 Lan Shao
Lan Shao- The Center for Translational Medicine, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
Rheumatoid arthritis (RA) is an autoimmune-mediated disease that is associated with significant cartilage damage and immunosenescence. Despite decades of research, the major signal pathways that initiate RA are still unclear. The DNA damage response (DDR) is a specific and hierarchical network that includes cell cycle checkpoints, DNA repair, and DNA-damage tolerance pathways. Recent studies suggest that this condition is associated with deficits in telomere maintenance and overall genomic instability in the T cells of RA patients. Analysis of the underlying mechanisms has revealed defects in DDR pathways. Particularly, the DNA repair enzyme, ataxia telangiectasia mutated (ATM), is downregulated, which leaves the damaged DNA breaks in RA-associated T cells unrepaired and pushes them to apoptosis, exhausts the T cell pool, and promotes the arthritogenesis effector function of T cells. This review discusses recent advancements and illustrates that risk factors for RA, such as viral infections, environmental events, and genetic risk loci are combat with DDR signals, and the impaired DDR response of RA-associated T cells, in turn, triggers disease-related phenotypes. Therefore, DDR is the dominant signal that converts genetic and environmental stress to RA-related immune dysfunction. Understanding the orchestration of RA pathogenesis by DDR signals would further our current knowledge of RA and provide novel avenues in RA therapy.
Introduction
The human genome is subjected to constant endogenous and environmental assaults. To maintain genomic integrity, cells have evolved a highly complex signal transduction network to recognize, signal, and repair DNA lesions. A chief component of DNA damage is PI3-kinase related protein kinases (PIKKs), including ataxia telangiectasia mutated (ATM), DNA-PKcs, and ATR(1). These enzymes orchestrate hierarchical pathways that protect genomic integrity, which is termed the DNA damage response (DDR). RPA-coated single-stranded DNA (ssDNA) activates ATR and double-stranded breaks (DSBs), which are particularly hazardous to the cells repaired by ATM and DNA-PK.When DSBs occur, the breaks are recognized by the MRN complex (MRE11A-Rad50-NBS1) and then ATM is recruited to the damage site (1, 2). Activated ATM orchestrates the DNA repair process by phosphorylating multiple proteins, such as H2AX and 53BP1, to form characteristic DNA foci (3). ATM also phosphorylates the substrates CHK2 and p53 to initiate cell-cycle arrest (4). This method of DNA repair is called the homologous recombination (HR) repair pathway. Alternatively, DSBs activate non-homologous end joining (NHEJ) repair pathway which mediated by DNA-PKcs (5). Upon detecting DNA damage, the cell follows two routes: activation of DDR-dominant cell cycle checkpoints and cell cycle arrest to facilitate repair, or if damaged beyond repair, induction of apoptosis.
Persistent DNA damage signals are considered a major contributor to cellular senescence and organismal aging (6). Immune aging is a central component of advancing age and leads to the loss of immune system-mediated protection against infections, and is a prominent risk factor for chronic inflammation and the pathogenesis of autoimmune diseases (7–9). Rheumatoid arthritis (RA) is an autoimmune disorder characterized by accelerated immune senescence (10–15). T cells from RA patients appear prematurely-aged and exhibit accumulation of CD28− effector T cells, shortened telomeres, DNA damage accumulation, as well as an excess production of proinflammatory cytokines, all of which are compatible with normal cellular senescence. Interestingly, however, T cells in RA patients lack an irreversible cell cycle arrest and are deficient in the characteristic DNA repair foci; these are considered cardinal features of cellular senescence. In contrast, RA-associated T cells have a suppressed DDR pathway and a disrupted G2/M cell cycle checkpoint (10). Consequently, cells are hyperproliferative, more vulnerable to apoptosis, and clonal expansion is decreased in naïve RA T cells (14). All those studies indicate that the DDR pathway is likely associated with RA pathogenesis-related T cell dysfunction.
In this review, we discuss the complex association between the DDR pathway and T cell pathogenesis in RA. Particularly, we focus on the mechanisms underlying the effects of RA risk factors on DDR pathway and the role of modified DDR signals in RA-mediated T cell dysfunction. Determination of the molecular role of the DDR pathway in RA-associated T cell evolution may facilitate our understanding and management of this disease.
RA Risk Factors Lead to Defects in DDR Signaling
T cells have a long life in circulation and are exposed to various environmental, pathogenic, and genetic stresses, which decrease their survival time. Consequently, cells require a common pathway to collectively combat endogenous and extracellular stresses. This usually occurs before the onset of RA, thus indicating a preclinical phase of the disease. Multiple high-risk factors for RA are also triggers for DDR signals (16, 17). This section discusses how cellular DDR pathways serve as the major sensors in T cells to combat endogenous and environmental stresses, and how they may be modified from their original course.
Genetic Factors
Multiple genetic foci have been identified as risk factors of RA. HLA-DRB10401 and HLA-DRB 0404 are the alleles most strongly associated with RA (18–20). Analyses of telomeric lengths in CD4+ T cells from RA patients showed that HLA-DR-B1*04 is sufficient to accelerate telomere shortening (21), suggesting that HLA-DR-B1*04 affects signals regulating telomere maintenance. Additionally, genome-wide association studies (GWASs) have identified more than 100 common single nucleotide polymorphisms (SNPs) for RA risk, including ATM, PTPN22, CTLA-4, TRAFs, PADI4, and STAT4 (22–26).
How are these genes involved in the regulation of DDR signals? ATM is the important element for DDR signals; ATM polymorphism contribute to RA development by affecting the efficiency of DDR repair. Moreover, ATM function is directly regulated by PTPN22 (27). The interaction between *T1858 allele of PTPN22 and polymorphism of *Pro allele of the p53 codon 72 strongly increase the autoimmune inflammatory (28). The key downstream target of CTLA-4 is Akt, which is also an upstream signal for ATM (29). Members of the TRAFs family are involved in DNA damage-induced NF-κB activation. After DNA damage, ATM is translocated to the cytosol and interacts with TRAF6 to form ATM-TRAF6-cIAP1 complex, which catalyzes the monoubiquitination of NEMO to activate genotoxic NF-κB activation (30). The PADI4, a citrullination enzyme, is critical for anti-citrullinated peptide antibodies (ACPA) production in RA. PADI4 has been reported to citrullinatic modification of multiply proteins in a p53/PADI4-dependent manner (31, 32). STAT4 is a strong responder to DDR signals. The STAT4 SNPs exert synergistic effects with DDR signals to mediate citrullination production in the T cells of RA patients (33). Further analysis suggests that more genetic risk factors for RA could be included in the network of DDR signals, functioning either upstream of DDR signals or playing important roles in DDR signaling by themselves.
Viral Infections
Viral infections, including the human T-cell leukemia virus type 1 (HTLV), hepatitis C virus (HCV), and cytomegalovirus (CMV) (34–36) are associated with RA development. It has long been known that viral infection pathways represent potent antiviral defense mechanisms that may be disabled upon viral penetration in the host cells. However, viruses also can harness DDR activation by taking control of specific host proteins in the DDR pathway to aid viral replication. Direct evidence regarding how the virus-modified DDR pathway in RA-associated T cells has yet not been obtained; however, T cells derived from RA patients mimic the biological effects of HCV infection in T cells, including cell susceptibility to apoptosis, attenuating the activation of ATM and MRE11A (37). HTLV-1 is a retrovirus associated with RA pathogenesis (38, 39). Upon entering T cells, HTLV-1 expresses Tax and the protein concentration of Tax is several fold higher in the blood of RA patients than in healthy donors (40). Tax is essential for viral replication through deregulation of DDR pathways. The dampened ATM kinase and reduced association of MDC1 with the repair foci have also been reported in Tax-positive cells, which may serve as the mechanism for insufficiency of ATM activity and DNA foci formation in RA-associated T cells (41). Moreover, Tax upregulates c-FLIP and inhibits the apoptosis caused by the CD95 death receptor, a phenomenon also observed in RA-associated T cells (42, 43). Recently, a study reported that the mitochondrial DNA damage activates cytosolic antiviral signaling by promoting interferon production after a herpes virus infection (44, 45). In RA patients, T cells chronically infected with CMV also express large amounts of IFN-γ (46, 47), suggesting that mitochondrial DNA damage signaling may exert synergistic effects with canonical DNA damage stress signaling to trigger antiviral immune response in RA-associated T cells.
Environmental Events
Constant environmental assaults are inflicted on human T cells inducing DNA damage. Several environmental factors affect either RA susceptibility, or the pathophysiology of RA, and several such environmental factors have been identified. Increased caffeine intake, a history of heavy smoking, and exposure to air pollution are all associated with increased rheumatoid factors. Caffeine reportedly uncouples the cell-cycle progression from DNA repair via the mechanism of directly repressing the G2/M checkpoint by inhibiting ATM kinase (48, 49). However, reports regarding the effects of caffeine are controversial. A cohort study comparing the effects of caffeinated, decaffeinated, and total coffee intake in women did not support the distinct association between caffeinated coffee or caffeine intake with RA pathogenesis (50).
Complex environmental carcinogens, such as tobacco smoke also serve as genotoxic agents that trigger H2AX phosphorylation and DDR signals (51). A mechanistic study reported that citrullinated antigens, the self-antigen for ACPA autoantibody of RA patients, are formed in individuals with a history of smoking via activation of peptidylarginine deiminase at the sites of inflammation (52). A subsequent study reported that nicotine in tobacco is a strong inducer of p53 gene mutations (52). Nicotine also promotes neutrophil extracellular traps (NETosis) production in the neutrophil of RA patients, which play an important role in RA-associated inflammation. The formation of NETosis need a systematic process for DNA which regulated by DDR signals (53, 54). Those studies suggest that DDR benefit the nicotine-regulated NETosis development and the DDR signal way in T cell and neutrophil of RA patients may be different.
Impaired DDR Signals Induce the Disease-Related Phenotypes in RA-Associated T Cells
Naïve T cells adapt to stress conditions via homeostatic replication to compensate for the loss of T cells, concurrent with a reduction in the diversity of the T cell repertoire (16–19). In RA disease, T cells adapt to the stress accompanied by modification of the DDR pathway, characterized by telomere shortening, accumulation of DNA damage and the expansion of CD4+CD28− end-differentiated T cells. These factors contribute to chronic, histolytic T cell genesis, which is critical for RA progression. This section discusses mechanisms underlying the induction of disease-related changes in T cell function in RA, owing to deficits in the DDR signal pathway.
The DDR Pathway and T Cell Aging
The T cells of RA patients exhibit premature aging and sensitivity to apoptosis, which are characteristics of lymphocytes in individuals with genetic progeroid syndromes. RA patients also recapitulate the features of genetic progeroid syndromes with defects in chromatin maintenance and cardiovascular diseases. The primary contributors to the pathogenesis of genetic progeroid syndromes are hypomorphic mutations in key DNA repair molecule, suggesting that T cells age in RA may be owing to impaired DNA repair. Further studies have confirmed that T cells from RA patients have the ATM-deficient naïve T cells, which fail to sense and repair DNA lesions induced by genotoxic stress (11). Comet assays revealed elongated T cell tails in RA patients compared with healthy controls. Reconstitution of ATM rescues the ability of DNA repair and clears DNA damage in RA-associated T cells (55). Mechanism Studies on DDR signals in RA patients reported that ATM transcripts regulate MRE11A, NBS1, RAD50, and p53 expression. ATM deficiency-mediated impairment of MRN complex in RA T cells fails to detect damaged DNA and subsequently attenuate the recruitment of ATM to damaged DNA foci, thereby leaving DBS unrepaired (11–13). Persistent DBS alternatively upregulates DNA-PKcs, in the NHEJ repair pathway. However, the first step in the NHEJ pathway is the detection of fragmented DNA by Ku70/Ku80, following recruit DNA-PKcs to DNA repair foci to form functional DNA-PK. In T cells of RA patients, DNA-PKcs are upregulated, accompanied with insufficient expression of ku70/ku80. Consequently, instead of repairing damaged DNA by forming DNA-PK, DNA-PKcs participate in the JNK-Bim pathway and induce spontaneous apoptosis in naïve T cells of RA patients (Figure 1) (12).
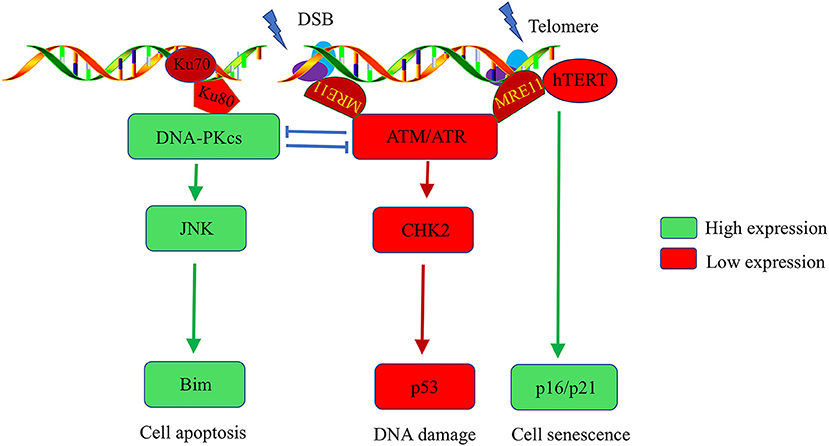
Figure 1. Abnormalities in the DNA damage repair pathway in T cells of rheumatoid arthritis patients. DNA stability is critical for the function of T cells. Naïve T cells from RA patients have ATM deficiency, which impairs the cells' ability to detect and repair DNA lesions. T cells alternatively upregulate DNA-PKcs. The activated DNA-PKcs-JNK-Bim axis eventually forces naïve T cells into apoptosis. Moreover, there is insufficient activation of the ATM kinase, hTERT, and MRE11A in RA-associated T cells after T cell receptor stimulation, which accelerates the G2/M checkpoint bypass and hyperproliferation. Unrepaired DNA breaks and telomere erosion finally induce T cell death and senescence. To compensate for the T cell pool, naive cells are forced into homeostatic clonal expansion, which facilitates the section of autoreactive T cell.
Moreover, ROS production is inadequate in T cells of RA patients after T cell receptor (TCR) stimulation, thereby blocking the ATM monomer and deterring its dimerization, which is a critical step for ATM kinase activation. ATM activity deficiency accelerates the G2/M checkpoint bypass and promotes apoptosis in RA-associated T cells after activation. Excessive loss of T cells in RA patients make a replicative stress to naive cells. To maintain the size of T cell pool, naive cells are forced to undergo homeostatic clonal expansion, thereby accelerating aging of the T cell repertoire and promoting oligoclonal expansion. Consequently, cell repertoire diversity is contracted which favor the autoreactive T cell selection and RA development (12).
The DDR Pathway and Telomere Intactness of T Cells
Telomeres are the most fragile sites to DNA damage stress in chromosomes. Telomere damage has several-fold more severe effects on the cell than does damage at other chromosomal sites (56). Telomere stability and the intactness of telomeres are critical for maintaining cell longevity and proliferation. Naive T cells from RA patients have shortened telomeres (21). Several potential mechanisms underlie non-age-dependent acceleration of telomeric erosion found in RA T cells. First, telomeric erosion may reflect an increased proliferative frequency of naive T cells in RA. Second, telomeric lengthening and maintenance are facilitated by telomerase, which is composed of a catalytic protein unit known as human telomerase reverse transcriptase (hTERT), and an RNA template complementary to the telomeric DNA (hTR) (57). T cells from RA patients have reduced hTERT expression and fail to upregulate telomerase activity after TCR stimulation. Insufficient telomerase also accelerates telomere shortening in T cells of RA patients (14). Moreover, telomeric shortening could be a consequence of deficits in DNA repair. In the G2 cell cycle phase, telomere overhang is recognized by ATM-regulate DDR signals that promote telomere shelterin-complex reconstitution, which is critical for telomere protection. ATM defects leave telomeres unprotected, subsequently trigger chromosome fusions and instability (58–60). Li et al. recently reported that reduced activity of MRE11A, a component in MRN complex, induces a cellular senescence module by promoting telomere uncapping and upregulation of cell senescence markers, such as p16, p21, and the cell surface receptor CD57 in RA-associated T cells (Figure 1). Moreover, MRE11A deficiency is directly implicated in the proinflammatory properties and tissue invasive in RA-associated T cells (13).
The DDR Pathway and T Cell Metabolism
Regulation of nutrient uptake and utilization in T cells is critical for signaling, differentiation, function, and the fate of T cells (61, 62). Metabolically, naive T cells from RA patients cannot supply enough energy to adapt to the demands of TCR stimulation. Insufficient induction of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), a rate-limiting glycolytic enzyme, leads to a deficient glycolysis flux in RA-associated T cells. Interestingly, PFKFB3 is also involved in DNA HR repair. Upon detecting the DSBs, PFKFB3 rapidly recruited to DNA repair foci via the MRN-ATM-γH2AX-MDC1 complex and participates in the DNA repair process (63), which indicating a strong connection between DDR signals and PFKFB3 regulated cellular metabolism.
In RA-associated T cells, reduced PFKFB3 decreased lactate/ATP levels and increased NADPH/biosynthetic precursor levels compared to T cells from healthy individuals (10, 64–66). The equivalentlowNADPHhigh condition in RA patients fails to appropriately balance mitochondrial ROS production and the cellular anti-oxidant machinery. Thus, ROS production is reduced in RA-associated T cells (10). Insufficient ROS prevents ATM activation and disrupts the G2/M checkpoint after T cells stimulation. Moreover, ATM is critical for mitochondrial biogenesis, ATM deficiency may be associated with aberrant mitochondria in T cells of RA patients (67, 68).
Concerning lipid metabolism, RA-associated T cells have increased fatty acid biosynthesis and decreased fatty acid oxidation. Usually, AMPK-mediated p53 activation negatively regulates lipid synthesis by suppressing mTOR (69). AMPK-p53 function is impaired in T cells of RA patients, which potentially explaining the enhanced mTOR activity, decreased fatty acid oxidation, and increased fatty acid synthesis observed in RA T cells (64, 70). How is metabolic dysfunction in those cells linked to RA pathogenesis? A recent study reported that an ATPlowpyruvatelow environment in RA patients induces adequate expression of TKS5, a podosome scaffolding protein, which triggers fatty acid biosynthesis and promotes T cell locomotion capabilities. These factors, together, promote the formation of tissue-invasive membrane structures in RA-associated T cells and facilitates the infiltration of inflammatory T cells in RA patients into non-lymphoid tissue sites (71).
Many key regulatory molecules in DDR pathways associated with RA development also play a role in cellular metabolism (72–74). Sirtuins are a class of NAD+-dependent deacetylases, which are critical for maintaining genomic homeostasis. Sirtuins control energy metabolism by regulating PGC-1α and forkhead transcription factors (FOXO) to supply the metabolites required for DNA repair (75, 76). The DNA damage repair enzymes, PARPs and SENPs, play important roles in metabolic regulation owing to their influence on mitochondrial function and oxidative metabolism (77, 78). Future studies are expected to elucidate the intrinsic associations between DDR signals and metabolic homeostasis in the T cells of RA patients.
The DDR Pathway and T Cell Differentiation
CD4+ T cells differentiate into specific subtypes with unique immune functions and secretion of specific cytokines; classical T cell subsets include Th1, Th2, Th17, and Treg (79). DDR signaling abnormalities in RA may accelerate synovial inflammation by affecting T cell differentiation. Hyperproliferative T cells in RA have biased differentiation patterns toward the Th1 and Th17 fate (10, 13). Therefore, Th1-associated cytokines IFN-γ and Th17-associated cytokines IL-17 are upregulated in the serum and synovial fluid of RA patients and serve as the primary inducer of the inflammatory response (80, 81). Both ATM and p53 reportedly regulate differentiation of T cells in RA (10, 82). The insufficient ATM-p53 function in T cells from RA patients shifts the differentiation of naïve CD4+ T cells toward the Th1 and Th17 effector lineages rather than Treg cells, thereby generating an arthrogenic effect of T cells and imposing a hyperinflammatory phenotype in the synovial tissue (10, 82). Moreover, ATM (-/-) mice have abnormal CD8, T cell immune responses with defective expansion, contraction, and differentiation bias to the effector-to-memory pathway. Mechanistic studies reported that CD8+ T cells with ATM deletion have hyperactive mTORC1 signaling, which results in inflammatory cytokine production and memory T cell development and acceleration of the autoimmune genesis in RA patients (83). Recently, a new subset of T peripheral helper (Tph) cell populations in RA patients with PD-1hiCXCR5− Bcl6low was identified, which infiltrating inflamed synovia by expressing the chemokine receptor, CCR2, and augmenting synovial B cell responses (84). Interestingly, p53 could repress the CXCR5 chemokine receptor gene via attenuation of NF-κB activity (85), and reduction of BCL6 enhance the DDR pathway. Those studies indicate that DDR pathway exert a strong influence in Tph formation and further studies are expected to examine the DDR signals in Tph subset of RA patients.
Perspective
Significant advancements have facilitated the understanding of the DDR pathway and RA; however, much remains to be learned. This review discussed studies on the molecular mechanisms underlying the effects of environmental, viral, and genetic risk factors of RA on DDR signaling and summarized how the key players in DDR signaling induce RA-related phenotypes such as apoptosis, aging, metabolic disorders, and an imbalance in the differentiation of T cells. Based on these studies, the present review suggests that DDR signaling plays an important role in the autoimmune pathogenesis of RA (Figure 2). To further clarify the role of DDR signaling in the development of RA-related autoimmunity, prospective studies on the preclinical and early phases of RA development are warranted. Those studies will provide a novel avenue to further the understanding of RA.
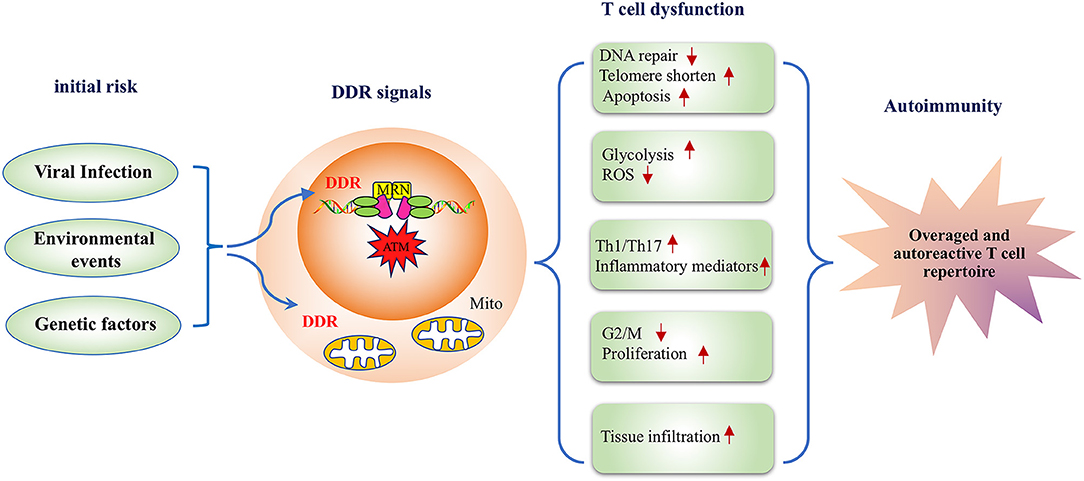
Figure 2. A model depicting the DNA damage response pathway inducing disease-related phenotypes in T cells in rheumatoid arthritis. RA risk factors, including viral infection, environmental events, and genetic factors lead to defective DDR signals in T cells, and the inadequate DDR responsiveness subsequently alters T cell functionality in multiple aspects. First, deficiency in DNA repair influences T cell survival resulting in a lower apoptotic threshold. Second, DDR is implicated in the metabolic regulation of RA-associated T cells. Moreover, an impaired DDR response biases T cell differentiation to inflammatory effector T cells. Therefore, DDR deficiency in RA-associated T cells represents a key mechanism for the pathogenesis of RA with transitional genetic and environmental factors to RA pathogenesis-related T cell dysfunction.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by the Guangdong Natural Science Foundation (grant no. 2017A030313859), Science and Technology Program of Guangzhou (grant no. 201707010120), and the Fundamental Research Funds for the Central Universities (grant no. 17ykjc 08) and 100 top talents program, Sun Yat-sen University.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Cornelia M. Weyand, and Dr. Yinyin Lin for discussion and manuscript revision.
Abbreviations
DDR, DNA damage response; RA, Rheumatoid arthritis; MRN, MRE11A-Rad50-NBS1; ATM, ataxia telangiectasia mutated; PIKK, phosphoinositide-3 kinase-like kinase; DSBs, DNA double strand breaks; ssDNA, single-stranded DNA; HR, Homologous recombination pathway; NHEJ, non-homologous end joining; GWASs, genome-wide association studies; SNPs, single nucleotide polymorphisms; ACPA, anticitrullinated protein antibodies; HTLV-1, Human T-cell leukemia virus type 1; HCV, hepatitis C virus; CMV, Cytomegalovirus; NETs, neutrophil extracellular traps; hTERT, telomerase reverse transcriptase; hTR, RNA template complementary to the telomeric DNA; TCR, T cell receptor; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; PGC, peroxisome proliferator-activated receptor-γ coactivator; PPP, pentose phosphate pathway; FOXO, forkhead transcription factors; Tregs, regulatory T cells.
References
1. Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature (2005) 434:605–11. doi: 10.1038/nature03442
2. Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science (2005) 308:551–4. doi: 10.1126/science.1108297
3. Campisi J, D'adda Di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. (2007) 8:729–40. doi: 10.1038/nrm2233
4. Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle (2007) 6:931–42. doi: 10.4161/cc.6.8.4180
5. Chung JH. The role of DNA-PK in aging and energy metabolism. FEBS J. (2018) 285:1959–72. doi: 10.1111/febs.14410
6. Pan MR, Li K, Lin SY, Hung WC. Connecting the dots: from DNA damage and repair to aging. Int J Mol Sci. (2016) 17:E685. doi: 10.3390/ijms17050685
7. Weyand CM, Goronzy JJ. Aging of the immune system. mechanisms and therapeutic targets. Ann Am Thorac Soc. (2016) 13:S422–8. doi: 10.1513/AnnalsATS.201602-095AW
8. Goronzy JJ, Weyand CM. Successful and maladaptive T cell aging. Immunity (2017) 46:364–78. doi: 10.1016/j.immuni.2017.03.010
9. Li Y, Goronzy JJ, Weyand CM. DNA damage, metabolism and aging in pro-inflammatory T cells: Rheumatoid arthritis as a model system. Exp Gerontol. (2018) 105:118–27. doi: 10.1016/j.exger.2017.10.027
10. Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, et al. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci Transl Med. (2016) 8:331ra338. doi: 10.1126/scitranslmed.aad7151
11. Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, Weyand CM. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J Exp Med. (2009) 206:1435–49. doi: 10.1084/jem.20082251
12. Shao L, Goronzy JJ, Weyand CM. DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO Mol Med. (2010) 2:415–27. doi: 10.1002/emmm.201000096
13. Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity (2016) 45:903–16. doi: 10.1016/j.immuni.2016.09.013
14. Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM. Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci USA (2009) 106:4360–5. doi: 10.1073/pnas.0811332106
15. Weyand CM, Fujii H, Shao L, Goronzy JJ. Rejuvenating the immune system in rheumatoid arthritis. Nat Rev Rheumatol. (2009) 5:583–8. doi: 10.1038/nrrheum.2009.180
16. Deane KD, Norris JM, Holers VM. Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am. (2010) 36:213–41. doi: 10.1016/j.rdc.2010.02.001
17. Oliver JE, Silman AJ. Risk factors for the development of rheumatoid arthritis. Scand J Rheumatol. (2006) 35:169–74. doi: 10.1080/03009740600718080
18. Karlson EW, Chang SC, Cui J, Chibnik LB, Fraser PA, De Vivo I, et al. Gene-environment interaction between HLA-DRB1 shared epitope and heavy cigarette smoking in predicting incident rheumatoid arthritis. Ann Rheum Dis. (2010) 69:54–60. doi: 10.1136/ard.2008.102962
19. Weyand CM, Goronzy JJ. Association of MHC and rheumatoid arthritis. HLA polymorphisms in phenotypic variants of rheumatoid arthritis. Arthritis Res. (2000) 2:212–6. doi: 10.1186/ar90
20. Diaz-Gallo LM, Ramskold D, Shchetynsky K, Folkersen L, Chemin K, Brynedal B, et al. Systematic approach demonstrates enrichment of multiple interactions between non-HLA risk variants and HLA-DRB1 risk alleles in rheumatoid arthritis. Ann Rheum Dis. (2018) 77:1454–62. doi: 10.1136/annrheumdis-2018-213412
21. Schonland SO, Lopez C, Widmann T, Zimmer J, Bryl E, Goronzy JJ, et al. Premature telomeric loss in rheumatoid arthritis is genetically determined and involves both myeloid and lymphoid cell lineages. Proc Natl Acad Sci USA (2003) 100:13471–6. doi: 10.1073/pnas.2233561100
22. Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol. (2014) 10:602–11. doi: 10.1038/nrrheum.2014.109
23. Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. (2010) 42:508–14. doi: 10.1038/ng.582
24. Bae SC, Lee YH. Association between the functional PTPN22 G788A (R263Q) polymorphism and susceptibility to autoimmune diseases: a meta-analysis. Cell Mol Biol. (2018) 64:46–51. doi: 10.14715/cmb/2018.64.5.7
25. Ding J, Eyre S, Worthington J. Genetics of RA susceptibility, what comes next? RMD Open (2015) 1:e000028. doi: 10.1136/rmdopen-2014-000028
26. Yamamoto K, Okada Y, Suzuki A, Kochi Y. Genetic studies of rheumatoid arthritis. Proc Jpn Acad Ser B Phys Biol Sci. (2015) 91:410–22. doi: 10.2183/pjab.91.410
27. Chang HH, Liu GY, Dwivedi N, Sun B, Okamoto Y, Kinslow JD, et al. A molecular signature of preclinical rheumatoid arthritis triggered by dysregulated PTPN22. JCI Insight (2016) 1:e90045. doi: 10.1172/jci.insight.90045
28. Gloria-Bottini F, Ammendola M, Saccucci P, Neri A, Magrini A, Bottini E. The effect of ACP1, ADA6 and PTPN22 genetic polymorphisms on the association between p53 codon 72 polymorphism and endometriosis. Arch Gynecol Obstet. (2016) 293:399–402. doi: 10.1007/s00404-015-3827-6
29. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol. (2003) 3:544–56. doi: 10.1038/nri1131
30. Xie P. TRAF molecules in cell signaling and in human diseases. J Mol Signal (2013) 8:7. doi: 10.1186/1750-2187-8-7
31. Tanikawa C, Ueda K, Nakagawa H, Yoshida N, Nakamura Y, Matsuda K. Regulation of protein Citrullination through p53/PADI4 network in DNA damage response. Cancer Res. (2009) 69:8761–9. doi: 10.1158/0008-5472.CAN-09-2280
32. Tanikawa C, Espinosa M, Suzuki A, Masuda K, Yamamoto K, Tsuchiya E, et al. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat Commun. (2012) 3:676. doi: 10.1038/ncomms1676
33. Wang B, Niu D, Lam TH, Xiao Z, Ren EC. Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Differ (2014) 21:521–32. doi: 10.1038/cdd.2013.132
34. Arleevskaya MI, Kravtsova OA, Lemerle J, Renaudineau Y, Tsibulkin AP. How rheumatoid arthritis can result from provocation of the immune system by microorganisms and viruses. Front Microbiol. (2016) 7:1296. doi: 10.3389/fmicb.2016.01296
35. Ball RJ, Avenell A, Aucott L, Hanlon P, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein-Barr virus and rheumatoid arthritis. Arthritis Res Ther. (2015) 17:274. doi: 10.1186/s13075-015-0755-6
36. Tanay A. Chikungunya virus and autoimmunity. Curr Opin Rheumatol. (2017) 29:389–93. doi: 10.1097/BOR.0000000000000396
37. Zhao J, Dang X, Zhang P, Nguyen LN, Cao D, Wang L, et al. Insufficiency of DNA repair enzyme ATM promotes naive CD4 T-cell loss in chronic hepatitis C virus infection. Cell Discov. (2018) 4:16. doi: 10.1038/s41421-018-0015-4
38. Hasunuma T, Sumida T, and Nishioka K. Human T cell leukemia virus type-I and rheumatoid arthritis. Int Rev Immunol. (1998) 17:291–307. doi: 10.3109/08830189809054407
39. Masuko-Hongo K, Kato T, Nishioka K. Virus-associated arthritis. Best Pract Res Clin Rheumatol. (2003) 17:309–18. doi: 10.1016/S1521-6942(03)00004-4
40. Zucker-Franklin D, Pancake BA, and Brown W. H. Prevalence of HTLV-I Tax in a subset of patients with rheumatoid arthritis. Clin Exp Rheumatol. (2002) 20:161–9. doi: 10.1111/j.1523-1755.2005.00238.x
41. Chandhasin C, Ducu RI, Berkovich E, Kastan MB, Marriott SJ. Human T-cell leukemia virus type 1 tax attenuates the ATM-mediated cellular DNA damage response. J Virol. (2008) 82:6952–61. doi: 10.1128/JVI.02331-07
42. Krueger A, Fas SC, Giaisi M, Bleumink M, Merling A, Stumpf C, et al. HTLV-1 Tax protects against CD95-mediated apoptosis by induction of the cellular FLICE-inhibitory protein (c-FLIP). Blood (2006) 107:3933–9. doi: 10.1182/blood-2005-06-2567
43. Grassmann R, Aboud M, Jeang KT. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene (2005) 24:5976–85. doi: 10.1038/sj.onc.1208978
44. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520:553–7. doi: 10.1038/nature14156
45. Kanneganti TD, Kundu M, Green DR. Innate immune recognition of mtDNA–an undercover signal? Cell Metab (2015) 21:793–4. doi: 10.1016/j.cmet.2015.05.019
46. Le Saux S, Weyand CM, Goronzy JJ. Mechanisms of immunosenescence: lessons from models of accelerated immune aging. Ann N Y Acad Sci. (2012) 1247:69–82. doi: 10.1111/j.1749-6632.2011.06297.x
47. Hertoghs KM, Moerland PD, Van Stijn A, Remmerswaal EB, Yong SL, Van De Berg PJ, et al. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J Clin Invest. (2010) 120:4077–90. doi: 10.1172/JCI42758
48. Ren L, Tan XJ, Xiong YF, Xu K, Zhou Y, Zhong H, et al. Transcriptome analysis reveals positive selection on the divergent between topmouth culter and zebrafish. Gene (2014) 552:265–71. doi: 10.1016/j.gene.2014.09.053
49. Sharif K, Watad A, Bragazzi NL, Adawi M, Amital H, Shoenfeld Y. Coffee and autoimmunity: more than a mere hot beverage! Autoimmun Rev. (2017) 16:712–21. doi: 10.1016/j.autrev.2017.05.007
50. Mikuls TR, Cerhan JR, Criswell LA, Merlino L, Mudano AS, Burma M, et al. Coffee, tea, and caffeine consumption and risk of rheumatoid arthritis: results from the Iowa Women's Health Study. Arthritis Rheum (2002) 46:83–91. doi: 10.1002/1529-0131(200201)46:1<83::AID-ART10042>3.0.CO;2-D
51. Tanaka T, Huang X, Halicka HD, Zhao H, Traganos F, Albino AP, et al. Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents. Cytometry A (2007) 71:648–61. doi: 10.1002/cyto.a.20426
52. Makrygiannakis D, Hermansson M, Ulfgren AK, Nicholas AP, Zendman AJ, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
53. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatology (Oxford) (2017) 56:644–53. doi: 10.1093/rheumatology/kew449
54. Mchugh J. Rheumatoid arthritis: nicotine exacerbates arthritis. Nat Rev Rheumatol. (2017) 13:132. doi: 10.1038/nrrheum.2017.3
55. Karaman A, Binici DN, Melikoglu MA. Comet assay and analysis of micronucleus formation in patients with rheumatoid arthritis. Mutat Res. (2011) 721:1–5. doi: 10.1016/j.mrgentox.2010.11.014
56. Shao L, Li QH, Tan Z. L-carnosine reduces telomere damage and shortening rate in cultured normal fibroblasts. Biochem Biophys Res Commun. (2004) 324:931–6. doi: 10.1016/j.bbrc.2004.09.136
57. Mceachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. (2000) 34:331–58. doi: 10.1146/annurev.genet.34.1.331
58. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature (2009) 461:1071–8. doi: 10.1038/nature08467
59. Verdun RE, Karlseder J. Replication and protection of telomeres. Nature (2007) 447:924–31. doi: 10.1038/nature05976
60. De Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. (2005) 19:2100–10. doi: 10.1101/gad.1346005
61. Chang CH, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol. (2016) 17:364–8. doi: 10.1038/ni.3415
62. Maciver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
63. Gustafsson NMS, Farnegardh K, Bonagas N, Ninou AH, Groth P, Wiita E, et al. Targeting PFKFB3 radiosensitizes cancer cells and suppresses homologous recombination. Nat Commun. (2018) 9:3872. doi: 10.1038/s41467-018-06287-x
64. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
65. Weyand CM, Goronzy JJ. Immunometabolism in early and late stages of rheumatoid arthritis. Nat Rev Rheumatol. (2017) 13:291–301. doi: 10.1038/nrrheum.2017.49
66. Weyand CM, Zeisbrich M, Goronzy JJ. Metabolic signatures of T-cells and macrophages in rheumatoid arthritis. Curr Opin Immunol. (2017) 46:112–20. doi: 10.1016/j.coi.2017.04.010
67. Valentin-Vega YA, Kastan MB. A new role for ATM: regulating mitochondrial function and mitophagy. Autophagy (2012) 8:840–1. doi: 10.4161/auto.19693
68. Franklin DA, He Y, Leslie PL, Tikunov AP, Fenger N, Macdonald JM, et al. p53 coordinates DNA repair with nucleotide synthesis by suppressing PFKFB3 expression and promoting the pentose phosphate pathway. Sci Rep. (2016) 6:38067. doi: 10.1038/srep38067
69. Goldstein I, Rotter V. Regulation of lipid metabolism by p53 - fighting two villains with one sword. Trends Endocrinol Metab. (2012) 23:567–75. doi: 10.1016/j.tem.2012.06.007
70. Yang Z, Goronzy JJ, Weyand CM. The glycolytic enzyme PFKFB3/ phosphofructokinase regulates autophagy. Autophagy (2014) 10:382–3. doi: 10.4161/auto.27345
71. Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, et al. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat Immunol. (2017) 18:1025–34. doi: 10.1038/ni.3808
72. Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. (2014) 20:967–77. doi: 10.1016/j.cmet.2014.10.008
73. Sahin E, Colla S, Liesa M, Moslehi J, Muller FL, Guo M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature (2011) 470:359–65. doi: 10.1038/nature09787
74. Wang Z, Long H, Chang C, Zhao M, Lu Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: a comprehensive overview. Cell Mol Life Sci. (2018) 75:3353–69. doi: 10.1007/s00018-018-2864-2
75. Brooks CL, Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer (2009) 9:123–8. doi: 10.1038/nrc2562
76. Chang HC, Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol Metab. (2014) 25:138–45. doi: 10.1016/j.tem.2013.12.001
77. Krishnakumar R, Kraus WL. PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell (2010) 39:736–49. doi: 10.1016/j.molcel.2010.08.014
78. Shao L, Zhou HJ, Zhang H, Qin L, Hwa J, Yun Z, et al. SENP1-mediated NEMO deSUMOylation in adipocytes limits inflammatory responses and type-1 diabetes progression. Nat Commun. (2015) 6:8917. doi: 10.1038/ncomms9917
79. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity (2008) 28:454–67. doi: 10.1016/j.immuni.2008.03.004
80. Sarkar S, Fox DA. Targeting IL-17 and Th17 cells in rheumatoid arthritis. Rheum Dis Clin North Am. (2010) 36:345–66. doi: 10.1016/j.rdc.2010.02.006
81. Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum (1999) 42:963–70. doi: 10.1002/1529-0131(199905)42:5<963::AID-ANR15>3.0.CO;2-E
82. Park JS, Lim MA, Cho ML, Ryu JG, Moon YM, Jhun JY, et al. p53 controls autoimmune arthritis via STAT-mediated regulation of the Th17 cell/Treg cell balance in mice. Arthritis Rheum (2013) 65:949–59. doi: 10.1002/art.37841
83. D'souza AD, Parish IA, Mckay SE, Kaech SM, Shadel GS. Aberrant CD8+ T-cell responses and memory differentiation upon viral infection of an ataxia-telangiectasia mouse model driven by hyper-activated Akt and mTORC1 signaling. Am J Pathol. (2011) 178:2740–51. doi: 10.1016/j.ajpath.2011.02.022
84. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542:110–4. doi: 10.1038/nature20810
85. Mitkin NA, Muratova AM, Sharonov GV, Korneev KV, Sviriaeva EN, Mazurov D, et al. p63 and p73 repress CXCR5 chemokine receptor gene expression in p53-deficient MCF-7 breast cancer cells during genotoxic stress. Biochim Biophys Acta Gene Regul Mech. (2017) 1860:1169–78. doi: 10.1016/j.bbagrm.2017.10.003
Keywords: DNA damage response, ataxia telangiectasia mutated, rheumatoid arthritis, high-risk factors, T cell dysfunction
Citation: Shao L (2018) DNA Damage Response Signals Transduce Stress From Rheumatoid Arthritis Risk Factors Into T Cell Dysfunction. Front. Immunol. 9:3055. doi: 10.3389/fimmu.2018.03055
Received: 24 July 2018; Accepted: 10 December 2018;
Published: 20 December 2018.
Edited by:
Laurence Morel, University of Florida, United StatesReviewed by:
Erika H. Noss, University of Washington, United StatesJason Weinstein, Rutgers Biomedical and Health Sciences, United States
Copyright © 2018 Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Shao, c2hhb2xhbkBtYWlsLnN5c3UuZWR1LmNu