 Min Li
Min Li Dongwei Zhu2
Dongwei Zhu2 Jie Tian
Jie Tian Shengjun Wang
Shengjun Wang- 1Department of Laboratory Medicine, The Affiliated People's Hospital, Jiangsu University, Zhenjiang, China
- 2Department of Immunology, Jiangsu Key Laboratory of Laboratory Medicine, School of Medicine, Jiangsu University, Zhenjiang, China
- 3Department of Laboratory Medicine, Affiliated Wuxi People's Hospital of Nanjing Medical University, Wuxi Children's Hospital, Wuxi, China
Emerging evidence suggests the promise of the use of myeloid-derived suppressor cells (MDSCs) in inflammatory disorders based on their unique immune-intervention properties. However, the roles of MDSCs in autoimmune arthritis are not completely understood. Indeed, their immunosuppressive functions in arthritic conditions remain controversial, with heterogeneity among MDSCs and differential effects among subpopulations receiving much attention. As a result, it is necessary to determine the roles of MDSC subpopulations in autoimmune arthritis to clarify their diagnostic and therapeutic potential. Interestingly, in the inflammation niche of autoimmune arthritis, each MDSC subpopulation can exhibit both alternatives of a given characteristic. Moreover, polymorphonuclear MDSCs (PMN-MDSCs) are likely to be more suppressive and stable compared with monocytic MDSCs (MO-MDSCs). Although various important cytokines associated with the differentiation of MDSCs or MDSC subpopulations from immature myeloid precursors, such as granulocyte colony-stimulating factor (G-CSF), have been largely applied in external inductive systems, their roles are not entirely clear. Moreover, MDSC-based clinical treatments in rheumatoid arthritis (RA) continue to represent a significant challenge, as also reported for other autoimmune diseases. In this review, we describe the effects and actions of MDSC subpopulations on the development of autoimmune arthritis and analyze several types of MDSC-based therapeutic strategies to provide comprehensive information regarding immune networks and a foundation for more effective protocols for autoimmune arthritis.
Introduction
Rheumatoid arthritis (RA) is a chronic systemic autoimmune disease characterized by multiple invasive and symmetrical joint inflammation and organic dysfunction (1). Abnormal interactions among inflammatory cells, such as Th17 cells, B cells, macrophages, dendritic cells (DCs), fibroblasts and osteoclasts, in inflamed tissues significantly contribute to the occurrence and evolution of autoimmune arthritis, mainly through molecular docking or the release of active products (2). Among CD4+ T cell subpopulations, CD4+CD25+Foxp3+ regulatory T cells (Tregs) represent a limited valuable subpopulation in RA treatment, and the balance of Th17 cells and Tregs is an important factor associated with the development of inflammation (1, 3). Myeloid-derived suppressor cells (MDSCs) comprise a group of highly heterogeneous cells derived from immature myeloid progenitors that are typically divided into two subpopulations: polymorphonuclear (PMN-MDSCs) and monocytic (MO-MDSCs) MDSCs (4–6). Although numerous reports have demonstrated the potent immunosuppression effects of MDSCs under abnormal conditions, the roles of MDSCs and their subpopulations in autoimmune arthritis remain unclear. As research about MDSCs in arthritis progresses, differences in the roles and actions of MDSC subpopulations have stimulated growing interest among researchers. In this review, we summarize the current research findings of MDSC subpopulations in autoimmune arthritis and evaluate the prospects of their application.
MDSCs
MDSCs are generally characterized by Gr-1+CD11b+ status in mice and CD11b+CD33+ status in humans. In normal mice, MDSCs reside in the bone marrow (BM) and spleen (~20–30 and 2–4%, respectively) (4–6). However, because of their complex compositions, MDSCs can not be completely defined exclusively by combinations of several surface molecules, which might explain part of the functional inconsistencies noted in the same disease models (7). As a result, it is necessary to define MDSC subpopulations that are highly suppressive and further delineate their corresponding immune regulation mechanisms within various local environments.
MDSC Subpopulations
Most reports involving mice classify MDSCs according to Ly6C and Ly6G expression and Gr-1 composition to define their morphologies and origins (8, 9). PMN-MDSCs and MO-MDSCs are characterized as CD11b+Ly6C+Ly6G+ and CD11b+Ly6C+Ly6G−, respectively (10). To detect the immune function of each subpopulation of MDSCs, some studies further divide Ly6C expression into Ly6Chi, Ly6Clo, and Ly6Cint (11–15). Other markers, such as CD115+, CD49+, and IL-4R+, have also been used to represent highly suppressive MDSC subpopulations. CD115 expression levels correlate with Treg expansion induced by MDSCs (16–18). IL-4R expression is closely associated with CD8+T cell inhibition (19), whereas CD49+ MDSCs reflect MO-MDSCs suppressive toward both antigen-specific and non-specific T cell proliferation (8, 20–22).
In humans, both PMN-MDSCs and MO-MDSCs from human peripheral blood (PB) inhibit the activation and expansion of autologous T cells. However, the marker profile of MDSC subpopulations is more complex than that in mice. Multiple combination types of surface markers, mostly discussed within specific types of cancer patients, were reported to select or define PMN-MDSCs and MO-MDSCs mainly sourced from human PBMCs (14, 23). Several combinations are selectively listed here, which have been determined from both nuclear morphology and suppressive activities. For PMN-MDSCs, the effective combinations of surface markers including CD11b+ CD15+ CD33low (24), CD11b+ CD14neg CD15+ (25), CD11b+ CD33+ CD14neg CD15+ (26), CD11b+ CD33+ CD66b+ VEGFR1+ CD14neg CD15+ (27), and CD11b+ CD33+ HLA-DRneg CD14negCD66b+CD15+ and so on (28), while CD14+HLA-DRneg/low (29), CD14+IL-4Ra+ (30) were the most frequently used for MO-MDSC identifications (31). Bronte V and colleagues reviewed the minimum requirements for definition and characterization of human PMN-MDSCs and MO-MDSCs in peripheral blood mononuclear cells (PBMCs) are, respectively CD11b+CD14−CD15+/CD66+ and CD11b+ CD14+CD15−HLA-DR−/low (32). Functionally, increased PD-1 expression on human PMN-MDSCs in multiple sclerosis (MS) was able to partially mediate T cell inhibition (33). High levels of S100 calcium-binding protein A9 (S100A9) on MDSCs were found to be consistent with NOS2 activity, representing a potential marker for human MO-MDSCs (34). These evidences suggest that several surface markers CD11b, CD14, CD15, CD66, and HLA-DR have been considered as applicable surface markers so far for the corresponding purification of PMN-MDSCs and MO-MDSCs. Whether other potential surface markers for human PMN/MO-MDSCs mentioned above such as CD62Llow, CD66bhi, IL-4Ra+, PD-1+, S100A9+ and high levels of vascular endothelial growth factor receptor 1 (VEGFR1) (35) are widely applicable or more effective for the purification of functional PMN/MO-MDSCs need to be further demonstrated.
Importantly, we can conclude that at the present, in the matter of possibly applicable surface markers for the precise purification of highly suppressive MDSC and/or MDSC subpopulations regardless from animal models or humans, most of these candidates were proposed only in specific disease settings; in other words, these markers are not universal and limited to one or several disease backgrounds. For example, expression of CD124 or CD115 on MDSCs was not significantly associated with MDSC-mediated suppression of CD8+ T cell responses in ET-4 lymphoma models, which disagreed with what was initially reported for C26-GM tumor cell lines (19, 36). Therefore, more evidence needs to be collected to support the common application of these markers.
MDSCs in Local Environments
MDSCs expand and accumulate within multiple pathological disorders and regions, such as tumor sites, inflammatory lesions, PB or the spleen, generally due to blockade of normal differentiation from hematopoietic stem cells (HSCs) to mature myeloid cells. Significant increases in MDSCs have also been observed in the PB of patients with cancers (4, 7, 8, 37) as well as multiple infections and autoimmune diseases (9, 38).
The systematic effects of MDSCs are controversial with regard to tumors and within the context of inflammatory disease progression. In general, murine MDSC-mediated immunosuppression involves secretion of active products, such as arginase1 (arg-1), nitric oxide (NO), reactive oxygen species (ROS), and inducible nitric oxide synthase (iNOS), and expression of vital signal molecules, such as programmed cell death protein 1 (PD-1). Effects include the following: direct or indirect suppression of CD4+T cell activation, proliferation or migration; cooperation with Tregs; and communication with multiple mature myeloid cells or tumor cells (37, 39). Arg-1, an enzyme that catalyzes hydrolysis of L-arginine (40), is more frequently utilized by PMN-MDSCs to block normal signal activation of T cells by directly inhibiting formation of the cyclin D3 (CD3 ζ) and cyclin-dependent kinase 4 complex due to arginine deficiency (41, 42). ROS, including superoxide anion and NO, also contribute to PMN-MDSC-mediated interruption of T cell proliferation (36, 43, 44). iNOS promotes decreased expression of signal transduction, activation of transcription 5 (STAT5) in MO-MDSCs and MHC II in inflammatory cells (36, 45, 46). Moreover, human MDSCs potently inhibit T cell proliferation in vitro (47–49). The pro-inflammatory effects of MDSCs are mainly mediated via the promotion of Th17 cell polarization, CD8+ T cell activation and their differentiation potentials into mature cells (50–54), which are primarily observed in animal models.
The local environment is one of the most important factors that regulates immune cell functions in vivo, and MDSCs are not excepted. On the one hand, different suppressive capacities and functional mechanisms of MDSCs have been demonstrated during disease development. A study of helminth infection reported that MDSCs from early-stage-infected mice were able to induce release of IFN-γ from T cells and inhibit T cell proliferation, and these activities were primarily mediated by NO. At later stages, the induction ability of IFN-γ released from T cells was lost, with arg-1 playing more important roles in the inhibition process than NO (55). Moreover, the role of MDSCs in T cell suppression was found to be reduced after the onset of collagen-induced arthritis (CIA) (14). These variations in MDSC function suggest the important influence of the local environment and are possibly related to the alternative activation phenotypes or variable expansion of MDSC subpopulations. On the other hand, MDSCs located at the periphery and center of lesions exhibit distinct behaviors that are not only related to functional changes in MDSCs themselves (56) but can also be attributed to the maturity or composition of contiguous immune cells involved in distinct local niches. This finding is similar to the discovery that Tregs in the synovial fluid (SF) of RA patients and mice with autoimmune arthritis display more mutual phenotypes and more potent immunosuppression compared with Tregs in PB (57, 58). The differentiation programs of Ly6Chi monocytes can also be switched from anti-inflammatory macrophages to inflammatory DCs within the context of inflammation in the colon (53). Given the wide interactions of immune cells with MDSCs at diseased sites, assessment of the compositions and functional states of immune cells around MDSCs within lesion sites is beneficial for accurate identification of the immune network and the development of more applicable MDSC-based therapies.
MDSCs in Autoimmune Arthritis
Similarly, the frequencies of MDSCs during the development of autoimmune arthritis are aberrant. Compared with healthy controls, MDSCs were increased in the spleen, paws, BM, and draining lymph nodes (DLNs) (popliteal or inguinal lymph nodes, periarticular lymph nodes) of experimental autoimmune arthritis models (10, 14, 59–63) and in the PB and SF of RA patients (61, 64). Most studies report that the frequencies of both PMN-MDSCs and MO-MDSCs in the spleen of experimental autoimmune arthritis models were decreased at the early stage but increased at the late stage, which might indicate a procession of myelopoiesis and functional roles during immunoregulation. Moreover, it has been reported that the number of circulating MDSCs in RA patients is positively correlated with Disease Activity Scores in 28 Joints (DAS28) and negatively correlated with Th17 cell frequencies.
Identification of MDSC Subpopulations in Autoimmune Arthritis
In an autoimmune arthritis mouse model, PMN-MDSCs have been identified as CD11b+Gr-1high/+ or CD11b+Ly6G+ Ly6C+/−/int/low, whereas MO-MDSCs have been identified as CD11b+Gr-1medium or CD11b+Ly6C+/highLy6G− (Table 1). CCR2, CD62L, and CD115 (14) expression on MO-MDSCs derived from experimental autoimmune arthritis models and tumors has also been reported (67). Low levels of other markers such as MHC II, F4/80, and CD11c on MDSCs are indicative of their myeloid source and immature features (14). Increased CD40 and CD86 expression on MO-MDSCs isolated from CIA mice compared with PMN-MDSCs has been observed, though no significant differences in surface expression of CD80, MHC II, or PD-L1 have been reported (10). In RA patients, MO-MDSCs are commonly marked by CD14+HLA-DR−/low (62), whereas PMN-MDSCs are identified by CD11b+ CD33+ HLA-DRlow/− CD14− CD15+ (61). CD11b, which is also expressed on neutrophils, monocytes, macrophages and NK cells, is the only common marker of MDSC subpopulations in humans and mice.
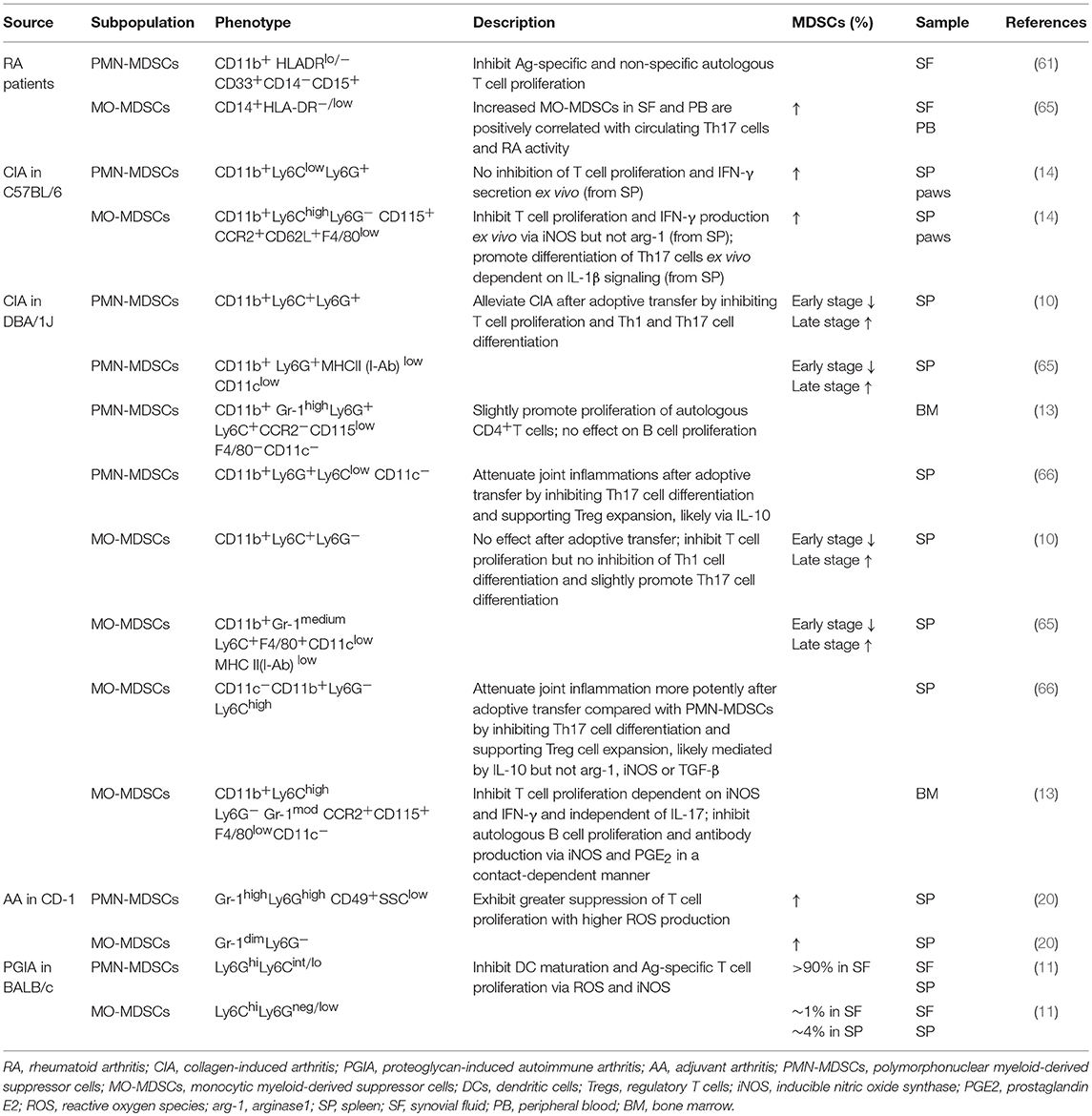
Table 1. MDSC subpopulations in autoimmune arthritis.
It was suggested that phenotypes of MDSC subpopulations vary within the context of evolving microenvironments, and similar phenotype characteristics have been noted for other precursors and mature myeloid cells. For example, inflammatory monocytes are characterized by CD11b+Ly6ChighLy6G−CD115+MHC II −F4/80+CD11c−, which is similar to MO-MDSCs (8, 14, 17, 18). In addition, PMN-MDSCs with variable phenotypes exhibit characteristics similar to those of CD11b+Ly6G+Ly6Clow F4/80−CD11c− granulocytes (8). As a result, both surface markers and key immune functions are necessary for identifying MDSC subpopulations. Moreover, standard methods for isolating MDSC subpopulations must be developed to facilitate comparison among studies.
Roles of MDSCs in Autoimmune Arthritis
Two different effects of MDSCs on immune responses during the development of autoimmune arthritis have been observed. Attenuation of arthritis has been attributed to MDSC-mediated disruptive effects on “vicious cycles of inflammation,” including suppression of T cell proliferation and the unfavorable impact on the responses of pro-inflammatory cells, such as CD8+T cells, Th17 cells and DCs. This mainly occurs via the mediators mentioned above, including arg-1, iNOS, and IL-10 (7, 9). iNOS plays a vital role in MDSC-mediated T cell suppression and more effectively inhibits antigen (Ag)-specific T cell proliferation compared with arg-1, as observed in mice with proteoglycan-induced autoimmune arthritis (PGIA). These data indicate the significant therapeutic potential of MDSCs for RA, which is consistent with their contributions to piperlongumine-attenuated CIA (60, 62). Undesirable pro-inflammatory effects of MDSCs have also been reported in animal models of RA and appear to facilitate responses by Th17 (65) and B (15) cells.
MDSC activities in distinct organs, such as the BM, PB, spleen or synovial membranes, have been reported (Table 2). Interestingly, in the spleens of CIA mice, MDSCs exhibit both inhibitory and promoting effects on Th17 cell differentiation, and it is unclear how these results can be reconciled. Nonetheless, the latter exhibited a stronger correlation with MO-MDSCs, in which IL-1 functions as a potentially critical supporter (14, 62), as previously demonstrated in experimental autoimmune encephalitis (52). MDSCs from the PB of RA patients also exhibit potential inhibitory activity against Th17 cell accumulation, and other reports have demonstrated a positive correlation between MDSC-PB and B cell proliferation. Further relationships between MDSC subpopulations and B cells in autoimmune arthritis are described below (see section MO-MDSC Involvement in Autoimmune Arthritis). More potent suppression of Ag-specific, as opposed to Ag-non-specific, T cell proliferation and responses by MDSCs from the spleen and SF of both RA patients and experimental autoimmune arthritis models have been reported (11, 61). MDSCs isolated from the SF of PGIA mice were also capable of suppressing DC maturation via iNOS and ROS and of inhibiting DC-dependent T cell proliferation with elevated expression of arg-1, NO and ROS. Some MDSCs derived from the BM of CIA mice showed enhanced transformation into mature myeloid cells compared with those obtained from normal mice (59). It is possible that MDSCs in peripheral and original tissues are less suppressive than are those in lymphoid organs and lesion sites, which is correlated with stimulation of chemokines, cytokines, and inflammatory cells, among others.
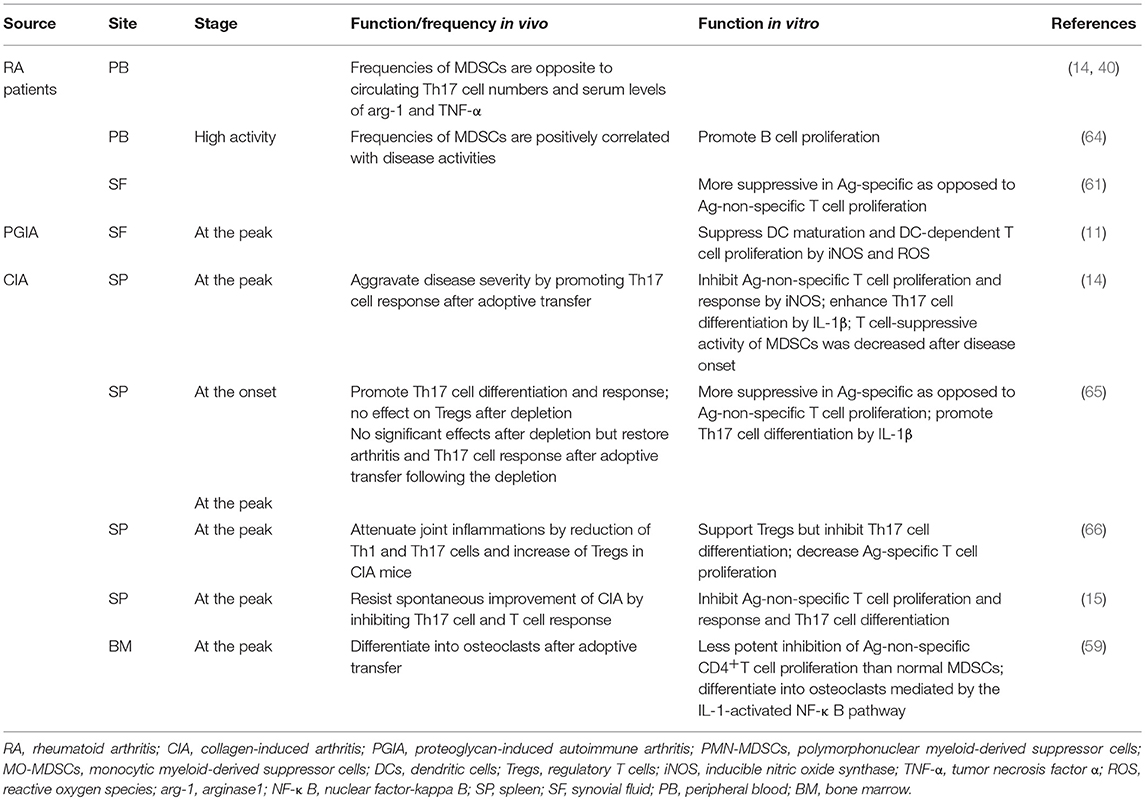
Table 2. Roles of MDSCs at different sites of autoimmune arthritis.
For MDSC migration within the context of autoimmune diseases, chemokine receptor 2 (CCR2) functions as a vital chemokine receptor for the migration of MO-MDSCs from the BM into the PB observed in arthritic mice (13). Monocyte chemotactic protein 1 (CCL2) is abundantly present in the synovium of patients with RA (68), whereas MO-MDSCs were absent from the blood, spleen, and lymph nodes of CCR2−/− mice that developed more severe CIA (13). CCL2 on mesenchymal stem/stromal cells (MSCs) also contributes to recruitment of MO-MDSCs to inflammatory sites during MSC-mediated suppression of autoimmunity (69). It has been reported that exposure to IFN-γ markedly upregulates CCR2 expression on MDSCs to retain these cells at inflammatory sites, and MDSCs generated from CCR2−/− mice fail to be mobilized (70). Therefore, IFN-γ and CCR2 may be important mediators during the activation of MDSCs as well as in migration into the periphery during arthritis development. Increased expression of CCR1 on PMN-MDSCs facilitates their entry into the circulation, as observed in systemic lupus erythematosus (SLE) mouse models (71). IL-10 also contributes to the accumulation of PMN-MDSCs in peripheral lymphoid organs during autoimmune disease (72). CXCR2 facilitates MDSC recruitment to allograft sites (73), and IL-6 and tumor necrosis factor alpha (TNF-α) are likely inducers of MDSC accumulation at inflamed tissues to resist aggravation of inflammatory responses (74). Although many other chemokines, such as CCL5, CXCL6, CXCL8, CCL15, CXCL12, have been reported to recruit MDSCs during cancer development (75), these processes in autoimmune diseases need to be further confirmed.
In summary, MDSCs in autoimmune arthritis function as a double-edged sword. More functional regulators of MDSC subpopulations and variability among MDSCs need to be explored to establish adjuvant treatment for MDSC transfer-based therapy targeting these key cytokines or cells.
Roles of MDSC Subpopulations in Autoimmune Arthritis
Under tumor (2) and transplant antagonism (76–78) conditions, Gr-1lowLy6Chigh MO-MDSCs are generally more suppressive than are Gr-1highLy6Clow PMN-MDSCs; however, circulating PMN-MDSCs are more abundant than MO-MDSCs (3:1 ratio) in naive mice (36). In mouse models of RA, PMN-MDSCs also predominate among MDSCs from the PB, DLNs, SF or paws compared with MO-MDSCs (10, 14, 59–63). Additionally, in the pathogenesis of autoimmune arthritis, both PMN-MDSCs and MO-MDSCs exhibit controversial roles and utilize relatively distinguishable pathways to inhibit immune responses, as previously demonstrated for tumors. In contrast, PMN-MDSCs are more suppressive than are MO-MDSCs (Table 1).
PMN-MDSC Involvement in Autoimmune Arthritis
Previous reports demonstrate that splenic PMN-MDSCs from CIA mice efficiently suppress anti-CD3- or CD28-stimulated T cell proliferation (10). In RA patients, MDSCs isolated from the SF, 95% of which are granulocytic MDSCs, more potently inhibit Ag (allogeneic peripheral blood mononuclear cells)-specific compared with Ag-non-specific autologous T cell proliferation (61). Similarly, SF cells from PGIA mouse models strongly suppress proliferation of DC-activated autologous T cells, but no effect on anti-CD3- or CD28-stimulated T cell proliferation was found (>90% were granulocyte-like MDSCs) (11). As a potent inhibitory cytokine in the synovial membranes of RA patients, IL-10 is associated with T cell anergy (79) and reductions in pro-inflammatory mediators, such as TNF-α and IL-1β (80). IL-10 is also increased in CIA mice and is partly involved in MDSC-mediated T cell inhibition (15). IL-10 also plays an important role during MDSC-mediated attenuation of joint inflammation in CIA mice via Treg cell expansion (66). As a result, inhibition of both Ag-specific and non-specific autologous CD4+T cell proliferation is generally involved in PMN-MDSC-mediated suppression of autoimmune arthritis. Regardless, PMN-MDSCs within lesions tend to more specifically execute the former, whereby the release of inhibitory cytokines might represent an important pathway in addition to commonly recognized arg-1- and ROS-mediated effects (Table 1).
PMN-MDSCs also correct the balance of CD4+T cell subpopulations. Both Th1 and Th17 cells are vital lymphocytes driving the progression of autoimmune arthritis (1) and significantly related to fibroblast and chondrocyte activation. In pilot clinical trials, the humanized anti-IL-17 monoclonal antibody LY2439821 provided an improved curative effect in RA patients (81). Significantly reduced ratios of IFN-γ+ (Th1) and IL-17A+ (Th17) cells were observed in the DLNs of CIA mice, and joint inflammation was alleviated after adoptive transfer of PMN-MDSCs (10). This effect in CIA mice was also observed in another study in which PMN-MDSCs promoted Treg cell expansion after injection in vivo (66). These results suggest the promising potential of PMN-MDSCs to correct the imbalance in CD4+T subpopulations as well as the vicious cycle in the synovial milieu of autoimmune arthritis.
Moreover, PMN-MDSCs efficiently inhibit DC maturation in mouse models of RA. DCs are the predominant antigen-presenting cells and function as an important stimulator in the attraction and subsequent activation of Th1, Th2, and Th17 cells in RA pathogenesis (82). In vitro, SF MDSCs potently suppressed DC maturation in a non-cell contact-dependent manner, as demonstrated by reduced expression of DC maturation surface markers CD86 and MHCII (11) and reduced maturation characteristics of SF-isolated DCs compared with those derived from the BM (11). Arg-1 plays vital roles in this process, which is also observed in other autoimmunity diseases (38, 83, 84) and tumors (23, 37). As a result, PMN-MDSC-mediated inhibition of the inflammation generated by monocyte-macrophage lineage cells in inflamed joints might explain such improvement in the signs and symptoms of autoimmune arthritis.
Nonetheless, the pro-inflammatory potential of PMN-MDSCs to develop into neutrophils should not be ignored. Levels of granulocyte colony-stimulating factor (G-CSF), a key factor in neutrophil development and recruitment (85), are significantly increased in the SF of RA patients and correlate positively with RA disease severity (86). Given the extraordinary number of neutrophils and PMN-MDSCs in the SF of RA patients (87, 88) and the destructive ability of neutrophils in joints (89), it is important to consider whether PMN-MDSCs consistently remain in a steady immunosuppressive state or grow and mature under the presence of factors, such as G-CSF, within inflamed joints to transform into neutrophils and promote arthritis development.
MO-MDSC Involvement in Autoimmune Arthritis
The therapeutic value of MO-MDSCs in autoimmune arthritis is primarily mediated via inhibition of CD4+T cells and B cells. CD11b+Ly6ChighLy6G− monocytes isolated from the BM are associated with a significant reduction in joint damage through effective suppression of CD4+T cell proliferation in vitro, which is dependent on iNOS and IFN-γ. These MO-MDSCs also inhibit autologous B cell responses via iNOS and prostaglandin E2 (PGE2) in a contact-dependent manner (13). Interestingly, a recent study of RA patients demonstrated that peripheral MDSCs promote autologous B cell proliferation. Although induction of B cell proliferation may decrease PMN-MDSCs, Crook et al. reported no effect of PMN-MDSCs on B cell proliferation. The differences between these two reports may be attributed to the targeted organs and sources of isolated MDSCs. In the former study, MDSCs were isolated from the BM of CIA mice; in the latter study, MDSCs were isolated from the PB of RA patients. These findings indicate the potentially powerful effect of lesion niches on MDSCs and the decreased suppressive effects of MDSCs obtained from the PB compared with BM, as observed in other diseases (56). Inhibition of B cell proliferation mediated by MO-MDSCs may constitute a self-resistance mechanism in excessively inflamed immune systems, though this hypothesis must be confirmed.
MO-MDSCs potently induce the development of Tregs in various inflammatory disorders, such as type 1 diabetes (T1D), transplantation models (73, 77, 90) and tumors (16, 76). CD14+HLA-DRlow/− MDSCs isolated from human PB alter the differentiation of monocyte-induced Th17 cells into Foxp3+ Tregs by secreting transforming growth factor-β (TGF-β) and retinoic acid (91). MO-MDSCs isolated from the spleen of CIA mice ameliorate joint inflammation by promoting Treg cell expansion and inhibiting Th1 and Th17 cell accumulation (66).
Furthermore, contradictory actions of MO-MDSCs have also been observed in the progression of autoimmune arthritis. Levels of CD14+HLA-DR−/low MDSCs in the PB or SF of RA patients were found to correlate with peripheral Th17 levels and RA status (14, 62), and IL-17 and IL-10 serum levels were significantly increased after adoptive transfer of MO-MDSCs into CIA mice. Slight acceleration in Th17 cell differentiation by MO-MDSCs was also observed in external co-culture systems (10). IL-1β is likely an important mediator of this progression (14), as observed in experimental models of SLE (71) and MS (multiple sclerosis) (33, 92). In addition, this effect might partially explain the remission of RA after treatment with an IL-1 antagonist (93). These results suggest that MO-MDSCs and PMN-MDSCs may regulate the balance of Th17 and Tregs in autoimmune arthritis, but whether the effect is beneficial for Th17 cells or Tregs in RA pathogenesis has not yet been determined.
Overall, the results described above do not completely support the notion that the pro-inflammatory effects of MO-MDSCs are mediated through inhibition of Ly6C+ classical monocytes. CD14++CD16− monocytes (classical monocytes), an important human monocyte subpopulation with CD11b expression, represent a main subpopulation of extravascular tissue monocytes (94, 95). Equivalent monocytes in mice are characterized by CD11b+Ly6C+CD62L+CD43−CCR2+ or Ly6C+CD11b+ CD11c−F4/80lowCD64int (96), and these monocytes are found in resting non-lymphoid as well as lymphoid tissues, such as the lung and lymph nodes, under steady states (97). Some markers on MO-MDSCs are co-expressed with classical monocytes, such as F4/80, CD115, CD62L, and CCR2 (8, 14, 62). IL-12 promotes Th1 cell proliferation and maturation, Th17 cell differentiation and B cell activation within the synovial milieu, and IL-12 is predominantly released by classical monocytes. As a result, IL-12 is not produced by classical monocytes, potentially accounting for the pro-inflammatory effects of MO-MDSCs described above.
Are PMN-MDSCs More Suppressive Than MO-MDSCs in Autoimmune Arthritis?
PMN-MDSCs and MO-MDSCs maintain immune homoeostasis of autoimmune arthritis through distinct effector molecules and pathways. However, it remains unclear whether any subpopulation becomes more suppressive as a result of the inflammatory niche. Adoptive transfer of PMN-MDSCs but not MO-MDSCs effectively relieves joint damage in CIA mice. Moreover, T cell proliferation and responses were reported to be more potently suppressed by PMN-MDSCs than by MO-MDSCs, which likely related to IL-10, TGF-β1, CCR5, and CXCR2 (10). Furthermore, PMN-MDSCs more effectively suppress Th17 cell differentiation than MO-MDSCs do (62). The increased therapeutic efficacy and suppressive potential of PMN-MDSCs were also demonstrated in asthmatic mice, which was likely due to Treg cell differentiation and arg-1 production (98). Granulocyte-macrophage colony-stimulating factor (GM-CSF) has a necessary function in the myelopoiesis of MO-MDSCs with great differentiation potential (99). It is abundant in the SF and synovial membranes of RA patients, which is vital for the activation of macrophages (100). As a result, it is important to determine whether MO-MDSCs are steadily maintained under the effects of cytokines, such as GM-CSF, involved in the inflamed joints of autoimmune arthritis or in secondary lymphoid organs, weakening them or rendering them unable to inhibit CD4+T cell immune responses. Consistent with the observation for external cultures of PMN-MDSCs and MO-MDSCs, MO-MDSCs mature and develop into CD11b+ DCs or F4/80+ macrophages in the presence of GM-CSF (36). These experimental data suggest that compared with MO-MDSCs, PMN-MDSCs likely exert a more suppressive function in the pathogenesis of autoimmune arthritis, as reflected by the strong inhibition of T cell proliferation, induction of Treg cell differentiation and a comparative steady state. In addition, interactions between these two subpopulations have also been reported during MDSC-mediated suppression of T cells, such as the production of peroxynitrite, a more suppressive mediator causing T cell unresponsiveness (44).
Therapeutic Potential of MDSC-Based Treatment
In humans, therapeutic advances in autoimmune diseases have set up therapeutic efficacy of biologic agents according to the pre-clinical/clinical applications of impressive cytokines and monoclonal antibodies for inhibiting inflammatory cytokines (2). Most of them exhibit effective therapeutic effects within partial autoimmune diseases. However, considering many unexpected side effects and unsatisfied efficacy appeared during clinical treatments such as granulocyte reduction, anemia, myelosuppression, injection site response, limited valid periods and relapses, another strategy based on immunosuppressive cell treatment has exhibited a promising prospect.
Adoptive Transfer of MDSCs in vivo
Adoptive Transfer of Directly Isolated MDSCs in vivo
To understand deeply the therapeutic value of MDSCs in RA patients, we analyzed partial adoptive transfer experiments of MDSCs and/or MDSC subpopulations into experimental animal models of RA. Some typical cases were filtered (Table 3). The results showed that arthritis was improved after total MDSC transfer via suppression of Th17 and Th1 cell accumulation and responses. However, some reports have also suggested aggravated effects, with increased numbers and enhanced responses of Th17 cells and even presentation of differentiation properties. Moreover, we found that injection points might be an important factor related to MDSC functions during adoptive transfer experiments (Table 3). By comparing the completely opposite results, alleviation and aggravation, by Chunqing Guo et al. and Zhang et al., respectively, we noticed that the injection point was before CIA establishment in the former and after in the latter. This finding indicates that the suppressive functions of transferred MDSCs might be more effective within non-strong inflammatory and complicated local environments. It is possible that distinct inflammatory environments stimulate the development of MDSC subpopulations to different extents. Guo et al. also reported that by promoting Th17 cell differentiation, adoptive transfer of MO-MDSCs prior to model establishment exhibits a pro-inflammatory property. Wang et al. suggested poor amelioration of arthritis after MO-MDSC transfer but effective improvement via PMN-MDSC transfer through inhibition of Th17 cell development. These data support the hypothesis mentioned above that MO-MDSCs tend to promote inflammation during autoimmune arthritis. In addition, it has been reported that the ratios of MO-MDSCs among total MDSCs increase steadily until the peak of arthritis, which is contrary to the observations for PMN-MDSCs (10). As a result, it is likely that the environment after arthritis onset is more suitable for MO-MDSC development with pro-inflammatory functions than for PMN-MDSCs, causing exacerbated symptoms. Moreover, it is also possible that seriously inflammatory local environments render MDSCs more changeable, as analyzed previously, resulting in greater difficulty in their suppression and perhaps promoting pro-inflammatory effects. In summary, MDSCs have the potential to regulate the immune imbalance that occurs in autoimmune arthritis, but the differential functions of MDSC subpopulations need to be elucidated.
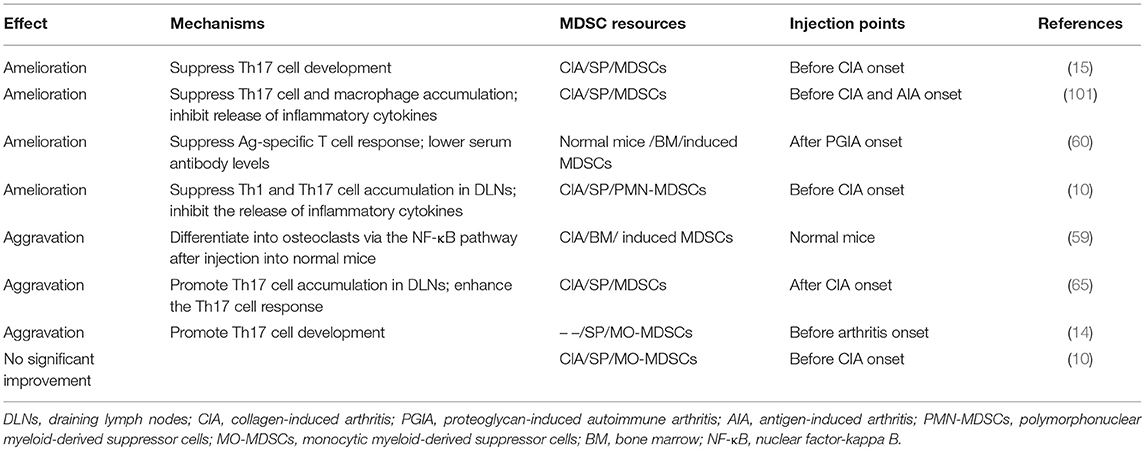
Table 3. Adoptive transfer experiments using MDSCs and/or MDSC subpopulations in experimental animal models of RA.
On account of the low frequency of MDSCs in healthy individuals which is unchangeable and the potential threat of immune rejections, it seems that the autologous application of MDSCs is more rational as the initial source of MDSCs than allogeneic transfer. However, the strategy is difficult to practice partially due to the unestablished perfect judgement methods for MDSC physiological and functional conditions, that is, the uncertainty of MDSC conditions in different RA patients and ambiguity clinical stages. Therefore, for autologous transfer of directly isolated MDSCs for RA treatment, it is necessary to resolve two difficulties in advance: making sure exact markers or methods for judgements of MDSC functional conditions and exploring corresponding optimal cultural systems of MDSCs in vitro to keep and strengthen their suppressive functions steadily.
Adoptive Transfer of Induced MDSCs in vitro
In addition, we want to emphasize another promising clinical application of MDSCs, namely, the induction of MDSCs within the context of autoimmune arthritis to meet the clinical needs for a large quantity of high-quality stable MDSCs. Some MDSC induction methods have been explored in vitro using DCs (102), embryonic stem cells (ESCs) (18), HSCs (18), PB mononuclear cells (103) or other normal myeloid progenitor cells (60, 104–106) via combinations of various cytokines. These methods have demonstrated therapeutic value in mouse models of infectious and autoimmune diseases. The initial/progenitor cells and stimulating cells mentioned above used in researches are mostly directly isolated from healthy individuals, which indicates it is allogeneic sourced MDSCs that act as a promising treatment for RA patients based on the adoptive transfer of induced MDSCs in vitro. Moreover, in my view, there is also a strong potential for MDSCs induced from autologous initial/progenitor cells for treatments of RA patients and it is undoubtedly more applicable.
As for the critical induction cytokines for MDSC development, it was reported that macrophage colony-stimulating factor (M-CSF) was specifically important for the differentiation of functional MO-MDSCs, whereas GM-CSF was more beneficial for PMN-MDSC development from mouse embryonic and HSCs (18). These induced MO-MDSCs displayed increased suppression of T cell proliferation and promotion of CD4+Foxp3+ Treg cell expansion compared with those directly isolated from tumors via NO and IL-10, suggesting the potential value of induced MDSCs. It is worth mentioning here in the matter of Treg cell subpopulations which are potentially synergetic with MDSCs in vivo that, besides CD4+ Tregs mentioned frequently in this review generally identified by CD4+ CD25 hiFoxp3+, it is likely that another Treg subpopulation, CD8+ Tregs, also act as an outstanding candidate capable of facilitating the suppressive functions mediated by MDSCs. It was widely reported that CD8+ Tregs were potently suppressive in tumors (107, 108), infectious diseases (109, 110), graft-vs.-host disease (GVHD) (111, 112) and autoimmune diseases such as SLE (113), MS (114), and experiment allergic encephalomyelitis (EAE) (115) mainly by strong inhibitions of CD4+T cell proliferations especially for Th1 cells and the pro-inflammatory cytokine productions via the releases of soluble cytokines or co-stimulator expression. In CIA models, CD8+ Tregs also showed a powerful suppression contributing to anti-CD3 mAb-improved arthritis and suggested a more potent capability than CD4+ Tregs in the suppression of IL-17 production. Importantly, one report in GVHD revealed a close connection between CD8+Foxp3+Tregs and MDSCs that the former could be induced to contribute the alleviation of GVHD mediated by MDSCs (109) as well as CD4+ Tregs. Whether this process or interactions between these two suppressors is true in RA patients is unclear at the present, but there is no doubt that the relationship between CD8+ Tregs and MDSCs is worthy to be further explored to perfect the immune regulatory system of MDSCs in autoimmune arthritis.
Furthermore, iNOS promotes prolonged survival of cardiac allografts mediated via BM-derived MDSCs (73). Addition of IL-13 to the combination of GM-CSF and G-CSF induced more suppressive BM-derived MDSCs and markedly decreased GVHD lethality, which was correlated with increased production of arg-1 by MDSCs (106). Moreover, MDSCs derived from normal BM cells promoted systemic exhaustion of immune systems conducive to allogenic graft survival under stimulation by IL-6 and GM-CSF (104). However, these induced MDSCs did not affect disease progression of autoimmune diabetes and exhibited paradoxical effects on antigen-specific CD8+ T cell proliferation in vitro compared with that observed in vivo. MDSCs induced by GM-CSF alone, without IL-6, from mouse BM cells induced Th17 cell differentiation independent of MDSC-T cell contact, and this effect was likely related to TGF-β and IL-6 production by MDSCs (51). It is possible that different in vivo environments caused the injected MDSCs or complex MDSC subgroups to exhibit different activation states, resulting in distinct immune effects. These results represent challenges in the use of MDSCs but also highlight their possible utility in clinical treatments. One report suggested that MDSCs induced from BM cells under IL-6 combined with GM-CSF and G-CSF were able to effectively ameliorate symptoms in RA mouse models, likely via NO-mediated inhibition of T cell responses (60), and PMN-MDSCs represented the majority of these cells. At present, there are few reports available on the function of induced MDSCs or MDSC subpopulations within the context of autoimmune arthritis. Regarding RA treatment, it may be more valuable to develop efficient induction systems for specific MDSC subpopulations based on their distinct actions within the context of arthritis.
Open Questions for Adoptive Transfer of MDSCs
As previously mentioned, the question of whether MDSCs differentiate into mature cells in vivo after adoptive transfer, even at sites with a large number of MDSCs, remains unanswered. Some reports have demonstrated the inability of transferred Ly6Chigh monocytes to develop into DCs under inflammatory conditions (53, 116–118). Nonetheless, osteoclasts that originated from macrophage/monocyte-lineage myeloid precursors similar to MO-MDSCs were activated by multiple inflammatory factors during bone destruction in RA (1). CD11b+Gr-1+ MDSCs reportedly developed into CtsK+ osteoclasts and aggravated bone destruction after transfer into the tibias of CIA mice (59). Another study reported MDSCs induction into osteoclasts in vitro under stimulation by various factors responsible for osteoclast differentiation, such as M-CSF and receptor activator for nuclear factor-κ B ligand (RANKL) (119). Accordingly, before these methods are applied in the clinic, it should be addressed whether transferred MDSCs, particularly MO-MDSCs, further differentiate in different inflamed niches of RA patients and whether there are any markers to suggest the dosage or frequency of MDSC injection to avoid overtreatment.
In addition, engineering MDSCs in vitro into suppressors that are more potent represents a feasible strategy for the treatment of autoimmune diseases. Induced MDSCs that express self-antigen derived from BM cells in vitro effectively ameliorated Ag-induced experimental autoimmune encephalomyelitis (120). Thus, stimulation of MDSCs or precursors using cytokines or antigens to render them more susceptible to or suppressive toward corresponding immune responses represents a promising approach. Whether this strategy is also suitable for RA is unknown, but it should be considered as a potential strategy.
Moreover, many other challenges remain unresolved. For example, how can proliferation be controlled to ensure survival after transfer in vivo? Will any side effects occur based on the use of a large number of cells or long-term use? How can these cells be generated in sufficient numbers and properly stored in vitro to maintain activity to meet the needs for clinical treatment.
Prospects for Exosome-Based Therapy
Given the limitations of cell-based therapy, exosome-based biotherapy has recently demonstrated promise for a new treatment strategy (121–124) with fewer side effects compared with drugs and better controllability compared with cell-based therapies. Exosomes, which are secreted by almost all cells and are composed of proteins, RNAs and DNAs under a lipid bilayer structure, are distributed throughout the body (125, 126). In autoimmune arthritis, exosomes are important in the maintenance and progression of arthritic inflammation via active molecule cargo (127, 128). For example, exosomes derived from DCs that express high levels of IL-4 cause remission in established CIA and relieve inflammation in the delayed-type hypersensitivity (DTH) model (129). MDSC-derived exosomes significantly benefit Treg cell expansion and inhibition of T cell responses (130) in alopecia areata models. Moreover, exosomes derived from PMN-MDSCs participate in inhibition of Th1 cell proliferation and promotion of Treg expansion in dextran sulfate sodium (DSS)-induced colitis (123). We also identified a pivotal role for PMN-MDSC-derived exosomes in the remission pathology of CIA mice, mainly by impeding Th17 cell generation.
On the one hand, exosomes retain some characteristics and functions of the donor cells and are easily remolded in vitro (125). Accumulating evidence indicates that proteins (126, 131, 132) as well as mRNAs and miRNAs (133, 134) within MDSC-derived exosomes obtained from different disorders are biologically active, and the molecular mechanisms involved are progressively being elucidated (133). Therefore, the use of exosomes directly collected from functional MDSC subpopulations or further reorganized after isolation may address some of the limitations of cell-based therapies. On the other hand, these methodologies may highlight therapeutic pathways for targeting exosomes derived from other cells to dramatically regulate MDSC activity during the pathogenesis of arthritis. Nevertheless, these effects are frequently reported under tumor conditions (135–139), and insufficient data have been reported from autoimmune arthritis microenvironments. Overall, exploring functional small RNAs or proteins within exosomes from MDSCs represents a promising approach for therapeutically targeting autoimmune diseases, and exosomes can be collected or further modified for application in clinical treatments. Regardless, issues regarding the properties of exosomes, such as their biogenesis, composition programming, targeted specificity, life span, and homogeneity, must be addressed prior to clinical application (130).
Other Immunosuppressive Cell Treatment Options for Autoimmune Diseases
Besides MDSC-based immunosuppressive cell treatments, other suppressive cells such as Tregs, tolerogenic DCs, have also shown promising therapeutic prospects in autoimmune diseases. In order to re-establish the badly damaged balance of T cell subpopulations, three extremely potential strategies for clinical use of Tregs was summarized that expansion of self-antigen-specific natural Tregs in vivo such as the promising injection with low-dose IL-2 (140–142), transfer of autogenous antigen-specific natural Tregs after antigen stimulating and propagation ex vivo, and transformation of antigen-specific conventional T cells into Tregs in vivo or ex vivo (143). Similarly, there are some critical problems necessarily to be resolved at the present. For instance, the methodology to transform stable Treg-like epigenetic changes targeting the third part, how to effectively stimulate and induce therapeutic Tregs with self-antigen for the second part, and how to avoid the potential side effects of cytokines promoting Treg cell proliferations after long-time use targing the first. Importantly, there is just one report adoptive transfer of autologous Tregs isolated from a patient with SLE after proliferation in vitro, which suggested no significant remission and a dynamic shift from Th1 to Th17 responses although activated Tregs was increased in inflamed skin (144). Therefore, further research and attempt were supposed to be conducted.
Recently, another DC subpopulation, tolerogenic DCs, has shown effectively regulatory effects in immune responses. Increasing evidence suggested that tolerogenic DCs could limit effector T cell responses (145) under signaling stimulations of IL-27 (146), IL-10 (147) or ligands of the aryl hydrocarbon receptor (AhR) (148, 149). In autoimmune arthritis, tolerogenic DCs were found a synergistic suppression with mesenchymal stem cells (MSC) in murine CIA models (150). Moreover, transfer of these cells into organ allograft animal models such as pancreatic islet, small intestine, renal and liver (151). However, it is still unclear that whether tolerogenic DCs was a specific subgroup or just represent a particular activation condition of DCs so far. Therefore, more evidence should be provided to completely recognize the nature, characteristics and roles in immune responses.
Notably, before clinical applications of these immunosuppressive cell treatments, length of curative effects, dose and frequency of injections were supposed to be extremely focused according to experience from preclinical and clinical cytokine treatments (2), which correlated closely with infectious risk and relapse problems. Otherwise, it is likely that internal interactions would be made use of in autoimmune diseases among these suppressive cells such as tolerogenic DCs and Foxp3+ Tregs (152, 153), MDSCs and Tregs (66, 73, 98), which suggests a considerable therapeutic strategy by combined transfer application of immunosuppressive cells for clinical treatments of autoimmune diseases.
Conclusion
Circumstantial findings substantiate the important and controversial roles of MDSC subpopulations in autoimmune arthritis and other inflammation-related diseases (116). Both MDSC subpopulations play important roles in regulating the proliferation, response and differentiation of CD4+ T cells during the progression of autoimmune arthritis. However, the maturation potential of these cells should not be ignored. Some reports demonstrate more potent inhibition of arthritis inflammation by PMN-MDSCs compared with MO-MDSCs, and the specific local microenvironment is important for MDSC subpopulations in autoimmune arthritis. Double-edged roles of MDSC subpopulations might depend on inflammatory systems or their various differentiation stages caused by dynamically altered microenvironments during arthritis progression. As exosomes derived from MDSCs are responsible for regulating immune function in immunocytes, exploring potent communication mechanisms of inhibitory MDSC subpopulations to discover more about MDSC-derived exosomes and target functional pathways for the clinical treatment of RNA represent important goals in the coming years.
Author Contributions
ML wrote the manuscript and discussed the content with the other coauthors. DZ, TW, XX and JT discussed the content with the other coauthors. SW conceived the topic and revised the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31470881, 31711530025, and 81771759), Natural Science Foundation of Jiangsu (Grant No. BK20150533), a project funded by the China Postdoctoral Science Foundation (Grant Nos. 2016M590423 and 2017T100336), Jiangsu Province's Key Medical Talents Program (Grant No. ZDRCB2016018), Jiangsu Province 333 Project (Grant No. BRA2017128), and Jiangsu Undergraduate Training Programs for Innovation (Grant No. 201710299044Z).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Eng J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
2. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. (2007) 7:429–42. doi: 10.1038/nri2094
3. Walter GJ, Evans HG, Menon B, Gullick NJ, Kirkham BW, Cope AP, et al. Interaction with activated monocytes enhances cytokine expression and suppressive activity of human CD4+CD45ro+CD25+CD127(low) regulatory T cells. Arth Rheum. (2013) 65:627–38. doi: 10.1002/art.37832
4. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
5. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
6. Tian J, Ma J, Ma K, Guo H, Baidoo SE, Zhang Y, et al. β-Glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur J Immunol. (2013) 43:1220–30. doi: 10.1002/eji.201242841
7. Nagaraj S, Collazo M, Corzo CA, Youn JI, Ortiz M, Quiceno D, et al. Regulatory myeloid suppressor cells in health and disease. Cancer Res. (2009) 69:7503–6. doi: 10.1158/0008–5472.CAN-09–2152
8. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
9. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 regulates the differentiation and function of myeloid-derived suppressor cells via targeting Runx1. J Immunol. (2015) 195:1301–11. doi: 10.4049/jimmunol.1500209
10. Wang W, Jiao Z, Duan T, Liu M, Zhu B, Zhang Y, et al. Functional characterization of myeloid-derived suppressor cell subpopulations during the development of experimental arthritis. Eur J Immunol. (2015) 45:464–73. doi: 10.1002/eji.201444799
11. Egelston C, Kurkó J, Besenyei T, Tryniszewska B, Rauch TA, Glant TT, et al. Suppression of dendritic cell maturation and T cell proliferation by synovial fluid myeloid cells from mice with autoimmune arthritis. Arthritis Rheum. (2012) 64:3179–88. doi: 10.1002/art.34494
12. Ochando JC, Shu HC. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res. (2012) 54:275. doi: 10.1007/s12026-012-8335-1
13. Crook KR, Jin M, Weeks MF, Rampersad RR, Baldi RM, Glekas AS, et al. Myeloid-derived suppressor cells regulate T cell and B cell responses during autoimmune disease. J Leukoc Biol. (2015) 97:573–82. doi: 10.1189/jlb.4A0314–139R
14. Guo C, Hu F, Yi H, Feng Z, Li C, Shi L, et al. Myeloid-derived suppressor cells have a proinflammatory role in the pathogenesis of autoimmune arthritis. Ann Rheum Dis. (2016) 75:278–85. doi: 10.1136/annrheumdis-2014–205508
15. Fujii W, Ashihara E, Hirai H, Nagahara H, Kajitani N, Fujioka K, et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J. Immunol. (2013) 191:1073–81. doi: 10.4049/jimmunol.1203535
16. Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ Immature Myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-Cell anergy in tumor-bearing host. Cancer Res. (2006) 66:1123. doi: 10.1158/0008–5472.CAN-05–1299
17. Yin B, Ma G, Yen CY, Zhou Z, Wang GX, Divino CM, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. J Immunol. (2010) 185:5828–34. doi: 10.4049/jimmunol.0903636
18. Zhou Z, French DL, Ma G, Eisenstein S, Chen Y, Divino CM, et al. Development and Function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells (2010) 28:620–32. doi: 10.1002/stem.301
19. Gallina G, Dolcetti L, Serafini P, Santo CD, Marigo I, Colombo MP, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. (2006) 116:2777–90. doi: 10.1172/JCI28828
20. Perfilyeva YV, Abdolla N, Ostapchuk YO, Tleulieva R, Krasnoshtanov VC, Belyaev NN. Expansion of CD11b(+)Ly6G(high) and CD11b(+)CD49d(+) myeloid cells with suppressive potential in mice with chronic inflammation and light-at-night-induced circadian disruption. Inflamm Res. (2017) 66:711–24. doi: 10.1007/s00011–017-1052–4
21. Haile LA, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. CD49d is a new marker for distinct myeloid-derived suppressor cell subpopulations in mice. J Immunol. (2010) 185:203–10. doi: 10.4049/jimmunol.0903573
22. Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. (2011) 11:802–7. doi: 10.1016/j.intimp.2011.01.003
23. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. (2011) 32:19. doi: 10.1016/j.it.2010.10.002
24. Eruslanov E, Neuberger M, Daurkin I, Perrin GQ, Algood C, Dahm P, et al. Circulating and tumor-infiltrating myeloid cell subsets in patients with bladder cancer. Int J Cancer (2012) 130:1109–19. doi: 10.1002/ijc.26123
25. Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. (2005) 65:3044–8. doi: 10.1158/0008-5472.CAN-04-4505
26. Liu CY, Wang YM, Wang CL, Feng PH, Ko HW, Liu YH, et al. Population alterations of L-arginase- and inducible nitric oxide synthase-expressed CD11b+/CD14(-)/CD15+/CD33+ myeloid-derived suppressor cells and CD8+ T lymphocytes in patients with advanced-stage non-small cell lung cancer. J Cancer Res Clin Oncol. (2010) 136:35–45. doi: 10.1007/s00432–009-0634–0
27. Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res. (2009) 69:1553–60. doi: 10.1158/0008–5472.CAN-08–1921
28. Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, Kleinschmidt-DeMasters BK, et al. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res. (2011) 17:6992–7002. doi: 10.1158/1078–0432.CCR-11–1107
29. Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, et al. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. (2007) 25:2546–53. doi: 10.1200/JCO.2006.08.5829
30. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol. (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
31. Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. (2012) 61:1155–67. doi: 10.1007/s00262–012-1294–5
32. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
33. Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A, et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol. (2012) 188:1136. doi: 10.4049/jimmunol.1101816
34. Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, et al. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology (2012) 136:176–83. doi: 10.1111/j.1365–2567.2012.03566.x
35. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology (2016) 5:e1004983. doi: 10.1080/2162402X.2015.1004983
36. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791. doi: 10.4049/jimmunol.181.8.5791
37. Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. (2015) 128:95–139. doi: 10.1016/bs.acr.2015.04.002
38. Crook KR, Liu P. Role of myeloid-derived suppressor cells in autoimmune disease. World J Immunol. (2014) 4:26–33. doi: 10.5411/wji.v4.i1.26
39. Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. (2008) 180:7898–906. doi: 10.4049/jimmunol.180.12.7898
40. Jiao Z, Hua S, Wang W, Wang H, Gao J, Wang X. Increased circulating myeloid-derived suppressor cells correlated negatively with Th17 cells in patients with rheumatoid arthritis. Scand J Rheumatol. (2013) 42:85–90. doi: 10.3109/03009742.2012.716450
41. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3ζ chain expression byl-Arginine. J Biol Chem. (2002) 277:21123–9. doi: 10.1074/jbc.M110675200
42. Rodriguez PC, Quiceno DG, Ochoa AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi: 10.1182/blood-2006–06-031856
43. Movahedi K, Guilliams M, Van dBJ, Van dBR, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111:4233–44. doi: 10.1182/blood-2007–07-099226
44. Dietlin TA, Hofman FM, Lund BT, Gilmore W, Stohlman SA, Rc VDV. Mycobacteria-induced Gr-1+ subsets from distinct myeloid lineages have opposite effects on T cell expansion. J Leuk Biol. (2007) 81:1205–12.doi: 10.1189/jlb.1006640
45. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol. (1998) 160:5729.
46. Harari O, Liao JK. Inhibition of MHC II Gene Transcription by nitric oxide and antioxidants. Curr Pharm Des. (2004) 10:893–8. doi: 10.2174/1381612043452893
47. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. (2004) 172:989–99. doi: 10.4049/jimmunol.172.2.989
48. Schmielau J, Finn OJ. Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res. (2001) 61:4756–60. Epub 2001/06/19.
49. Tian X, Ma J, Wang T, Tian J, Zhang Y, Mao L, et al. Long non-coding RNA HOXA transcript antisense RNA myeloid-specific 1-HOXA1 axis downregulates the immunosuppressive activity of myeloid-derived suppressor cells in lung cancer. Front Immunol. (2018) 9:473. doi: 10.3389/fimmu.2018.00473
50. Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol. (2013) 43:2930–42. doi: 10.1002/eji.201343349
51. Chatterjee S, Das S, Chakraborty P, Manna A, Chatterjee M, Choudhuri SK. Myeloid derived suppressor cells (MDSCs) can induce the generation of Th17 response from naive CD4+ T cells. Immunobiology (2013) 218:718–24. doi: 10.1016/j.imbio.2012.08.271
52. Yi H, Guo C, Yu X, Zuo D, Wang XY. Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol. (2012) 189:4295–304. doi: 10.4049/jimmunol.1200086
53. Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. (2012) 209:139–55. doi: 10.1084/jem.20101387
54. Nagaraj S, Gabrilovich DI. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. (2008) 68:2561–3. doi: 10.1158/0008–5472.CAN-07–6229
55. Brys L, Beschin A, Raes G, Ghassabeh GH, Noel W, Brandt J, et al. Reactive oxygen species and 12/15-lipoxygenase contribute to the antiproliferative capacity of alternatively activated myeloid cells elicited during helminth infection. J Immunol. (2005) 174:6095–104. doi: 10.4049/jimmunol.174.10.6095
56. Corzo CA, Thomas C, Lu L, Cotter MJ, Je-In Y, Cheng P, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
57. Kleer IMD, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4+CD25bright Regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. (2004) 172:6435–43. doi: 10.4049/jimmunol.172.10.6435
58. van Amelsfort JM, Jacobs KM, Bijlsma JW, Lafeber FP, Taams LS. CD4(+)CD25(+) regulatory T cells in rheumatoid arthritis: differences in the presence, phenotype, and function between peripheral blood and synovial fluid. Arthritis Rheum. (2004) 50:2775–85. doi: 10.1002/art.20499
59. Zhang H, Huang Y, Wang S, Fu R, Guo C, Wang H, et al. Myeloid-derived suppressor cells contribute to bone erosion in collagen-induced arthritis by differentiating to osteoclasts. J Autoimmunity (2015) 65:82–9. doi: 10.1016/j.jaut.2015.08.010
60. Kurko J, Vida A, Ocsko T, Tryniszewska B, Rauch TA, Glant TT, et al. Suppression of proteoglycan-induced autoimmune arthritis by myeloid-derived suppressor cells generated in vitro from murine bone marrow. PLoS ONE (2014) 9:e111815. doi: 10.1371/journal.pone.0111815
61. Kurko J, Vida A, Glant TT, Scanzello CR, Katz RS, Nair A, et al. Identification of myeloid-derived suppressor cells in the synovial fluid of patients with rheumatoid arthritis: a pilot study. BMC Muscul Disord. (2014) 15:281. doi: 10.1186/1471–2474-15–281
62. Sun J, Xu P, Du X, Zhang Q, Zhu Y. Piperlongumine attenuates collagen-induced arthritis via expansion of myeloid-derived suppressor cells and inhibition of the activation of fibroblast-like synoviocytes. Mol Med Rep. (2015) 11:2689–94. doi: 10.3892/mmr.2014.3001
63. Baddack U, Hartmann S, Bang H, Grobe J, Loddenkemper C, Lipp M, et al. A chronic model of arthritis supported by a strain-specific periarticular lymph node in BALB/c mice. Nat Commun. (2013) 4:1644. doi: 10.1038/ncomms2625
64. Chen W, Hu FL, Liu HJ, Xu LL, Li YN, Li ZG. Myeloid-derived suppressor cells promoted autologous B cell proliferation in rheumatoid arthritis. Beijing Da Xue Xue Bao Yi Xue Ban (2017) 49:819–23. doi: 10.3969/j.issn.1671-167X.2017.05.013
65. Zhang H, Wang S, Huang Y, Wang H, Zhao J, Gaskin F, et al. Myeloid-derived suppressor cells are proinflammatory and regulate collagen-induced arthritis through manipulating Th17 cell differentiation. Clin Immunol. (2015) 157:175–86. doi: 10.1016/j.clim.2015.02.001
66. Park MJ, Lee SH, Kim EK, Lee EJ, Baek JA, Park SH, et al. Interleukin-10 produced by myeloid-derived suppressor cells is critical for the induction of Tregs and attenuation of rheumatoid inflammation in mice. Sci Rep. (2018) 8:3753. doi: 10.1038/s41598-018-21856-2
67. Wang Y, Tian J, Wang S. The potential therapeutic role of myeloid-derived suppressor cells in autoimmune arthritis. Semin Arthritis Rheum. (2016) 45:490–5. doi: 10.1016/j.semarthrit.2015.07.003
68. Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, Bosch FVD, et al. Modulation of CCR2 in rheumatoid arthritis: A double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. (2014) 58:1931–9. doi: 10.1002/art.23591
69. Lee HJ, Ko JH, Jeong HJ, Ko AY, Kim MK, Wee WR, et al. Mesenchymal stem/stromal cells protect against autoimmunity via CCL2-dependent recruitment of myeloid-derived suppressor cells. J Immunol. (2015) 194:3634–45. doi: 10.4049/jimmunol.1402139
70. Qin J, Arakawa Y, Morita M, Fung JJ, Qian S, Lu L. C-C Chemokine receptor type 2 (CCR2)-dependent migration of myeloid-derived suppressor cells in protection of islet transplants. Transplantation (2017) 101:1. doi: 10.1097/TP.0000000000001529
71. Ji J, Xu J, Zhao S, Liu F, Qi J, Song Y, et al. Myeloid-derived suppressor cells contribute to systemic lupus erythematosus by regulating differentiation of Th17 cells and Treg. Clin Sci. (2016) 130:1453. doi: 10.1042/CS20160311
72. Wegner A, Verhagen J, Wraith DC. Myeloid-derived suppressor cells mediate tolerance induction in autoimmune disease. Immunology (2017) 151:26–42. doi: 10.1111/imm.12718
73. Zhao Y, Shen XF, Cao K, Ding J, Kang X, Guan WX, et al. Dexamethasone-induced myeloid-derived suppressor cells prolong allo cardiac graft survival through iNOS- and glucocorticoid receptor-dependent mechanism. Front Immunol. (2018) 9:282. doi: 10.3389/fimmu.2018.00282
74. Jin Y, Wang C, Min H, Xia M, Zhou J, Zhang Y. Circulating CD14+HLA-DR-/low myeloid-derived suppressor cells in leukemia patients with allogeneic hematopoietic stem cell transplantation: novel clinical potential strategies for the prevention and cellular therapy of graft-versus-host disease. Cancer Med. (2016) 5:1654–69. doi: 10.1002/cam4.688
75. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208. doi: 10.1016/j.it.2016.01.004
76. Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. (2010) 120:2486–96. doi: 10.1172/JCI41628
77. Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, et al. Monocytic Myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4 + Foxp3 + Treg Expansion. Am J Transpl. (2013) 13:3123–31. doi: 10.1111/ajt.12461
78. MacDonald KP, Rowe V, Clouston AD, Welply JK, Kuns RD, Ferrara JL, et al. Cytokine expanded myeloid precursors function as regulatory antigen-presenting cells and promote tolerance through IL-10-producing regulatory T cells. J Immunol. (2005) 174:1841–50. doi: 10.4049/jimmunol.174.4.1841
79. Katsikis PD, Chu CQ, Brennan FM, Maini RN, Feldmann M. Immunoregulatory role of interleukin 10 in rheumatoid arthritis. J Exp Med. (1994) 179:1517–27. doi: 10.1084/jem.179.5.1517
80. Becker JC, Brabletz T, Czerny C, Termeer C, Bröcker EB. Tumor escape mechanisms from immunosurveillance: induction of unresponsiveness in a specific MHC-restricted CD4+; human T cell clone by the autologous MHC class II+ melanoma. Int Immunol. (1993) 5:1501–8.
81. Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, et al. LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: A phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum. (2010) 62:929–39. doi: 10.1002/art.27334
82. Moret FM, Hack CE, Km WJ, Radstake TR, Lafeber FP, van Roon JA. Thymic stromal lymphopoietin (TSLP): a novel proinflammatory mediator in rheumatoid arthritis that potently activates CD1c+ myeloid dendritic cells to attract and stimulate T cells. Arthritis Rheum. (2014) 66:1176–84. doi: 10.1002/art.38338
83. Bowen JL, Olson JK. Innate immune CD11b+Gr-1+ cells, suppressor cells, affect the immune response during Theiler's virus-induced demyelinating disease. J Immunol. (2009) 183:6971–80. doi: 10.4049/jimmunol.0902193
84. Moline-Velazquez V, Cuervo H, Vila-Del Sol V, Ortega MC, Clemente D, de Castro F. Myeloid-derived suppressor cells limit the inflammation by promoting T lymphocyte apoptosis in the spinal cord of a murine model of multiple sclerosis. Brain Pathol. (2011) 21:678–91. doi: 10.1111/j.1750–3639.2011.00495.x
85. Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, et al. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood (2008) 112:5193–201. doi: 10.1182/blood-2008–02-139535
86. Nakamura H, Ueki Y, Sakito S, Matsumoto K, Yano M, Miyake S, et al. High serum and synovial fluid granulocyte colony stimulating factor (G-CSF) concentrations in patients with rheumatoid arthritis. Clin Exp Rheum. (2000) 18:713–8. doi: 10.1016/b978-0-444-53717-1.00822-2
87. Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. (2001) 167:1601–8. doi: 10.4049/jimmunol.167.3.1601
88. Edwards SW, Hallett MB. Seeing the wood for the trees: the forgotten role of neutrophils in rheumatoid arthritis. Immunol Today (1997) 18:320–4. doi: 10.1016/S0167–5699(97)01087–6
89. Németh T, Mócsai A, Lowell CA. Neutrophils in animal models of autoimmune disease. Semin Immunol. (2016) 28:174–86. doi: 10.1016/j.smim.2016.04.001
90. Turnquist HR, Zhao Z, Rosborough BR, Liu Q, Castellaneta A, Isse K, et al. IL-33 expands suppressive CD11b+ Gr-1(int) and regulatory T cells, including ST2L+ Foxp3+ cells, and mediates regulatory T cell-dependent promotion of cardiac allograft survival. J Immunol. (2011) 187:4598–610. doi: 10.4049/jimmunol.1100519
91. Hoechst B, Gamrekelashvili J, Manns MP, Greten TF, Korangy F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood (2011) 117:6532. doi: 10.1182/blood-2010–11-317321
92. Mildner A, Mack M, Schmidt H, Bruck W, Djukic M, Zabel MD, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain (2009) 132(Pt 9):2487–500. doi: 10.1093/brain/awp144
93. Ruscitti P, Cipriani P, Cantarini L, Liakouli V, Vitale A, Carubbi F, et al. Efficacy of inhibition of IL-1 in patients with rheumatoid arthritis and type 2 diabetes mellitus: two case reports and review of the literature. J Med Case Rep. (2015) 9:123. doi: 10.1186/s13256–015-0603-y
94. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood (2011) 118:50–61. doi: 10.1182/blood-2011–01-326827
95. Cros J, Cagnard N, Woollard K, Patey N, Zhang SY, Senechal B, et al. Human CD14 monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity (2010) 33:375. doi: 10.1016/j.immuni.2010.08.012
96. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. (2014) 9:591–604. doi: 10.1016/j.celrep.2014.09.032
97. Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal differentiation of classical monocytes as they survey steady state tissues and transport antigen to lymph nodes. Immunity (2013) 39:599–610. doi: 10.1016/j.immuni.2013.08.007
98. Zheng YN, Yu HP, Chen X, Deng HJ, Fan HZ, Gong YX, et al. Effect and mechanism of lipopolysaccharides-induced myeloid-derived suppressor cells on airway inflammation in asthmatic mice. Zhonghua Yi Xue Za Zhi (2012) 92:3147–50. doi: 10.3760/cma.j.issn.0376-2491.2012.44.016
99. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood (2009) 113:3190–7. doi: 10.1182/blood-2008–07-168575
100. Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. (2015) 6:e1887. doi: 10.1038/cddis.2015.246
101. Zhang L, Zhang Z, Zhang H, Wu M, Wang Y. Myeloid-derived suppressor cells protect mouse models from autoimmune arthritis via controlling inflammatory response. Inflammation (2014) 37:670–7. doi: 10.1007/s10753–013-9783-z
102. Greifenberg V, Ribechini E, Rössner S, Lutz MB. Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur J Immunol. (2009) 39:2865–76. doi: 10.1002/eji.200939486
103. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol. (2010) 185:2273–84. doi: 10.4049/jimmunol.1000901
104. Drujont L, Carretero-Iglesia L, Bouchet-Delbos L, Beriou G, Merieau E, Hill M, et al. Evaluation of the therapeutic potential of bone marrow-derived myeloid suppressor cell (MDSC) adoptive transfer in mouse models of autoimmunity and allograft rejection. PLoS ONE (2014) 9:e100013. doi: 10.1371/journal.pone.0100013
105. Haile LA, Von WR, Gamrekelashvili J, KrüGer C, Bachmann O, Westendorf AM, et al. Myeloid-derived suppressor cells in inflammatory bowel disease: a new immunoregulatory pathway. Gastroenterology (2008) 135:871–81, 881.e1–5. doi: 10.1053/j.gastro.2008.06.032
106. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116:5738. doi: 10.1182/blood-2010–06-287839
107. Kiniwa Y, Miyahara Y, Wang HY, Peng W, Peng G, Wheeler TM, et al. CD8+ Foxp3+ Regulatory T cells mediate immunosuppression in prostate cancer. Clin Cancer Res. (2007) 13:6947–58. doi: 10.1158/1078–0432.CCR-07–0842
108. Chaput N, Louafi S, Bardier A, Charlotte F, Vaillant JC, Menegaux F, et al. Identification of CD8+CD25+Foxp3+ suppressive T cells in colorectal cancer tissue. Gut (2009) 58:520–9. doi: 10.1136/gut.2008.158824
109. Janikashvili N, Trad M, Gautheron A, Samson M, Lamarthee B, Bonnefoy F, et al. Human monocyte-derived suppressor cells control graft-versus-host disease by inducing regulatory forkhead box protein 3-positive CD8+ T lymphocytes. J Allergy Clin Immunol. (2015) 135:1614–24.e4. doi: 10.1016/j.jaci.2014.12.1868
110. Boer MC, van Meijgaarden KE, Joosten SA, Ottenhoff TH. CD8+ Regulatory T Cells, and Not CD4+ T Cells, dominate suppressive phenotype and function after in vitro live Mycobacterium bovis-BCG activation of human cells. PLoS ONE (2014) 9:e94192. doi: 10.1371/journal.pone.0094192
111. Zheng J, Liu Y, Liu Y, Liu M, Xiang Z, Lam KT, et al. Human CD8+ regulatory T cells inhibit GVHD and preserve general immunity in humanized mice. Sci Transl Med. (2013) 5:168ra9. doi: 10.1126/scitranslmed.3004943
112. Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol. (2012) 189:464–74. doi: 10.4049/jimmunol.1200886
113. Zhang L, Bertucci AM, Ramsey-Goldman R, Harsha-Strong ER, Burt RK, Datta SK. Major pathogenic steps in human lupus can be effectively suppressed by nucleosomal histone peptide epitope-induced regulatory immunity. Clin Immunol. (2013) 149:365–78. doi: 10.1016/j.clim.2013.08.008
114. Liu GZ, Fang LB, Hjelmstrom P, Gao XG. Increased CD8+ central memory T cells in patients with multiple sclerosis. Mult Scler. (2007) 13:149–55. doi: 10.1177/1352458506069246
115. Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, et al. Regulatory functions of CD8+CD28– T cells in an autoimmune disease model. J Clin Invest. (2003) 112:1037. doi: 10.1172/JCI17935
116. Kim YJ, Chang SY, Ko HJ. Myeloid-derived suppressor cells in inflammatory bowel disease. Int Res. (2015) 13:105–11. doi: 10.5217/ir.2015.13.2.105
117. Zhu B, Kennedy JK, Wang Y, Sandoval-Garcia C, Cao L, Xiao S, et al. Plasticity of Ly-6Chi myeloid cells in T cell regulation. J Immunol. (2011) 187:2418. doi: 10.4049/jimmunol.1100403
118. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. (2009) 9:393–407. doi: 10.1038/nri2550
119. Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone (2007) 40:251–64. doi: 10.1016/j.bone.2006.09.023
120. Borges TJ, Wieten L, van Herwijnen MJ, Broere F, van der Zee R, Bonorino C, et al. The anti-inflammatory mechanisms of Hsp70. Front Immunol. (2012) 3:95. doi: 10.3389/fimmu.2012.00095
121. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials (2014) 35:2383. doi: 10.1016/j.biomaterials.2013.11.083
122. Chen W, Jiang J, Xia W, Jian H. Tumor-related exosomes contribute to tumor-promoting microenvironment: an immunological perspective. J Immunol Res. (2017) 2017:1073947. doi: 10.1155/2017/1073947
123. Wang Y, Tian J, Tang X, Rui K, Tian X, Ma J, et al. Exosomes released by granulocytic myeloid-derived suppressor cells attenuate DSS-induced colitis in mice. Oncotarget (2016) 7:15356–68. doi: 10.18632/oncotarget.7324
124. Jiang XC, Gao JQ. Exosomes as novel bio-carriers for gene and drug delivery. Int J Pharm. (2017) 521:167–75. doi: 10.1016/j.ijpharm.2017.02.038
125. Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim Biophys Acta (2012) 1820:940–8. doi: 10.1016/j.bbagen.2012.03.017
126. Burke MC, Oei MS, Edwards NJ, Ostrandrosenberg S, Fenselau C. Ubiquitinated proteins in exosomes secreted by myeloid-derived suppressor cells. J Proteome Res. (2014) 13:5965–72. doi: 10.1021/pr500854x
127. Cloutier N, Tan S, Boudreau LH, Cramb C, Subbaiah R, Lahey L, et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: the microparticle-associated immune complexes. Embo Mol Med. (2013) 5:235–49. doi: 10.1002/emmm.201201846
128. Zhang HG, Liu C, Su K, Su K, Yu S, Zhang L, et al. A membrane form of TNF-alpha presented by exosomes delays T cell activation-induced cell death. J Immunol. (2006) 176:7385–93. doi: 10.4049/jimmunol.176.12.7385
129. Kim SH, Bianco NR, Shufesky WJ, Morelli AE, Robbins PD. Effective treatment of inflammatory disease models with exosomes derived from dendritic cells genetically modified to express IL-4. J Immunol. (2007) 179:2242–9. doi: 10.4049/jimmunol.179.4.2242
130. Ller M. Janus-Faced Myeloid-Derived suppressor cell exosomes for the good and the bad in cancer and autoimmune disease. Front Immunol. (2018) 9:137. doi: 10.3389/fimmu.2018.00137
131. Geisasteggiante L, Dhabaria A, Edwards N, Ostrandrosenberg S, Fenselau C. Top-down analysis of low mass proteins in exosomes shed by murine myeloid-derived suppressor cells. Int J Mass Spectr. (2015) 378:264–9. doi: 10.1016/j.ijms.2014.08.035
132. Burke M, Choksawangkarn W, Edwards N, Ostrandrosenberg S, Fenselau C. Exosomes from myeloid-derived suppressor cells carry biologically active proteins. J Proteome Res. (2014) 13:836–43. doi: 10.1021/pr400879c
133. Geisasteggiante L, Belew AT, Clements VK, Edwards NJ, Ostrandrosenberg S, Elsayed NM, et al. Differential content of proteins, mRNAs, and miRNAs suggests that MDSC and their exosomes may mediate distinct immune suppressive functions. J Proteome Res. (2017) 17:486–98. doi: 10.1021/acs.jproteome.7b00646
134. Deng Z, Rong Y, Teng Y, Zhuang X, Samykutty A, Mu J, et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene (2017) 36:639–51. doi: 10.1038/onc.2016.229
135. Diao J, Yang X, Song X, Chen S, He Y, Wang Q, et al. Exosomal Hsp70 mediates immunosuppressive activity of the myeloid-derived suppressor cells via phosphorylation of Stat3. Med Oncol. (2015) 32:35. doi: 10.1007/s12032–014-0453–2
136. Ridder K, Sevko A, Heide J, Dams M, Rupp AK, Macas J, et al. Extracellular vesicle-mediated transfer of functional RNA in the tumor microenvironment. Oncoimmunology (2015) 4:e1008371. doi: 10.1080/2162402X.2015.1008371
137. Mignot G, Chalmin F, Ladoire S, Rébé C, Ghiringhelli F. Tumor exosome-mediated MDSC activation. Am J Pathol. (2011) 178:1403–4. doi: 10.1016/j.ajpath.2010.11.078
138. Xiang X, Poliakov AC, Liu Y, Deng Z, Wang J, Cheng Z, et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int J Cancer (2009) 124:2621–33. doi: 10.1002/ijc.24249
139. Wang J, Kim DV, Nathan DB, Ken M, Elke DB, Els VV, et al. The bone marrow microenvironment enhances multiple myeloma progression by exosome-mediated activation of myeloid-derived suppressor cells. Oncotarget (2015) 6:43992–4004. doi: 10.18632/oncotarget.6083
140. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-Cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Eng J Med. (2011) 365:2067–77. doi: 10.1056/NEJMoa1105143
141. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 Combination Therapy in Patients With Type 1 diabetes augments tregs yet transiently impairs β-Cell function. Diabetes (2012) 61:2340. doi: 10.2337/db12–0049
142. Hippen KL, Riley JL, June CH, Blazar BR. Clinical perspectives for regulatory T cells in transplantation tolerance. Semin Immunol. (2011) 23:462–8. doi: 10.1016/j.smim.2011.07.008
143. Miyara M, Ito Y, Sakaguchi S. TREG-cell therapies for autoimmune rheumatic diseases. Nat Rev Rheumatol. (2014) 10:543–51. doi: 10.1038/nrrheum.2014.105
144. Dall'Era M, Pauli ML, Remedios K, Taravati K, Sandoval PM, Putnam AL, et al. Adoptive regulatory T cell therapy in a patient with systemic lupus erythematosus. Arthritis Rheumatol. (2018). doi: 10.1002/art.40737. [Epub ahead of print].
145. Yogev N, Frommer F, Lukas D, Kautz-Neu K, Karram K, Ielo D, et al. Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD-1 receptor(+) regulatory T cells. Immunity (2012) 37:264–75. doi: 10.1016/j.immuni.2012.05.025
146. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. (2013) 14:1054–63. doi: 10.1038/ni.2695
147. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. (2001) 19:683–765. doi: 10.1146/annurev.immunol.19.1.683
148. Murray IA, Morales JL, Flaveny CA, Dinatale BC, Chiaro C, Gowdahalli K, et al. Evidence for ligand-mediated selective modulation of aryl hydrocarbon receptor activity. Mol Pharmacol. (2010) 77:247–54. doi: 10.1124/mol.109.061788
149. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Ann Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
150. Li R, Zhang Y, Zheng X, Peng S, Yuan K, Zhang X, et al. Synergistic suppression of autoimmune arthritis through concurrent treatment with tolerogenic DC and MSC. Sci Rep. (2017) 7:43188. doi: 10.1038/srep43188
151. Sun G, Shan J, Li Y, Zhou Y, Guo Y, Wu W, et al. Adoptive infusion of tolerogenic dendritic cells prolongs the survival of pancreatic islet allografts: a systematic review of 13 mouse and rat studies. PLoS ONE (2012) 7:e52096. doi: 10.1371/journal.pone.0052096
152. Quintana FJ, Wekerle H. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2010) 107:20768–73. doi: 10.1073/pnas.1009201107
Keywords: autoimmune arthritis, myeloid-derived suppressor cells, PMN-MDSCs, MO-MDSCs, immunosuppression
Citation: Li M, Zhu D, Wang T, Xia X, Tian J and Wang S (2018) Roles of Myeloid-Derived Suppressor Cell Subpopulations in Autoimmune Arthritis. Front. Immunol. 9:2849. doi: 10.3389/fimmu.2018.02849
Received: 12 July 2018; Accepted: 19 November 2018;
Published: 04 December 2018.
Edited by:
Laurence Morel, University of Florida, United StatesReviewed by:
Mi-La Cho, Catholic University of Korea, South KoreaNona Janikashvili, Tbilisi State Medical University, Georgia
Copyright © 2018 Li, Zhu, Wang, Xia, Tian and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengjun Wang, c2p3anNAdWpzLmVkdS5jbg==
Jie Tian, dGpqODUwOTEzQDE2My5jb20=