 Laís Teodoro da SilvaBruna Tereso SantilloAlexandre de AlmeidaAlberto Jose da Silva Duarte
Laís Teodoro da SilvaBruna Tereso SantilloAlexandre de AlmeidaAlberto Jose da Silva Duarte Telma Miyuki Oshiro*
Telma Miyuki Oshiro*- Laboratorio de Investigacao em Dermatologia e Imunodeficiencias, Hospital das Clinicas HCFMUSP, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil
Harnessing dendritic cells (DC) to treat HIV infection is considered a key strategy to improve anti-HIV treatment and promote the discovery of functional or sterilizing cures. Although this strategy represents a promising approach, the results of currently published trials suggest that opportunities to optimize its performance still exist. In addition to the genetic and clinical characteristics of patients, the efficacy of DC-based immunotherapy depends on the quality of the vaccine product, which is composed of precursor-derived DC and an antigen for pulsing. Here, we focus on some factors that can interfere with vaccine production and should thus be considered to improve DC-based immunotherapy for HIV infection.
Introduction
Although antiretroviral therapy has deeply improved the quality of life of HIV-infected individuals, some problems must be overcome, such as viral resistance, drug toxicities, therapeutic failure, and lack of drug access to viral reservoirs (1–4), which impair treatment effectiveness and patient adherence and hinder the discovery of functional or sterilizing cures.
In this context, harnessing dendritic cells (DC) to treat HIV infection is a promising strategy that has been extensively studied in recent years (5–21). The rationale for using DC is based on their essential role in the immune system, priming a specific immune response (22, 23). This strategy was initially tested for cancer treatment (24) and then for infectious diseases (5, 25), and more recently, tolerogenic DC have been evaluated for treating autoimmune diseases (26, 27).
Particularly in HIV infection, DC are qualitatively and quantitatively impaired in the host. In fact, HIV is able to evade innate immune sensing by DC, leading to suboptimal maturation that results in a poor antiviral adaptive immune response (28). Thus, the administration of properly sensitized DC can drive the immune response to a specific target, improving the anti-HIV-specific response.
More recent approaches have proposed a strategy using DC to reactivate HIV reservoirs (29, 30) together with a potent antiretroviral drug, which could finally promote the discovery of a long-awaited sterilizing cure. This timely and paramount approach encourages further studies on this type of intervention.
While many studies have demonstrated the high potential of DC-based strategies to stimulate an anti-HIV immune response in vitro (31–33), a systematic review of currently published trials concluded that in general, clinical outcomes have been modest, and the expected success rates have not been achieved (34). This outcome suggests that the full potential of this technique has not yet been realized, and opportunities to improve the efficacy of this strategy remain.
In many clinical trials, the patients' vaccine responses are very heterogeneous. Among patients enrolled in the same study and treated with the same strategy, some patients have good clinical outcomes, while others do not present a vaccine response (6, 20), which could be due to the individual characteristics of each patient and/or differences in the final vaccine product.
In this context, the efficacy of DC-based immunotherapy depends mainly on two factors: (i) the general condition of the patient, which is determined by genetic factors and clinical status; and (ii) the vaccine product, generally composed of monocyte-derived DC (MoDC) and the antigen used to pulse them (Figure 1).
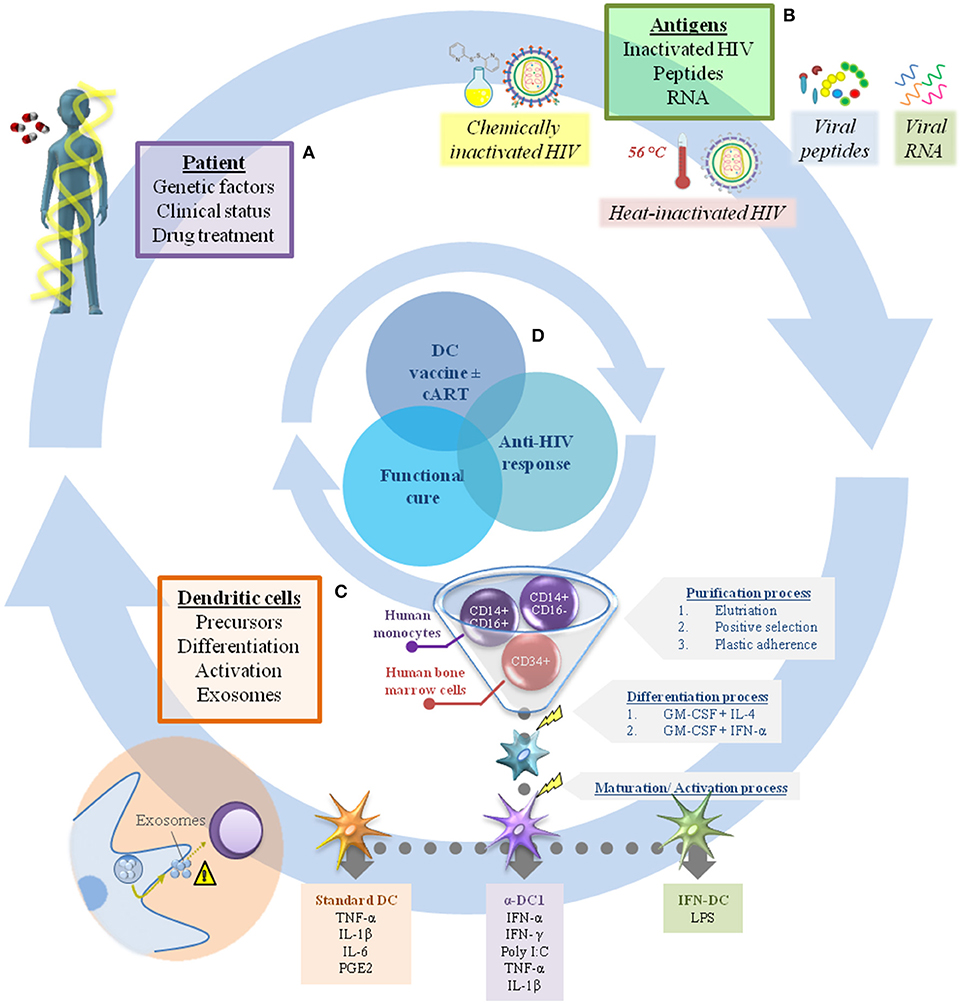
Figure 1. Challenges in dendritic cell immunotherapy for HIV infection. There are many factors that should be considered in the production of DC-based vaccines to achieve a sufficient immune response against HIV combined with viral load control. In general, these can include elements related to the individual patient (A), such as genetic factors, clinical status, and drug treatment (cART interruption or not after receiving the immunization). In addition, the range of antigens available to pulse DC is extensive, making it a challenge to choose the best one (B). The factors related to the vaccine product (B,C) are just as important, including the choice of appropriate DC precursors (CD34+ cells or monocytes) and their differentiation/activation protocols (e.g., standard DC, α-DC1, IFN-DC), while also taking into account the potential of DC to produce exosomes (considering their role in regulation of the immune response) (C). In this context, proper assembly of each individual gear could achieve viral infection control and make possible the “functional cure” (D).
Host genetic and clinical determinants will not be addressed in the present review. Instead, this review will focus on some factors that can interfere with vaccine production and should be taken into account to improve DC-based immunotherapy for HIV infection.
Clinical Trials
Clinical trials performed thus far have been phase I or phase II trials enrolling from four up to fifty-two patients, who received from one to thirty million DC per dose (5–21). To date, four clinical trials recruited treatment-naïve (5, 6, 11) or untreated (13) HIV-1-infected subjects, while 13 other studies enrolled patients on combined antiretroviral therapy (cART) in which the drug treatment was either interrupted (7, 8, 10, 15–17, 19–21) or not interrupted (9, 12, 14, 18) after patients received the immunization. Analytical treatment interruption is useful to evaluate the effects of DC immunization on viral replication (Table 1).
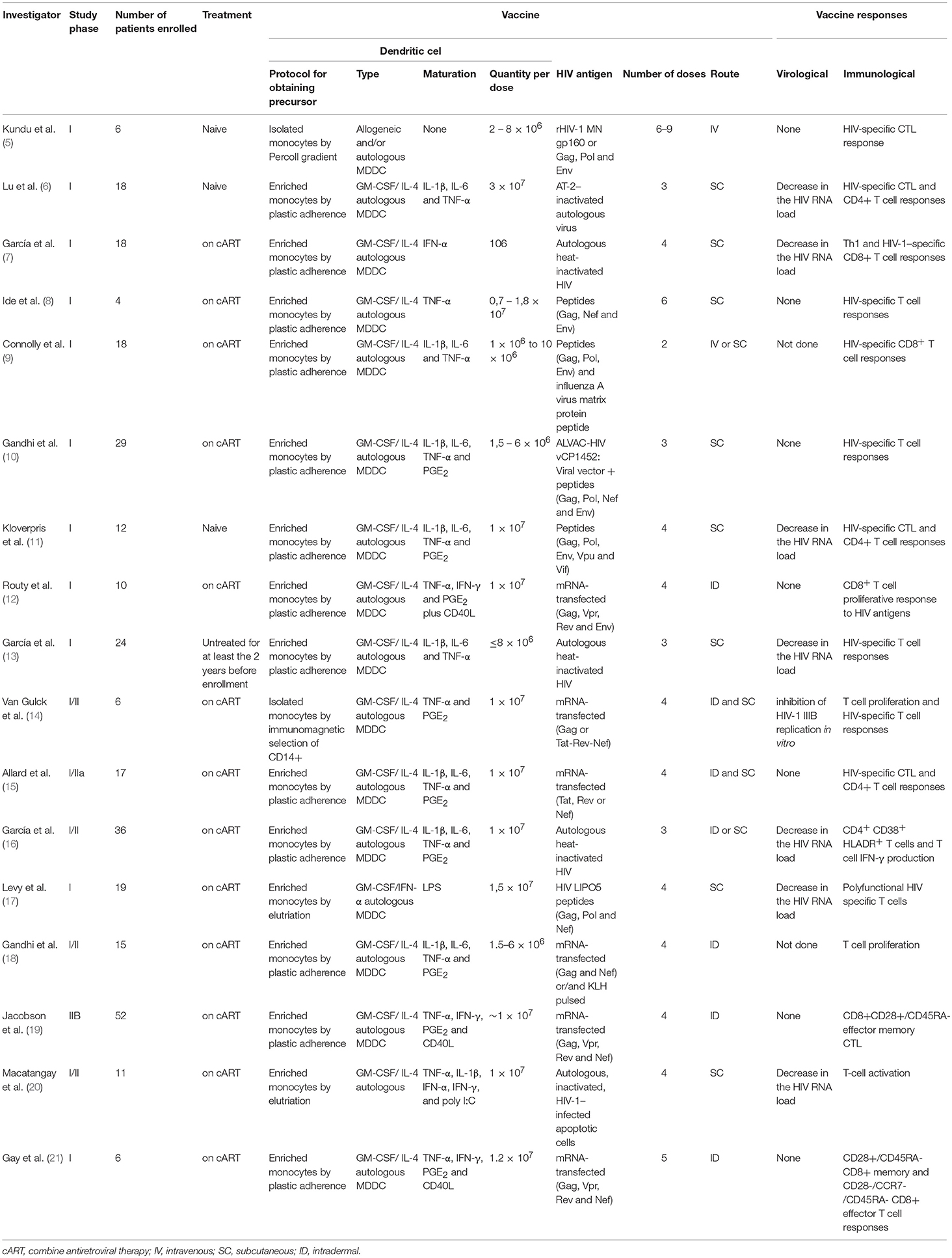
Table 1. Currently published clinical trials on the use of DC-based vaccines for HIV infection.
Overall, immunotherapy trials present high variability in terms of the protocol used to obtain DCs, the number of doses, patient profiles and the immunization route. In this regard, the only commonality between currently published DC-based HIV vaccines is that the DC used in all protocols have been derived from monocytes because they are easy to obtain (Figure 1). However, despite the variability in study design, DC immunotherapy has been shown to be well-tolerated and safe, with only minor and transient side effects, including fever (8, 9, 14), enlargement of local lymph nodes (8, 13, 16), mild local redness (14–16) and flu-like symptoms (7, 13, 16).
Clinical outcomes were also highly variable between studies. In some, decreased plasma viral loads were observed in HIV-infected vaccinated individuals, but specific immune responses were usually transitory (6, 7, 11, 13, 16, 17). When the effects of immunotherapy on activation markers were monitored, CD38, and human leukocyte antigen (HLA)–DR expression increased on T cells (16, 20). In addition, eight out of seventeen trials showed that some individuals exhibited HIV-specific T cell responses that were not associated with decreased viral load or virologic control (5, 8, 10, 12, 15, 19–21).
These different outcomes may have been influenced by the protocols used to generate the vaccine products and their quality (MoDC maturation cocktail, HIV antigen used to pulse MoDC, quantities of cells inoculated per dose and number of doses administered) as well as patients' individual characteristics (e.g., CD4+ T cell nadir (35), HLA alleles (36), and polymorphisms in genes involved in immune modulation (37–41).
The combination of factors discussed above may have affected the immune responses of vaccinated patients, which could explain why some of these individuals did not respond to immunotherapy (40).
Challenges in MoDC Preparation
Cell Precursors
Myeloid DC can be detected at a reduced frequency in peripheral blood. For immunotherapeutic protocols in which large numbers of cells are required, DC may alternatively be differentiated from precursors, such as CD34+ hematopoietic progenitor cells and CD14+ monocytes present in peripheral blood (42, 43).
Considering that the number of peripheral blood DC is low and that differentiation techniques require complex generation methods, only a small number of clinical trials, all related to cancer, have been published using DC generated from CD34+ cells (44, 45); thus, MoDC are the most commonly used cells in a wide range of clinical applications (5–21, 46).
Monocytes are highly plastic cells that can alter their phenotype according to signals present in the microenvironment; for example, they may differentiate into MoDC under inflammatory conditions (43). MoDC have a high capacity for antigen presentation and naive T lymphocyte stimulation, similar to DC generated from CD34+ cells (47).
Three circulating monocyte subsets have been described in human blood: classical (CD14++CD16−), intermediate (CD14++CD16+), and non-classical (CD14+CD16++) (48). Increased numbers of inflammatory CD16+ monocytes are found in HIV-infected individuals (49, 50) and can act as targets for HIV entry through the highly expressed CCR5 (an HIV co-receptor); these monocytes may be more permissive to productive HIV infection than other monocyte subtypes (51).
While MoDC generated from CD16+ monocytes secrete increased amounts of TGF-β1, MoDC generated from CD16− monocytes produce more of the IL-12p70 cytokine (52). In this context, considering that all three monocyte subtypes can be differentiated into MoDC in vitro and that the MoDC generated have distinct phenotypic and functional abilities (53), selection of the monocytic precursor may substantially influence vaccine performance. In fact, it was shown that “CD16+” MoDC-stimulated T cells produce more IL-4 than lymphocytes co-cultured with MoDC obtained from CD16− monocytes; therefore, “CD16+” MoDC can polarize the naive T cell response toward the Th2 phenotype (53). In the context of anti-HIV therapy, obtaining MoDC that secrete IL-12p70 is desirable for inducing IFN-γ-producing T lymphocytes (Th1 profile) (54). In the future, developing a clinical-scale procedure to enrich the non-classical monocyte subset could be a promising option.
Another important point to consider is the technique used to acquire peripheral blood cells. To obtain a large number of monocytes, leukapheresis is first performed, followed by an additional step to isolate or enrich the monocyte population [elutriation (55) or positive purification by CD14+ microbeads or adherence to plastic (56)].
During leukapheresis, a higher centrifuge speed yields residual platelets (57), which may subsequently attach to the monocytes and induce the production of cytokines (IL-1α and TNF-α) (58), pre-activating monocytes that could impair their differentiation into DC after in vitro stimulation. In addition, if leukapheresis itself leads to platelet activation, HIV-infected patients, even those receiving cART, may exhibit basal activation of these blood cells (59–61), which interfere with monocyte functionality and induce DC activation in vitro, affecting DC-mediated T lymphocyte polarization (62, 63).
Furthermore, if the monocytes are obtained by plastic adherence, the adhesion capacity of the platelets can reduce their yield, which subsequently reduces the yield of MoDC.
Considering the factors discussed above, vaccine product quality can be directly influenced by the first stages of DC acquisition, such as the leukapheresis process and the monocyte subsets used.
MoDC Differentiation/Activation Protocols
MoDC-based immunotherapy requires custom conditions for producing mature MoDC capable of stimulating an appropriate immune response. For this reason, protocols should be guided by factors that contribute to viability, migration, co-stimulatory molecule expression, cytokine secretion, antigen presentation and T cell stimulation (64).
Although IL-4 and GM-CSF are used for MoDC differentiation in most studies, different concentrations or cytokine arrangements have been used in clinical trials (16, 17, 20), resulting in different vaccine products with variable performance once these cells present high plasticity.
Another important point to consider is maturation/activation stimuli. Correct insight is fundamental because the product has the potential to “educate” MoDC behavior. The commonly used maturation cocktail for MoDC comprises the proinflammatory cytokines TNF-α, IL-1ß, and IL-6 combined with PGE2, which was established as the “gold standard” for MoDC maturation (the so-called “standard DC” or “sDC”) (65). sDC upregulate major histocompatibility complex (MHC) class I and II molecules, co-stimulatory molecules and CCR7 but fail to induce IL-12p70 production, probably due to PGE2 (66–68). The removal of PGE2 from these cocktails generates MoDC with similar profiles but low CCR7 expression and subsequent decreased migration to the lymphoid organs (69). The combination of different cytokines induces distinct responses, reflecting the complexity involved in establishing an effective protocol. In fact, sDC (with or without PGE2) have been adopted in most MoDC-based HIV immunotherapy protocols (Figure 1).
To improve the performance of sDC, alternative strategies have been developed. For example, type I and II interferons have been used to supplement standard activation stimuli to obtain polarized DC, called alpha-type-I polarized DC (α-DC1), driving a potent Th1 response (54). Recently, α-DC1 were used in a clinical trial for the treatment of HIV-infected individuals after stimulation with autologous HIV-infected apoptotic cells (ApB-DC vaccine). Although safe and immunogenic, only a modest decrease in the HIV-1 RNA load set point was observed after vaccination, and this was not sustained after cART discontinuation. Suboptimal DC function, evidenced by low IL-12 production, was attributed to this modest outcome (20).
Recently, a cancer research group developed “self-differentiated myeloid-derived antigen-presenting cells reactive against tumors DC” (Smart-DC), which are generated via the genetic reprogramming of monocytes. For production, monocytes are transduced with lentiviral vectors co-expressing GM-CSF and IL-4 and a melanoma self-antigen, allowing their self-differentiation into DC, which express typical DC surface molecules and stimulate antigen-specific CTL responses (70). This interesting and innovative approach could be a potential option for future antiviral immunotherapy applications.
Another promising strategy is a preclinical evaluation of an mRNA-electroporated MoDC-based therapeutic vaccine against HIV-1-encoding activation signals (TriMix: CD40L + CD70 + caTLR4 -activated form of TLR4) combined with rationally selected antigen sequences of Gag, Pol, Vif and Nef (HTI—HIV T cell immunogen). In vitro assays demonstrated MoDC with appropriate maturation profiles and the ability to induce T cell responses, especially CD8+ T cells (71).
Overall, these studies aim to reach the same goal of developing a protocol that can induce the best MoDC capable of eliciting a sufficiently potent immune response that is reproducible in vivo and also controls viral replication. Several combinations of differentiation and activation factors are available, but the search for an ideal MoDC continues, and a gold standard protocol for generating successful MoDC-based therapeutic vaccines for HIV-infected individuals has not been established.
Exosomes
Exosomes have emerged as potential modulators of the immune response in a DC-based immunotherapy context. As a type of extracellular vesicle, exosomes are endocytic-originating small particles (30-100 nm in diameter) composed of lipids that are released by cells into the extracellular environment by the fusion of internal multivesicular compartments. Exosomes participate in intercellular communication via the transfer of a variety of molecules, such as lipids, proteins, DNA, mRNA, and microRNA (72–74).
Many cells, such as neurons, tumors and immune cells, are capable of releasing exosomes. In particular, DC-derived exosomes can express class I and II MHCs, adhesion and co-stimulatory molecules, enabling their ability to directly activate CD8 and CD4 T cells (75–77).
Interestingly, in the context of HIV infection, the exosome dissemination pathway converges to capture and transfer HIV particles via mature DC, suggesting that HIV exploits this pathway to mediate T lymphocyte transinfection (78). Additionally, exosomes derived from HIV-infected DC can transmit HIV to T cells (79) and are capable of inducing the activation of resting primary CD4+ T lymphocytes as well as reactivating the HIV-1 reservoir (80). These findings illustrate the close relationship between exosomes and HIV in the DC therapy context.
Considering that whole HIV particles are used in some DC-based immunotherapy protocols, the role of exosomes in DC performance should be considered. After pulsing DC with HIV, DC-derived exosomes were shown to induce the apoptosis of neighboring CD4 T lymphocytes, which has the potential to impair specific anti-HIV immune responses (81). In line with this, preliminary data suggested that exosomes may play a role in modulating the immune response during anti-HIV DC-based immunotherapy. Using gene expression analysis of an exosome marker, it was hypothesized that low exosomal release is more beneficial for DC-based immunotherapy responses than high exosomal release (82), which is an important point that should be considered when using whole viral particles to pulse DC.
Although systematic analyses have not considered the role of exosomes in anti-HIV DC-based clinical trial outcomes, studies have demonstrated a relevant function for these vesicles in immune response regulation. In the context of cancer research, if on one hand tumor-derived exosomes can directly activate specific immune responses and improve anti-cancer responses (83), on the other hand these exosomes can create an immunosuppressive pro-tumorigenic microenvironment, which allows the disease to progress (84). Similarly, in an anti-HIV context, this duality may also be present and should be considered in future trials.
Antigens and Challenges in Dc-Loading Strategies
A fundamental aspect of DC-based immunotherapy is the selection of the antigen to be incorporated, a decision that must consider the safety and efficacy originating from the effects of the antigen's interaction with DC during the pathogenesis of infection in addition to knowledge of HIV structure.
Although whole particles (inactivated or attenuated) advantageously have greater epitope diversity, they have pathogenic potential due to the virus-cell interaction.
Attenuated viral particles represent one of the most potent known immunogens (85). Research on and development of an anti-HIV vaccine has previously incorporated attenuation procedures, but this approach was abandoned for safety reasons (86). With modern technologies, research examining this process has resumed, and such a strategy potentially represents another DC-based immunotherapy option (87, 88).
The deleterious effects arising from the interaction of viral particles with DC can be minimized by using killed whole HIV particles as an antigen. The consequences of such an approach will depend on the methodology used for chemical or heat inactivation (89–91), which may or may not alter the virus structure and will also influence the type of immune response induced (92).
When using HIV fragments as antigens, the risk of deleterious effects on the individual is lower (although persistent) relative to that posed by using the entire viral particle. However, an even more significant challenge associated with using HIV fragments is the selection of which one to use. The lack of immune protection correlates in HIV infection imposes an unprecedented degree of difficulty on this definition when determining the composition of the product for intervention (93, 94).
Groups have studied the immunogenic potential of DC transfected with mRNA encoding HIV proteins in vitro (71, 95, 96) and in vivo (12, 14, 15, 18, 19, 21). Some advantages of using RNA as an antigen include the absence of a biological risk of infection and the possibility of designing a sequence restricted to targeting MHC class I or II molecules, thus activating immune responses to CD8+ T cells or both CD4+ and CD8+ T cells, respectively.
Another strategy is the use of the HIV fragments Gag, Tat, Rev, Nef and Vpr, which are commonly employed because they are more conserved than other proteins and are known to induce a T cell immune response (97). However, Nef interferes with DC functionality (98); for this reason, reduced quantities of Nef RNA are used in immunotherapeutic protocols. Similarly, the Vpr gene is truncated to remove its ability to impair DC expression of co-stimulatory molecules and production of the cytokine IL-12 (12, 99).
In clinical trials, both consensus viral sequences for a cohort of patients (14, 15, 18) or mRNA constructs personalized for each individual (12, 19, 21) have been used as immunogens. Although researchers have observed the induction of HIV-specific T cell responses in vivo as increases in T cell proliferation (12, 14) and enhancements in effector/memory CD8+ T cell responses (15, 19, 21), there has been no sustained impact on patient viral load.
Another strategy is loading DC with DNA encoding HIV antigens. The use of DNA constructs as antigen vectors may overcome difficulties associated with MHC haplotype and peptide mismatches. Additionally, DNA can express antigens with natural posttranslational modifications (100). Moreover, DNA vaccines present several advantages, such as safety, potential to elicit both humoral and cellular immune responses and low cost (101). In this sense, plasmid DNA can be efficiently combined with DC to induce a specific immune response, as demonstrated in vitro (33, 102), representing a promising strategy to improve DC-based vaccines.
Summary
Overall, this review highlights some emerging factors that should be considered to improve the production of vaccines for anti-HIV DC-based immunotherapy protocols. Approaches to improve anti-HIV immunotherapy are complex and challenging. HIV infection is characterized by a chronic immune activation state, a consequence of intense immune stimulation and sustained inflammation, which promotes massive immune cell loss (103, 104). Given that immunotherapy aims to induce the patient's immune response, treatment requires an equilibrium between stimulating a specific immune response to fight the virus and limiting the immune activation state to avoid “adding fuel to the fire.” Additionally, depending on the patient's clinical status, DC precursors as well as effector cells may be committed, which could interfere with the performance of vaccine products.
Author Contributions
TO conceived the study. AdA, BS, LdS and TO discussed, wrote, and edited the manuscript, and BS also contributed to figure construction. AD provided intellectual guidance. All authors have read and approved the final manuscript.
Funding
This work was supported by the Sao Paulo Research Foundation—FAPESP, Brazil (grant number 2017/22131-0 and 2016/25212-9). BS is a recipient of Coordenacao de Aperfeicoamento de Pessoal de Nível Superior—Brasil (CAPES)–Finance Code 001, and LdS is a recipient of FAPESP (grant number 2018/12460-0).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Autran B, Carcelain G, Li TS, Blanc C, Mathez D, et al. Positive effects of combined antiretroviral therapy on CD4(+) T cell homeostasis and function in advanced HIV disease. Science (1997) 277:112–6. doi: 10.1126/science.277.5322.112
2. Dalod M, Harzic M, Pellegrin I, Dumon B, Hoen B, Sereni D, et al. Evolution of cytotoxic T lymphocyte responses to human immunodeficiency virus type 1 in patients with symptomatic primary infection receiving antiretroviral triple therapy. J Infect Dis. (1998) 178:61–69. doi: 10.1086/515587
3. Ogg GS, Jin X, Bonhoeffer S, Moss P, Nowak M, et al. Decay kinetics of human immunodeficiency virus-specific effector cytotoxic T lymphocytes after combination antiretroviral therapy. J. Virol. (1999) 73:797–800.
5. Kundu SK, Engleman E, Benike C, Shapero MH, Dupuis M, van Schooten WC, et al. A pilot clinical trial of HIV antigen-pulsed allogeneic and autologous dendritic cell therapy in HIV-infected patients. AIDS Res Hum Retroviruses. (1998) 14:551–60. doi: 10.1089/aid.1998.14.551
6. Lu W, Arraes L C, Ferreira W T, Andrieu J. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat Med. (2004) 10:1359–65. doi: 10.1038/nm1147
7. Garcia F, Lejeune M, Climent N, Gil C, Alcami J, Morente V, et al. Therapeutic immunization with dendritic cells loaded with heat-inactivated autologous HIV-1 in patients with chronic HIV-1 infection. J Infect Dis. (2005) 191:1680–5. doi: 10.1086/429340
8. Ide F, Nakamura T, Tomizawa M, Kawana-Tachikawa A, Odawara T, Hosoya N, et al. Peptide-loaded dendritic-cell vaccination followed by treatment interruption for chronic HIV-1 infection: A phase 1 trial. J Med Virol. (2006) 78:711–8. doi: 10.1002/jmv.20612
9. Connolly NC, Whiteside TL, Wilson C, Kondragunta V. Therapeutic immunization with human immunodeficiency virus type 1 (HIV-1) peptide-loaded dendritic cells is safe and induces immunogenicity in HIV-1-Infected individuals. Clin Vacc Immunol. (2008) 15:284–92. doi: 10.1128/cvi.00221-07
10. Gandhi RT, O'Neill D, Bosch RJ, Chan E. A randomized therapeutic vaccine trial of canarypox-HIV-pulsed dendritic cells vs. canarypox-HIV alone in HIV-1-infected patients on antiretroviral therapy. Vaccine (2009) 27:6088–94. doi: 10.1016/j.vaccine.2009.05.016
11. Kloverpris H, Karlsson I, Bonde J, Thorn M, Vinner L, Pedersen A, et al. Induction of novel CD8(+) T-cell responses during chronic untreated HIV-1 infection by immunization with subdominant cytotoxic T-lymphocyte epitopes. AIDS (2009) 23:1329–40. doi: 10.1097/QAD.0b013e32832d9b00
12. Routy JP, Boulassel MR, Yassine-Diab B, Nicolette C, Healey D, Jain R, et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin Immunol. (2010) 134:140–7. doi: 10.1016/j.clim.2009.09.009
13. Garcia F, Climent N, Assoumou L, Gil C, Gonzalez N, Alcami J, et al. A therapeutic dendritic cell-based vaccine for HIV-1 infection. J Infect Dis. (2011) 203:473–8. doi: 10.1093/infdis/jiq077
14. Van Gulck E, Vlieghe E, Vekemans M, Van Tendeloo VF I A, et al. mRNA-based dendritic cell vaccination induces potent antiviral T-cell responses in HIV-1-infected patients. AIDS (2012) 26:F1–F12. doi: 10.1097/QAD.0b013e32834f33e8
15. Allard SD, De Keersmaecker B, de Goede AL, Verschuren E, et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin Immunol. (2012) 142:252–68. doi: 10.1016/j.clim.2011.10.010
16. Garcia F, Climent N, Guardo AC, Gil C, Leon A, Autran B, et al. (2013). A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci Trans Med. 5:166ra2. doi: 10.1126/scitranslmed.3004682
17. Levy Y, Thiebaut R, Montes M, Lacabaratz C, Sloan L, King B, et al. Dendritic cell-based therapeutic vaccine elicits polyfunctional HIV-specific T-cell immunity associated with control of viral load. Eur J Immunol. (2014) 44:2802–10. doi: 10.1002/eji.201344433
18. Gandhi RT, Kwon DS, Macklin EA, Shopis JR, McLean AP, McBrine N, et al. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mRNA encoding HIV-1 gag and Nef: results of a randomized, placebo-controlled clinical trial. JAIDS (2016) 71:246–53. doi: 10.1097/qai.0000000000000852
19. Jacobson JM, Routy JP, Welles S, DeBenedette M, Tcherepanova I, Angel JB, et al. Dendritic cell immunotherapy for HIV-1 infection using autologous HIV-1 RNA: a randomized, double-blind, placebo-controlled clinical trial. JAIDS (2016) 72:31–8. doi: 10.1097/qai.0000000000000926
20. Macatangay BJC, Riddler SA, Wheeler ND, Spindler J, Lawani M, Hong F, et al. Therapeutic vaccination with dendritic cells loaded with autologous HIV type 1-infected apoptotic cells. J Infect Dis. (2016) 213:1400–9. doi: 10.1093/infdis/jiv582
21. Gay CL, DeBenedette MA, Tcherepanova IY, Gamble A, Lewis WE, Cope AB, et al. Immunogenicity of AGS-004 dendritic cell therapy in patients treated during acute HIV infection. AIDS Res Hum Retroviruses. (2018) 34:111–22. doi: 10.1089/aid.2017.0071
22. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
23. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YT, et al. Immunobiology of dendritic cells. Ann Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
24. Nestle FO, Alijagic S, Gilliet M, Sun YS, Grabbe S, Dummer R, Burg G, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. (1998) 4:328–32. doi: 10.1038/nm0398-328
25. Chen M, Li YG, Zhang DZ, Wang ZY, Zeng WQ, Shi XF, et al. Therapeutic effect of autologous dendritic cell vaccine on patients with chronic hepatitis B: A clinical study. World J Gastroenterol. (2005) 11:1806–8.
26. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase 1 (Safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34:2026–32. doi: 10.2337/dc11-0472
27. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci Trans Med. (2015) 7:aaa9301. doi: 10.1126/scitranslmed.aaa9301
28. Miller E, Bhardwaj N. Dendritic cell dysregulation during HIV-1 infection. Immunol Rev. (2013) 254:170–89. doi: 10.1111/imr.12082
29. van Montfort T, Speijer D, Berkhout B. Dendritic cells as natural latency reversing agent: A wake-up call for HIV-1. Virulence (2017) 8:1494–7. doi: 10.1080/21505594.2017.1356535
30. van der Sluis RM, van Montfort T, Pollakis G, Sanders RW, Speijer D, Berkhout B, et al. Dendritic cell-induced activation of latent HIV-1 provirus in actively proliferating primary T lymphocytes. Plos Pathogens (2013) 9:e1003259. doi: 10.1371/journal.ppat.1003259
31. Lu W, Andrieu JM. In vitro human immunodeficiency virus eradication by autologous CD8(+) T cells expanded with inactivated-virus-pulsed dendritic cells. J Virol. (2001) 75:8949–56. doi: 10.1128/jvi.75.19.8949-8956.2001
32. Huang XL, Fan Z, Colleton BA, Buchli R, Li H, Hildebrand WH, et al. Processing and presentation of exogenous HLA class I peptides by dendritic cells from human immunodeficiency virus type 1-infected persons. JVirol. (2005) 79:3052–62. doi: 10.1128/jvi.79.5.3052-3062.2005
33. Apostólico JdS, Lunardelli VAS, Yamamoto MM, Souza HFS, Cunha-Neto E, Boscardin SB, et al. Dendritic cell targeting effectively boosts T cell responses elicited by an HIV multiepitope DNA vaccine. Front Immunol. (2017) 8:101. doi: 10.3389/fimmu.2017.00101
34. Coelho AV, de Moura RR, Kamada AJ, da Silva RC, Guimarães RL, Brandão LA, et al. Dendritic cell-based immunotherapies to fight hiv: how far from a success story? a systematic review and meta-analysis. Int J Mol Sci. (2016) 17:E1985. doi: 10.3390/ijms17121985
35. Lange CG, Lederman MM, Medvik K, Asaad R, Wild M, Kalayjian R. et al. Nadir CD4+ T-cell count and numbers of CD28+ CD4+ T-cells predict functional responses to immunizations in chronic HIV-1 infection. AIDS (2003) 17:2015–23. doi: 10.1097/00002030-200309260-00002
36. Corbet S, Nielsen HV, Vinner L, Lauemoller S, Therrien D, Tang S, et al. Optimization and immune recognition of multiple novel conserved HLA-A2, human immunodeficiency virus type 1-specific CTL epitopes. J Gen Virol. (2003) 84:2409–21. doi: 10.1099/vir.0.19152-0
37. Reis EC, da Silva LT, da Silva WC, Rios A, Duarte AJ, Oshiro TM, et al. Host genetics contributes to the effectiveness of dendritic cell-based HIV immunotherapy. Hum Vaccin Immunother. (2018) 14:1995–2002. doi: 10.1080/21645515.2018.1463942
38. Moura R, Pontillo A, D'Adamo P, Pirastu N, Coelho AC, Crovella S. Exome analysis of HIV patients submitted to dendritic cells therapeutic vaccine reveals an association of CNOT1 gene with response to the treatment. J Int AIDS Soc. (2014) 17:18938. doi: 10.7448/ias.17.1.18938
39. Pontillo A, Da Silva RC, Moura R, Crovella S. Host genomic HIV restriction factors modulate the response to dendritic cell-based treatment against HIV-1. Hum Vaccin Immunother. (2014) 10:512–8. doi: 10.4161/hv.27125
40. Segat L, Brandao LAC, Guimares RL, Pontillo A, Athanasakis E, de Oliveira RM, et al. Polymorphisms in innate immunity genes and patients response to dendritic cell-based HIV immuno-treatment Vaccine. (2010) 28:2201–6. doi: 10.1016/j.vaccine.2009.12.056
41. Ronald M, Plana M, Garcia F, Zupin L, Kuhn L, Crovella S. Genome-wide scan in two groups of HIV-infected patients treated with dendritic cell-based immunotherapy. Immunol Res. (2016) 64:1207–15. doi: 10.1007/s12026-016-8875-x
42. Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman J C. Dendritic cells as the terminal stage of monocyte differentiation. J Immunol. (1998) 160:4587–95.
43. Zhou LJ, Tedder TF. CD14(+) blood monocytes can differentiate into functionally mature CD83(+) dendritic cells. Proc Natl Acad Sci USA. (1996) 93:2588–92. doi: 10.1073/pnas.93.6.2588
44. Lotze MT, Hellerstedt B, Stolinski L, Tueting T, Wilson C, Kinzler D, et al. The role of interleukin-2, interleukin-12, and dendritic cells in cancer therapy. Cancer J Sci Am. (1997) 3:S109–14.
45. Mackensen A, Herbst B, Chen JL, Kohler G, Noppen C, Herr W, et al. Phase I study in melanoma patients of a vaccine with peptide-pulsed dendritic cells generated in vitro from CD34(+) hematopoietic progenitor cells. Int J Cancer (2000) 86:385–92. doi: 10.1002/(sici)1097-0215(20000501)86:3 < 385::aid-ijc13>3.0.co;2-t
46. Fong L, Engleman EG. Dendritic cells in cancer immunotherapy. Annu Rev Immunol. (2000) 18:245–73. doi: 10.1146/annurev.immunol.18.1.245
47. Sallusto F, Lanzavecchia A. Efficient presentation of soluble-antigen by cultured human dendritic cells is maintained by granulocyte-macrophage colony-stimulating factor plus interleukin-4 and down-regulated by tumor-necrosis-factor-alpha. J Exp Med. (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
48. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart D N, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116:E74–80. doi: 10.1182/blood-2010-02-258558
49. Ansari AW, Meyer-Olson D, Schmidt RE. Selective expansion of pro-inflammatory chemokine CCL2-loaded CD14(+)CD16(+) monocytes subset in HIV-infected therapy naive individuals. J Clin Immunol. (2013) 33:302–6. doi: 10.1007/s10875-012-9790-0
50. Thieblemont N, Weiss L, Sadeghi H M, Estcourt C, HaeffnerCavaillon N. CD14(low)CD16(high): a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. (1995) 25:3418–24. doi: 10.1002/eji.1830251232
51. Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, et al. The CD16(+) monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. (2007) 178:6581–9. doi: 10.4049/jimmunol.178.10.6581
52. Gogolak P, Rethi B, Szatmari I, Lanyi A, Dezso B, Nagy L, et al. Differentiation of CD1a(-) and CD1a(+) monocyte-derived dendritic cells is biased by lipid environment and PPAR gamma. Blood (2007) 109:643–52. doi: 10.1182/blood-2006-04-016840
53. Sanchez-Torres C, Garcia-Romo GS, Cornejo-Cortes MA, Rivas-Carvalho A, Sánchez-Schmitz G. CD16(+) and CD16(-) human blood monocyte subsets differentiate in vitro to dendritic cells with different abilities to stimulate CD4(+) T cells. Int Immunol. (2001) 13:1571–81. doi: 10.1093/intimm/13.12.1571
54. Mailliard RB, Wankowicz-Kalinska A, Cai Q, Wesa A, Hilkens C, et al. alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res. (2004) 64:5934–7. doi: 10.1158/0008-5472.can-04-1261
55. Erdmann M, Dorrie J, Schaft N, Strasser E, Hendelmeier M, Kampgen E, et al. Effective clinical-scale production of dendritic cell vaccines by monocyte elutriation directly in medium, subsequent culture in bags and final antigen loading using peptides or RNA transfection. J Immunother. (2007) 30:663–74. doi: 10.1097/CJI.0b013e3180ca7cd6
56. Elkord E, Williams PE, Kynaston H, Rowbottom AW. Human monocyte isolation methods influence cytokine production from in vitro generated dendritic cells. Immunology (2005) 114:204–12. doi: 10.1111/j.1365-2567.2004.02076.x
57. Gutensohn K, Maerz M, Kuehnl P. Alteration of platelet-associated membrane glycoproteins during extracorporeal apheresis of peripheral blood progenitor cells. J Hematother. (1997) 6:315–21. doi: 10.1089/scd.1.1997.6.315
58. Aiura K, Clark BD, Dinarello CA, Margolis NH, Kaplanski G, Burke JF, et al. Interaction with autologous platelets multiplies interleukin-1 and tumor necrosis factor production in mononuclear cells. J Infect Dis. (1997) 175:123–9. doi: 10.1093/infdis/175.1.123
59. Corrales-Medina VF, Simkins J, Chirinos JA, Serpa JA, Horstman LL, Jy W, et al. Increased levels of platelet microparticles in Hiv-infected patients with good response to highly active antiretroviral therapy. JAIDS (2010) 54:217–9. doi: 10.1097/QAI.0b013e3181c8f4c9
60. Kiebala M, Singh MV, Piepenbrink MS, Qiu X, Kobie JJ, Maggirwar SB. Platelet Activation in human immunodeficiency virus type-1 patients is not altered with cocaine abuse. PLoS ONE (2015) 10:130061. doi: 10.1371/journal.pone.0130061
61. Holme PA, Muller F, Solum NO, Brosstad F, Frøland SS, Aukrust P. Enhanced activation of platelets with abnormal release of RANTES in human immunodeficiency virus type 1 infection. Faseb Journal. (1998) 12:79–89
62. Kissel K, Berber S, Nockher A, Santoso S, Bein G, Hackstein H. Human platelets target dendritic cell differentiation and production of proinflammatory cytokines. Transfusion (2006) 46:818–27. doi: 10.1111/j.1537-2995.2006.00802.x
63. Strasser EF, Eckstein R. Optimization of leukocyte collection and monocyte isolation for dendritic cell culture. Transfus Med Rev. (2010) 24:130–9. doi: 10.1016/j.tmrv.2009.11.004
64. Bryant CE, Sutherland S, Kong B, Papadimitrious MS, Fromm PD, Hart DNJ. Dendritic cells as cancer therapeutics. Semin. Cell Dev. Biol. (2018) 17:S1084-9521(17)30495-0. doi: 10.1016/j.semcdb.2018.02.015
65. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. (1997) 27:3135–42. doi: 10.1002/eji.1830271209
66. Lee AW, Truong T, Bickham K, Fonteneau JF, Larsson M, Da Silva I, et al. A clinical grade cocktail of cytokines and PGE(2) results in uniform maturation of human monocyte-derived dendritic cells: implications for immunotherapy. Vaccine (2002) 20:A8–22. doi: 10.1016/s0264-410x(02)00382-1
67. Kalinski P, Hilkens CMU, Snijders A, Snijdewint FG. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E-2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. (1997) 159:28–35.
68. Hansen M, Hjorto GM, Donia M, Met O, Larsen N, et al. Comparison of clinical grade type I polarized and standard matured dendritic cells for cancer immunotherapy. Vaccine (2013) 31:639–46. doi: 10.1016/j.vaccine.2012.11.053
69. Scandella E, Men Y, Gillessen S, Forster R, Groettrup M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived denctritic cells. Blood (2002) 100:1354–61. doi: 10.1182/blood-2002-11-0017
70. Sundarasetty BS, Chan L, Darling D, Giunti G, Farzaneh F, Schenck S, et al. Lentivirus-induced 'Smart' dendritic cells: Pharmacodynamics and GMP-compliant production for immunotherapy against TRP2-positive melanoma. Gene Ther. (2015) 22:707–20. doi: 10.1038/gt.2015.43
71. Guardo AC, Joe PT, Miralles L, Bargallo ME, Mothe B, Krasniqi A, et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). AIDS (2017) 31:321–32. doi: 10.1097/qad.0000000000001276
72. Brouwers JF, Aalberts M, Jansen JWA, van Niel G, Wauben MH, Stout TA, et al. Distinct lipid compositions of two types of human prostasomes. Proteomics. (2013) 13:1660–6. doi: 10.1002/pmic.201200348
73. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide S. Vesicle formation during reticulocyte maturation - association of plasma-membrane activities with released vesicles (Exosomes). J Biol Chem. (1987) 262:9412–20.
74. Zhang X, Yuan X, Shi H, Wu LJ, Qian H, Xu WR. Exosomes in cancer: small particle, big player. J Hematol Oncol. (2015) 8:83. doi: 10.1186/s13045-015-0181-x
75. Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4(+) T cells by dendritic cell-derived exosomes. Nat Immunol. (2002) 3:1156–62. doi: 10.1038/ni854
76. Morelli AE, Larregina AT, Shufesky WJ, Sullivan ML, Stolz DB, Papworth GD, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood (2004) 104:3257–66. doi: 10.1182/blood-2004-03-0824
77. De Toro J, Herschlik L, Waldner C, Mongini C. Emerging roles of exosomes in normal and pathological conditions: new insights for diagnosis and therapeutic applications. Front Immunol. (2015) 6:203. doi: 10.3389/fimmu.2015.00203
78. Izquierdo-Useros N, Naranjo-Gomez M, Archer J, Hatch SC, Erkizia I, Blanco J, et al. Capture and transfer of HIV-1 particles by mature dendritic cells converges with the exosome-dissemination pathway. Blood (2009) 113:2732–41. doi: 10.1182/blood-2008-05-158642
79. Kulkarni R, Prasad A. Exosomes derived from HIV-1 infected dcs mediate viral trans-infection via fibronectin and galectin-3. Sci Rep. (2017) 7:8. doi: 10.1038/s41598-017-14817-8
80. Chiozzini C, Arenaccio C, Olivetta E, Anticoli S, Manfredi F, Ferrantelli F, et al. Trans-dissemination of exosomes from HIV-1-infected cells fosters both HIV-1 trans-infection in resting CD4(+) T lymphocytes and reactivation of the HIV-1 reservoir. Arch Virol. (2017) 162:2565–77. doi: 10.1007/s00705-017-3391-4
81. Subra C, Simard S, Mercier S, Bancila AA, Lambert A, et al. Dendritic cells pulsed with HIV-1 release exosomes that promote apoptosis in CD4+ T lymphocytes. J Clin Cell Immunol. (2011) S7:001. doi: 10.4172/2155-9899.S7-001
82. Ellwanger JH, Crovella S, Dos Reis EC, Pontillo A, Chies JA. Exosomes are possibly used as a tool of immune regulation during the dendritic cell-based immune therapy against HIV-I. Med Hypotheses (2016) 95:67–70. doi: 10.1016/j.mehy.2016.09.005
83. Pitt JM, Andre F, Amigorena S, Soria JC, Eggermont A, Kroemer G, et al. Dendritic cell-derived exosomes for cancer therapy. J Clin Invest. (2016) 126:1224–32. doi: 10.1172/jci81137
84. Barros FM, Carneiro F, Machado JC, Melo SA. Exosomes and immune response in cancer: friends or foes? Front Immunol. (2018) 9:730. doi: 10.3389/fimmu.2018.00730
85. Plotkin S. History of vaccination. Proc Natl Acad Sci USA. (2014) 111:12283–7. doi: 10.1073/pnas.1400472111
86. Almond N, Stott J. Live attenuated SIV - a model of a vaccine for AIDS. Immunol Lett. (1999) 66:167–170. doi: 10.1016/s0165-2478(98)00153-9
87. Whitney JB, Ruprecht RM. Live attenuated HIV vaccines: pitfalls and prospects. Curr Opin Infect Dis. (2004) 17:17–26. doi: 10.1097/01.qco.0000113644.29255.92
88. Yuan Z, Wang NX, Kang GB, Niu W, Li QS, Guo JT. Controlling multicycle replication of live-attenuated HIV-1 using an unnatural genetic switch. ACS Synth Biol. (2017) 6:721–31. doi: 10.1021/acssynbio.6b00373
89. Gil C, Climent N, Garcia F, Hurtado C, Nieto-Marquez S, Leon A, et al. Ex vivo production of autologous whole inactivated HIV-1 for clinical use in therapeutic vaccines. Vaccine (2011) 29:5711–24. doi: 10.1016/j.vaccine.2011.05.096
90. Rossio JL, Esser MT, Suryanarayana K, Schneider D, Bess JW, Vasquez GM, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. (1998) 72:7992–8001.
91. Rutebemberwa A, Bess JW, Brown B, Arroyo M, Eller M, et al. Evaluation of aldrithiol-2-inactivated preparations of HIV type 1 subtypes A, B, and D as reagents to monitor T cell responses. AIDS Res Hum Retroviruses. (2007) 23:532–42. doi: 10.1089/aid.2006.0136
92. Kang CY, Gao Y. Killed whole-HIV vaccine; employing a well established strategy for antiviral vaccines. AIDS Res Ther. (2017) 14:4. doi: 10.1186/s12981-017-0176-5
93. Foster JL, Garcia JV. Role of Nef in HIV-1 replication and pathogenesis. Adv Pharmacol. (2007) 55:389–409. doi: 10.1016/S1054-3589(07)55011-8
94. Chirmule N, Pahwa S. Envelope glycoproteins of human immunodeficiency virus type 1: profound influences on immune functions. Microbiol Rev. (1996) 60:386.
95. Van Gulck E, Cools N, Atkinson D, Bracke L, Vereecken K, Vekemans M, et al. Interleukin-12p70 expression by dendritic cells of HIV-1-infected patients fails to stimulate gag-specific immune responses. Clin Dev Immunol. (2012) 2012:184979. doi: 10.1155/2012/184979
96. Tcherepanova I, Harris J, Starr A, Cleveland J, Ketteringham H, Calderhead D, et al. Multiplex RT-PCR amplification of HIV genes to create a completely autologous DC-Based immunotherapy for the treatment of HIV infection. PLoS ONE (2008) 3:e0001489. doi: 10.1371/journal.pone.0001489
97. Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreno J, et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J Trans Med. (2011) 9:208. doi: 10.1186/1479-5876-9-208
98. Quaranta MG, Mattioli B, Giordani L, Viora M. The immunoregulatory effects of HIV-1 Nef on dendritic cells and the pathogenesis of AIDS. Faseb J. (2006) 20:2198–208. doi: 10.1096/fj.06-6260rev
99. Majumder B, Janket ML, Schafer EA, Schaubert K, Huang XL, Kan-Mitchell J, et al. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-Cell activation: implications for viral immune escape. J Virol. (2005) 79:7990–8003. doi: 10.1128/jvi.79.13.7990-8003.2005
100. Kulikova EV, Kurilin VV, Shevchenko JA, Obleukhova IA, Khrapov EA, Boyarskikh UA, et al. Dendritic cells transfected with a DNA construct encoding tumour-associated antigen epitopes induce a cytotoxic immune response against autologous tumour cells in a culture of mononuclear cells from colorectal cancer patients. Scand J Immunol. (2015) 82:110–7. doi: 10.1111/sji.12311
101. Santoro Rosa DJD, Apostólico S, Boscardin SB. DNA vaccines: how much have we accomplished in the last 25 Years? J Vaccines Vaccin. (2015) 6:283. doi: 10.4172/2157-7560.1000283
102. Li JY, Valentin A, Beach RK, Alicea C, Felber BK, Pavlakis GN. et al. DNA is an efficient booster of dendritic cell-based vaccine. Hum Vaccin Immunother. (2015) 11:1927–35. doi: 10.1080/21645515.2015.1020265
103. Brenchley J M, Price D A, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
Keywords: dendritic cells, HIV, immunotherapy, therapeutic vaccine, clinical trial
Citation: da Silva LT, Santillo BT, de Almeida A, Duarte AJdS and Oshiro TM (2018) Using Dendritic Cell-Based Immunotherapy to Treat HIV: How Can This Strategy be Improved? Front. Immunol. 9:2993. doi: 10.3389/fimmu.2018.02993
Received: 27 July 2018; Accepted: 04 December 2018;
Published: 18 December 2018.
Edited by:
Daniela Santoro Rosa, Federal University of São Paulo, BrazilReviewed by:
Juliana Maricato, Federal University of São Paulo, BrazilIrina Caminschi, Monash University, Australia
Copyright © 2018 da Silva, Santillo, de Almeida, Duarte and Oshiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Telma Miyuki Oshiro, dGVsbWEub3NoaXJvQGhjLmZtLnVzcC5icg==