 Hongbing Yang
Hongbing Yang Zoë Wallace
Zoë Wallace Lucy Dorrell
Lucy Dorrell- 1Nuffield Department of Medicine, University of Oxford, Oxfordshire, United Kingdom
- 2Immunocore Ltd., Oxon, United Kingdom
- 3Oxford NIHR Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
HIV cannot be cured by current antiretroviral therapy (ART) because it persists in a transcriptionally silent form in long-lived CD4+ cells. Leading efforts to develop a functional cure have prioritized latency reversal to expose infected cells to immune surveillance, coupled with enhancement of the natural cytolytic function of immune effectors, or “kick and kill.” The most clinically advanced approach to improving the kill is therapeutic immunization, which aims to augment or re-focus HIV-specific cytolytic T cell responses. However, no vaccine strategy has enabled sustained virological control after ART withdrawal. Novel approaches are needed to overcome the limitations of natural adaptive immune responses, which relate to their specificity, potency, durability, and access to tissue reservoirs. Adoptive T cell therapy to treat HIV infection was first attempted over two decades ago, without success. Since then, progress in the field of cancer immunotherapy, together with recognition of the similarities in tumor microenvironments and HIV reservoirs has reignited interest in the application of T cell therapies to HIV eradication. Advances in engineering of chimeric antigen receptor (CAR)-transduced T cells have led to improved potency, persistence and latterly, resistance to HIV infection. Immune retargeting platforms have incorporated non-neutralizing and broadly neutralizing antibodies to generate Bispecific T cell Engagers (BiTEs) and Dual-Affinity Re-Targeting proteins (DARTs). T cell receptor engineering has enabled the development of the first bispecific Immune-mobilizing monoclonal T Cell receptors Against Viruses (ImmTAV) molecules. Here, we review the potential for these agents to provide a better “kill” and the challenges ahead for clinical development.
Introduction
Long-lived cells that harbor replication-competent HIV are responsible for viral persistence during antiretroviral therapy (ART). Within the first few days of infection, HIV-1 inserts into its host cell genome, primarily in CD4+ T cells that are transitioning from an activated to a quiescent state (1–4). Other cell populations of lymphoid, myeloid, and stromal origins also contribute to HIV reservoirs; however, the mechanisms involved are less well-understood, in part due to the challenges of sampling tissues (5). The persistence of these reservoirs has been explained by clonal proliferation of T cells and the existence of sanctuary sites, which may be both the cause and consequence of inadequate antiretroviral drug penetration and ineffective adaptive immune responses (6–11). The lack of a unique and reliable marker of these diverse reservoirs has significantly encumbered the development of strategies to provide either a sterilizing cure, in which all forms of HIV are eliminated from the body, or a functional cure (remission), defined by long-term control of HIV and preserved immune competence after ART withdrawal (12, 13). Only one person has achieved a sterilizing cure and cases of sustained antiretroviral-free HIV control remain rare (14, 15). However, advances in quantification and characterization of viral reservoirs, coupled with an unprecedented expansion in cancer immunotherapeutics to counteract immune exhaustion have opened up new possibilities for their application to the eradication of HIV reservoirs (16–18). In addition, it is possible to limit seeding of reservoirs and viral escape by initiating ART during primary infection, which may improve the chance of achieving a functional cure (19–21).
How Can Immune Retargeting Therapies Overcome the Failure of Natural Immune Responses to Eliminate HIV Reservoir Cells?
Several barriers to clearance of HIV reservoirs by the host immune response need to be considered when assessing the potential benefits and limitations of T cell retargeting agents: (1) low or absent viral antigen expression during ART; (2) viral heterogeneity resulting from mutational escape; (3) T cell dysfunction and/or exhaustion; (4) the inaccessibility of reservoir cells (22–25).
Viral latency is a state of reversible non-productive infection within a cell, which permits immune evasion. The rationale for using latency-reversing agents, the kick in “kick and kill,” is to initiate viral gene transcription and protein synthesis, thus removing protection from immune surveillance (26–28). However, recent developments in characterizing the molecular composition of the reservoir suggest that this may be overly simplistic. The vast majority of latently infected cells harbor defective proviruses, yet they may be transcriptionally active; translation of open reading frames with intact gag and nef sequences can lead to protein synthesis and susceptibility to killing by CD8+ T cells in vitro (29–31). Boosting of CD8+ T cells by therapeutic vaccination, with or without latency reversal, has not been successful in reducing viral reservoirs. This may reflect targeting of irrelevant epitopes, persistent T cell dysfunction and limited potency of LRAs (32–35). Furthermore, cells harboring intact and inducible proviruses may be inherently resistant to CD8+ T cell killing (36). Individuals who spontaneously control HIV have smaller latent reservoirs and display functionally superior CD8+ T cell responses, providing a model for functional cure (37, 38). However, loss of controller/non-progressor status is frequent, possibly due to ongoing viral replication in tissue sites that are inaccessible to cytolytic T cells (39–41).
In this review, we discuss the potential for T cell retargeting therapies to bring about a functional cure by overcoming the hurdles outlined above, namely, overcoming low antigen expression through affinity enhancement of antigen receptors, mobilizing sufficient numbers of effectors targeting conserved or non-escaped viral epitopes, recruiting functionally intact cells, and exploiting technologies to optimize tissue penetration and persistence (Figure 1). In addition, we examine the safety implications and the challenges for delivering these therapies to patients. Although adoptive T cell therapy, with or without TCR gene transfer, was the forerunner of these technologies and new adapted approaches are showing promise, this is beyond the scope of the discussion and is comprehensively covered elsewhere (42, 43).
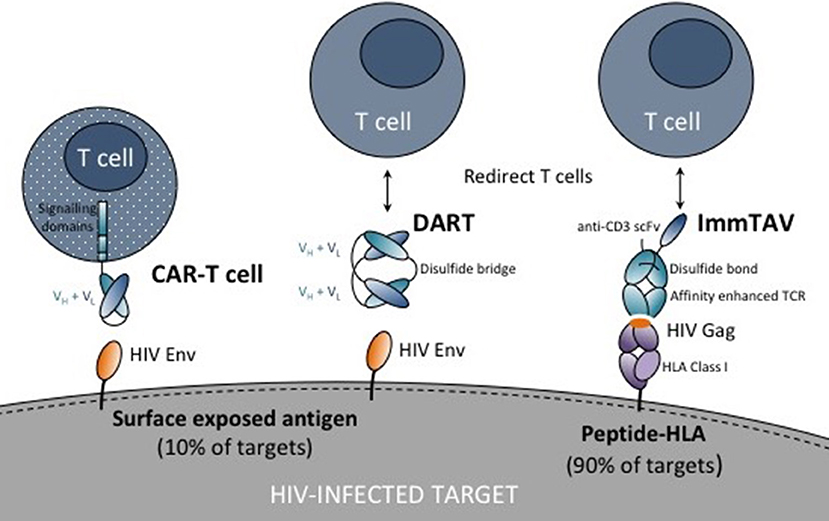
Figure 1. Schematic showing chimeric antigen receptor (CAR) T cell, dual affinity retargeting (DART) and immune-mobilizing monoclonal T cell receptor against viruses (ImmTAV) antigen recognition domains (antibodies or T cell receptors shown as blue ovals) and their respective targets on HIV-infected cells. The CAR is fused to one or more intracellular signaling domains. DARTs and ImmTAVs initiate signaling in T cells through cell surface CD3 via an anti-CD3 single chain variable fragment (scFv) which is fused to the antibody/TCR by a flexible linker (black line).
Chimeric Antigen Receptor (CAR) T Cells
CAR technology has evolved over more than two decades. It provides a means to re-programme T cells to recognize cell surface proteins through gene transfer of synthetic chimeric antigen receptors (CAR) (monoclonal antibodies) fused to a T cell activation domain. While the repertoire of potential CAR targets is smaller than that of T cell receptors, antigen recognition is not HLA-restricted, which is an advantage over conventional adoptive T cell therapy. Furthermore, CARs exploit healthy T cells that do not display the immune exhaustion phenotype typical of HIV-specific T cells in chronic infection.
The first anti-HIV CAR comprised the extracellular region of CD4 fused to a CD3ζ signaling domain (CD4ζ-CAR), conferring specificity for HIV-infected cells through binding of CD4 to the envelope protein, gp120. However, despite evidence of antiviral efficacy in vitro, CD4ζ-CAR T cells infusions had minimal impact on plasma viraemia in ART-naïve subjects or on viral reservoirs in ART-suppressed patients (44, 45). Modest cytolytic function in vivo, limited persistence due to poor proliferative capacity and susceptibility to HIV infection may have contributed to these disappointing results. Improvements in the technology were achieved through successive modifications in the signaling domains to enhance anti-tumor activity: second- and third-generation CARs were produced by addition of one or more costimulatory domains such as 4-1BB, CD28 and ICOS (46–48). Second-generation CD19-specific CARs achieved spectacular response rates in the treatment of B cell malignancies (albeit with high levels of toxicity) leading to their approval by the US Food and Drug Administration and paving the way for the development of more effective HIV-specific CARs (49–51). A re-engineered CD4 CAR containing the 4-1BB domain showed significantly enhanced antiviral potency in vitro and greater capacity to proliferate and prevent HIV spread in a humanized mouse model than the first-generation version (52). In addition, a large number of broadly neutralizing antibodies (bNAbs), which target regions of vulnerability in the viral envelope, have since been identified as potential CAR candidates (53).
Achieving sustained virological control after ART cessation will likely require repeated infusions of CAR T cells or strategies to prolong their persistence in vivo. One approach is to engineer the expression of CD4ζ CAR in hematopoietic stem cells (HSC). Modified HSC-derived CAR T cells were shown to engraft, proliferate, and display antiviral activity in a humanized mouse model of HIV infection; however, in a non-human primate model only transient and partial control of viral replication was observed after SHIV challenge and withdrawal of ART (54, 55).
A further consideration is to protect modified T cells from HIV infection. The safety and efficacy of zinc finger nuclease disruption of CCR5, the co-receptor for viral entry into CD4+ T cells has been demonstrated in a clinical trial: modified autologous CD4+ T cells had a mean half-life of 48 weeks during ART and were maintained for longer than unmodified CD4+ T cells after ART withdrawal (56). To generate functional HIV-resistant CAR T cells, site-directed megaTAL nuclease-mediated CCR5 gene disruption has been successfully combined with targeted delivery of an adeno-associated virus CAR cassettes to the CCR locus using homology-directed recombination/repair (HDR), which achieves highly efficient integration (57). Simultaneous CCR5 gene disruption and expression of single chain variable fragments (scFv) of HIV-specific broadly neutralizing antibodies (bNAb) coupled to a second-generation co-stimulatory domain in primary human T cells endowed them with the capacity to suppress viral replication in vitro (57, 58). However, feasibility of manufacture for clinical application has yet to be demonstrated.
B cell follicles in lymphoid tissue are a sanctuary site for HIV replication, which occurs preferentially in T follicular helper cells, due to exclusion of CD8+ T cells that lack expression of the follicular homing chemokine receptor CXCR5 (59). Haran et al. successfully co-expressed CXCR5 with an antiviral bispecific CAR comprising rhesus CD4 domains fused to mannose-binding lectin, which targets carbohydrate residues on the viral envelope. SIV CAR/CXCR5-expressing T cells were able to suppress SIV replication in vitro and to migrate to B cell follicles in lymphoid tissue explants (60).
Two clinical trials of CAR T cell therapy in HIV-positive ART-treated subjects are ongoing or due to recruit: one is evaluating a bNAb (VRC01)-based CAR, VC-CAR-T (NCT03240328) and the other, a CD4 CAR modified for HIV resistance by ZFN disruption of CCR5 and conjugation of the C34 peptide to the CXCR4 N-terminus (NCT03617198) (61).
Bispecific Antibodies
Bispecific antibodies (bsAb) have emerged as a more scalable alternative to CAR T cell therapy. Originally developed to overcome the limitations of monoclonal antibodies for cancer therapy, the first generation bsAb platform, bispecific T cell engagers (BiTEs), comprised a tumor-specific variable fragment and a CD3-specific fragment joined by a single peptide linker. This format enabled redirection of effector T cells to eliminate tumor cells bearing the target antigen (62). Blinatumomab (CD19xCD3) was the first BiTE to be licensed by the FDA for clinical use, initially in the treatment of acute lymphoblastic leukemia [reviewed in (63)]. The BiTe format was improved by addition of a disulphide bond to stabilize the variable domains of the two antigen-binding moieties; the resulting Dual-Affinity Re-Targeting protein (DART) has extended storage and serum stability (64). CD19xCD3 DARTs showed enhanced potency in directing B cell lysis over a single-chain format molecule with the same specificities, likely due to a higher association rate and target affinity (65).
HIV-specific BiTes and DARTs have recently been developed and tested in vitro. The BiTEs consisted of a scFV derived from either of the HIV gp120-specific bNAbs B12 and VRC01, the scFv of the antibody 17b, with or without two CD4 extracellular domains, CD4 (1+2); all were fused to an anti-human CD3ε scFV (66). The parent antibodies target conserved epitopes in Env. These constructs were tested for their capacity to suppress HIV replication in peripheral blood mononuclear cells or macrophages cultured with autologous CD8+ T cells. The B12 BiTe was the most potent, achieving >90% HIV inhibition at a concentration 10-fold lower than the other BiTes. Of note, the CD4(1+2) BiTE promoted infection of CD8+/CD4- T cells in vitro, possibly due to effects on gp120 conformation, whereas the other constructs did not.
Sung et al. reported the development of an HIVxCD3 DART comprising a non-neutralizing mAb against Env (A32 or 7B2) and a humanized anti-CD3 mAb. These HIVxCD3 DARTs were able to redirect ex vivo polyclonal CD8+ T cells from both HIV-seronegative and ART-suppressed HIV-seropositive donors to kill HIV-infected CD4+ T cells. Reduction in virus recovery even without the addition of CD8+ T cells (infected CD4+ T cells and DARTs only) was observed, which suggested that the DARTs could also recruit cytotoxic CD4+ T cells. The DARTs did not induce T cell activation or lysis in the absence of Env (67). Sloan et al. also developed a panel of HIVxCD3 DARTs based on the bNAbs PGT121 and PGT145, in addition to the nNAbs A32 and 7B2. These DARTs redirected CD8+ T cell killing of HIV-infected resting primary cells with low Env expression. Using ex vivo PBMCs from patients on suppressive ART a combination of two HIV DARTs was able to significantly reduce viral RNA in culture supernatants after 14 days in 3/4 subjects, suggesting that a subset of HIV reservoir cells may express enough Env to be targeted (68). In a third study, exposure of PBMC from ART-treated subjects to an HIV DART based on the VRC07 bNAb (VRC07-αCD3) was reported to upregulate cell surface Env. This was accompanied by a reduction in CD4+ T cells containing HIV DNA, suggesting that the DART both activated HIV reservoir cells and targeted them for cytolysis. In addition, short-term safety of a VRC07-rhesus CD3 DART was demonstrated in simian-human immunodeficiency virus-infected ART-treated macaques; importantly, no viral rebound was observed despite the possibility of viral reactivation by the DART (69). Based on pre-clinical work of Sung and Sloan, MacroGenics registered the first clinical trial of an HIV DART, MGD014, which will assess safety, tolerability and effects of the latent reservoir in ART-treated subjects (NCT03570918). A critical question is whether a combination of DARTs may be needed in order to achieve clinical benefit, given that viral escape driven by the selective pressure of anti-Env bNAbs was observed in a clinical trial in involving bNAb monotherapy followed by analytical ART interruption (70).
Bispecific T Cell Receptors
A limitation of CAR T cells and bsAb therapies is their specificity for cell surface antigens. TCRs have the capacity to recognize intracellular proteins in the form of cell surface HLA class I-bound peptides. Soluble bispecific agents that combine T cell receptors (TCRs) with anti-CD3 effector function exploit both the vastly greater array range of potential targets (up to 9-fold more than antibodies) and the scalability of bsAb. Furthermore, picomolar affinity for the desired peptide-HLA (pHLA) complex can be achieved through targeted mutations in the complementarity-determining regions using phage display technology, generating molecules with higher potency than typical affinity-enhanced antibodies [(71), reviewed in (72)]. A stable soluble molecule is generated by truncation of the transmembrane domains and incorporation of a disulphide bond between the alpha and beta chains. The TCR is fused to an anti-CD3 scFV for redirection of T cells (as per BiTes and DARTS). Immune-mobilizing monoclonal TCRs Against Cancer antigens (ImmTAC) were shown to recruit non-cancer antigen-specific T cells to form an immunological synapse resulting in T cell activation and specific tumor cell lysis in vitro and in vivo (73, 74). The first ImmTAC to enter clinical trials is targeted to the melanoma antigen, gp100 and is in pivotal trials for the treatment of uveal melanoma (75) (NCT03070392).
The ImmTAC platform technology has been applied to HIV, specifically to the Gag protein, which is both highly conserved and abundant in infected cells, in contrast to Env. The first Immune-mobilizing monoclonal TCR Against Viruses (ImmTAV) was designed to target the HLA-A*0201-restricted Gag p17 epitope, SLYNTVATL (SL9) and known escape mutants (76). We reported that the HIV-specific ImmTAV molecule was able to redirect CD8+ T cells to kill Gag+ cells at nanomolar concentrations and low effector/target ratios. As with the bsAbs, the ImmTAV molecules were able to redirect CD8+ T cells from both HIV-seronegative and HIV-positive ART-suppressed donors to eliminate reservoir cells harboring inducible viruses (77).
The potential for ImmTAC and ImmTAV molecules to overcome low antigen expression was demonstrated by using tagged versions of the relevant TCRs to quantify pHLA complexes on cancer cells and virus-infected cells, respectively. These analyses showed that as few as 10 epitopes per cell could be quantified (77, 78). However, with such high TCR affinities the risk of off-target reactivity to self-peptide mimics must be carefully assessed in order to define a therapeutic window and avoid serious toxicity (79). Furthermore, conventional non-human toxicology testing is of limited predictive value, since TCR binding to peptide targets only occurs in the context of HLA class I molecules. This has been addressed by the development of comprehensive in silico and ex vivo human tissue analyses for pre-clinical safety evaluation (72). To date, ImmTAC molecules have been developed only for HLA-A02-restricted targets as this family of alleles is dominant in most ethnic groups across the world and is the best characterized. However, there is potential to extend the platform to other HLA alleles, which would broaden population coverage further.
Future Directions
Over the past 5 years, the kick and kill concept has been tested in several clinical trials, with disappointing results. However, these studies have helped to refine our understanding of the dynamics and distribution of latent HIV reservoirs and have highlighted that each component of the strategy needs to be optimized. The licensing of CAR T cell and BiTe therapies is a reflection of astonishing progress in T cell-based immunotherapies for cancer in recent years. Forthcoming trials of HIV-specific versions of these agents will seek to assess whether clinically meaningful reductions in cellular reservoirs can be achieved with an “upgraded” kill. These trials could provide valuable insight into the levels of antigen required to sensitize cells to elimination by affinity-enhanced antibodies and TCRs, with and without latency reversal. The presence of archived CD8+ T cell escape mutants is perhaps the most obvious challenge for T cell retargeting therapy. Engineering specificity toward functionally constrained epitopes may mitigate against both past and future viral escape; however, we anticipate that combinations of agents will be needed, in an analogous fashion to combination ART. Furthermore, the efficiency of killing will depend on the functional capacity of non-HIV-specific T cells, which may be compromised in chronic HIV infection even after long-term ART (77, 80). This may require correction by immune checkpoint receptor blockade or other forms of immunomodulation.
Long-term follow-up of trial participants will be necessary to ascertain the requirement for repeated or continued administration of T cell retargeting therapy, a key consideration for its utility in clinical practice. Safety is also paramount since the risk/benefit ratio for people with well-controlled HIV is much higher than for individuals with cancer, who may have few treatment options. Early data suggested that gene-edited CD4+ cell infusions are well-tolerated. The risk of cytokine release syndrome (CRS) with CAR T cell administration in virologically suppressed patients is expected to be lower than that observed during treatment of malignancies, since Env is a non-self antigen and CRS is related to high (tumor) antigenic load. Furthermore, HIV BiTes and DARTs are based on monoclonal antibodies with an established safety profile. The exceptionally high affinity of ImmTAVs for pHLA will require cautious dose escalations in first-in-human trials. However, a major advantage of soluble agents over cellular therapies is that they are cleared within hours, enabling exposure in vivo to be tightly controlled. Human CD3 scFv has a short binding half-life (minutes) due to a nanomolar binding affinity. Experience with blinatumomab indicates this molecule does not cause off-target activation. On the other hand, on-target activation of CD4+ T cells by CD3 scFv could have the potential to spread infection, which highlights the need to protect uninfected cells by administration with effective ART, ideally including an integrase inhibitor. Lastly, retargeting therapies should be affordable and accessible to patients across the world. The soluble “single vial” bispecific agents have an advantage over cellular therapies with respect to cost and feasibility of manufacture at scale. A possible model for sustainable low-cost production is the recently announced strategic partnership between the International AIDS Vaccine Initiative and the Serum Institute of India, which aims to ensure global access to monoclonal antibody therapies (81).
Author Contributions
HY, ZW, and LD contributed to the literature search and drafting of manuscript. ZW prepared the illustration.
Conflict of Interest Statement
HY has received research funding from Immunocore Ltd. ZW and LD are employees of Immunocore Ltd.
References
1. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science (1997) 278:1295–300.
2. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci USA. (1997) 94:13193–7.
3. Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature (2014) 512:74–7. doi: 10.1038/nature13594
4. Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat Med. (2018) 24:923–6. doi: 10.1038/s41591-018-0026-6
5. Kandathil AJ, Sugawara S, Balagopal A. Are T cells the only HIV-1 reservoir? Retrovirology (2016) 13:86. doi: 10.1186/s12977-016-0323-4
6. Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. (2009) 15:893. doi: 10.1038/nm.1972
7. Josefsson L, von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Killian M, et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci USA. (2013) 110:E4987–96. doi: 10.1073/pnas.1308313110
8. Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, et al. HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol. (2016) 90:8968–83. doi: 10.1128/JVI.00674-16
9. Buzón MJ, Codoñer FM, Frost SDW, Pou C, Puertas MC, Massanella M, et al. Deep molecular characterization of HIV-1 dynamics under suppressive HAART. PLoS Pathog. (2011) 7:e1002314. doi: 10.1371/journal.ppat.1002314
10. Lorenzo-Redondo R, Fryer HR, Bedford T, Kim E-Y, Archer J, Kosakovsky Pond SL, et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature (2016) 530:51–6. doi: 10.1038/nature16933
11. McMichael AJ, Borrow P, Tomaras GD, Goonetilleke N, Haynes BF. The immune response during acute HIV-1 infection: clues for vaccine development. Nat Rev Immunol. (2010) 10:11–23. doi: 10.1038/nri2674
12. Badia R, Ballana E, Castellví M, García-Vidal E, Pujantell M, Clotet B, et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat Commun. (2018) 9:2739. doi: 10.1038/s41467-018-05157-w
13. Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, Spivak AM, Nell RA, Tomescu C, et al. CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci Transl Med. (2018) 10:eaar6759. doi: 10.1126/scitranslmed.aar6759
14. Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood (2011) 117:2791–9. doi: 10.1182/blood-2010-09-309591
15. Henrich TJ, Hanhauser E, Marty FM, Sirignano MN, Keating S, Lee T-H, et al. Antiretroviral-free HIV-1 remission and viral rebound following allogeneic stem cell transplantation: a report of two cases. Ann Intern Med. (2014) 161:319–27. doi: 10.7326/M14-1027
16. Mumprecht S, Schürch C, Schwaller J, Solenthaler M, Ochsenbein AF. Programmed death 1 signaling on chronic myeloid leukemia–specific T cells results in T-cell exhaustion and disease progression. Blood (2009) 114:1528–36. doi: 10.1182/blood-2008-09-179697
17. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
18. Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. (2013) 94:41–53. doi: 10.1189/jlb.1212631
19. Jain V, Hartogensis W, Bacchetti P, Hunt PW, Hatano H, Sinclair E, et al. Antiretroviral therapy initiated within 6 months of hiv infection is associated with lower T-cell activation and smaller hiv reservoir size. J Infect Dis. (2013) 208:1202–11. doi: 10.1093/infdis/jit311
20. Ananworanich J, Schuetz A, Vandergeeten C, Sereti I, de Souza M, Rerknimitr R, et al. Impact of multi-targeted antiretroviral treatment on gut t cell depletion and HIV reservoir seeding during acute HIV infection. PLoS ONE (2012) 7:e33948. doi: 10.1371/journal.pone.0033948
21. Ananworanich J, Chomont N, Eller LA, Kroon E, Tovanabutra S, Bose M, et al. HIV DNA set point is rapidly established in acute HIV infection and dramatically reduced by early ART. EBioMed. (2016) 11:68–72. doi: 10.1016/j.ebiom.2016.07.024
22. Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, et al. T-bet and eomes are differentially linked to the exhausted phenotype of CD8+ T cells in hiv infection. PLoS Pathog. (2014) 10:e1004251. doi: 10.1371/journal.ppat.1004251
23. Migueles SA, Weeks KA, Nou E, Berkley AM, Rood JE, Osborne CM, et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J Virol. (2009) 83:11876–89. doi: 10.1128/JVI.01153-09
24. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature (2015) 517:381–5. doi: 10.1038/nature14053
25. Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS (2013) 8:165–9. doi: 10.1097/COH.0b013e32835fc601
26. Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity (2012) 36:491–501. doi: 10.1016/j.immuni.2012.01.014
27. Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV (2014) 1:e13–21. doi: 10.1016/S2352-3018(14)70014-1
28. Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, et al. The depsipeptide romidepsin reverses HIV-1 latency in vivo. PLoS Pathog. (2015) 11:e1005142. doi: 10.1371/journal.ppat.1005142
29. Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat Med. (2016) 22:1043–9. doi: 10.1038/nm.4156
30. Imamichi H, Dewar RL, Adelsberger JW, Rehm CA, O'Doherty U, Paxinos EE, et al. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc Natl Acad Sci USA. (2016) 113:8783–8. doi: 10.1073/pnas.1609057113
31. Pollack RA, Jones RB, Pertea M, Bruner KM, Martin AR, Thomas AS, et al. Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host Microbe (2017) 21:494–506.e4. doi: 10.1016/j.chom.2017.03.008
32. Casazza JP, Bowman KA, Adzaku S, Smith EC, Enama ME, Bailer RT, et al. Therapeutic vaccination expands and improves the function of the HIV-specific memory T-cell repertoire. J Infect Dis. (2013) 207:1829–40. doi: 10.1093/infdis/jit098
33. Achenbach CJ, Assoumou L, Deeks SG, Wilkin TJ, Berzins B, Casazza JP, et al. Effect of therapeutic intensification followed by HIV DNA prime and rAd5 boost vaccination on HIV-specific immunity and HIV reservoir (EraMune 02): a multicentre randomised clinical trial. Lancet HIV (2015) 2:e82–91. doi: 10.1016/S2352-3018(15)00026-0
34. Hancock G, Morón-López S, Kopycinski J, Puertas MC, Giannoulatou E, Rose A, et al. Evaluation of the immunogenicity and impact on the latent HIV-1 reservoir of a conserved region vaccine, MVA.HIVconsv, in antiretroviral therapy-treated subjects. J Int AIDS Soc. (2017) 20:21171. doi: 10.7448/IAS.20.1.21171
35. Leth S, Schleimann MH, Nissen SK, Højen JF, Olesen R, Graversen ME, et al. Combined effect of Vacc-4x, recombinant human granulocyte macrophage colony-stimulating factor vaccination, and romidepsin on the HIV-1 reservoir (REDUC): a single-arm, phase 1B/2A trial. Lancet HIV (2016) 3:e463–72. doi: 10.1016/S2352-3018(16)30055-8
36. Huang SH, Ren Y, Thomas AS, Chan D, Mueller S, Ward AR, et al. Latent HIV reservoirs exhibit inherent resistance to elimination by CD8+ T cells. J Clin Invest. (2018) 128:876–89. doi: 10.1172/JCI97555
37. Graf EH, Mexas AM, Yu JJ, Shaheen F, Liszewski MK, Di Mascio M, et al. Elite suppressors harbor low levels of integrated HIV DNA and high levels of 2-LTR circular HIV DNA compared to HIV+ patients on and off HAART. PLoS Pathog. (2011) 7:e1001300. doi: 10.1371/journal.ppat.1001300
38. Buzon MJ, Yang Y, Ouyang Z, Sun H, Seiss K, Rogich J, et al. Susceptibility to CD8 T cell-mediated killing influences the reservoir of latently HIV-1 infected CD4 T cells. J Acquir Immune Defic Syndr. (2014) 65:1–9. doi: 10.1097/QAI.0b013e3182a1bc81
39. Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med. (2015) 21:132–9. doi: 10.1038/nm.3781
40. Leon A, Perez I, Ruiz-Mateos E, Benito JM, Leal M, Lopez-Galindez C, et al. Rate and predictors of progression in elite and viremic HIV-1 controllers. AIDS (2016) 30:1209–20. doi: 10.1097/QAD.0000000000001050
41. Grabar S, Selinger-Leneman H, Abgrall S, Pialoux G, Weiss L, Costagliola D. Loss of long-term non-progressor and HIV controller status over time in the French Hospital Database on HIV - ANRS CO4. PLoS ONE (2017) 12:e0184441. doi: 10.1371/journal.pone.0184441
42. Patel S, Jones RB, Nixon DF, Bollard CM. T-cell therapies for HIV: Preclinical successes and current clinical strategies. Cytotherapy (2016) 18:931–42. doi: 10.1016/j.jcyt.2016.04.007
43. Sung JA, Patel S, Clohosey ML, Roesch L, Tripic T, Kuruc JD, et al. HIV-Specific, ex vivo expanded t cell therapy: feasibility, safety, and efficacy in ART-suppressed HIV-infected individuals. Mol Theor. (2018) 26:2496–506. doi: 10.1016/j.ymthe.2018.08.015
44. Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific t-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Theor. (2002) 5:788–97. doi: 10.1006/mthe.2002.0611
45. Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+and CD8+ T cells in human immunodeficiency virus–infected subjects. Blood (2000) 96:785–93.
46. Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia (2004) 18:676–84. doi: 10.1038/sj.leu.2403302
47. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest (2011) 121:1822–6. doi: 10.1172/JCI46110
48. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. (2013) 6:33. doi: 10.1186/1756-8722-6-33
49. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
50. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
51. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
52. Leibman RS, Richardson MW, Ellebrecht CT, Maldini CR, Glover JA, Secreto AJ, et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. (2017) 13:e1006613. doi: 10.1371/journal.ppat.1006613
53. Ali A, Kitchen SG, Chen ISY, Ng HL, Zack JA, Yang OO. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J Virol. (2016) 90:6999–7006. doi: 10.1128/JVI.00805-16
54. Zhen A, Rezek V, Youn C, Rick J, Lam B, Chang N, et al. Stem-cell based engineered immunity against HIV infection in the humanized mouse model. J Vis Exp. (2016) 113:e54048. doi: 10.3791/54048
55. Zhen A, Peterson CW, Carrillo MA, Reddy SS, Youn CS, Lam BB, et al. Long-term persistence and function of hematopoietic stem cell-derived chimeric antigen receptor T cells in a nonhuman primate model of HIV/AIDS. PLoS Pathog. (2017) 13:e1006753. doi: 10.1371/journal.ppat.1006753
56. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. (2014) 370:901–10. doi: 10.1056/NEJMoa1300662
57. Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med. (2015) 7:307ra156. doi: 10.1126/scitranslmed.aac5530
58. Hale M, Mesojednik T, Romano Ibarra GS, Sahni J, Bernard A, Sommer K, et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol Ther. (2017) 25:570–9. doi: 10.1016/j.ymthe.2016.12.023
59. Hufert FT, van Lunzen J, Janossy G, Bertram S, Schmitz J, Haller O, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS (1997) 11:849–57.
60. Haran KP, Hajduczki A, Pampusch MS, Mwakalundwa G, Vargas-Inchaustegui DA, Rakasz EG, et al. Simian immunodeficiency virus (SIV)-specific chimeric antigen receptor-T cells engineered to target b cell follicles and suppress SIV replication. Front Immunol. (2018) 9:492. doi: 10.3389/fimmu.2018.00492
61. Liu B, Zou F, Lu L, Chen C, He D, Zhang X, et al. Chimeric antigen receptor T cells guided by the single-chain fv of a broadly neutralizing antibody specifically and effectively eradicate virus reactivated from latency in CD4(+) T lymphocytes isolated from HIV-1-infected individuals receiving suppressive. J Virol. (2016) 90:9712–24. doi: 10.1128/JVI.00852-16
63. Nagorsen D, Kufer P, Baeuerle PA, Bargou R. Blinatumomab: a historical perspective. Pharmacol Ther. (2012) 136:334–42. doi: 10.1016/j.pharmthera.2012.07.013
64. Johnson S, Burke S, Huang L, Gorlatov S, Li H, Wang W, et al. Effector cell recruitment with novel fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. (2010) 399:436–49. doi: 10.1016/j.jmb.2010.04.001
65. Moore PA, Zhang W, Rainey GJ, Burke S, Li H, Huang L, et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood (2011) 117:4542–51. doi: 10.1182/blood-2010-09-306449
66. Brozy J, Schlaepfer E, Mueller CKS, Rochat M-A, Rampini SK, Myburgh R, et al. Antiviral activity of HIV gp120-targeting bispecific T cell engager antibody constructs. J Virol. (2018) 92:e00491–18. doi: 10.1128/JVI.00491-18
67. Sung JAM, Pickeral J, Liu L, Stanfield-Oakley SA, Lam C-YK, Garrido C, et al. Dual-affinity re-targeting proteins direct T cell–mediated cytolysis of latently HIV-infected cells. J Clin Invest. (2015) 125:4077–90. doi: 10.1172/JCI82314
68. Sloan DD, Lam C-YK, Irrinki A, Liu L, Tsai A, Pace CS, et al. Targeting HIV reservoir in infected CD4 T cells by dual-affinity re-targeting molecules (DARTs) that Bind HIV envelope and recruit cytotoxic T cells. PLoS Pathog. (2015) 11:e1005233. doi: 10.1371/journal.ppat.1005233
69. Pegu A, Asokan M, Wu L, Wang K, Hataye J, Casazza JP, et al. Activation and lysis of human CD4 cells latently infected with HIV-1. Nat Commun. (2015) 6:8447. doi: 10.1038/ncomms9447
70. Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu C-L, Lorenzi JCC, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature (2016) 535:556–60. doi: 10.1038/nature18929
71. Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, et al. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. (2005) 23:349–54. doi: 10.1038/nbt1070
72. Oates J, Hassan NJ, Jakobsen BK. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol Immunol. (2015) 67:67–74. doi: 10.1016/j.molimm.2015.01.024
73. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med. (2012) 18:980–7. doi: 10.1038/nm.2764
74. McCormack E, Adams KJ, Hassan NJ, Kotian A, Lissin NM, Sami M, et al. Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1- and LAGE-1-positive tumors. Cancer Immunol Immunother. (2013) 62:773–85. doi: 10.1007/s00262-012-1384-4
75. Sato T, Nathan PD, Hernandez-Aya L, Sacco JJ, Orloff MM, Visich J, et al. Redirected T cell lysis in patients with metastatic uveal melanoma with gp100-directed TCR IMCgp100: overall survival findings. J Clin Oncol. (2018) 36:9521. doi: 10.1200/JCO.2018.36.15_suppl.9521
76. Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. (2008) 14:1390–95. doi: 10.1038/nm.1779
77. Yang H, Buisson S, Bossi G, Wallace Z, Hancock G, So C, et al. Elimination of latently HIV-infected cells from antiretroviral therapy-suppressed subjects by engineered immune-mobilizing T-cell receptors. Mol Ther. (2016) 24:1913–25. doi: 10.1038/mt.2016.114
78. Bossi G, Gerry AB, Paston SJ, Sutton DH, Hassan NJ, Jakobsen BK. Examining the presentation of tumor-associated antigens on peptide-pulsed T2 cells. Oncoimmunology (2013) 2:e26840. doi: 10.4161/onci.26840
79. Raman MCC, Rizkallah PJ, Simmons R, Donnellan Z, Dukes J, Bossi G, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep. (2016) 6:18851. doi: 10.1038/srep18851
80. Booiman T, Wit FW, Girigorie AF, Maurer I, De Francesco D, Sabin CA, et al. Terminal differentiation of T cells is strongly associated with CMV infection and increased in HIV-positive individuals on ART and lifestyle matched controls. PLoS ONE (2017) 12:e0183357. doi: 10.1371/journal.pone.0183357
81. International AIDS Vaccine Initiative. IAVI Serum Institute of India to Develop Manufacture Globally Affordable Accessible Antibody Products for HIV. (2018) Available online at: https://www.iavi.org/newsroom/press-releases/2018/
Keywords: HIV reservoirs, T cells, CAR (chimeric antigen receptor) T cells, dual affinity re-targeting (DART), T cell receptor (TCR), clinical trial, kick and kill
Citation: Yang H, Wallace Z and Dorrell L (2018) Therapeutic Targeting of HIV Reservoirs: How to Give T Cells a New Direction. Front. Immunol. 9:2861. doi: 10.3389/fimmu.2018.02861
Received: 02 October 2018; Accepted: 20 November 2018;
Published: 04 December 2018.
Edited by:
Julia G. Prado, IrsiCaixa, SpainReviewed by:
Paul Goepfert, University of Alabama at Birmingham, United StatesLisa A. Chakrabarti, Institut Pasteur, France
Copyright © 2018 Yang, Wallace and Dorrell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucy Dorrell, bHVjeS5kb3JyZWxsQG5kbS5veC5hYy51aw==