 Jia-Xin Shen
Jia-Xin Shen Jing Liu
Jing Liu Guo-Jun Zhang
Guo-Jun Zhang- 1Chang Jiang Scholar's Laboratory, Shantou University Medical College, Shantou, China
- 2Department of Hematology, The First Affiliated Hospital of Shantou University Medical College, Shantou, China
- 3Department of Physiology, Shantou University Medical College, Shantou, China
- 4The Cancer Center and the Department of Breast-Thyroid Surgery, Xiang'an Hospital of Xiamen University, Xiamen, China
The human Interleukin-33 (IL-33), a member of the IL-1 family, is the cytokine as a cell endogenous alarmin, released by damaged or necrotic barrier cells (endothelial and epithelial cells). The signal transduction of IL-33 relies on recognition and interaction with specific receptor ST2, mainly expressed in immune cells. In both innate and adoptive immunity, IL-33 regulates the homeostasis in response to stress from within/out the microenvironment. Various, even negative biofunctions of IL-33 pathways have now been widely verified in pathogenesis among immunological mechanisms, like Th2-related immune-stimuli, inflammation/infection-induced tissue protectors. A larger versatility in studies of IL-33 on malignancies now focuses on: (1) promoting myeloid-derived suppressor cells (MDSC), (2) intervention toward CD8+ T, Natural Killer (NK) cell infiltration, group 2 innate lymphoid cells (ILC2) proliferation, dendritic cells (DC) activation, and (3) inhibiting tumor growth and/or further metastasis as an immunoadjuvant. Although IL-33 functioned pro-tumorigenically in various cancers, for some cancer types the findings so far are controversial. This review begins from a summarized introduction of IL-33, to its remarkable implications and molecular transduction pathway in malignant neoplasms, ends with latest inspiration for IL-33 in treatment.
Introduction
Cytokines are central mediators between cells in the inflammatory tumor microenvironment, in which Interleukin-33 (IL-33) is considered as an alarmin released after cellular damage. IL-33 was discovered as a member of the IL-1 family of cytokines. The IL-1 gene family contains 11 members (IL-1α, IL-1β, IL-1RA, IL-18, IL-36RA, IL-36α, IL37, IL-36β, IL-36γ, IL-38, IL-33), which induces a complex network of pro-inflammatory cytokines, and regulates and initiates inflammatory responses, via expressing integrins on leukocytes and endothelial cells (1). IL-33 gene is constitutively located on the short arm of chromosome 9 at 9p24.1. Sequencing alignment toward human and murine IL-33 revealed two evolutionary conserved domains; a chromatin-binding motif, and a cleavage site for inflammatory proteases and apoptotic caspases (Figure 1A). The discovery of two ST2-binding sites further confirmed an exerted binding of IL-33 to a heterodimer, formed by the specific primary receptor ST2 and the IL-1 receptor accessory protein (2, 3).
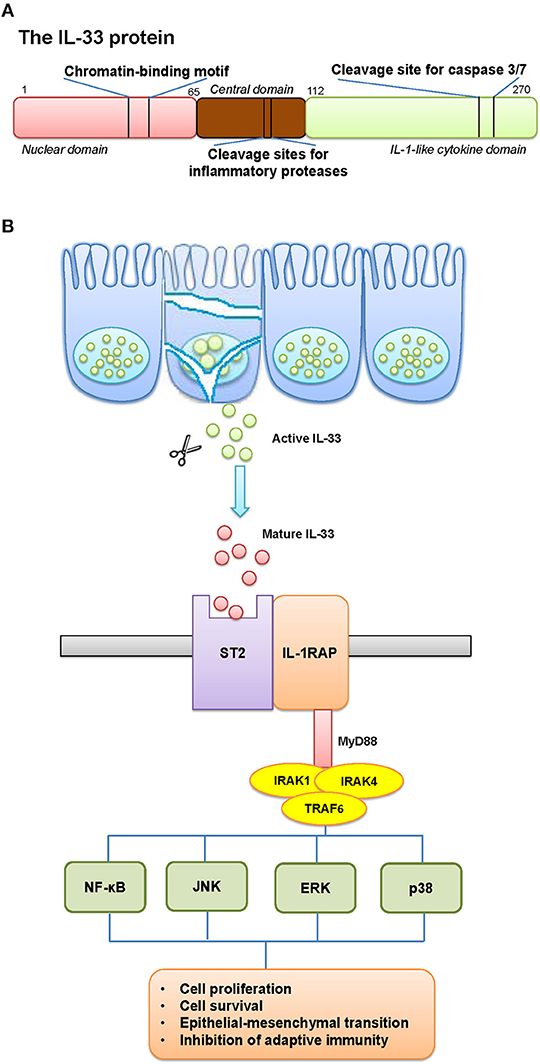
Figure 1. The schematic elucidation of IL-33. (A) The schematic structure of IL-33 protein and its critical domains. (B) The pathway involved IL-33 and its receptor. (A) IL-33 gene is constitutively located on the short arm of chromosome 9 at 9p24.1. The structure of IL-33 protein contains two evolutionary conserved domains: a chromatin-binding motif, and a cleavage site for inflammatory proteases and apoptotic caspases. (B) The release mechanisms of IL-33 can occur by mechanical and oxidative stress, necrotic cell death, or cell activation that functions as an alarmin. IL-33 is rapidly released from cells during necrosis or tissue injury, and signals through a cell surface receptor complex, ST2 (IL-1 receptor-like 1, IL1RL1) and IL1RAcP (IL-1 receptor accessory protein), to initiate inflammatory pathways in immune cells. This clustered domain recruits signaling adaptor and kinases (including MyD88, IRAK1/4, TRAF6) in order to activate the transcription factors in tumor cells. The possible results would be a generated cancer-related inflammatory microenvironment, with tumor-promoting effects.
The full-length IL-33 contains 270 amino acids in human and 266 in mice, which harbors a homeodomain-like helix-turn-helix domain presumably allowing to bind to DNA (4). The release of IL-33 can be associated with mechanical and oxidative stress, necrotic cell death, or cell activation through ATP signaling in the absence of cell death (5). IL-33 is rapidly released from cells during necrosis or tissue injury, and signals through a cell surface receptor complex, ST2 (IL-1 receptor-like 1, IL1RL1) and IL1RAcP (IL-1 receptor accessory protein), to initiate inflammatory pathways in immune cells, such as type-2 innate lymphoid cells (ILC2), mast cells and natural killer (NK) cells (6). Although advances have been made, mechanisms regulating the alarmin activity of IL-33 remain poorly understood (Figure 1B).
Human nervous tissues, barrier structures with widespread of endothelial, epithelial and fibroblast-like cells that are exposed to the environment, were indicated high level of constitutive expression of IL-33. However, inducible IL-33 by inflammation catches more attention of researchers from classical cases with chronic obstructive pulmonary disease, inflammatory responses to suffered graft-vs.-host diseases after bone marrow transplantation. Most recently, tissue fibrosis, mucosal healing, and wound repairmen were as well found to be possible initiatives of IL-33 during inflammation. There are outcomes of cross-talks and interactions within DNA, mRNA, and protein levels. Strict regulation starts from nuclear localization and chromosome association of IL-33, where nuclear IL-33 functions as a transcriptional repressor when overexpressed in chronically in?amed tissues from patients with rheumatoid arthritis and Crohn's disease (7, 8).
The Roles of IL-33 in Malignancies
The multiple roles of IL-33 in malignancies were summarized in Figure 2. Most of the current studies on IL-33's multiple roles in cancers focus on tumor microenvironment, tumorigenesis and tumor-associated inflammatory responses. In head-and-neck-squamous cancers, cancer associated fibroblasts was found releasing IL-33, sequentially leading migration and invasion through epithelial-to-mesenchymal transition (9). Data from tongue cancer patients witnesses a worse prognosis with higher level of IL-33 or ST2. Well-designed tissue comparison assays showed an elevated IL-33 and IL1RL1 or ST2 in tissue of both human breast cancer and non-small-cell-lung cancer (NSCLC), compared to adjacent non-tumor tissues. Similarly, high level of serum IL-33 also indicated a poor prognosis in patients with these two types of cancers (10–13). Related mechanism includes genomic instability and mutation, epigenetic modification, apoptosis resistance to cancer-initiated cells and increases of cancer metastasis.
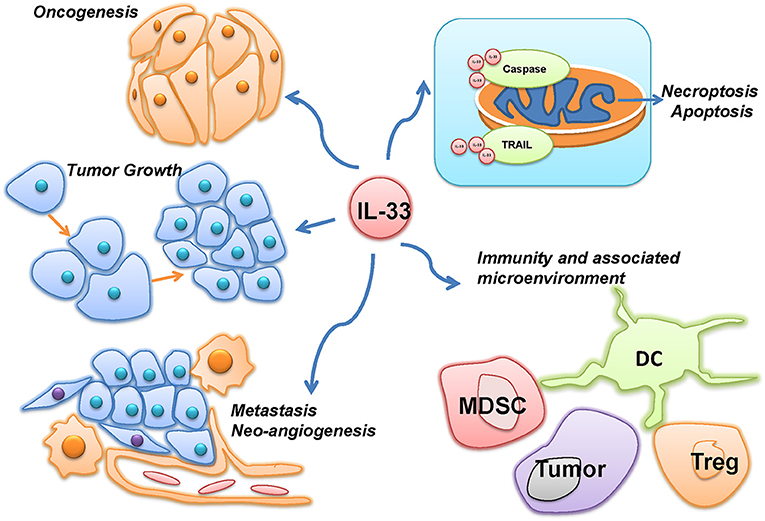
Figure 2. The multiple roles of IL-33 in malignancies. IL-33 was reported to be involved in the different hallmarks of cancer, such as oncogenesis, tumor growth, metastasis, neo-angiogenesis, and evading programmed cell death. However, as an immune-associated factor, IL-33 was also demonstrated to affect the immunity and associated microenvironment of cancer through immune cells, such as myeloid-derived suppressor cells (MDSC), Dendritic cells (DC), and Regulatory T cells (Treg).
Immunity and Associated Microenvironment
The process of tumor development can trigger anti-tumor immune responses. The type 1 immune response is a critical component of cell-mediated immunity, which includes tumor-induced IFN-γ-producing Th1 cells, cytotoxic T lymphocytes, NK T cells, and γδ T cells, to limit tumor growth and metastasis (14). Since inflammation is another important component in malignancies, it drove more studies on how IL-33 plays roles in improving cancerous surveillance and immunity against tumor. Gao et al. successfully gained the increased innate immunity of CD8(+) and NK cells by overexpressing IL-33 in tumor-bearing mice. The metastatic potential in models of B16 melanoma and Lewis lung carcinoma were significantly attenuated by transgenic expression of IL-33 (15). When depleting CD8(+) T cells and NK cells, pulmonary metastasis was significantly increased, indicating that IL-33 can mediate the anti-tumor immunity of CD8(+) T cells and NK cells.
However, other researches considered IL-33 promoted cancer progression through diminishing innate anti-tumor immunity and increasing intra-tumor accumulation of immunosuppressive cells in transgenic mice with breast cancer. Jovanovic et al. detected a time-dependent increase of endogenous IL-33 at both mRNA and protein levels in 4T1 breast cancer model and pulmonary metastasis during cancer progressions. In IL-33-treated mice, intra-tumor NKp46(+) NKG2D(+) and NKp46(+) FasL(+) cells were markedly reduced, while PBS-treated ST2-deficient mice had increased frequencies of these tumoricidal NK cells compared to that in untreated wild-type mice (16). Similarly, IL-33 increased IFN-γ by CD8(+) T and NK cells in tumor tissues, thereby inducing the microenvironment accessible to tumor eradication (17).
The tumor microenvironment, in which the tumor exists, constantly interacts with tumor cells. The developing tumor itself can promote anti-tumor immune responses by recruiting cytotoxic T lymphocytes, T helper 1 cells, and NK cells; so as to influence the microenvironment by releasing extracellular signals, promoting tumor angiogenesis and/or peripheral immune tolerance (14, 18). At the same time, the immune cells involved can affect the growth and evolution of tumor cells (19). Immune suppression specific to tumor and inflammatory stimuli may therefore display a cancer-initiated action, where cytokines are the central mediators (20, 21). IL-33 was proved to be a robust inducer of T helper 2 cells (releasing cytokines like IL-4/5/6/10/13) in tumor microenvironment, but a suppressor to the secretion of T helper 1 cells (cytokines like IL-12, IFN-γ) (22). Recent data revealed the role of IL-33 in several cancers, indicating dual functions as a damage-associated molecular pattern, or nuclear factors mediating gene expression (23).
Oncogenesis
Environmental factors (nicotiana or alcoholic use), and infections (hepatitis B virus), were indicated as triggering factors for minor changes in lungs, heart and livers (24). These were later proved to initiate a higher rate of neoplastic growth, followed by chronic inflammation to targeted organs and/or tissues. Therefore, the tumor-associated inflammatory pathways to cancer development are research hotspots.
Tumors often locally accumulated Treg cells, preventing tumor clearance. In intestinal tumors from APCMin/+ mice, researchers found that a high level of cytokine IL-33 were preferentially expanded in KLRG1+ GATA3+ Treg cells, sequentially to activate E-cadherin ablation and increase β-catenin signals in epithelial cells (25, 26). In another remarkable study of patients with metastatic colon cancer, higher expression of IL-33 in cancer tissues was significantly associated with poorer survival. Similarly, the study pointed out that IL-33 activated core stem cell genes via ST2 signaling pathway, recruited macrophages and stimulated stem-like characters to promote carcinogenesis of colon cancer cells (27, 28). Another new function of IL-33 discovered from non-basophilic leukemia, a rare subtype of acute myeloblastic leukemia, indicated IL-33 enhanced the basophilic differentiation of MYB-GATA1 expression, demonstrating a new role of IL-33 in leukemic cells and CD34-positive primary cells (29).
Tumor Growth
IL-33 is a cytokine implicated in mutual modulation of not only anti-tumor immunity but tumor growth. Inflammatory responses can be triggered by tumor growth through releasing danger signals and expression of tumor antigens (14). Nevertheless, tumors progress by enlisting and driving the dominance of immune suppressive cell types such as Treg and myeloid-derived suppressor cells, as well as myeloid cells that produce cancer-promoting factors (18, 30, 31). Moreover, IL-33 also promoted the growth and metastasis of solid cancers, such as gastric cancer, colorectal cancer, ovarian cancer, and breast cancer (32, 33). Wang et al. reported blocking IL-33 activities restricted tumor growth of NSCLC xenografts, indicating IL-33 blockade as a novel therapeutics for NSCLC patients (34).
Conversely, other studies pointed out anti-cancer activities of IL-33 to inhibit tumor progression in cellular levels and animal models. Qin et al. showed that recombinant IL-33 dramatically repressed the leukemia growth and prolonged the survival of leukemia-bearing mice by increasing IFN-γ production of leukemia-reactive CD8+ T cells (35). Tumor expression of IL-33 was also reported to inhibit tumor growth and favor tumor eradication by modifying the tumor microenvironment through CD8+ T cells (17). Put together, it is proposed that IL-33 might exert the anti-cancer activities of suppressing tumor growth under certain circumstances.
Metastasis and Neo-Angiogenesis
Cancer metastasis is one of the severe outcomes in most patients with malignancies. It is characterized by rapid and uncontrolled proliferation with high ATP demands, which requires a high rate of glucose uptake. In the surface of NSCLC cells, IL-33/ST2 pathway upregulated membrane glucose transporter 1 to enhance their glucose uptake and glycolysis to meet the ATP switch of metastasis (36).
Saranchova et al. described a new mechanism of immune-surveillance in cancer. They found metastatic carcinomas expressed low levels IL-33 and antigen processing machinery (APM), compared to syngeneic primary tumors. Supplementation of IL-33 in metastatic tumors restrained tumor growth rates and frequencies of circulating tumor cells by upregulating APM and functionality of major histocompatibility complex (MHC)-molecules, indicating the inhibition function of IL-33 to primary tumors in cancer immune-surveillance and losing that function during metastatic transition as immune escape (37). Evidences pointed out IL-33 as a possible inducer and prognostic marker of cancer metastasis by way of mechanisms like immune regulation, with intensive cytotoxic activities of NK cells and increased systemic Th1 and Th17 cytokines (38).
Other malignancies witness proof of IL-33 to metastatic process. The soluble form of the IL-33 receptor (sST2), resisting IL-33-induced angiogenesis, is downregulated in metastatic cells compared with low-metastatic colorectal cancer cells (39). Wang et al. determined that IL-33 overexpression enhances robust outgrowth and metastasis in vitro/in vivo, while genetic knockdown of IL-33 limited the progression of NSCLC (40). In lines with these findings, epithelial ovarian cancer knocked down of IL-33 gene had reduced metastatic potential, while ectopic IL-33 promoted the migratory and invasive capacity. Underlying mechanisms clarified sST2 might block the ERK and JNK signaling pathways, where IL-33 in return, was regarded as a prognosis markers and targets for ovarian cancers (41). In gastric cancer cells, IL-33 promoted cancer migration and invasion through stimulating the secretion of MMP-3 and IL-6 via aberrant activation of ST2-ERK1/2 pathway (42). Taken together, IL-33 may promote cancer development and metastasis through different pathways.
Cancer Cell Death
Different modes of cell death regulate inflammation by modulating professional phagocyte activation (43). The topic concerning IL-33 initiated cancer cell death is still emerging. It is likely to influence a range of diseases, but evidence for IL-33 limited to only a few examples.
Apoptosis is one of the classically understood processes of cell death that denotes a specific caspase-8–dependent programmed cellular death (44). Ye et al. released a recent study that IL-33 prevent cancer cells against platinum-induced apoptosis via the JNK pathway in gastric cancer cells (45). Similarly, in colon cancer cells, IL-33 stimulated cell sphere formation and prevented chemotherapy-induced tumor apoptosis (28). One exception was noticed in MIA PaCa-2, a pancreatic cancer cell line, that not only colonies and proliferations rate, but relative caspase-3 activities were attenuated in the presence of IL-33. Subsequent anti-proliferative effect on cancer cells even correlated with down-regulated anti-apoptotic molecule FLIP and up-regulated pro-apoptotic molecule TRAIL (46). Since unidentified mechanisms of IL-33 in basic research, we need further experiments to clarify the possibly opposite effects in different situations, like inflammatory stages, cell types or even the concentration of IL-33.
Based on signaling pathway studies, IL-33 may also be involved in co-activation of receptor-interacting protein 1/3 (RIP1/3) kinases, subsequently to damage, press and chronic inflammation (47, 48). This discovery shed light on another versatility of IL-33, an atypical cell death named necroptosis. Unlike apoptosis, this caspase-8-independence cell death allows the cell to bypass caspase activation (49, 50). In liver malignancies, inflammatory stimuli are predominantly driven to tissue-resident Kupffer cell from by bacterial and viral infections. Necroptosis of Kupffer cells released the alarmin IL-33 to trigger basophil IL-4, which successively recruited macrophages proliferation. This ultimately activated the macrophages and thereby achieved to liver homeostasis (51). While in cervical cancer, IL-33 related necroptosis could work as a possible pioneer for immunotherapy in cervical cancer. This HPV-induced cancer was found a rather low level of cytokine, indicating a stimulated releasing of IL-1α for an induced necroptotic cell death (52). Still, potential mechanisms on these studies need further clarified.
Therapeutic Strategies for IL-33 in Cancers
Understanding of direct and indirect effects of IL-33 would be important for profound therapeutic implications, especially in the realm of cancer immunotherapy. By using what scientists learn about the immune system, several synthetic molecules are created to attack a tumor more precisely and effectively (53). Current cancer immunotherapies include cytokines, monoclonal antibodies, and lymphocytes that will enhance existing antitumor immune responses (54). Cytokine-based immunotherapy has been extensively investigated in the treatment of malignancies. These immune-modulating effects allow interleukin-related treatment to provoke an immune response to chronic rheumatic diseases and attenuate disease progression of pathogenic conditions. However, only a few agents, such as interferon and IL-2, have proven to have sufficient clinical benefits to justify their more widespread use (55). Many preclinical studies demonstrated the antitumor effects of Th1 cytokines, to which IL-33 belongs, while clinical efficacy still limited.
Since IL-33 dually drives the immune system in either responsiveness/activation or tolerance/inhibition, IL-33 administration to the status of immune system dynamics controversially direct immune response and determine outcome (56). Any underlying natural bimodal homeostatic dynamic will be critical principal determinant of clinical efficacy. It is suspected that cancer therapy will be to analyze accurately the patient's underlying tumor immune response in a serial manner, then appropriately and accurately synchronize therapy with immune fluctuation (57). This time-dependent dynamic aspect of the immune response in the cancer patient has been largely overlooked in the past and has prevented us from being able to observe how our in vivo therapeutic approaches are influencing the immune response to produce the observed clinical effects.
IL-33 has been possibly considered as an immune adjuvant for vaccine therapy. Indeed, IL-33 functions as a promoter to memory T cell immunity in transgenic melanoma mice, thereby mediating a microenvironment that favors tumor rejection. Based on insights from Villarreal, this immunoadjuvant effects in an HPV-associated model is critical for protective immunity (58). Besides, increased IL-33 in pathological settings including tumor immunotherapy, viral infection and graft-vs.-host diseases, suggest that IL-33 overexpression might elicit potent antitumor immunity (59). Thus far, mouse models provide most of the information at hand. To what degree these observations are applicable to real patients is still unclear. Experimental evidences are currently lacking that clearly indicate the suitability of such an approach.
As a tumor biomarker, IL-33 could be versatile serving as a therapeutic target in patients. In breast cancers, estrogen receptor expressions account for most clinical cases. Thanks to hormone therapy with tamoxifen, prognoses of ER-positive breast cancer are significantly improved. While on the other hand, endocrine resistance to tamoxifen led to commonly-found compromised efficacy of hormone therapy where IL-33 played crucially. Knockdown of IL-33 is likely to correct this problem and resistance to tamoxifen-induced tumor growth inhibition could therefore be reversed (60). Similar cases derived from previous studies in lung and gastric cancer, in metastatic prostate carcinomas, and glioblastoma (61). These findings point out new connection betweenIL-33 and cancer pathogenesis and pinpoint IL-33 promising to optimize therapy in clinical practice.
Future Perspectives
The role of IL-33 in inflammatory diseases has been widely discussed since decades ago (62). This review highlights the remarkable span and diversity of its modulatory potency in tumors. Interestingly, our perspectives in IL-33 has now extended beyond its previous identification as an inducer of immune responses to that of a potency in chronic inflammation and timely activation by malignancies. However, many questions remain unraveled, such as the regulatory elements, the bioactive forms, and the inner homeostasis of IL-33. A study of network map insights on IL-33-mediated crosstalk in the pathogenesis of acute and chronic inflammatory diseases (63). It revealed various roles of classical signaling modules like ERK1/2, NK-κB, and PI3K/AKT, etc. that played in disease development, which in no doubt left us with new inspirations to tumor-related development. In therapeutic practices, IL-33-targeted antibody has only been put into clinical trials for asthma. Other approaches were newly recommended, but cautions should be exercised due to the many immune responses and the potential for driving cancer development.
Author Contributions
JL and G-JZ contributed conception and design of the study. JL and J-XS organized the database, searched literatures, structured, and drafted the manuscript carefully. JL and G-JZ revised the original manuscript critically. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 81501539 and 81320108015) and the Natural Science Foundation of Guangdong Province (Nos. 2015A030310211 and 2016A030312008). G-JZ is a recipient of the Chang Jiang Scholar's award granted by the Ministry of Education of China. Special appreciation would be given to Miss Yang Wu for figure editing.
References
1. Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood (2011) 117:3720–32. doi: 10.1182/blood-2010-07-273417
2. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
3. Baekkevold ES, Roussigne M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
4. Scott IC, Majithiya JB, Sanden C, Thornton P, Sanders PN, Moore T, et al. Interleukin-33 is activated by allergen- and necrosis-associated proteolytic activities to regulate its alarmin activity during epithelial damage. Sci Rep. (2018) 8:3363. doi: 10.1038/s41598-018-21589-2
5. Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity (2013) 39:1003–18. doi: 10.1016/j.immuni.2013.11.010
6. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Ann Rev Immunol. (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
7. Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
8. Bessa J, Meyer CA, de Vera Mudry MC, Schlicht S, Smith SH, Iglesias A, et al. Altered subcellular localization of IL-33 leads to non-resolving lethal inflammation. J Autoimmun. (2014) 55:33–41. doi: 10.1016/j.jaut.2014.02.012
9. Chen SF, Nieh S, Jao SW, Wu MZ, Liu CL, Chang YC, et al. The paracrine effect of cancer-associated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J Pathol. (2013) 231:180–9. doi: 10.1002/path.4226
10. Ishikawa K, Yagi-Nakanishi S, Nakanishi Y, Kondo S, Tsuji A, Endo K, et al. Expression of interleukin-33 is correlated with poor prognosis of patients with squamous cell carcinoma of the tongue. Auris Nasus Larynx (2014) 41:552–7. doi: 10.1016/j.anl.2014.08.007
11. Liu J, Shen JX, Hu JL, Huang WH, Zhang GJ. Significance of interleukin-33 and its related cytokines in patients with breast cancers. Front Immunol. (2014) 5:141. doi: 10.3389/fimmu.2014.00141
12. Kim JY, Lim SC, Kim G, Yun HJ, Ahn SG, Choi HS. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene (2015) 34:4928–38. doi: 10.1038/onc.2014.418
13. Yang ZP, Ling DY, Xie YH, Wu WX, Li JR, Jiang J, et al. The association of serum IL-33 and sST2 with breast cancer. Dis Markers (2015) 2015:516895. doi: 10.1155/2015/516895
14. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
15. Gao K, Li X, Zhang L, Bai L, Dong W, Gao K, et al. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. (2013) 335:463–71. doi: 10.1016/j.canlet.2013.03.002
16. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer J. (2014) 134:1669–82. doi: 10.1002/ijc.28481
17. Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. J Immunol. (2015) 194:438–45. doi: 10.4049/jimmunol.1401344
18. Chen LJ, Zheng X, Shen YP, Zhu YB, Li Q, Chen J, et al. Higher numbers of T-bet(+) intratumoral lymphoid cells correlate with better survival in gastric cancer. Cancer Immunol Immunother. (2013) 62:553–61.
19. Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine (2017) 89:127–35. doi: 10.1016/j.cyto.2016.01.021
20. Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol. (2013) 33(Suppl 1):S79–84. doi: 10.1007/s10875-012-9847-0
21. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
22. Cui G, Florholmen J. Polarization of cytokine profile from Th1 into Th2 along colorectal adenoma-carcinoma sequence: implications for the biotherapeutic target? Inflamm Allergy Drug Targets (2008) 7:94–7. doi: 10.2174/187152808785107589
23. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. doi: 10.1016/j.coi.2014.09.004
24. Mathurin P, Lucey MR. Alcohol, liver disease, and transplantation: shifting attitudes and new understanding leads to changes in practice. Curr Opin Organ Transplant. (2018) 23:175–9. doi: 10.1097/MOT.0000000000000517
25. Meinicke H, Bremser A, Brack M, Akeus P, Pearson C, Bullers S, et al. Tumour-associated changes in intestinal epithelial cells cause local accumulation of KLRG1+ GATA3+ regulatory T cells in mice. Immunology (2017) 152:74–88. doi: 10.1111/imm.12750
26. He Z, Chen L, Souto FO, Canasto-Chibuque C, Bongers G, Deshpande M, et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc (Min/+) mice. Sci Rep. (2017) 7:5520. doi: 10.1038/s41598-017-05716-z
27. O'Donnell C, Mahmoud A, Keane J, Murphy C, White D, Carey S, et al. An antitumorigenic role for the IL-33 receptor, ST2L, in colon cancer. Br J Cancer (2016) 114:37–43. doi: 10.1038/bjc.2015.433
28. Fang M, Li Y, Huang K, Qi S, Zhang J, Zgodzinski W, et al. IL33 promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer Res. (2017) 77:2735–45. doi: 10.1158/0008-5472.CAN-16-1602
29. Ducassou S, Prouzet-Mauleon V, Deau MC, Brunet de la Grange P, Cardinaud B, Soueidan H, et al. MYB-GATA1 fusion promotes basophilic leukaemia: involvement of interleukin-33 and nerve growth factor receptors. J Pathol. (2017) 242:347–57. doi: 10.1002/path.4908
30. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
31. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
32. Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, et al. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinogenesis (2017) 56:272–87. doi: 10.1002/mc.22491
33. Schwartz C, O'Grady K, Lavelle EC, Fallon PG. Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol. (2016) 46:1091–100. doi: 10.1002/eji.201545780
34. Wang K, Shan S, Yang Z, Gu X, Wang Y, Wang C, et al. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget (2017) 8:8571–82. doi: 10.18632/oncotarget.19786
35. Qin L, Dominguez D, Chen S, Fan J, Long A, Zhang M, et al. Exogenous IL-33 overcomes T cell tolerance in murine acute myeloid leukemia. Oncotarget (2016) 7:61069–80. doi: 10.18632/oncotarget.11179
36. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi: 10.1038/nrc2981
37. Saranchova I, Han J, Huang H, Fenninger F, Choi KB, Munro L, et al. Discovery of a metastatic immune escape mechanism initiated by the loss of expression of the tumour biomarker interleukin-33. Sci Rep. (2016) 6:30555. doi: 10.1038/srep30555
38. Jovanovic I, Radosavljevic G, Mitrovic M, Juranic VL, McKenzie AN, Arsenijevic N, et al. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur J Immunol. (2011) 41:1902–12. doi: 10.1002/eji.201141417
39. Akimoto M, Maruyama R, Takamaru H, Ochiya T, Takenaga K. Soluble IL-33 receptor sST2 inhibits colorectal cancer malignant growth by modifying the tumour microenvironment. Nat Commun. (2016) 7:13589. doi: 10.1038/ncomms13589
40. Wang C, Chen Z, Bu X, Han Y, Shan S, Ren T, et al. IL-33 signaling fuels outgrowth and metastasis of human lung cancer. Biochem Biophys Res Commun. (2016) 479:461–8. doi: 10.1016/j.bbrc.2016.09.081
41. Tong X, Barbour M, Hou K, Gao C, Cao S, Zheng J, et al. Interleukin-33 predicts poor prognosis and promotes ovarian cancer cell growth and metastasis through regulating ERK and JNK signaling pathways. Mol Oncol. (2016) 10:113–25. doi: 10.1016/j.molonc.2015.06.004
42. Yu XX, Hu Z, Shen X, Dong LY, Zhou WZ, Hu WH. IL-33 Promotes gastric cancer cell invasion and migration via ST2-ERK1/2 pathway. Digest Dis Sci. (2015) 60:1265–72. doi: 10.1007/s10620-014-3463-1
43. Mora J, Schlemmer A, Wittig I, Richter F, Putyrski M, Frank AC, et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J Mol Cell Biol. (2016) 8:426–38. doi: 10.1093/jmcb/mjw006
44. Seifert L, Miller G. Molecular pathways: the necrosome-a target for cancer therapy. Clin Cancer Res. (2017) 23:1132–6.
45. Ye XL, Zhao YR, Weng GB, Chen YC, Wei XN, Shao JP, et al. IL-33-induced JNK pathway activation confers gastric cancer chemotherapy resistance. Oncol Rep. (2015) 33:2746–52. doi: 10.3892/or.2015.3898
46. Fang Y, Zhao L, Xiao H, Cook KM, Bai Q, Herrick EJ, et al. IL-33 acts as a foe to MIA PaCa-2 pancreatic cancer. Medical Oncol. (2017) 34:23. doi: 10.1007/s12032-016-0880-3
47. Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature (2011) 471:368–72. doi: 10.1038/nature09857
48. Trichonas G, Murakami Y, Thanos A, Morizane Y, Kayama M, Debouck CM, et al. Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA. (2010) 107:21695–700. doi: 10.1073/pnas.1009179107
49. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. (2014) 15:135–47. doi: 10.1038/nrm3737
50. Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol. (2011) 12:79–88. doi: 10.1038/nri3131
51. Bleriot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity (2015) 42:145–58. doi: 10.1016/j.immuni.2014.12.020
52. Schmidt SV, Seibert S, Walch-Ruckheim B, Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, et al. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1alpha release, and efficient paracrine dendritic cell activation. Oncotarget (2015) 6:8635–47. doi: 10.18632/oncotarget.3249
53. Kojima R, Scheller L, Fussenegger M. Nonimmune cells equipped with T-cell-receptor-like signaling for cancer cell ablation. Nat Chem Biol. (2018) 14:42–9. doi: 10.1038/nchembio.2498
54. Qu X, Tang Y, Hua S. Immunological approaches towards cancer and inflammation: a cross talk. Front Immunol. (2018) 9:563. doi: 10.3389/fimmu.2018.00563
55. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood (2010) 115:3508–19. doi: 10.1182/blood-2009-09-241398
56. Eissmann MF, Dijkstra C, Wouters MA, Baloyan D, Mouradov D, Nguyen PM, et al. Interleukin 33 signaling restrains sporadic colon cancer in an interferon-gamma-dependent manner. Cancer Immunol Res. (2018) 6:409–21. doi: 10.1158/2326-6066.CIR-17-0218
57. Poh AR, Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. (2018) 8:49. doi: 10.3389/fonc.2018.00049
58. Villarreal DO, Wise MC, Walters JN, Reuschel EL, Choi MJ, Obeng-Adjei N, et al. Alarmin IL-33 acts as an immunoadjuvant to enhance antigen-specific tumor immunity. Cancer Res. (2014) 74:1789–800. doi: 10.1158/0008-5472.CAN-13-2729
59. Lu B, Yang M, Wang Q. Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J Mol Med. (2016) 94:535–43. doi: 10.1007/s00109-016-1397-0
60. Hu H, Sun J, Wang C, Bu X, Liu X, Mao Y, et al. IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem Biophys Res Commun. (2017) 485:643–50. doi: 10.1016/j.bbrc.2017.02.080
61. Wasmer MH, Krebs P. The role of IL-33-dependent inflammation in the tumor microenvironment. Front Immunol. (2016) 7:682. doi: 10.3389/fimmu.2016.00682
62. Foo YL, Jean-PG Heth RT. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
Keywords: IL-33, cytokine, cancer, immunology, therapy
Citation: Shen J-X, Liu J and Zhang G-J (2018) Interleukin-33 in Malignancies: Friends or Foes? Front. Immunol. 9:3051. doi: 10.3389/fimmu.2018.03051
Received: 01 June 2018; Accepted: 10 December 2018;
Published: 20 December 2018.
Edited by:
Fang-Ping Huang, The University of Hong Kong, Hong KongReviewed by:
Remo Castro Russo, Universidade Federal de Minas Gerais, BrazilAngela Bonura, Italian National Research Council, Italy
Copyright © 2018 Shen, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Liu, amxpdTEyQHN0dS5lZHUuY24=
Guo-Jun Zhang, Z2p6aGFuZ0B4YWgueG11LmVkdS5jbg==