS, Yagita H, Blazar BR, Ahmed R. Opposing effects of CD70 costimulation during acute and chronic lymphocytic choriomeningitis virus infection of mice. J Virol. (2011) 85:6168–74. doi: 10.1128/JVI.02205-10
40. Matter M, Odermatt B, Yagita H, Nuoffer JM, Ochsenbein AF. Elimination of chronic viral infection by blocking CD27 signaling. J Exp Med. (2006) 203:2145–55. doi: 10.1084/jem.20060651
41. Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity (2010) 32:79–90. doi: 10.1016/j.immuni.2009.11.012
42. Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. (2005) 11:748–56. doi: 10.1038/nm1257
43. Au-Yeung BB, Smith GA, Mueller JL, Heyn CS, Jaszczak RG, Weiss A, et al. IL-2 modulates the TCR signaling threshold for CD8 but not CD4 T cell proliferation on a single-cell level. J Immunol. (2017) 198:2445–56. doi: 10.4049/jimmunol.1601453
44. Gerlach C, van Heijst JW, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. (2010) 207:1235–46. doi: 10.1084/jem.20091175
45. Knudson KM, Goplen NP, Cunningham CA, Daniels MA, Teixeiro E. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep. (2013) 4:554–65. doi: 10.1016/j.celrep.2013.07.008
46. Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity (2010) 32:67–78. doi: 10.1016/j.immuni.2009.10.010
47. Ochel A, Cebula M, Riehn M, Hillebrand U, Lipps C, Schirmbeck R, et al. Effective intrahepatic CD8+ T-cell immune responses are induced by low but not high numbers of antigen-expressing hepatocytes. Cell Mol Immunol. (2016) 13:805–15. doi: 10.1038/cmi.2015.80
48. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med. (2016) 213:1819–34. doi: 10.1084/jem.20150598
49. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol. (2012) 12:749–61. doi: 10.1038/nri3307
50. Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. (2013) 25:556–63. doi: 10.1016/j.coi.2013.09.003
51. Navarro MN, Feijoo-Carnero C, Arandilla AG, Trost M, Cantrell DA. Protein kinase D2 is a digital amplifier of T cell receptor-stimulated diacylglycerol signaling in naive CD8(+) T cells. Sci Signal. (2014) 7:ra99. doi: 10.1126/scisignal.2005477
52. Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, et al. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. (2015) 34:2008–24. doi: 10.15252/embj.201490252
53. Balyan R, Gund R, Ebenezer C, Khalsa JK, Verghese DA, Krishnamurthy T. Modulation of naive CD8 T cell response features by ligand density, affinity, and continued signaling via internalized TCRs. J Immunol. (2017) 198:1823–37. doi: 10.4049/jimmunol.1600083
54. Nayar R, Schutten E, Bautista B, Daniels K, Prince AL, Enos M, et al. Graded levels of IRF4 regulate CD8+ T cell differentiation and expansion, but not attrition, in response to acute virus infection. J Immunol. (2014) 192:5881–93. doi: 10.4049/jimmunol.1303187
55. Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. (2005) 3:e356. doi: 10.1371/journal.pbio.0030356
56. van Heijst JW, Gerlach C, Swart E, Sie D, Nunes-Alves C, Kerkhoven RM, et al. Recruitment of antigen-specific CD8+ T cells in response to infection is markedly efficient. Science (2009) 325:1265–9. doi: 10.1126/science.1175455
57. Cukalac T, Chadderton J, Handel A, Doherty PC, Turner SJ, Thomas PG, et al. Reproducible selection of high avidity CD8+ T-cell clones following secondary acute virus infection. Proc Natl Acad Sci USA. (2014) 111:1485–90. doi: 10.1073/pnas.1323736111
58. Krummey SM, Chen CW, Guasch SA, Liu D, Wagener M, Larsen CP, et al. Enhanced requirement for TNFR2 in graft rejection mediated by low-affinity memory CD8+ T cells during heterologous immunity. J Immunol. (2016) 197:2009–15. doi: 10.4049/jimmunol.1502680
59. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1(+) effector CD8(+) T cells lose KLRG1, differentiate into all memory T cell lineages, and convey enhanced protective immunity. Immunity (2018) 48:716–29 e8. doi: 10.1016/j.immuni.2018.03.015
60. Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. (2002) 195:1541–8. doi: 10.1084/jem.20020369
61. Carrio R, Rolle CE, Malek TR. Non-redundant role for IL-7R signaling for the survival of CD8+ memory T cells. Eur J Immunol. (2007) 37:3078–88. doi: 10.1002/eji.200737585
62. Day EK, Carmichael AJ, ten Berge IJ, Waller EC, Sissons JG, Wills MR. Rapid CD8+ T cell repertoire focusing and selection of high-affinity clones into memory following primary infection with a persistent human virus: human cytomegalovirus. J Immunol. (2007) 179:3203–13. doi: 10.4049/jimmunol.179.5.3203
63. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest. (2018) 128:721–33. doi: 10.1172/JCI96957
64. Klarenbeek PL, Remmerswaal EB, ten Berge IJ, Doorenspleet ME, van Schaik BD, Esveldt RE, et al. Deep sequencing of antiviral T-cell responses to HCMV and EBV in humans reveals a stable repertoire that is maintained for many years. PLoS Pathog. (2012) 8:e1002889. doi: 10.1371/journal.ppat.1002889
65. Kedzierska K, Turner SJ, Doherty PC. Conserved T cell receptor usage in primary and recall responses to an immunodominant influenza virus nucleoprotein epitope. Proc Natl Acad Sci USA. (2004) 101:4942–7. doi: 10.1073/pnas.0401279101
66. Iancu EM, Corthesy P, Baumgaertner P, Devevre E, Voelter V, Romero P, et al. Clonotype selection and composition of human CD8 T cells specific for persistent herpes viruses varies with differentiation but is stable over time. J Immunol. (2009) 183:319–31. doi: 10.4049/jimmunol.0803647
67. Xin A, Masson F, Liao Y, Preston S, Guan T, Gloury R, et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat Immunol. (2016) 17:422–32. doi: 10.1038/ni.3410
68. Nayar R, Enos M, Prince A, Shin H, Hemmers S, Jiang JK, et al. TCR signaling via Tec kinase ITK and interferon regulatory factor 4 (IRF4) regulates CD8+ T-cell differentiation. Proc Natl Acad Sci USA. (2012) 109:E2794–802. doi: 10.1073/pnas.1205742109
69. Kallies A, Xin A, Belz GT, Nutt SL. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity (2009) 31:283–95. doi: 10.1016/j.immuni.2009.06.021
70. Man K, Miasari M, Shi W, Xin A, Henstridge DC, Preston S, et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat Immunol. (2013) 14:1155–65. doi: 10.1038/ni.2710
71. Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity (2010) 33:229–40. doi: 10.1016/j.immuni.2010.08.002
72. Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science (2003) 302:1041–3. doi: 10.1126/science.1090148
73. Posnett DN, Engelhorn ME, Houghton AN. Antiviral T cell responses: phalanx or multipronged attack? J Exp Med. (2005) 201:1881–4. doi: 10.1084/jem.20050928
74. Lauer GM, Barnes E, Lucas M, Timm J, Ouchi K, Kim AY, et al. High resolution analysis of cellular immune responses in resolved and persistent hepatitis C virus infection. Gastroenterology (2004) 127:924–36. doi: 10.1053/j.gastro.2004.06.015
75. Edwards BH, Bansal A, Sabbaj S, Bakari J, Mulligan MJ, Goepfert PA. Magnitude of functional CD8+ T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J Virol. (2002) 76:2298–305. doi: 10.1128/jvi.76.5.2298-2305.2002
76. Dalod M, Dupuis M, Deschemin JC, Sicard D, Salmon D, Delfraissy JF, et al. Broad, intense anti-human immunodeficiency virus (HIV) ex vivo CD8(+) responses in HIV type 1-infected patients: comparison with anti-Epstein-Barr virus responses and changes during antiretroviral therapy. J Virol. (1999) 73:7108–16.
77. Letourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, et al. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor alpha subunit CD25. Proc Natl Acad Sci USA. (2010) 107:2171–6. doi: 10.1073/pnas.0909384107
78. McNeel DG. TCR diversity - a universal cancer immunotherapy biomarker? J Immunother Cancer (2016) 4:69. doi: 10.1186/s40425-016-0175-4
Keywords: immunity, CD8 T cell, affinity, memory, T cell receptor, differentiation, effector
Citation: Kavazović I, Polić B and Wensveen FM (2018) Cheating the Hunger Games; Mechanisms Controlling Clonal Diversity of CD8 Effector and Memory Populations. Front. Immunol. 9:2831. doi: 10.3389/fimmu.2018.02831
Received: 28 September 2018; Accepted: 16 November 2018;
Published: 29 November 2018.
Edited by:
Weiguo Cui, Bloodcenter of Wisconsin, United StatesReviewed by:
Shahram Salek-Ardakani, Pfizer, United StatesSara Hamilton, University of Minnesota Twin Cities, United States
Copyright © 2018 Kavazović, Polić and Wensveen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix M. Wensveen, RmVsaXguV2Vuc3ZlZW5AbWVkcmkudW5pcmkuaHI=