 Edoardo Marrani
Edoardo Marrani Jane C. Burns
Jane C. Burns Rolando Cimaz
Rolando Cimaz- 1Rheumatology Unit, Department of Neurosciences, Psychology, Drug Research and Child Health, Meyer Children's Hospital, University of Florence, Florence, Italy
- 2Department of Pediatrics, Kawasaki Disease Research Center, Rady Children's Hospital, University of California, San Diego, San Diego, CA, United States
The exact classification of Kawasaki disease (KD) has been debated. Infectious disease specialists have claimed it as an infection with a classic immune responses to an as yet unidentified pathogen that localizes to the coronary arteries. Others have favored an autoreactive hypothesis that KD is triggered by an antigen that shares homology with structures in the vascular wall, and molecular mimicry resulting in an immune response directed to that tissue. Rheumatologists have classified it as a systemic vasculitis, while some immunologists have stressed the robust nature of the innate immune response that causes both systemic inflammation as well as damage to the coronary arterial wall and questioned whether KD falls within the spectrum of autoinflammatory diseases. This review will describe the evidences available up to now regarding these hypotheses.
Kawasaki Disease (KD) is an acute, self-limited vasculitis and is the most common cause of acquired heart disease in children of developed countries (1). The diagnosis of KD is clinical and it is based on a combination of fever and five clinical features (Figure 1).
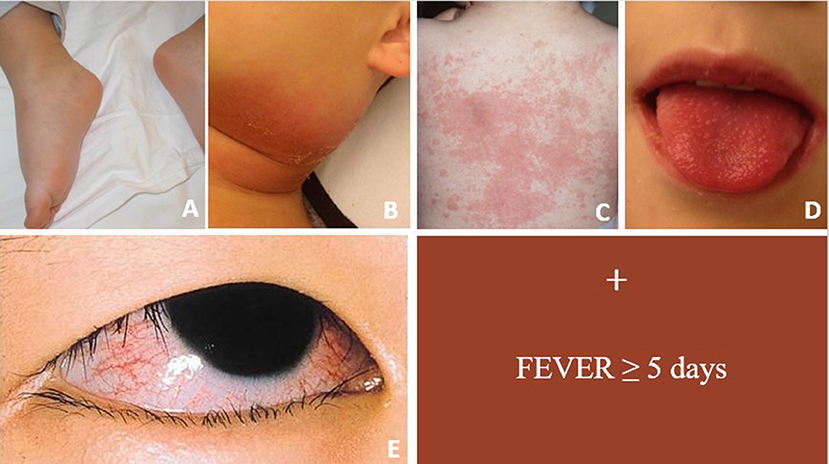
Figure 1. Figure showing main clinical signs of acute KD. For complete form ≥4 clinical criteria are required in conjunction with fever ≥5 days. (A) Erythema and edema of the hands and feet; (B) unilateral cervical lymphadenopathy; (C) polymorphous rash; (D) oropharyngeal changes; (E) bilateral bulbar conjunctival injection without exudate.
While in most cases the acute phase subsides within 2 weeks, even in untreated patients, up to 25% of all KD patients are at risk for significant coronary arteritis and subsequent development of coronary artery aneurysms (CAA) (2). Treatment with high dose intravenous immunoglobulin (IVIG) has been associated with a reduction of this risk to 3–6% and is recommended for all patients (3, 4).
A subset of patients with KD (10–20%) develops recrudescent or persistent fever at least 36 h after the end of the IVIG infusion (5). IVIG-resistant patients have a greater risk of cardiac complications. Furthermore, due to a limited knowledge of the exact mechanism of IVIG resistance, treatment of these patients remains challenging (6).
For decades, the exact classification of Kawasaki disease (KD) has been debated. Infectious disease specialists have claimed it as an infection with classic innate and adaptive immune responses to an as yet unidentified pathogen that localizes to the coronary arteries (7). Others have favored an autoreactive hypothesis that KD is triggered by an antigen that shares homology with structures in the vascular wall and the molecular mimicry results in an immune response directed to that tissue (1). Rheumatologists have classified it as a systemic vasculitis, thus lumping KD with other, usually chronic vasculitides of unknown etiology (8). Some immunologists have stressed the robust nature of the innate immune response that causes both systemic inflammation as well as damage to the coronary arterial wall and questioned whether KD falls within the spectrum of autoinflammatory diseases (9). Still others have labeled KD as autoimmune. Understanding the nature of the immune process that underlies KD will have direct implications for the direction that research initiatives should take and will influence how we attempt to modulate the immune system. The therapeutic approach for an autoimmune disease differs from an antigen-driven process, which, in turn, differs from a superantigen driven process. Therefore, we review here the evidence that supports different conclusions about the disease classification of KD.
Evidence for Response to a Pathogen
The inflammation of the coronary arteries is the most striking feature of KD and it is responsible for the long-term sequelae, with children having giant aneurysms showing a relatively poor prognosis decades after the acute phase (10). The natural history of coronary damage in KD has been tracked by studies of tissues obtained at autopsy or at cardiac transplantation Thus, our knowledge of KD pathology is based on only the most severe cases and is therefore, incomplete. In the acute phase of fatal cases, a necrotizing arteritis is found in medium-sized vessels characterized by a neutrophilic and macrophage infiltration and destruction of the internal elastic lamina (11, 12). There is some evidence that this process may originate in the vasa vasorum. This innate immune response is followed by a subacute phase dominated by an adaptive immune response, in which infiltration of CD8+ cytotoxic T cells, plasma cells, and macrophages predominates. Myofibroblasts that may derive from endothelial or epithelial to mesenchymal transition can lead to luminal myofibroblast proliferation (LMP) (12). Progressive luminal obstruction by LMP with or without layering of thrombus along the aneurysmal wall are responsible for the long-term complications of KD, mainly coronary artery stenosis and acute coronary syndromes (13).
The myocardium is inflamed in all children with KD based on right endomyocardial biopsies during the acute disease (14, 15). In the convalescent phase, the myocardium may show diffuse, bridging fibrosis that may be related to low levels of persistent inflammation or chronic microvascular ischemia, as shown in pediatric patients (16) and in the long-term studies in adults (15).
Thus, the pathology of KD supports the concept of an early and robust innate immune response with infiltration of neutrophils into the arterial wall followed by an adaptive immune response with participation of T and B cells. Recently, Martin et al. studied the plasmablast expansion in KD. Plasmablasts are B cells (CD19+ CD20- CD27+ CD38+) transitioning to plasma cells that circulate in the peripheral blood cell compartment. KD patients showed a plasmablast response similar to children with infectious diseases, thus suggesting that circulating plasmablasts during KD produce antibodies that specifically target the agent responsible for the KD (17). Moreover, electron microscopy studies on bronchial epithelium of KD showed intracytoplasmic inclusion bodies (18) and oligoclonal IgA antibodies (19) suggesting a response to a pathogen entering through the upper respiratory tract. Rowley at al. synthesized artificial versions of these IgA antibodies and identified inclusion bodies suggestive of vesicles with virus-like particles (20).
However, gene expression profiling revealed that patients with acute KD have a unique gene expression pattern that is clearly divergent from patients with viral or bacterial diseases (21). In particular, Interferon type 1-stimulated gene expression was notably low in KD patients, when compared to patients infected with viral pathogens (22). Moreover, recently Wright et al. proposed a unique 13-transcript signature test as a tool to differentiate KD patients from those with bacterial, mycobacterial, and viral infections. This suggests that the host response of KD differs in important ways from the response of children infected with these other classes of agents (23).
Evidence for an autoantibody or T Cell Driven Autoimmune Process
Autoimmunity is, by definition, a self-directed inflammatory response caused by dysfunction of mechanisms of tolerance (24). KD has been classified by some investigators, along with other vasculitides, as having an autoimmune origin (8). However, a considerable amount of evidence has progressively led to discrediting a role for autoimmunity in KD pathogenesis. First, the self-limited nature of KD and the low rate of recurrence argue against a primary autoimmune response, as autoimmune diseases typically manifest a chronic relapsing course. Furthermore, KD patients lack high autoimmunity burden or familial aggregation of autoimmune disease, as reported for other rheumatic diseases (25–27). National databases in high-prevalence populations from Japan and Korea have not reported any significant association with other autoimmune conditions, despite more than 20 years of observational studies (28, 29). Only a few studies have specifically addressed the association between KD and autoimmune diseases, such as autoimmune thyroiditis or celiac disease. Stagi et al. reported an increased prevalence of celiac disease in an Italian cohort of KD patients while the rate of autoimmune thyroiditis rate was similar between cases and controls (30). More recently, a cohort study conducted in a larger Brazilian population failed to show any association between celiac disease and KD (31), and only few case reports have been published regarding the co-occurrence of vitiligo in KD (32). Moreover, serological analysis has failed to demonstrate the consistent presence of disease-specific autoantibodies, with conflicting results regarding the presence of antinuclear antibodies (ANA), anticardiolipin (aCL), antineutrophil cytoplasmic antibodies (ANCA), and Anti-Human Cardiac Myosin Autoantibodies (33–37).
Therefore, it is unlikely that they play a major pathogenic role. Anti-endothelial cell antibodies (AECAs) have been more widely reported in KD (35, 36, 38, 39).
Although AECAs have been described in almost all primary systemic vasculitides, these autoantibodies are also present in several other diseases characterized by vascular involvement, including systemic lupus erythematosus, anti-phospholipid syndrome, rheumatoid arthritis, systemic sclerosis, and solid organ transplantation (40). So, it is not clear whether these are a primary driver for the inflammatory process or a secondary antibody response following endothelial cell destruction with the release of neo-antigens that could be the target of an antibody response. Moreover, an experimental model suggests that although AECAs from KD directly affect EC function in vitro and are associated with the development of some KD features, they are not able to initiate the vasculitic process or to induce coronary artery vasculitis (41).
Intriguingly, after IVIG administration, expansion of a natural regulatory T cell population (CD4+, FOXP3+, CD25highnTreg) is seen (42). The expansion of this population of nTreg requires presentation of the Fc region of IgG in a conventional MHC-restricted, TcR-mediated fashion by antigen presenting cells. These regulatory CD4+ cells may down-regulate the acute vasculitis by suppressing pro-inflammatory T cells in the lymph nodes and in secondary lymphoid organs and not directly in the inflamed coronary arteries. Thus, the resolution of the inflammatory process seems to be sustained both by secretion of IL-10 by tolerogenic myeloid dendritic cells (CD11c+ CD11b+ CD14+ CD4+ tmDCs) (43) and by the expansion of this population of Fc-specific nTreg cells (42).The selection of these Fc-specific Treg cells and their important role in immune regulation clearly differentiate KD from autoimmune diseases. Indeed, in children with a new onset autoimmune disease, failure to expand specific Treg populations may be associated with failure to abate the inflammatory process and consequently is responsible for the chronic course of these diseases. A defect in this Treg-mediated suppression of the pro-inflammatory T cell response has been documented only in KD patients with coronary artery abnormalities and in children with autoimmune disorders (42).
Evidence for an Autoinflammatory Process
In 1999 McDermott et al. coined the term “autoinflammatory disease” (AIDs) to describe a group of rare diseases characterized by apparently unprovoked, recurrent systemic inflammation without any evident involvement of autoimmune pathways and no demonstrated causative infectious agents (44). AIDs are typically associated with aberrant activation of innate immunity due to genetic abnormalities in specific pathways of the inflammatory response (45). The “classical” monogenic AID are now known to include tumor necrosis factor (TNF) receptor–associated periodic fever syndrome, familial Mediterranean fever (FMF), hyperimmunoglobulinaemia D with periodic fever syndrome (HIDS), and cryopyrin-associated periodic syndromes (CAPS) (46). Irrespective of the specific underlying pathways, these syndromes are characterized by exaggerated Interleukin-1β (IL-1β) production. Active IL-1β initiates the inflammatory cascade and attracts monocytes and neutrophils causing tissue damage. This cytokine is also responsible for antigen-driven CD8+ T cell differentiation, proliferation, memory, and migration into tissues (45). Clinically, these syndromes are characterized by bouts of fever with a characteristic frequency and constellation of symptoms, including cutaneous rashes, lymphadenopathy, mucosal ulcerations, ocular findings, abdominal complaints, serositis, and musculoskeletal involvement (46). The overlap between the clinical presentation of KD and the prominent features of AIDs have suggested an autoinflammatory origin of KD, with activation of similar pathways that trigger innate immune responses.
The suggestion of an autoinflammatory origin of KD is further supported by gene expression data. Global gene expression profiling conducted on a large KD cohort by Hoang et al. (21) revealed that the overwhelming signature for acute KD involved signaling pathways of the innate immune system. Most importantly, activation of the IL-1 signaling pathway was identified as an essential driver of disease pathogenesis. Common features of the top 3 pathways for KD were the abundance of transcripts related to the NLRP3 inflammasome, IL-1α and IL-1β, and caspase 1. When compared to children with various infectious diseases, only patients with KD showed up-regulation of two key receptors IL1-receptor (IL1R) and IL-1 receptor accessory protein (IL1RAP). IL1R and IL1RAP are expressed on the cell surface, where IL1R directly binds to circulating IL1, while IL1RAP interacts with IL-1R for signal transduction and inflammatory cascade activation. Furthermore, studies have documented that IVIG treatment in vitro increases IL-1 receptor antagonist (IL-1Ra) expression in human monocytes and reduces in vivo IL-1β secretion by peripheral blood mononuclear cell in patients without CAA, when compared to KD patients developing CAA (47, 48).
In the Lactobacillus casei cell-wall extract (LCWE)-induced mouse model of KD (49), but not in the murine model induced by CAWS (water-soluble extracellular polysaccharide fraction obtained from the culture supernatant of Candida albicans) (50), IL-1β and IL-1α have been shown to induce myocarditis and aneurysm formation (51, 52). Even if the precise mechanism remains to be clarified, IL-1β signaling is likely to amplify intra-mural inflammatory processes, to prolong neutrophil survival and to enhance uncontrolled proliferation of luminal myofibroblasts. Moreover, CD8+ T cell infiltration of coronary artery wall and optimal effector function of pre-committed CD4+ T cells are also favored by IL-1β (53). The myocardial inflammation and aneurysm formation in the animal model were halted more effectively by treatment with IL-1 inhibitors compared to IVIG or TNF blocking agents (52). This experimental model has led the way to clinical use of IL-1 blocking agents in KD patients unresponsive to IVIG. At present, only few cases have been published (54–59) and the results from two clinical trials, currently ongoing in Europe and US, are awaited to elucidate the potential role of this therapeutic approach in KD (9).
However, several limitations to this “autoinflammatory hypothesis” for KD pathogenesis exist. As a first point, KD is not associated with alteration of single gene, as reported for AIDs, and genetic studies suggest a polygenic influence on disease susceptibility (60). Moreover, AIDs typically show a recurrent pattern, with a strict periodicity or with a variable interval between attacks. In KD, the disease is typically monophasic, even in absence of treatment, suggesting the activation of mechanisms of immune regulation that eventually halt the inflammatory response and induce immune memory.
Summary: Classifying KD as an Immunizing Event
The available data as summarized above do not fit well with classic autoimmune or autoinflammatory diseases, as summarized in Table 1. The immune response is clearly influenced by genetic determinants that most likely shape the host response to one or more conventional antigens. However, the focused immunological assault on the coronary arteries remains a mystery. The rarity of KD in children under 6 months of age might be explained by the protective effect of maternal immunoglobulins and breast milk as there is a reduced incidence of KD in breastfed infants (61). Moreover, from epidemiologic studies we know that recurrent KD is rare and is influenced by host genetics with Japanese children having recurrence rates of 3–4%, while lower rates have been cited for children of European descent (62). Thus, the lack of recurrence seems to suggest that KD is usually an “immunizing” event associated with the development of immunological memory that protects the host from KD recurrences.
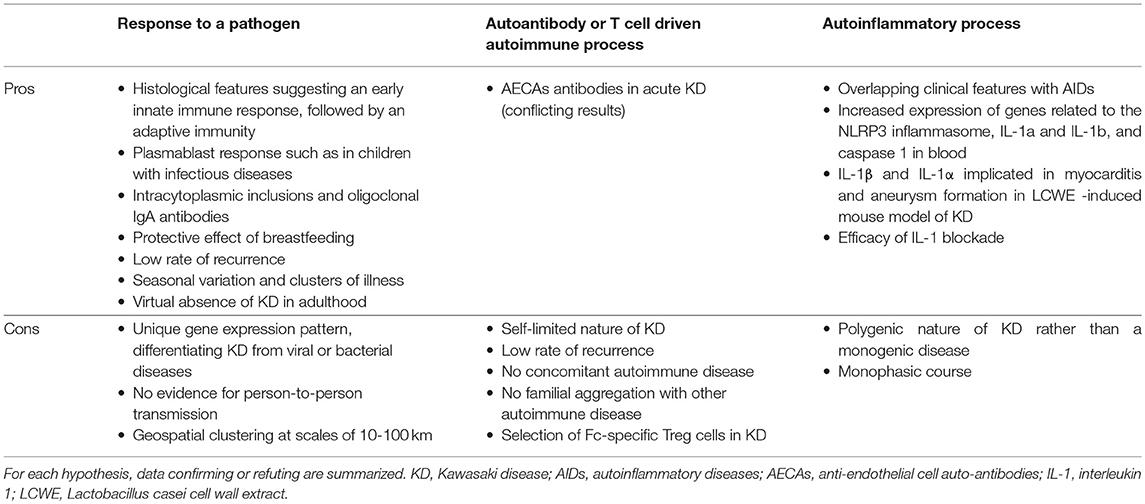
Table 1. Table representing the principal hypotheses for the etiopathogenesis of KD.
Until the environmental trigger(s) is identified, the possibility of molecular mimicry, such as in rheumatic fever and streptococcal infection, cannot be excluded. If there is a genetically determined immunological defect in KD patients, it must be highly restricted to the response to a specific antigen since these children go on to be ostensibly immunologically normal with normal responses to vaccines and the panoply of childhood infectious diseases.
Genetic and immunological data from KD patients and data from the experimental mouse models of KD vasculitis have converged on the complex interplay between actors of both the innate and adaptive immune systems. However, as reported above, the exact mechanisms that lead to activation of the inflammatory cascade are still debated. According to available data, KD can be depicted as an acute inflammatory disease with genetic predisposition. The inter-ethnic differences in incidence rates, with a far higher incidence in Asian populations (63), the increased risk in first-degree relatives (64), and the increased severity of the disease in some populations support these findings. Epidemiological studies have found that KD shows seasonal variation and clusters of illness have been described (65, 66). Furthermore, the virtual absence of KD in adulthood suggests that the agent is widely distributed in the environment and thus genetically susceptible individuals will encounter the trigger for KD before they reach adulthood (7).
In conclusion, the weight of the evidence suggests that KD patients make an innate and adaptive immune response to one or more traditional antigens that confers lasting immunity against subsequent exposures in most patients. The autoreactive and autoimmune hypotheses lack evidence. The molecular mimicry hypothesis will be difficult to discard until the inciting antigens have been identified. The final classification of KD awaits identification of the inciting agent or agents.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Burns JC, Glodé MP. Kawasaki syndrome. Lancet (2004) 364:533–44. doi: 10.1016/S0140-6736(04)16814-1
2. Suzuki A, Kamiya T, Kuwahara N, Ono Y, Kohata T, Takahashi O, et al. Coronary arterial lesions of Kawasaki disease: cardiac catheterization findings of 1100 cases. Pediatr Cardiol. (1986) 7:3–9. doi: 10.1007/BF02315475
3. Tremoulet AH, Best BM, Song S, Wang S, Corinaldesi E, Eichenfield JR, et al. Resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatr. (2008) 153:117–121.e3. doi: 10.1016/j.jpeds.2007.12.021
4. McCrindle BW, Rowley AH, Newburger JW, Burns JC, Bolger AF, Gewitz M, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a scientific statement for health professionals from the american heart association. Circulation (2017) 135:e927–999. doi: 10.1161/CIR.0000000000000484
5. Bar-Meir M, Kalisky I, Schwartz A, Somekh E, Tasher D, Israeli Kawasaki group. Prediction of resistance to intravenous immunoglobulin in children with Kawasaki disease. J Pediatric Infect Dis Soc. (2018) 7:25–9. doi: 10.1093/jpids/piw075
6. Phuong LK, Curtis N, Gowdie P, Akikusa J, Burgner D. Treatment options for resistant Kawasaki disease. Pediatr Drugs (2018) 20:59–80. doi: 10.1007/s40272-017-0269-6
7. Rowley AH. Is Kawasaki disease an infectious disorder? Int J Rheum Dis. (2018) 21:20–5. doi: 10.1111/1756-185X.13213
8. Mahr A, de Menthon M. Classification and classification criteria for vasculitis. Curr Opin Rheumatol. (2015) 27:1–9. doi: 10.1097/BOR.0000000000000134
9. Burns JC, Koné-Paut I, Kuijpers T, Shimizu C, Tremoulet A, Arditi M. Review: found in translation: international initiatives pursuing interleukin-1 blockade for treatment of acute Kawasaki disease. Arthritis Rheumatol. (2017) 69:268–76. doi: 10.1002/art.39975
10. Miura M, Kobayashi T, Kaneko T, Ayusawa M, Fukazawa R, Fukushima N, et al. Association of severity of coronary artery aneurysms in patients with Kawasaki disease and risk of later coronary events. JAMA Pediatr. (2018) 172:1–8. doi: 10.1001/jamapediatrics.2018.0030
11. Takahashi K, Oharaseki T, Naoe S, Wakayama M, Yokouchi Y. Neutrophilic involvement in the damage to coronary arteries in acute stage of Kawasaki disease. Pediatr Int. (2005) 47:305–10. doi: 10.1111/j.1442-200x.2005.02049.x
12. Orenstein JM, Shulman ST, Fox LM, Baker SC, Takahashi M, Bhatti TR, et al. Three linked vasculopathic processes characterize Kawasaki disease: a light and transmission electron microscopic study. PLoS ONE (2012) 7:e38998. doi: 10.1371/journal.pone.0038998
13. Tsuda E, Hirata T, Matsuo O, Abe T, Sugiyama H, Yamada O. The 30-year outcome for patients after myocardial infarction due to coronary artery lesions caused by Kawasaki disease. Pediatr Cardiol. (2011) 32:176–82. doi: 10.1007/s00246-010-9838-y
14. Yonesaka S, Nakada T, Sunagawa Y, Tomimoto K, Naka S, Takahashi T, et al. Endomyocardial biopsy in children with Kawasaki disease. Acta Paediatr Jpn Overseas Ed. (1989) 31:706–11. doi: 10.1111/j.1442-200X.1989.tb01384.x
15. Numano F, Shimizu C, Jimenez-Fernandez S, Vejar M, Oharaseki T, Takahashi K, et al. Galectin-3 is a marker of myocardial and vascular fibrosis in Kawasaki disease patients with giant aneurysms. Int J Cardiol. (2015) 201:429–37. doi: 10.1016/j.ijcard.2015.07.063
16. Yonesaka S, Takahashi T, Eto S, Sato T, Otani K, Ueda T, et al. Biopsy-proven myocardial sequels in Kawasaki disease with giant coronary aneurysms. Cardiol Young. (2010) 20:602–9. doi: 10.1017/S1047951109991132
17. Martin M, Wrotniak BH, Hicar M. Suppressed plasmablast responses in febrile infants, including children with Kawasaki disease. PLoS ONE (2018):13:e0193539. doi: 10.1371/journal.pone.0193539
18. Rowley AH, Baker SC, Shulman ST, Garcia FL, Fox LM, Kos IM, et al. RNA-containing cytoplasmic inclusion bodies in ciliated bronchial epithelium months to years after acute Kawasaki disease. Ratner A, ed. PLoS ONE (2008) 3:e1582. doi: 10.1371/journal.pone.0001582
19. Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute kawasaki disease. J Immunol. (2001) 166:1334–43. doi: 10.4049/jimmunol.166.2.1334
20. Rowley AH, Shulman ST, Garcia FL, Guzman-Cottrill JA, Miura M, Lee HL, et al. Cloning the arterial IgA antibody response during acute kawasaki disease. J Immunol. (2005) 175:8386–91. doi: 10.4049/jimmunol.175.12.8386
21. Hoang LT, Shimizu C, Ling L, Naim AN, Khor CC, Tremoulet AH, et al. Global gene expression profiling identifies new therapeutic targets in acute Kawasaki disease. Genome Med. (2014) 6:1–13. doi: 10.1186/s13073-014-0102-6
22. Popper SJ, Watson VE, Shimizu C, Kanegaye JT, Burns JC, Relman DA. Gene transcript abundance profiles distinguish Kawasaki disease from adenovirus infection. J Infect Dis. (2009) 200:657–66. doi: 10.1086/603538
23. Wright VJ, Herberg JA, Kaforou M, Shimizu C, Eleftherohorinou H, Shailes H, et al. Diagnosis of Kawasaki disease using a minimal whole-blood gene expression signature. JAMA Pediatr. (2018) 172:e182293. doi: 10.1001/jamapediatrics.2018.2293
24. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med. (2006) 3:1242–8. doi: 10.1371/journal.pmed.0030297
25. Niewold TB, Wu SC, Smith M, Morgan GA, Pachman LM. Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics. (2011) 127:e1239–46. doi: 10.1542/peds.2010-3022
26. Prahalad S, McCracken CE, Ponder LA, Angeles-Han ST, Rouster Stevens KA, Vogler LB, et al. Familial autoimmunity in the childhood arthritis and rheumatology research alliance registry. Pediatr Rheumatol Online J. (2016) 14:14. doi: 10.1186/s12969-016-0075-7
27. Tronconi E, Miniaci A, Pession A. The autoimmune burden in juvenile idiopathic arthritis. Ital J Pediatr. (2017) 43:56. doi: 10.1186/s13052-017-0373-9
28. Makino N, Nakamura Y, Yashiro M, Sano T, Ae R, Kosami K, et al. Epidemiological observations of Kawasaki disease in Japan, 2013–2014. Pediatr Int. (2018) 60:581–7. doi: 10.1111/ped.13544
29. Kim GB, Park S, Eun LY, Han JW, Lee SY, Yoon KL, et al. Epidemiology and clinical features of kawasaki disease in South Korea, 2012–2014. Pediatric Infect Dis J. (2017) 36:2012–4. doi: 10.1097/INF.0000000000001474
30. Stagi S, Simonini G, Ricci L, de Martino M, Falcini F. Coeliac disease in patients with Kawasaki disease. Is there a link? Rheumatology (2006) 45:847–50. doi: 10.1093/rheumatology/kel007
31. Dos Santos Domingues A, Selleski N, Uenishi RH, Medeiros Ribeiro de Magalhães C, Gandolfi L, Pratesi CB. The possible link between coeliac and Kawasaki diseases in Brazil: a cross-sectional study. BMJ Open (2018) 8:e018803.doi: 10.1136/bmjopen-2017-018803
32. Cho HK, Eun LY, Song J-S, Kang WH, Ro BI. A case of vitiligo after Kawasaki's disease. Ann Dermatol. (2009) 21:75. doi: 10.5021/ad.2009.21.1.75
33. Jiang Z-G, Liu L, Yang C-Y, Wu J. Value of anti-neutrophil cytoplasmic antibody in diagnosis of Kawasaki disease. Zhongguo Dang Dai Er Ke Za Zhi. (2012) 14:45–7.
34. Zhao J-M, Wang X-H. Clinical significance of anti-neutrophil cytoplasmic antibodies and anti-endothelial cell antibodies in children with Kawasaki disease. Zhongguo Dang Dai Er Ke Za Zhi. (2014) 16:740–4.
35. Guzman J, Fung M, Petty RE. Diagnostic value of anti-neutrophil cytoplasmic and anti-endothelial cell antibodies in early Kawasaki disease. J Pediatr. (1994) 124:917–20. doi: 10.1016/S0022-3476(05)83180-4
36. Nash MC, Shah V, Reader JA, Dillon MJ. Anti-neutrophil cytoplasmic antibodies and anti-endothelial cell antibodies are not increased in Kawasaki disease. Br J Rheumatol. (1995) 34:882–7. doi: 10.1093/rheumatology/34.9.882
37. Cunningham MW, Meissner HC, Heuser JS, Pietra BA, Kurahara DK, Leung DY. Anti-human cardiac myosin autoantibodies in Kawasaki syndrome. J Immunol. (1999) 163:1060–5.
38. Tizard EJ, Baguley E, Hughes GR, Dillon MJ. Antiendothelial cell antibodies detected by a cellular based ELISA in Kawasaki disease. Arch Dis Child. (1991) 66:189–92. doi: 10.1136/adc.66.2.189
39. Guilpain P, Mouthon L. Antiendothelial Cells Autoantibodies in Vasculitis-Associated Systemic Diseases. Clin Rev Allergy Immunol. (2008) 35:59–65. doi: 10.1007/s12016-007-8069-3
40. Legendre P, Régent A, Thiebault M, Mouthon L. Anti-endothelial cell antibodies in vasculitis: a systematic review. Autoimmun Rev. (2017) 16:146–53. doi: 10.1016/j.autrev.2016.12.012
41. Grunebaum E, Blank M, Cohen S, Afek A, Kopolovic J, Meroni PL, et al. The role of anti-endothelial cell antibodies in Kawasaki disease - In vitro and in vivo studies. Clin Exp Immunol. (2002) 130:233–40. doi: 10.1046/j.1365-2249.2002.02000.x
42. Franco A, Touma R, Song Y, Shimizu C, Tremoulet AH, Kanegaye JT, et al. Specificity of regulatory T cells that modulate vascular inflammation. Autoimmunity (2014) 47:95–104. doi: 10.3109/08916934.2013.860524
43. Burns JC, Song Y, Bujold M, Shimizu C, Kanegaye JT, Tremoulet AH, et al. Immune-monitoring in Kawasaki disease patients treated with infliximab and intravenous immunoglobulin. Clin Exp Immunol. (2013) 174:337–44. doi: 10.1111/cei.12182
44. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
45. Lucherini OM, Rigante D, Sota J, Fabiani C, Obici L, Cattalini M, et al. Updated overview of molecular pathways involved in the most common monogenic autoinflammatory diseases. Clin Exp Rheumatol. (2018) 36 (Suppl. 1):3–9.
46. Rigante D. The fresco of autoinflammatory diseases from the pediatric perspective. Autoimmun Rev. (2012) 11:348–56. doi: 10.1016/j.autrev.2011.10.008
47. Leung DY, Cotran RS, Kurt-Jones E, Burns JC, Newburger JW, Pober JS. Endothelial cell activation and high interleukin-1 secretion in the pathogenesis of acute Kawasaki disease. Lancet (1989) 2:1298–302. doi: 10.1016/S0140-6736(89)91910-7
48. Fury W, Tremoulet AH, Watson VE, Best BM, Shimizu C, Hamilton J, et al. Transcript abundance patterns in Kawasaki disease patients with intravenous immunoglobulin resistance. Hum Immunol. (2010) 71:865–73. doi: 10.1016/j.humimm.2010.06.008
49. Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. (1985) 28:652–9. doi: 10.1002/art.1780280609
50. Ohno N. Murine model of Kawasaki disease induced by mannoprotein-beta-glucan complex, CAWS, obtained from Candida albicans. Jpn J Infect Dis. (2004) 57:S9–10.
51. Lee Y, Wakita D, Dagvadorj J, Shimada K, Chen S, Huang G, et al. IL-1 Signaling Is critically required in stromal cells in kawasaki disease vasculitis mouse model: role of both IL-1α and IL-1β. Arterioscler Thromb Vasc Biol. (2015) 35:2605–616. doi: 10.1161/ATVBAHA.115.306475
52. Wakita D, Kurashima Y, Crother TR, Noval Rivas M, Lee Y, Chen S, et al. Role of Interleukin-1 signaling in a mouse model of kawasaki disease-associated abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. (2016) 36:886–97. doi: 10.1161/ATVBAHA.115.307072
53. Jain A, Song R, Wakeland EK, Pasare C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun. (2018) 9:3185. doi: 10.1038/s41467-018-05489-7
54. Cohen S, Tacke CE, Straver B, Meijer N, Kuipers IM, Kuijpers TW. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann Rheum Dis. (2012) 71:2059–61. doi: 10.1136/annrheumdis-2012-201658
55. Shafferman A, Birmingham JD, Cron RQ. High dose anakinra for treatment of severe neonatal Kawasaki disease: a case report. Pediatr Rheumatol. (2014) 12:26. doi: 10.1186/1546-0096-12-26
56. Sánchez-Manubens J, Gelman A, Franch N, Teodoro S, Palacios JR, Rudi N, et al. A child with resistant Kawasaki disease successfully treated with anakinra: a case report. BMC Pediatr. (2017) 17:102. doi: 10.1186/s12887-017-0852-6
57. Guillaume M-P, Reumaux H, Dubos F. Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol Young. (2018) 28:739–42. doi: 10.1017/S1047951117002864
58. Kone-Paut I, Cimaz R, Herberg J, Bates O, Carbasse A, Saulnier JP, et al. The use of interleukin 1 receptor antagonist (anakinra) in Kawasaki disease: a retrospective cases series. Autoimmun Rev. (2018) 17:768–74. doi: 10.1016/j.autrev.2018.01.024
59. Blonz G, Lacroix S, Benbrik N, Warin-Fresse K, Masseau A, Trewick D, et al. Severe late-onset kawasaki disease successfully treated with anakinra. J Clin Rheumatol. (2018). doi: 10.1097/RHU.0000000000000814. [Epub ahead of print].
60. Onouchi Y. The genetics of Kawasaki disease. Int J Rheum Dis. (2018) 21:26–30. doi: 10.1111/1756-185X.13218
61. Yorifuji T, Tsukahara H, Doi H. Breastfeeding and risk of Kawasaki disease: a nationwide longitudinal survey in Japan. Pediatrics (2016) 137:e20153919. doi: 10.1542/peds.2015-3919
62. Maddox RA, Holman RC, Uehara R, Callinan LS, Guest JL, Schonberger LB, et al. Recurrent Kawasaki disease: USA and Japan. Pediatr Int. (2015) 57:1116–20. doi: 10.1111/ped.12733
63. Okubo Y, Nochioka K, Sakakibara H, Testa M, Sundel RP. National survey of pediatric hospitalizations due to Kawasaki disease and coronary artery aneurysms in the USA. Clin Rheumatol. (2017) 36:413–419. doi: 10.1007/s10067-016-3512-6
64. Uehara R, Yashiro M, Nakamura Y, Yanagawa H. Kawasaki disease in parents and children. Pediatr Infect Dis J. (2003) 92:694–7. doi: 10.1111/j.1651-2227.2003.tb00602.x
65. Uehara R, Belay ED. Epidemiology of Kawasaki disease in Asia, Europe, and the United States. J Epidemiol. (2012) 22:79–85. doi: 10.2188/jea.JE20110131
Keywords: Kawasaki disease, etiopathogenesis, pediatric vasculitis, intravenous immune globulin (IVIg), coronary aneurysm
Citation: Marrani E, Burns JC and Cimaz R (2018) How Should We Classify Kawasaki Disease? Front. Immunol. 9:2974. doi: 10.3389/fimmu.2018.02974
Received: 25 July 2018; Accepted: 03 December 2018;
Published: 14 December 2018.
Edited by:
Randy Q. Cron, University of Alabama at Birmingham, United StatesReviewed by:
Jillian M. Richmond, University of Massachusetts Medical School, United StatesSophie Anne Georgin-Lavialle, Université Pierre et Marie Curie, France
Copyright © 2018 Marrani, Burns and Cimaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rolando Cimaz, cm9sYW5kby5jaW1hekBtZXllci5pdA==