 Joseph M. Gaballa
Joseph M. Gaballa Manuel Bonfim Braga Neto
Manuel Bonfim Braga Neto Guilherme Piovezani Ramos
Guilherme Piovezani Ramos Adebowale O. Bamidele
Adebowale O. Bamidele Michelle M. Gonzalez
Michelle M. Gonzalez Mary R. Sagstetter
Mary R. Sagstetter Olga F. Sarmento
Olga F. Sarmento William A. Faubion Jr.*
William A. Faubion Jr.*- Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN, United States
T cell lineage decisions are critical for the development of proper immune responses to pathogens as well as important for the resolution of inflammatory responses. This differentiation process relies on a combination of intrinsic and extrinsic factors converging upon epigenetic regulation of transcriptional networks relevant to specific T cell lineages. As these biochemical modifications represent therapeutic opportunities in cancer biology and autoimmunity, implications of writers and readers of epigenetic marks to immune cell differentiation and function are highly relevant. Given the ready adoption of histone methyltransferase inhibitors in the clinic, we focus this review on the role of three histone modifying complexes: PRC-1, PRC-2, and G9A in modulating T cell fate decisions. Furthermore, we explore the role of long non-coding RNAs in regulating these processes, and discuss recent advances and challenges of implementing epigenetic therapies into clinical practice.
Background
The immune system comprises a large number of cell types that have the ability to respond to external environmental cues and adopt a wide variety of cell fates. These lineage decisions are critical for the development of proper immune responses to pathogens as well as resolution of inflammatory responses. As part of the adaptive immune system, T cells have the capacity to respond to the external environment by modulating the expression of lineage specific factors which are critical for protecting against a wide variety of pathogens. For the development of distinct T cell lineages, naive CD4+ T cells must convert the extrinsic instructions provided by encounters with antigen-presenting cells into cell-intrinsic changes (1). These intrinsic changes are largely facilitated by transcription factors that directly induce or repress gene networks and drive T cell differentiation (2). Emerging data demonstrates that lineage specific transcription factors recruit epigenetic complexes to regulate gene expression over multiple rounds of cell division, and their roles are indispensable for maintaining T cell homeostasis.
Deregulation of epigenetic pathways is a feature of many cancers, autoimmune diseases, and neurodegenerative disorders (3–5). The reversible nature of epigenetic modifications makes them attractive targets for pharmacological intervention, and indeed drugs targeting histone-modifying complexes, such as Enhancer of Zeste Homolog 2 (EZH2), are currently being evaluated in patients for treatment of malignancy (6) and immune-mediated conditions (7, 8). While recent clinical trials have demonstrated a favorable safety profile of selective inhibition of EZH2 (6), a comprehensive understanding of the role that epigenetic modifying complexes play in the development and function of different immune cell types is relevant to the development and safety of epigenetic therapeutics. Here we review the role of three histone modifying complexes: PRC-1, PRC-2, and G9A in modulating T cell fate decisions. Furthermore, we explore the role of long non-coding RNAs in regulating these processes, and discuss recent advances and challenges associated with implementing epigenetic therapies in clinical practice.
PRC1, PRC2, G9a, and Long Non-coding RNAs
PRC1
The Polycomb-Group proteins, Polycomb Repressive Complex 1 (PRC1) and 2 (PRC2), mediate post-translational modifications (PTMs) of histones required for cell differentiation and development through the regulation of chromatin structure and gene expression. PRC1 is a multimeric protein complex containing the core proteins RING1A/B, and Polycomb-group ring finger (PCGF) proteins such as Bmi-1 (PCGF4) and Mel-18 (PCGF2). PRC1 functions to mono-ubiquitinate lysine 119 on histone H2A (H2AKub119), an epigenetic mark that is associated with transcriptional repression (9). Bmi-1 specifically is highly enriched in pericentric heterochromatin which is required for chromatin compaction and silencing (10). Although Ring1A/B is the catalytic subunit of PRC1, knockdown of Bmi-1 results in a significant loss of H2A ubiquitylation, demonstrating the important role that it plays in facilitating the enzymatic function of PRC1 (11). In the canonical or hierarchical model of Polycomb (PcG)-mediated transcription regulation, PRC1 is primarily described as the maintenance complex which silences target genes previously marked by the initiator complex, PRC2. More recently, a histone-independent role of Bmi-1 in driving NF-κB signaling has been reported (12). An interesting story is also evolving related to a PRC2-independent role for PRC1 in the maintenance of 3D genome structure through association with super-enhancers (13, 14). No immune cell specific data has yet emerged related to these exciting areas of investigation.
PRC2
PRC2 modulates chromatin dynamics via the tri-methylation of lysine 27 on histone 3 (H3K27Me3), which is associated with transcriptional repression. EZH2, ubiquitously expressed by many mammalian cell-types, is the enzymatic subunit of PRC2 which contains other supporting non-catalytic proteins namely Suppressor of Zeste (SUZ12), embryonic ectoderm development (EED), Adipocyte Binding Protein 2 (AEBP2) and Retinoblastoma protein Associated protein 46 and 48 (RpAp46/48) (15). H3K27me3 recruits protein complexes involved in chromatin compaction and is associated with inactive genes (16). Histone-independent functions of PRC2 have also been reported to play important roles in regulating transcription factor stability and T cell receptor-mediated signaling (17–20). While EZH2 has a role in normal cellular and tissue function, studies involving EZH2 overexpression or genetic mutations show that EZH2 is critical in the development and progression of a variety of cancers (21–29). EZH2 is most frequently associated with the silencing of tumor suppressor genes, and decreased expression of PRC-target genes are associated with poor prognosis (30, 31). Thus, derepression of these genes using selective EZH2 enzymatic inhibitors or disruptors of PRC2 stability are likely to improve clinical outcomes, and are currently being explored in preclinical or clinical studies for cancer therapy (32–38).
G9a
The histone methyltransferase G9a and the related G9a-like protein (GLP) form a heterodimeric complex to catalyze mono and di-methylation of lysine 9 on histone 3 (H3K9me1 & H3K9me2) at euchromatin in vivo (39). G9a and GLP are encoded by the EHMT2 and EHMT1 genes, respectively, both of which contain a SET domain necessary for the methylation of lysine residues. G9a has been shown to play a larger role in H3K9me2 methylation in vivo, but levels of H3K9me1 and H3K9me2 are severely reduced in both G9a and GLP knockout models (39). Furthermore, G9a has been shown to promote gene activation through a methyltransferase-independent fashion in different settings, including type II cytokine production in helper T cells, possibly by acting as a scaffold to recruit transcriptional machinery (40, 41). G9a/GLP-mediated H3K9me2 has been associated with cognition and adaptive behavior, germ cell development and meiosis, embryo development, cocaine-induced plasticity, tumor cell growth and metastasis, and more recently the immune response reviewed below (39, 42).
Long Non-coding RNAs
Non-coding RNA's have emerged as an exciting new frontier of gene regulation in the immune system. It is now known that 75–90% of the human genome transcriptome is comprised of non-coding RNAs (43, 44). Long non-coding RNAs are defined as transcripts with minimal coding potential that are composed of more than 200 nucleotides; an arbitrary cutoff that distinguishes them from microRNAs (< 200 nucleotides). Over 15,000 lncRNA genes have been annotated, although only 159 lncRNAs have known function1,2 (45), highlighting a critical gap in knowledge in the field. They can be classified based on their position relative to protein coding genes as intergenic, intronic and antisense (46). Like mRNAs, long non-coding RNA's undergo transcription by RNA polymerase II, are 5′ capped, spliced and polyadenylated. However, distinct from mRNA, they lack canonical ORFs (and, therefore have minimal protein-coding potential), tend to be shorter in size, have lower expression levels, fewer exons and can localize to the nucleosome, chromatin or cytoplasm. For example, long intergenic non-coding RNAs localize primarily in the nucleus, in contrast to mRNAs which are primarily localized in the cytoplasm where they undergo translation (47). Furthermore, lncRNAs function by interacting with DNA, RNA, or proteins and the majority modulate transcription in cis (affecting nearby genes), although they can also modulate in trans (targeting distant genes), acting as scaffolds, molecular decoys and guides for epigenetic modifying complexes. Interestingly, lncRNAs can both activate and suppress target genes by a variety of mechanisms and are expressed in a cell-type and stage-specific manner (48, 49). They have been shown to play key roles in autoimmunity, cancer and infection (50–52). A recent comprehensive transcriptomic profiling of T cells demonstrated unique lncRNA signatures for specific T cell phenotypes signifying the relevance of lncRNA to cell and stage specific function (49). Thus, lncRNAs may represent exciting precise therapeutic targets.
PRC1, PRC2, G9A, and lncRNAs in the Adaptive Immune System
The development of T cells, an integral component of the adaptive immune system, occurs in the thymus where thymocytes mature into distinct T cell lineages defined by either CD4 or CD8 co-receptor expression. CD4+ T cells and CD8+ T cells are known to possess conventional alpha beta (αβ) T cell receptors (TCR), which recognize antigen-derived peptides bound by major histocompatibility complex (MHC) class II or I molecules, respectively. Upon antigen recognition and inflammatory environmental cues, naïve CD4+ T cells differentiate into distinct effector T helper (Th) subsets by expressing lineage-specific transcriptional programs. Th1, Th2, and Th17 cells mediate protective anti-pathogenic responses against bacteria and viruses via the secretion of distinct IFN-γ, IL-4, and IL-17 effector cytokines, respectively (53). Post-infection, Tregs, a regulatory component of the immune system, are recruited to inhibit effector T cell functions and reestablish homeostasis. Tregs can be generated from the thymus (natural Tregs) or induced in the periphery (pTreg) or in vitro (iTreg) from naïve CD4+ T cells via a FOXP3-driven transcriptome (54–56). Nonetheless, persistent activation of these effector T cell subsets has been associated with the pathogenesis of autoimmune disorders such as inflammatory bowel disease (IBD), rheumatoid arthritis (RA) and psoriasis (57).
PRC1, PRC2, G9a, and a variety of lncRNAs influence T helper cell differentiation and maintenance by epigenetically regulating transcriptional programs associated with different T cell subsets. Given their significant influence in the pathogenicity of diseases as stated above, we focus here on the role of these molecules in the differentiation and maintenance of Th1, Th2, Treg, and Th17 phenotypes (Figure 1, Table 1).
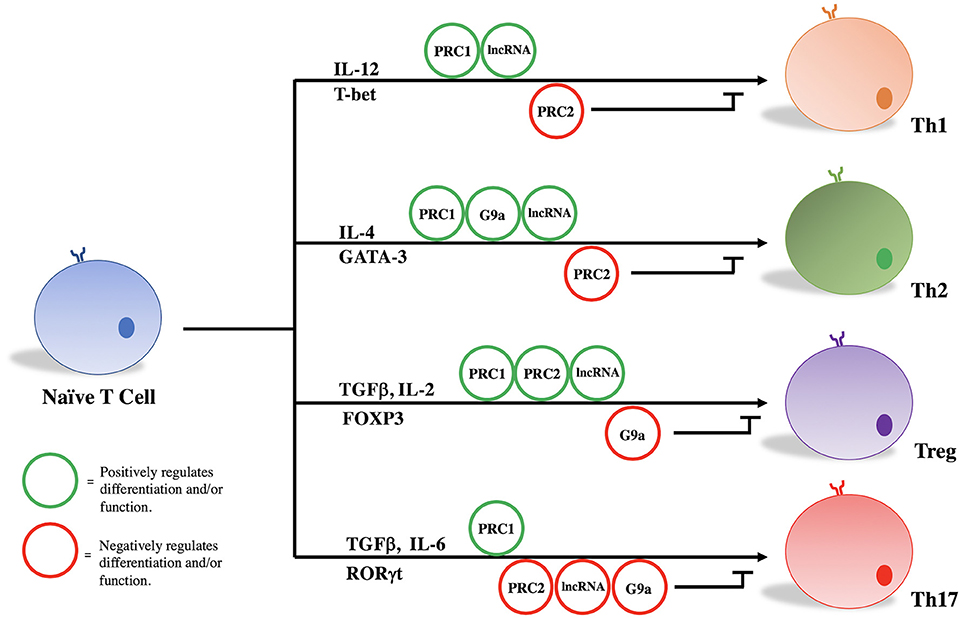
Figure 1. PRC1, PRC2, G9a, and lncRNAs regulate T cell differentiation and function.
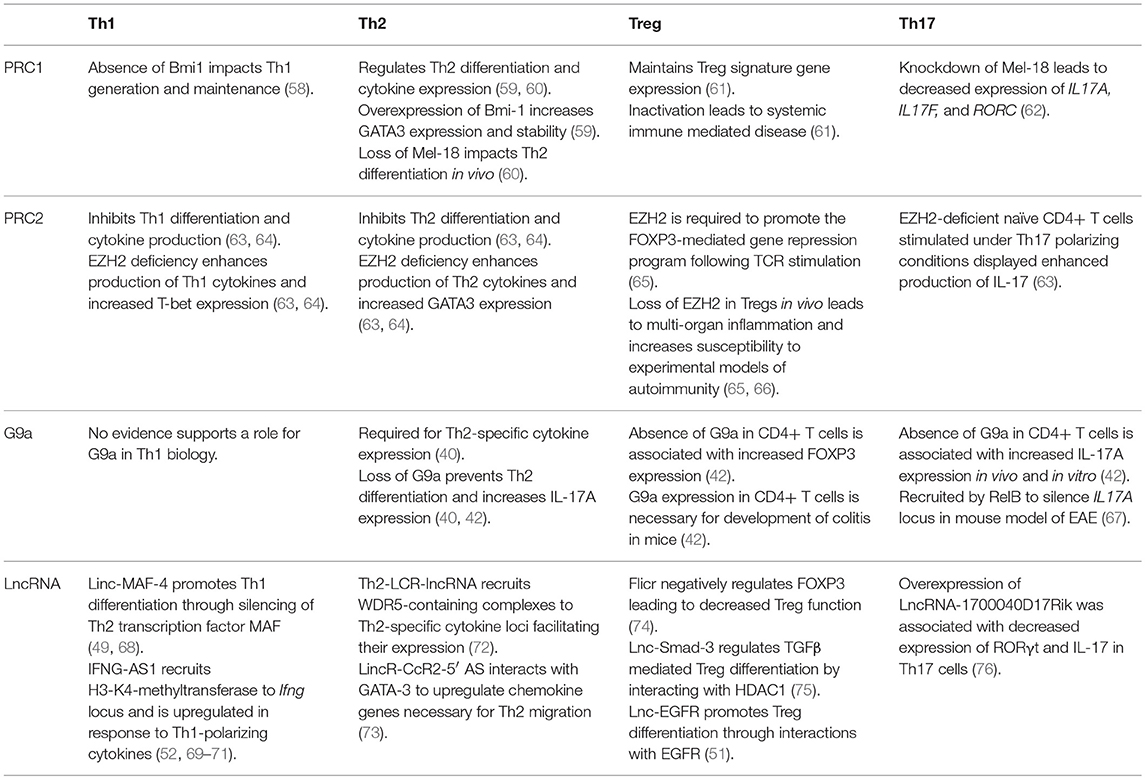
Table 1. Roles of PRC1, PRC2, G9a, and annotated lncRNAs in the development and function of Th1, Th2, Treg, and Th17 cells.
Treg/Th17
Treg and Th17 cells appear to share precursor lineage as demonstrated by in vitro study and murine lineage tracing experiments (77, 78). While TGFβ signaling is required for both effector cell types, IL-6 appears principally responsible for ultimate derivation of Th17 cells (79–81). Ultimately, lineage-specific transcription factors (FOXP3 and RORγt) drive the Treg or Th17 transcriptional program, respectively. FOXP3 and RORγt are known to reciprocally regulate one another, and the delicate balance between suppressive Tregs and effector Th17 cells has proven critical for maintaining immune homeostasis (78). Epigenetic modifying complexes, namely PRC2 and G9a, play key roles in orchestrating the Treg and Th17 transcriptional programs, and disruption of these epigenetic networks are characterized by the development of autoimmunity in murine models of human disease and human inflammatory bowel disease (66, 82, 83).
We and others have demonstrated that mice lacking EZH2 in natural FOXP3+ Tregs developed spontaneous multi-organ inflammation and were more susceptible to experimental models of autoimmunity (65, 66). In addition to decreased frequency of EZH2-depleted Tregs observed in certain murine tissues, DuPage et al. showed that EZH2 was required to promote the FOXP3-mediated gene repression program upon TCR activation as a number of FOXP3-bound genes were de-repressed in the absence of EZH2 (65). In support of the failure of EZH2-deleted Tregs to maintain the expression of Treg-specific signature genes, EZH2-deleted Tregs displayed impaired suppression of effector T cells in vitro (65, 66). Translating these findings from mice to human relevance, Crohn's disease (CD)-lamina propria CD4+ T cells were transcriptionally different from healthy controls (66). Specifically, normally repressed FOXP3-target genes were upregulated in CD CD4+ T cells and approximately 50% of these differentially expressed genes (DEGs) were EZH2 targets. Moreover, CD4+ T cells displayed a Th1/Th17 effector-like phenotype in contrast to that of healthy controls. Thus, loss of EZH2 function and consequently Treg dysfunction may drive pathophysiological mechanisms of particular autoimmune disorders.
In G9a deficient CD4+ T cells stimulated under Treg or Th17 promoting conditions, a significant increase in FOXP3-expressing and IL-17A-expressing cells is observed. In undifferentiated T cells, G9a normally functions as a mediator of H3K9me2 on loci associated with driving Treg and Th17 phenotypes (42). Loss of G9a-mediated H3K9me2 increases chromatin accessibility to transactivating factors and increases responsiveness to TGFβ (42). Much more work is required to define the molecular underpinnings of G9a's effects on Treg development, but some consistency is emerging regarding Th17 biology. G9a was shown to be recruited by RelB, a non-canonical NF-κB family member, to silence the IL17A locus and prevent Th17-mediated autoimmunity in an in vivo model of experimental autoimmune encephalomyelitis (EAE) (67). This work is consistent with effects seen in other T cell subsets, namely Th2 cells, in which loss of G9a leads to abnormal IL-17 expression (42). How these effects influence the balance between Treg and Th17 phenotypes is yet to be determined. Thus, G9a may become a viable target for therapeutic intervention of human Th17 mediated diseases.
Three lncRNAs (Flicr, Lnc-Smad-3, and LncEGFR) have been shown to influence Treg function. Flicr is selectively expressed in both human and mouse T regulatory cells and negatively regulates FOXP3 in cis leading to decreased Treg function and heightened autoimmunity (74). Mechanistically, Flicr modifies chromatin accessibility in the FOXP3 locus, specifically non-coding sequence 3 (CNS3) and accessible region 5 (AR5), leading to decreased expression of FOXP3. In vivo, knockdown of Flicr decreased the incidence of autoimmune diabetes in mice (74).
Lnc-Smad-3 was recently shown to modulate TGFβ-mediated Treg polarization both in human and murine assays (75). Mechanistically, lnc-Smad3 prevents the histone deacetylase HDAC1 to bind to the SMAD3 promoter region, which renders the chromatin compact and inaccessible to Ash1l, an H3K4 methyltransferase that promotes SMAD3 activation and transcription. From a disease relevance standpoint, these results suggest a potential role for this long non-coding RNA in the pathogenesis of autoimmune diseases, such as rheumatoid arthritis (75).
Lnc-EGFR was shown to stimulate Treg differentiation by a forward-feedback loop (51). Mechanistically, lnc-EGFR binds to EGFR using its R1 domain, preventing interaction with c-CBL and ubiquitination. In turn, EGFR activates ERK1/2 and AP-1, which then leads to increased expression of lnc-EGFR and FOXP3, perpetuating increased Treg differentiation. The authors found this to be a critical pathway for hepatocellular carcinoma (51).
LncRNA-1700040D17Rik was found to be deregulated in CD4+ cells derived from a mouse model of autoimmune encephalitis and have been shown to play a role in differentiation of Th17 cells. In vitro, overexpression of lncRNA-1700040D1Rik decreased expression of RORγt and IL-17 in Th17 cells, although the precise mechanism is yet to be known (76). These findings suggest a potential role for this long non-coding RNA in the pathogenesis of multiple sclerosis.
Th1/Th2
Studies investigating the impact of G9a on Th1 biology have shown that the absence of G9a has little effect on Th1 responses in vitro nor in vivo, however, it is a critical component of the Th2 regulatory machinery (40). Lehnertz et al. demonstrated G9a to be necessary for expression of lineage-specific Th2-associated cytokines such as IL-4, and that loss of G9a in CD4+ T cells prevents Th2 cell differentiation. Mice with targeted CD4+ T cell deletions of G9a were susceptible to helminth infection by Trichuris muris due to the inability to express Th2-associated cytokines. Consistent with previous work (42), the absence of G9a in CD4+ T cells also resulted in the upregulation of IL-17A in vivo. Interestingly, whereas repression of IL-17A appears to be associated with G9a methyltransferase activity (42), Th2 gene regulation by G9a is independent of enzymatic activity, and thought to be related to G9a functioning as a scaffolding protein (40, 41).
The role of PRC1 in regulating T cell lineage fate decisions is best illustrated by the influence it has on the Th1/Th2 axis of development. Both Bmi1 (PCGF 4) and Mel-18 (PCGF 2) have been shown to physically interact with GATA3, a lineage specific transcription factor for Th2 differentiation, in a Ring finger dependent manner (59, 60). Mel-18 has been shown to regulate GATA3 transcription, and knockout of mel-18 severely impacts Th2 differentiation in vivo (60). Bmi-1 regulates Th2 cell differentiation by acting as an inhibitor of GATA3 degradation and regulator of its stability. Bmi-1 overexpression in itself leads to an increase in GATA3 expression and an increase in Th2 cell differentiation under a Th2 specific cytokine milieu. Comparatively little data exist regarding the role of PRC1 in Th1 cell development/function; however adoptive transfer of CD4+ T cells from Bmi1−/− mice into nude mice showed impaired generation and maintenance of memory Th1 cells through Bmi1-mediated repression of Noxa, a pro-apoptotic gene (58).
The role of EZH2 in modulating effector T function was recently illuminated by Yang et al. who showed that EZH2-deficient naïve CD4+ T cells stimulated under Th1, Th2 or Th17 polarizing conditions displayed enhanced production of IFN-γ, IL-13 or IL-17 cytokines, respectively (63). Moreover, Tumes et al. also showed that EZH2 deficiency in naïve CD4+ T cells led to the upregulation of Th1 and Th2-associated cytokines with concomitant increase in lineage-specific transcription factors T-bet and Gata3, respectively (64). However, in vivo studies have revealed that EZH2 plays a dichotomous role in the differentiation and senescence of CD4+ T cells (63). For example, in an in vivo model of Listeria monocytogenes infection known to induce a Th1 response, CD4+ T-specific EZH2 deleted mice displayed impaired clearance of infection due to decreased survival of memory Th1 cells (84). Additionally, OVA-specific EZH2-deficient Th2 cells were pathogenic in a mouse model of allergic asthma due to an accumulated and exaggerated immune response from memory Th2 cells (64). Taken together, EZH2 inhibits effector cytokine production in naïve CD4+ T cells, and loss of EZH2 enhances differentiation to effector Th cells as well as effector Th cell plasticity. Based on evidence from in vivo studies in mice in the context of EZH2 deletion in T cells, effector Th cell dysfunction is consistent across all disease models, evidently through impaired clearance of pathogens or aggravated autoimmunity (potentiated tissue destruction). Additionally, H3K27me3-independent functions of EZH2 have been reported in T cells expressing conventional αβ-TCRs (17, 18). Vasanthakumar et al. demonstrated that EZH2 prevents NKT cell expansion through methylation, ubiquitination and subsequent degradation of the transcription factor promyelocytic leukemia zinc finger (PLZF) (17). In vivo studies have demonstrated that an increase in the frequency of NKT cells in the thymus and spleen occurs as a result of CD4+ T-specific EZH2 deletion, which may contribute to the perturbed immunity seen in murine studies previously mentioned (63, 64, 84).
Two lncRNAs, MAF-4, and IFNG-AS1 (also called NeST or Tmevpg1), have been shown to influence Th1 biology by recruiting different epigenetic modifying complexes. Linc-MAF-4 is selectively expressed in Th1 cells and promotes Th1 differentiation through epigenetic silencing of the Th2 transcription factor MAF. Downregulation of linc-MAF-4 in human CD4+ cells skewed differentiation toward a Th2 phenotype. Mechanistically, linc-MAF-4 promotes a cis chromatin looping conformation, leading to the recruitment of chromatin remodelers EZH2 and LSD1 that place repressive H3K27me3 marks on the promoter region of MAF-4 silencing its expression (49). Recently, linc-MAF-4 was shown to be involved in the pathogenesis of multiple sclerosis by promoting Th1 cell differentiation (68). Thus, far, linc-MAF-4 has not been studied in vivo.
IFNG-AS1 is expressed in CD4+ Th1, CD8+, and natural killer cells (52, 69). It is upregulated in CD4+ cells in response to Th1-differentiating cytokine stimuli and plays a critical role in transcription of Ifng. This has been demonstrated both in vitro and in vivo. Mechanistically, it has been shown to recruit the H3K4-methyltranferase complex to the Ifng locus, leading to placement of activating marks at the promoter region. It has been associated with the pathogenesis of Hashimoto's thyroiditis (70), ulcerative colitis (71), and the immune response to viral infections in vivo (52).
Two lncRNAs, Th2-LCR-lncRNA and lincR-CcR2-5′AS, have been shown to influence the development and function of Th2 cells. Th2-LCR-lncRNA is selectively expressed in human Th2 cells and is transcribed in the RAD50 locus and epigenetically regulates expression of IL-4, IL-5 and IL-13 (72). Mechanistically, Th2-LCR-lncRNA recruits WDR5-containing complexes to targeted cytokine loci, enhancing transcription. Knockdown of human Th2-LCR-lncRNA in vitro causes major loss of expression of IL-4, IL-5 and IL-13 in Th2 cells through loss of H3K4me3 activating marks (72). Unfortunately, Th2-LCR-lncRNA is not conserved in mice, complicating in vivo studies.
LincR-CcR2-5′AS is selectively expressed in mouse Th2 cells and upregulates CCR1, CCR2, CCR3 and CCR5 chemokine genes in a GATA3-dependent fashion (73). Interestingly, knockdown of this lincRNA not only affected neighboring genes CCR2 and CCR3, but also affected nearly 1,200 genes some of which were located in distant loci, suggesting it can act in both cis and trans. Although the precise mechanism is yet to be fully understood, in vitro knock down of lincR-CcR2-5′AS did not result in chromatin accessibility or modification of H3K4me3, suggesting that it does not act through recruitment of histone-modifying enzymes or chromatin structure modifications.
Future Perspectives: Epigenetic Modulation of T cells in Clinical Practice
Epigenetic mechanisms of disease are in theory inducible and reversible through environmental manipulation, however, some epigenetic features have been shown to be maintained after cellular division as a result of self-enforcing feedback mechanisms (85). The heritable, yet reversible nature of epigenetic therapy makes this a promising option for treatment. Persistence of epigenetic maintenance of engineered modifications has been shown to be stable up to 40 days post modification induction in vivo (86). Most epigenetic drugs currently in use inhibit DNA methyltransferase and histone deacetylase activity, and have been shown to reverse immune suppression and thus sensitize the host immune system in combination with anti-cancer therapies. Several anti-cancer mechanisms have been reported, such as enhancing antigen processing and presenting machinery pathways, inhibiting immune checkpoints, and enhancing chemokine production. For patients, there are three treatment options available: therapies reported to affect DNA methylation, inhibitors of histone post-translational modifications, and compounds interfering with non-coding RNA regulation (87). Repurposing drugs and screening for new compounds that display converse effects to treatment autoimmune disease is an exciting new option for autoimmune illnesses.
Distinct DNA methylation profiles have been demonstrated in CD8+ and CD4+ T cells isolated from patients experiencing autoimmune diseases (88–90). Epigenetic based therapeutics currently being employed for the clinic for non-inflammatory conditions, such as arrhythmias (procainamide), hypertension (hydralazine), and neoplasia (5-azacytidine), have been shown to induce auto-reactive pathology (7, 8). However, the 5-azacytidine derivative 5-aza-2'deoxycytidine, which is also a DNA methyltransferase inhibitor used in hematological malignancies, has been shown to have a positive outcome when administered in animal models of diabetes (91), colitis (92), multiple sclerosis (93), and graft-versus-host-disease (GvHD) (94). We need a better understanding of the implications of DNA methylation, the pharmokinetics of available compounds, and synergistic effects of combination therapy with immunomodulatory drugs already in practice for autoimmune diseases to allow us to develop and implement novel therapies. As of now, we are lacking a therapeutic arsenal to target global hypomethylation, which is most often associated with lymphocytes recovered from patients experiencing some of the most common autoimmune diseases.
The ubiquitous expression of EZH2 and the opposing role it plays in different cell-types makes EZH2 a delicate therapeutic target. Recent identification of PRC2- and H3K27me3-independent EZH2 functions in oncogenesis indicates that a complete suppression of all oncogenic functions of EZH2 is required to combat cancer. Anti-EZH2 therapy inhibits methylation at key repression/silencing associated histone marks, and these compounds have emerged as a promising therapy for cancer treatment, especially for B cell non-Hodgkin's lymphoma. However, we have observed that systemic anti-EZH2 therapy leads to mucosal hypersensitivity in mice. One complicating factor is that EZH2 is also utilized by PRC1 in the nucleus, therefore more study needs to be undertaken to dissect the specific roles these complexes play in inflammation before on can determine whether histone methyltransferase inhibitors can be co-opted for anti-inflammatory therapy. Of note, cytosolic forms of PRC2 have been shown in murine models to be necessary for TCR-mediated activation of signaling pathways that drive T cell proliferation and autoimmunity. Thus, pharmacologic targeting of cytosolic PRC2 may represent a more precise therapeutic approach to suppressing autoimmunity caused by excessive T cell activation (19, 20).
From a translational standpoint, several studies have demonstrated that long non-coding RNAs can be used as biomarkers in malignancy and autoimmune diseases (95–97). Potential lncRNA-targeted therapeutic approaches include silencing by antisense base pairing (e.g., targeting lncUBE3ATS, which silences paternal UBE3A in Angelman's syndrome) or by targeting molecules that are necessary for lncRNA transcription, such as transcription factors (98, 99). The cell type specific expression of lncRNAs makes them excellent targets for therapeutic intervention, as off-target effects are minimized. One option being pursued in cancer therapies is to directly target HOTAIR; a primarily trans-acting long-non coding RNA that promotes gene silencing through recruitment of PRC2 and LSD1 complexes, resulting in trimethylation of H3K27 and demethylation of H3K4, respectively (100–102). Knocking down HOTAIR provides compelling evidence for therapeutic targeting in cancer. Arresting glioblastoma multiform cell migration and invasion through this approach is a case in point (103). To overcome the limitation of genetic targeting, peptide nucleic acids have been developed which disrupt complex function. This approach has had positive results in inhibiting NF-κB activity in addition to decreasing ovarian and breast cancer properties such as reduced tumor formation and survival (104). The potential for this approach in inflammatory diseases is still to be determined.
Precision medicine has brought about the advent of using CRISPR/Cas9 to target this gene editing tool to target epigenetic modifying enzymes to precise locus specific locations on the genome instead of the DNA endonucleases the technology originally utilized (105). This technique can be exploited to recruit enzymes that impact the methylation of the DNA, enzymes that post-translationally modify the histones, and proteins which interfere with non-coding RNA regulation. Further, it has been recently reported CRISPR/Cas9 technology can be rapidly delivered via a non-viral delivery technique capable of integrating large DNA sequences (106). These new developments will allow us flexible and precise epigenetic manipulation toward creating therapeutically epi-engineered primary human immune cells without the off-target effects associated with systemic epigenetic therapies.
Author Contributions
WF contributed conception and design of the manuscript. JG, MB, GR, AB, MG, MS, and OS wrote sections of the manuscript. JG wrote the first draft of the manuscript. All authors provided critical revision and final approval of the manuscript.
Funding
Supported by grants 5R01AI089714-08, 5R01AI089714-08S1, 30DK084567, and CCFA #401661.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^ GENCODE, v27 Release. Available online at: https://www.gencodegenes.org/human/release_27.html
2. ^ Long Non-coding RNA Database v2.,0 (lncRNAdb). Available online at: http://www.lncrnadb.org/
References
1. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. (2009) 9:91–105. doi: 10.1038/nri2487
2. Kanno Y, Vahedi G, Hirahara K, Singleton K, O'Shea JJ. Transcriptional and epigenetic control of T helper cell specification: molecular mechanisms underlying commitment and plasticity. Annu Rev Immunol. (2012) 30:707–31. doi: 10.1146/annurev-immunol-020711-075058
3. Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. (2009) 8:1056–72. doi: 10.1016/S1474-4422(09)70262-5
4. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. (2007) 8:286–98. doi: 10.1038/nrg2005
5. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res. (2010) 20:170–9. doi: 10.1101/gr.100289.109
6. Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. (2018) 19:649–59. doi: 10.1016/S1470-2045(18)30145-1
7. Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors. 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest. (1993) 92:38–53. doi: 10.1172/JCI116576
8. Deng C, Lu Q, Zhang Z, Rao T, Attwood J, Yung R, et al. Hydralazine may induce autoimmunity by inhibiting extracellular signal-regulated kinase pathway signaling. Arthritis Rheum. (2003) 48:746–56. doi: 10.1002/art.10833
9. Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature (2004) 431:873–8. doi: 10.1038/nature02985
10. Abdouh M, Hanna R, El Hajjar J, Flamier A, Bernier G. The polycomb repressive complex 1 protein BMI1 is required for constitutive heterochromatin formation and silencing in mammalian somatic cells. J Biol Chem. (2016) 291:182–97. doi: 10.1074/jbc.M115.662403
11. Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, et al. RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3. Cell (2012) 148:664–78. doi: 10.1016/j.cell.2011.12.029
12. Okuyama Y, Tanaka Y, Jiang JJ, Kamimura D, Nakamura A, Ota M, et al. Bmi1 Regulates IkappaBalpha Degradation via Association with the SCF Complex. J Immunol. (2018) 201:2264–72. doi: 10.4049/jimmunol.1701223
13. King HW, Fursova NA, Blackledge NP, Klose RJ. Polycomb repressive complex 1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res. (2018); 28:1494–150. doi: 10.1101/gr.237180.118
14. Chan HL, Beckedorff F, Zhang Y, Garcia-Huidobro J, Jiang H, Colaprico A, et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat Commun. (2018) 9:3377. doi: 10.1038/s41467-018-05728-x
15. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature (2011) 469:343–9. doi: 10.1038/nature09784
16. Spivakov M, Fisher AG. Epigenetic signatures of stem-cell identity. Nat Rev Genet. (2007) 8:263–71. doi: 10.1038/nrg2046
17. Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N, et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. (2017) 18:619–31. doi: 10.15252/embr.201643237
18. Koubi M, Poplineau M, Vernerey J, N'Guyen L, Tiberi G, Garciaz S, et al. Regulation of the positive transcriptional effect of PLZF through a non-canonical EZH2 activity. Nucleic Acids Res. (2018) 46:3339–50. doi: 10.1093/nar/gky080
19. Dobenecker MW, Park JS, Marcello J, McCabe MT, Gregory R, Knight SD, et al. Signaling function of PRC2 is essential for TCR-driven T cell responses. J Exp Med. (2018) 215:1101–13. doi: 10.1084/jem.20170084
20. Su IH, Dobenecker MW, Dickinson E, Oser M, Basavaraj A, Marqueron R, et al. Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell (2005) 121:425–36. doi: 10.1016/j.cell.2005.02.029
21. Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. (2010) 116:5247–55. doi: 10.1182/blood-2010-04-280149
22. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature (2002) 419:624–9. doi: 10.1038/nature01075
23. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. (2003) 22:5323–35. doi: 10.1093/emboj/cdg542
24. Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. (2006) 24:268–73. doi: 10.1200/JCO.2005.01.5180
25. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. (2010) 42:181–5. doi: 10.1038/ng.518
26. Bodor C, O'Riain C, Wrench D, Matthews J, Iyengar S, Tayyib H, et al. EZH2 Y641 mutations in follicular lymphoma. Leukemia (2011) 25:726–9. doi: 10.1038/leu.2010.311
27. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA. (2010) 107:20980–5. doi: 10.1073/pnas.1012525107
28. Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. (2011) 117:2451–9. doi: 10.1182/blood-2010-11-321208
30. Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. (2008) 647:21–9. doi: 10.1016/j.mrfmmm.2008.07.010
31. Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, et al. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. (2007) 67:10657–63. doi: 10.1158/0008-5472.CAN-07-2498
32. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. (2012) 8:890–6. doi: 10.1038/nchembio.1084
33. Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. (2012) 3:1091–6. doi: 10.1021/ml3003346
34. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492:108–12. doi: 10.1038/nature11606
35. Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. (2012) 109:21360–5. doi: 10.1073/pnas.1210371110
36. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. (2013) 110:7922–7. doi: 10.1073/pnas.1303800110
37. Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. (2014) 13:842–54. doi: 10.1158/1535-7163.MCT-13-0773
38. Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. (2013) 9:643–50. doi: 10.1038/nchembio.1331
39. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. (2011) 25:781–8. doi: 10.1101/gad.2027411
40. Lehnertz B, Northrop JP, Antignano F, Burrows K, Hadidi S, Mullaly SC, et al. Activating and inhibitory functions for the histone lysine methyltransferase G9a in T helper cell differentiation and function. J Exp Med. (2010) 207:915–22. doi: 10.1084/jem.20100363
41. Scheer S, Zaph C. The lysine methyltransferase G9a in immune cell differentiation and function. Front Immunol. (2017) 8:429. doi: 10.3389/fimmu.2017.00429
42. Antignano F, Burrows K, Hughes MR, Han JM, Kron KJ, Penrod NM, et al. Methyltransferase G9A regulates T cell differentiation during murine intestinal inflammation. J Clin Invest. (2014) 124:1945–55. doi: 10.1172/JCI69592
43. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. (2012) 22:1775–89. doi: 10.1101/gr.132159.111
44. Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. (2012) 22:1760–74. doi: 10.1101/gr.135350.111
45. Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. (2018) 19:143–57. doi: 10.1038/nrm.2017.104
46. Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. (2017) 18:962–72. doi: 10.1038/ni.3771
47. Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. (2013) 493:231–5. doi: 10.1038/nature11661
48. Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science (2013) 341:789–92. doi: 10.1126/science.1240925
49. Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, Curti S, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol. (2015) 16:318–25. doi: 10.1038/ni.3093
50. Aune TM, Crooke PS III, Patrick AE, Tossberg JT, Olsen NJ, Spurlock CF III. Expression of long non-coding RNAs in autoimmunity and linkage to enhancer function and autoimmune disease risk genetic variants. J Autoimmun. (2017) 81:99–109. doi: 10.1016/j.jaut.2017.03.014
51. Jiang R, Tang J, Chen Y, Deng L, Ji J, Xie Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. (2017) 8:15129. doi: 10.1038/ncomms15129
52. Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell (2013) 152:743–54. doi: 10.1016/j.cell.2013.01.015
53. Maynard CL, Weaver CT. Intestinal effector T cells in health and disease. Immunity (2009) 31:389–400. doi: 10.1016/j.immuni.2009.08.012
54. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. (2001) 2:301–6. doi: 10.1038/86302
55. Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, et al. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. (2009) 182:3461–8. doi: 10.4049/jimmunol.0802535
56. Weiss JM, Bilate AM, Gobert M, Ding YMA, Curotto de Lafaille P, Lafaille JJ. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med. (2012) 209:1723–42S1. doi: 10.1084/jem.20120914
57. Cho JH, Feldman M. Heterogeneity of autoimmune diseases: pathophysiologic insights from genetics and implications for new therapies. Nat Med. (2015) 21:730–8. doi: 10.1038/nm.3897
58. Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, et al. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med. (2008) 205:1109–20. doi: 10.1084/jem.20072000
59. Hosokawa H, Kimura MY, Shinnakasu R, Suzuki A, Miki T, Koseki H, et al. Regulation of Th2 cell development by Polycomb group gene bmi-1 through the stabilization of GATA3. J Immunol. (2006) 177:7656–64. doi: 10.4049/jimmunol.177.11.7656
60. Kimura M, Koseki Y, Yamashita M, Watanabe N, Shimizu C, Katsumoto T, et al. Regulation of Th2 cell differentiation by mel-18, a mammalian polycomb group gene. Immunity (2001) 15:275–87. doi: 10.1016/S1074-7613(01)00182-0
61. Gonzalez M, Sagstetter M, Svingen P, Bamidele AO, Sarmento OF, Sun Z, et al. The epigenetic complex PRC-1 maintains T regulatory cell lineage instability. AGA Abstracts (2017) 152:S79. doi: 10.1016/S0016-5085(17)30612-1
62. Hod-Dvorai R, Jacob E, Boyko Y, Avni O. The binding activity of Mel-18 at the Il17a promoter is regulated by the integrated signals of the TCR and polarizing cytokines. Eur J Immunol. (2011) 41:2424–35. doi: 10.1002/eji.201141620
63. Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep. (2015) 5:10643. doi: 10.1038/srep10643
64. Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity (2013) 39:819–32. doi: 10.1016/j.immuni.2013.09.012
65. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42:227–38. doi: 10.1016/j.immuni.2015.01.007
66. Sarmento OF, Svingen PA, Xiong Y, Sun Z, Bamidele AO, Mathison AJ, et al. The role of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) in the pathobiological mechanisms underlying inflammatory bowel disease (IBD). J Biol Chem. (2017) 292:706–22. doi: 10.1074/jbc.M116.749663
67. Xiao X, Shi X, Fan Y, Wu C, Zhang X, Minze L, et al. The costimulatory receptor OX40 inhibits interleukin-17 expression through activation of repressive chromatin remodeling pathways. Immunity (2016) 44:1271–83. doi: 10.1016/j.immuni.2016.05.013
68. Zhang F, Liu G, Wei C, Gao C, Hao J. Linc-MAF-4 regulates Th1/Th2 differentiation and is associated with the pathogenesis of multiple sclerosis by targeting MAF. FASEB J. (2017) 31:519–25. doi: 10.1096/fj.201600838R
69. Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM. Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol. (2012) 189:2084–8. doi: 10.4049/jimmunol.1200774
70. Peng H, Liu Y, Tian J, Ma J, Tang X, Rui K, et al. The long noncoding RNA IFNG-AS1 promotes T helper type 1 cells response in patients with hashimotos thyroiditis. Sci Rep. (2015) 5:17702. doi: 10.1038/srep17702
71. Padua D, Mahurkar-Joshi S, Law IK, Polytarchou C, Vu JP, Pisegna JR, et al. A long noncoding RNA signature for ulcerative colitis identifies IFNG-AS1 as an enhancer of inflammation. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G446–57. doi: 10.1152/ajpgi.00212.2016
72. Spurlock CF III, Tossberg JT, Guo Y, Collier SP, Crooke PS III, Aune TM. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat Commun. (2015) 6:6932. doi: 10.1038/ncomms7932
73. Hu G, Tang Q, Sharma S, Yu F, Escobar TM, Muljo SA, et al. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol. (2013) 14:1190–8. doi: 10.1038/ni.2712
74. Zemmour D, Pratama A, Loughhead SM, Mathis D, Benoist C. Flicr, a long noncoding RNA, modulates Foxp3 expression and autoimmunity. Proc Natl Acad Sci USA. (2017) 114:E3472–E80. doi: 10.1073/pnas.1700946114
75. Xia M, Liu J, Liu S, Chen K, Lin H, Jiang M, et al. Ash1l and lnc-Smad3 coordinate Smad3 locus accessibility to modulate iTreg polarization and T cell autoimmunity. Nat Commun. (2017) 8:15818. doi: 10.1038/ncomms15818
76. Guo W, Lei W, Yu D, Ge Y, Chen Y, Xue W, et al. Involvement of lncRNA-1700040D17Rik in Th17 cell differentiation and the pathogenesis of EAE. Int Immunopharmacol. (2017) 47:141–9. doi: 10.1016/j.intimp.2017.03.014
77. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. (2009) 10:1000–7. doi: 10.1038/ni.1774
78. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature (2008) 453:236–40. doi: 10.1038/nature06878
79. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
80. Mangan PR, Harrington LE, OQuinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
81. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. (2007) 8:967–74. doi: 10.1038/ni1488
82. Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut (2003) 52:65–70. doi: 10.1136/gut.52.1.65
83. Luo A, Leach ST, Barres R, Hesson LB, Grimm MC, Simar D. The microbiota and epigenetic regulation of T helper 17/regulatory T cells: in search of a balanced immune system. Front Immunol. (2017) 8:417. doi: 10.3389/fimmu.2017.00417
84. Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, et al. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood (2014) 124:737–49. doi: 10.1182/blood-2013-12-544106
85. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17:487–500. doi: 10.1038/nrg.2016.59
86. Saunderson EA, Stepper P, Gomm JJ, Hoa L, Morgan A, Allen MD, et al. Hit-and-run epigenetic editing prevents senescence entry in primary breast cells from healthy donors. Nat Commun. (2017) 8:1450. doi: 10.1038/s41467-017-01078-2
87. Jenuwein T, Allis CD. Translating the histone code. Science. (2001) 293:1074–80. doi: 10.1126/science.1063127
88. Maltby VE, Graves MC, Lea RA, Benton MC, Sanders KA, Tajouri L, et al. Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics (2015) 7:118. doi: 10.1186/s13148-015-0152-7
89. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. (1990) 33:1665–73. doi: 10.1002/art.1780331109
90. Zhang Y, Zhao M, Sawalha AH, Richardson B, Lu Q. Impaired DNA methylation and its mechanisms in CD4(+)T cells of systemic lupus erythematosus. J Autoimmun. (2013) 41:92–9. doi: 10.1016/j.jaut.2013.01.005
91. Zheng Q, Xu Y, Liu Y, Zhang B, Li X, Guo F, et al. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J Mol Med. (2009) 87:1191–205. doi: 10.1007/s00109-009-0530-8
92. Lal G, Zhang NW, van der Touw DY, Ju W, Bottinger EP, Bromberg JS. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. (2009) 182:259–73. doi: 10.4049/jimmunol.182.1.259
93. Chan MW, Chang CB, Tung CH, Sun J, Suen JL, Wu SF. Low-dose 5-aza-2-deoxycytidine pretreatment inhibits experimental autoimmune encephalomyelitis by induction of regulatory T cells. Mol Med. (2014) 20:248–56. doi: 10.2119/molmed.2013.00159
94. Sanchez-Abarca LI, Gutierrez-Cosio S, Santamaria C, Caballero-Velazquez T, Blanco B, Herrero-Sanchez C, et al. Immunomodulatory effect of 5-azacytidine (5-azaC): potential role in the transplantation setting. Blood (2010) 115:107–21. doi: 10.1182/blood-2009-03-210393
95. Teschendorff AE, Lee SH, Jones A, Fiegl H, Kalwa M, Wagner W, et al. HOTAIR and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med. (2015) 7:108. doi: 10.1186/s13073-015-0233-4
96. Serghiou S, Kyriakopoulou A, Ioannidis JP. Long noncoding RNAs as novel predictors of survival in human cancer: a systematic review and meta-analysis. Mol Cancer. (2016) 15:50. doi: 10.1186/s12943-016-0535-1
97. Li Z, Li X, Jiang C, Qian W, Tse G, Chan MTV, et al. Long non-coding RNAs in rheumatoid arthritis. Cell Prolif. (2018) 51:1–6. doi: 10.1111/cpr.12404
98. Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature (2015) 518:409–12. doi: 10.1038/nature13975
99. Sheik Mohamed J, Gaughwin PM, Lim B, Robson P, Lipovich L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA (2010) 16:324–37. doi: 10.1261/rna.1441510
100. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science (2010) 329:689–93. doi: 10.1126/science.1192002
101. Loewen G, Jayawickramarajah J, Zhuo Y, Shan B. Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol. (2014) 7:90. doi: 10.1186/s13045-014-0090-4
102. Gao JZ, Li J, Du JL, Li XL. Long non-coding RNA HOTAIR is a marker for hepatocellular carcinoma progression and tumor recurrence. Oncol Lett. (2016) 11:1791–8. doi: 10.3892/ol.2016.4130
103. Zhou X, Ren Y, Zhang J, Zhang C, Zhang K, Han L, et al. HOTAIR is a therapeutic target in glioblastoma. Oncotarget (2015) 6:8353–65. doi: 10.18632/oncotarget.3229
104. Ozes AR, Wang Y, Zong X, Fang F, Pilrose J, Nephew KP. Therapeutic targeting using tumor specific peptides inhibits long non-coding RNA HOTAIR activity in ovarian and breast cancer. Sci Rep. (2017) 7:894. doi: 10.1038/s41598-017-00966-3
105. Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell (2013) 154:442–51. doi: 10.1016/j.cell.2013.06.044
Keywords: epigenetics, EZH2, G9a, long non-coding RNAs, PRC1, PRC2, T cell
Citation: Gaballa JM, Braga Neto MB, Ramos GP, Bamidele AO, Gonzalez MM, Sagstetter MR, Sarmento OF and Faubion WA Jr (2018) The Role of Histone Methyltransferases and Long Non-coding RNAs in the Regulation of T Cell Fate Decisions. Front. Immunol. 9:2955. doi: 10.3389/fimmu.2018.02955
Received: 06 September 2018; Accepted: 30 November 2018;
Published: 13 December 2018.
Edited by:
Keiko Ozato, National Institutes of Health (NIH), United StatesReviewed by:
Avinash Bhandoola, National Institutes of Health (NIH), United StatesJonathan Kaye, Cedars-Sinai Medical Center, United States
Remy Bosselut, National Cancer Institute (NCI), United States
Vishal Nehru, National Institutes of Health (NIH), United States
Copyright © 2018 Gaballa, Braga Neto, Ramos, Bamidele, Gonzalez, Sagstetter, Sarmento and Faubion. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William A. Faubion Jr, ZmF1Ymlvbi53aWxsaWFtQG1heW8uZWR1