Abstract
Apoptosis is a form of cell death by which the body maintains the homeostasis of the internal environment. Apoptosis is an initiative cell death process that is controlled by genes and is mainly divided into endogenous pathways (mitochondrial pathway), exogenous pathways (death receptor pathway), and apoptotic pathways induced by endoplasmic reticulum (ER) stress. The homeostasis imbalance in ER results in ER stress. Under specific conditions, ER stress can be beneficial to the body; however, if ER protein homeostasis is not restored, the prolonged activation of the unfolded protein response may initiate apoptotic cell death via the up-regulation of the C/EBP homologous protein (CHOP). CHOP plays an important role in ER stress-induced apoptosis and this review focuses on its multifunctional roles in that process, as well as its role in apoptosis during microbial infection. We summarize the upstream and downstream pathways of CHOP in ER stress induced apoptosis. We also focus on the newest discoveries in the functions of CHOP-induced apoptosis during microbial infection, including DNA and RNA viruses and some species of bacteria. Understanding how CHOP functions during microbial infection will assist with the development of antimicrobial therapies.
Introduction
The endoplasmic reticulum (ER) is an important organelle in eukaryotic cells and plays important roles in protein synthesis, modification and processing, folding, assembly, and the transportation of nascent peptide chains (1, 2). The ER has a strong homeostasis system and the stability of the internal environment is the basis for the ER to achieve its functions (3). Some physiological and pathological conditions, including changes in temperature and pH, the accumulation of damaged DNA, contamination with toxic effluents, and infection with viruses and bacteria can cause ER stress (4). ER stress can be divided into three types, including the unfolded protein response (UPR), the ER overload response, and sterol regulatory elements combined with protein-mediated regulatory responses (5). ER stress usually refers to the UPR, which occurs when the misfolded or unfolded proteins in the ER increase and activate the stress signal that transmits to the nucleus through the ER membrane. Upon ER stress, cells mainly elicit two responses: one leading to cellular survival and the other leading to apoptosis (6). Using the survival pathway, cells conquer such disadvantageous effects and maintain homeostasis through the UPR, inhibiting the transcription of mRNA, enhancing the folding capacity of the ER, and ERAD (ER-assisted degradation) to restore homeostasis (7). Under chronic or overwhelming ER stress, the normal functions of the ER fail to recover, resulting in cellular dysfunction and apoptosis (8).
Many ER stress related diseases have been reported in clinical populations (9, 10). When ER stress occurs with high intensity, or it is prolonged, homeostasis is not restored and apoptosis is induced by ER-related molecules. ER-induced apoptosis occurs via three primary pathways, including the IRE1/ASK1/JNK pathway, the caspase-12 kinase pathway, and the C/EBP homologous protein (CHOP)/GADD153 pathway (11, 12). The IRE1/ASK1/JNK pathway is important for apoptosis in the ER and has been found active during many diseases, such as osteoporosis, urothelial carcinoma (13, 14). The caspase-12 kinase pathway is also involved in many diseases, neonatal hypoxic-ischemic encephalopathy, parkinson's disease, etc. (15–17). The CHOP pathway plays an important role in ER stress-induced apoptosis due to pathogenic microbial infections, neurological diseases and neoplastic diseases.
The Structure and Characterization of CHOP
The CHOP protein was first identified during methyl methanesulfonate research involving UV irradiation and alkylation (18). CHOP belongs to the family of CCAAT/enhancer binding proteins (C/EBPs) and is involved in the regulation of genes that encode proteins involved in proliferation, differentiation and expression, and energy metabolism. CHOP is a 29 kD protein with 169 (human) or 168 (rodents) amino acid residues. It contains two functional domains, including an N-terminal transcriptional activation domain and a C-terminal basic-leucine zipper (bZIP) domain (19). Deletion mutant analyses showed that the bZIP domain plays a crucial role in CHOP-induced apoptosis (20). Research showed that CHOP-deficient cells were resistant to ER stress-induced apoptosis (11, 21). CHOP deficient mouse experiments revealed that CHOP-induced apoptosis is relevant to many diseases that cause ER stress (22, 23). Apoptosis caused by the CHOP pathway is becoming the focus of an increasing number of researchers (24–26).
The collective findings of CHOP research have indicated that this factor plays an important role in ER stress-mediated apoptosis. Here, we summarize the recent findings on the functions of CHOP during ER stress-induced apoptosis and microbial infection.
The Functions of CHOP in ER Stress-induced Apoptosis
Upstream Regulatory Pathway of CHOP
There are a series of precise mechanisms in cells, which ensure the correct folding and assembly of intracellular proteins. Thus, only correctly folded proteins can be transported out of the ER to perform their functions. During normal physiology, CHOP is ubiquitously expressed at very low levels (27). However, pathological conditions or microbial infection caused ER stress is overwhelming, the expression of CHOP rises sharply and apoptosis is activated, and this process can occur in a wide variety of cells (27, 28). Those processes are mainly regulated by three factors, including protein kinase RNA-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring protein 1 (IRE1) (11).
PERK
PERK is a transmembrane protein and an important sensor that participates in the UPR by attenuating protein translation and regulating oxidative stress (29). Unfolded proteins in the ER stimulate PERK oligomerization and autophosphorylation, and can phosphorylate eukaryotic translation initiation factor 2α (eIF2α) (29). Phosphorylation of eIF2α promotes the transcription of ATF4, which converges on the promoters of target genes, including CHOP, GADD34, and ATF3 (1, 30, 31). Research shows that PERK−/−and ATF4−/−cells and eIF2α (Ser51Ala) knock-in cells fail to induce CHOP during ER stress (32). The PERK/ATF4/CHOP signaling pathway is considered to play a pivotal function in inducing cell apoptosis, both in vitro and in vivo (33–35). However, research shows that CHOP may not fully induce cell death, and that CHOP and ATF4 cooperation is required for the induction of cell death (31).
ATF6
ATF6 is a transmembrane protein. Under ER stress, ATF6 translocates to the Golgi compartment where it is cleaved and activated (36). When ATF6 is activated, it translocates to the nucleus as a homo-or heterodimer and interacts with ATF/cAMP response elements and ER stress-response elements (1). Such complexes bind the promoters of several genes involved in UPR (such as CHOP, GRP78, XBP1) and induce target-gene transcription (37, 38). Along with XBP1(s), ATF6 contributes to the augmentation of ER size and ER protein-folding capacity through target genes. ATF6 can activate the transcription of both CHOP and XBP-1, while XBP-1 can also regulate the expression of CHOP. Thus, ATF6 can cooperate with XBP-1 to activate CHOP (39).
IRE1
IRE1 is a transmembrane protein containing two functional domains, including an N-terminal luminal sensor domain and a C-terminal cytosolic effector (40). IRE1 contains protein kinase and endoribonuclease activities. Unfolded proteins in the ER stimulate IRE1α oligomerization and autophosphorylation, which activates the endoribonuclease activity (41). Upon activation, IRE1α splices the substrate precursor, XBP-1, mRNA introns to produce a mature and active XBP-1 protein (42, 43). The active protein then binds the promoters of several genes involved in UPR and ERAD, and regulates gene expression (such as CHOP) to restore protein homeostasis. Thus, CHOP expression can be upregulated by XBP1(s) (44–46). IRE1α can stimulate the activation of the apoptotic-signaling kinase-1 (ASK1), which then activates the downstream kinases, Jun-N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38 MAPK), which cause apoptosis (40). The P38 MAP kinase family phosphorylates Ser78 and Ser81 of CHOP, which induces cell apoptosis (47, 48). Moreover, during tunicamycin-induced apoptosis, the JNK inhibitor, SP600125, could suppress CHOP upregulation and subsequent death receptor 5 (DR5) expression, indicating that JNK activation is also involved in the modulation of CHOP (49). JNK and p38 MAPK can also promote the phosphorylation and activation of the pro-apoptotic protein, BAX, to regulate cell apoptosis (50). Therefore, CHOP can cooperate with JNK and p38 MAPK to regulate cell apoptosis (the upstream regulatory pathways of CHOP are summarized in Figure 1).
Figure 1
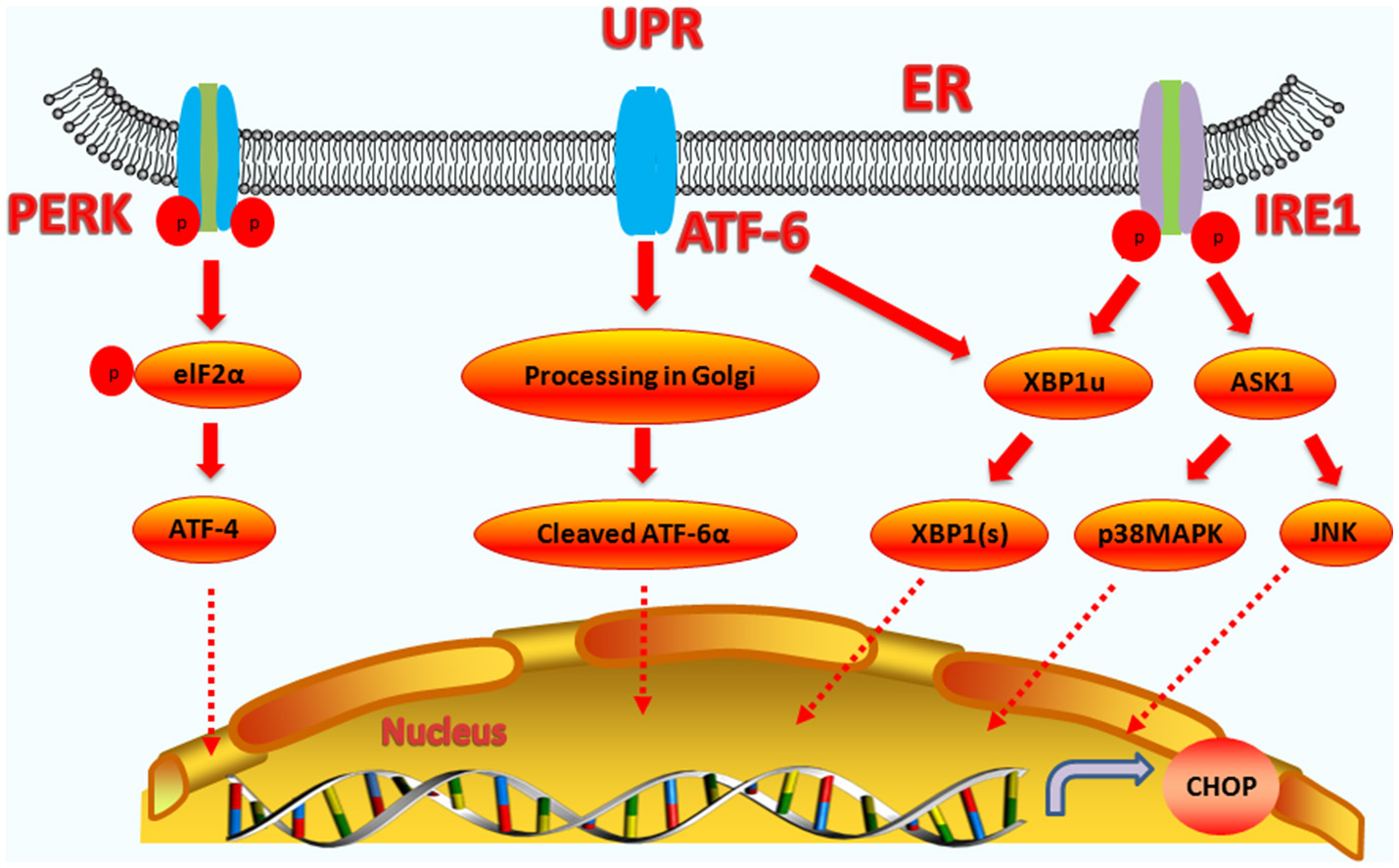
Upstream regulatory pathway of CHOP. The three signaling branches of UPR lead to CHOP transcription. Activated PERK phosphorylates eIF2α, which results in the translation of ATF4. Subsequently, ATF4 translocates to the nucleus, which increases the expression of CHOP and ATF3. CHOP and ATF3 then bind to the promoters downstream genes. ATF6 translocates to Golgi apparatus, where it is activated by proteolysis. Activated ATF6 transcriptionally upregulates CHOP expression. Additionally, ATF6 can regulate XBP-1 to activate CHOP. On one hand, the activation of IRE1α processes unspliced XBP1 mRNA to create activated XBP1(s), which enters the nucleus and controls the expression of CHOP. On the other hand, IRE1α activates apoptotic signaling kinase 1 (ASK1), which in turn phosphorylates p38MAPK and JNK to activate CHOP.
Downstream Regulatory Pathway of CHOP
CHOP Induces Apoptosis Through Mitochondria-Dependent Pathway
A variety of upstream pro-apoptotic signals act on the mitochondrial membrane. Active BCL2-family proteins form protein channels in the mitochondrial membrane and the opening of the mitochondrial PT hole enables the apoptotic active substances (such as cytochrome c, Smac, etc.) to be released into the cytoplasm (51). Such events cause downstream caspase-family proteins to activate and act on the corresponding substrates leading to apoptosis.
As a transcription factor, CHOP can regulate the expression of many anti-apoptotic and pro-apoptotic genes, including genes encoding the BCL2-family proteins, GADD34, TRB-3, and DOCs (52, 53). In the CHOP-induced apoptotic pathway, CHOP regulates the BCL2 protein family. The BCL2 family consists of 25 members that share up to four conserved motifs known as BCL2 homology domains (BH1–4) (54). The BCL2 protein family can be divided into two categories: anti-apoptotic proteins and proapoptotic proteins (55). Anti-apoptotic proteins mainly include BCL2, BCL-XL, MCL-1, and BCL-W, while the proapoptotic proteins can be divided into two categories: multidomain and BH3-only domain proteins (56, 57). Multidomain proteins include BAK, BAX, and BOK, while the BH3-only domain proteins include BID, BIM, BAD, BIK, NOXA, and PUMA (58, 59). The BH3-only proteins regulate cell apoptosis mainly by inhibiting the expression of the BCL2 anti-apoptotic protein, or promoting the expression of multidomain proteins, such as BAX (60).
Under ER stress, CHOP can function as either a transcriptional activator or repressor. It forms heterodimers with other C/EBP family transcription factors via bZIP-domain interactions to inhibit the expression of genes responsive to C/EBP family transcription factors, while enhancing the expression of other genes containing a specific 12–14 bp cis-acting element (19). CHOP can downregulate the expressions of BCL2, BCL-XL, and MCL-1, and upregulate the expression of BIM, causing increased BAK and BAX expression (60, 61). After BAX-BAK oligomerization, the oligomers cause the release of apoptotic factors such as cytochrome c (Cyt-C) and apoptosis-inducing factor (AIF) through mitochondria permeabilization, eventually cause cell death (62).
TRB3 is an intracellular pseudokinase that modulates the activity of the signal transduction cascade and is highly regulated in many cells (63, 64). Research has shown that under conditions of hypoxia and ER stress in non-cardiac cells, TRB3 increases its expression (65). During ER stress, TRB3 is upregulated by the ER stress-inducible transcriptional factor, ATF4-CHOP (66). CHOP interacts with TRB3, which contributes to the induction of apoptosis (67, 68). The binding site of CHOP overlaps the amino acid response elements in the TRB3 promoter, and the specific regions in CHOP and TRB3 are responsible for their interaction (69). The expression of TRB3 can inhibit AKT activity and has a pro-apoptotic capacity (70, 71). AKT directly modulates the expression of caspase-3 and caspase-9, as well as the mitochondrial pro-apoptotic proteins, BAX, and BAD (72). TRB3 can also inhibit the anti-apoptotic activity of AKT by inhibiting the phosphorylation of the Ser473 and Thr308 sites of AKT (63, 73). The upregulation of TRB3 expression can be accompanied by the activation of caspase-3, thereby enhancing apoptosis. Interfering with the expression of TRB3 partially attenuates caspase-3 activity (74, 75), and therefore, CHOP also regulates apoptosis by up-regulating the expression of the TRB3 gene, and directly or indirectly affecting the activity of caspase (the endogenous pathway-induced apoptosis pathways are summarized in Figure 2).
Figure 2

The functions of CHOP in endogenous-pathway-induced apoptosis. CHOP triggers the intrinsic apoptotic pathway through the inhibition of BCL-2, BCL-XL, MCL-1, and the upregulation of BIM, which regulates BAX-BAK-mediated mitochondrial outer membrane permeabilization. This leads to cytochrome c release and the caspase cascade. Bag5 can reduce CHOP expression and increase Bcl-2 gene expression. CHOP can also regulate the apoptosis of cells by up-regulating the expression of the TRB3 gene, preventing Akt phosphorylation, which inhibits the activity of caspases-3/9.
CHOP Induces Apoptosis Through Death-Receptor Pathway
Cell death induced by ER stress also can be mediated through exogenous pathways. Death receptor-mediated apoptosis occurs via death ligands (Fas, TNF, and TRAIL) combined with death receptors. The receptor protein, Fas-associated death domain protein, is recruited to form a death-inducing signal complex, which activates the cytosolic caspase-8, which then activates the downstream caspase to induce apoptosis.
The PERK-ATF4-CHOP pathway can induce apoptosis by binding to the death receptor pathway and upregulating the expression of death receptor 4 (DR4) and DR5. CHOP regulates DR4 or DR5, or both DR4 and DR5 to induce apoptosis is dependent on the different cell types and stimulus. The TRAIL-R1/DR4 death receptors can be activated by ER stress. CHOP interacts with the phosphorylated transcription factor JUN to form a complex that binds to the promoter region of DR4 in lung cancer cells (76). In giant brain neuronal cells (GCN5), the N-terminal domain of CHOP interacts with phosphorylated JUN to form a complex that regulates the expression of DR4 and DR5 (76). CHOP also upregulates the expression of DR5 by binding to the 5′-region of the DR5 gene (77). ATF3 is also involved in mediating DR5 production. In a colon cancer cell model in which the p53 gene is deleted, the ATF3 gene involved in ER is also involved in mediating DR5 production (78).
Research shows that the CHOP-DR5 signaling sensitizes several chemically challenged cancer cells to extrinsic apoptosis mediated by reactive oxygen species (ROS), in vitro (79, 80). If the ER stress is not reversible, PERK-CHOP function will persist, permitting DR5 mRNA to rise. The accumulation of DR5 in the ER and Golgi can drive ligand-independent multimerization of the long splice variants of death receptor 5 (DR5L). DR5L accelerates the formation of the death-inducing signaling complex (DISC) and activates caspase-8 (81). The activation of caspase-8 can also cleave BID located in the cytoplasm into tBID. tBID has powerful pro-apoptotic activity and can act on the mitochondrial membranes with BAK and BAX, which cause Cyt-C release. Subsequently, this leads to apoptosis via the exogenous and the endogenous pathways (82) (the exogenous pathway-induced apoptosis pathways are summarized in Figure 3).
Figure 3
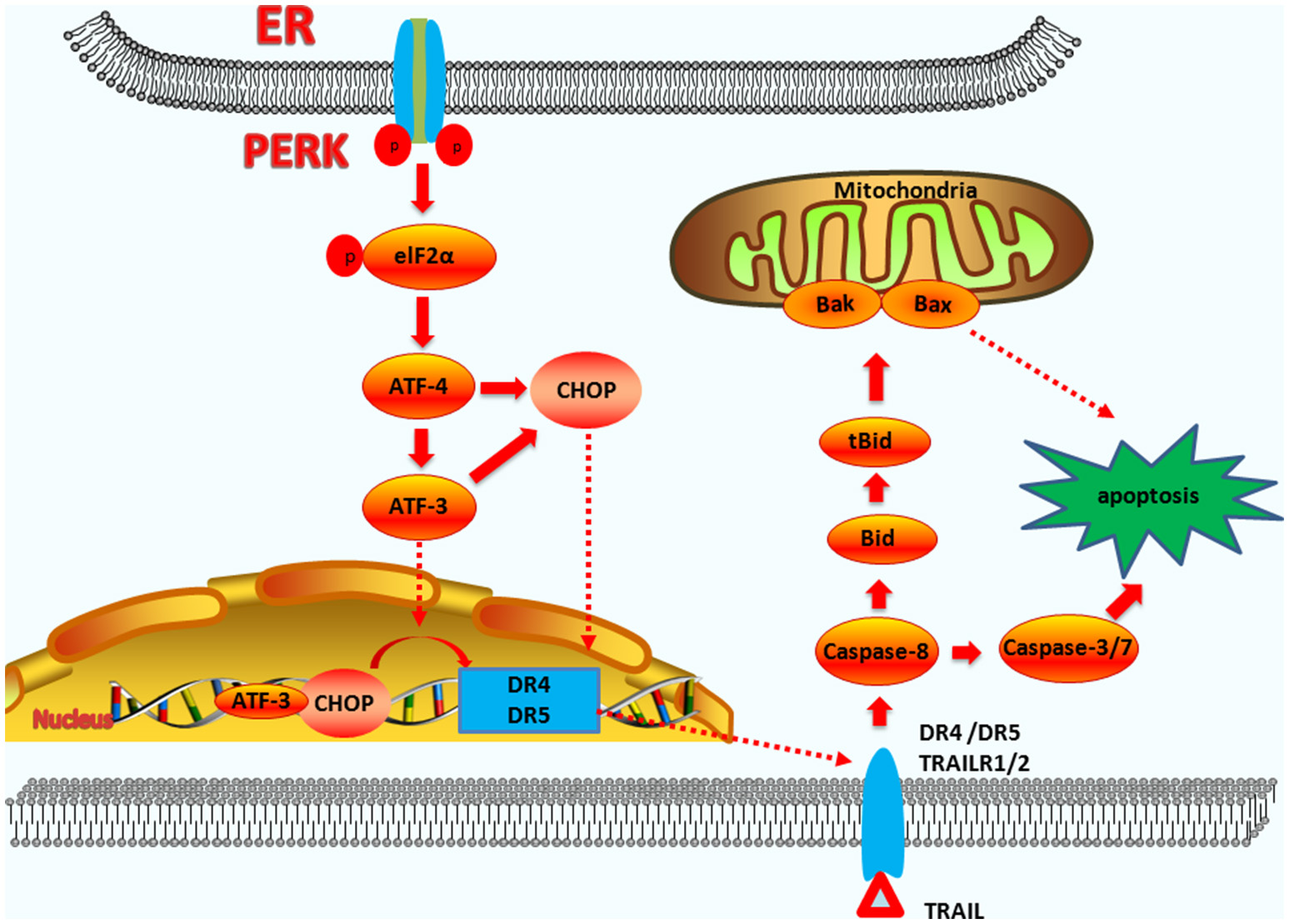
The functions of CHOP in exogenous-pathway-induced apoptosis. CHOP triggers the extrinsic apoptotic pathway through the upregulation of DR4 and DR5. PERK can induce ATF4 expression, which results in CHOP and ATF3 expression. CHOP and ATF3 then bind to the promoters of the DR4 and DR5 genes, upregulating their expression. TRAIL combining with DR4 and DR5 regulates the caspase 8-mediated cascade, which activates caspase3/7 to lead apoptosis. On the other hand, Bid is cleavaged into tBid. tBid then regulates the BAX-BAK-mediated mitochondrial apoptosis pathways.
CHOP Induces Apoptosis Through Other Pathways
In addition to mediating apoptosis through the endogenous and exogenous pathways, CHOP also mediates apoptosis through other pathways. CHOP can increase the expression of the ERO1α (ER reductase) gene, which catalyzes the oxidation of protein disulfide isomerase (PDI), resulting in the production of H2O2 in the ER. The highly oxidized state of the ER (22, 83) results in H2O2 leakage into the cytoplasm, induces the production of ROS and a series of apoptotic and inflammatory reactions (84–86).
High ROS concentrations in the lumen of the ER activate the IP3R1 calcium ion release channel, allowing calcium ions to enter the cytosol. Cytoplasmic calcium promotes ROS by activating the calcium-sensing kinase, CaMKII (calcium-dependent protein kinase), and NOX2, a subunit of NADPH oxidase on the cell membrane. Those factors then promote the transcription of CHOP, resulting in apoptosis (87). This is a positive feedback pathway. The CHOP-CaMKII pathway also activates JNK, which is involved in ER stress-induced apoptosis by inducing the expression of Fas, NOX2, and oxidative stress (88, 89). In some models of diabetes, the deletion of CHOP can inhibit the apoptosis of pancreatic beta cells, and provides a protective mechanism to reduce the production of ERO1α, which subsequently reduces markers of oxidative stress and the expression of antioxidant genes (90). Moreover, ROS scavengers can attenuate the expression of PERK/eIF2α/CHOP pathway-related proteins (91).
The CHOP gene also directly activates GADD34 (DNA damage protein), which combines with phosphatase 1 protein (PP1) and dephosphorylates eIF2α, resulting in protein translation recovery and increases of ER stress and cell apoptosis (92, 93). Under ER stress, cells with GADD34 mutations can significantly reduce protein complex formation compared to normal cells.
The overexpression of CHOP can lead to cell cycle arrest and result in cell apoptosis. At the same time, CHOP-induced apoptosis can also trigger cell death by inhibiting the expression of cell cycle regulatory protein, p21. The p21 protein not only inhibits the G1 phase of the cell cycle, but also has a close relationship with the activity of pre-apoptotic factors. Thus, the relationship between CHOP and p21 may explain changes in the state of the cell from adapting to ER stress to pre-apoptotic activity (94, 95).
Under most conditions, CHOP can directly bind to the promoters of downstream related genes, while under specific condition, CHOP can cooperate with other transcription factors to affect apoptosis. Recent studies have shown that Bcl-2-associated athanogene 5 (Bag5) is over-expressed in prostate cancer and inhibits ER stress-induced apoptosis. Overexpression of Bag5 results in decreased CHOP and BAX expression, and increased Bcl-2 gene expression (96). Bag5 overexpression inhibited ER stress-induced apoptosis in the UPR by suppressing PERK-eIF2-ATF4 and enhancing the IRE1-Xbp1 activity (97).
CHOP has also been reported to regulate the expression of BH3-only proteins by interacting with FOXO3A (in neuronal cells treated with tunicamycin) and the AP-1 complex protein, cJUN, leading to its phosphorylation. Knockdown of CHOP prevented the dephosphorylation of its downstream target FOXO3a (Thr32) (58).
Synoptically, CHOP-dependent apoptosis is mainly mediated by directly or indirectly altering the expression of pro-apoptotic or anti-apoptotic genes. Collectively, the apoptotic pathways are mediated by the downstream targets of CHOP; however, the molecular interaction mechanisms by which this occurs remain to be understood.
CHOP Functions During Apoptosis and Autophagy
Autophagy is a lysosomal degradation system involving the degradation and recycling of obsolete, damaged, or harmful cytoplasmic materials, and organelles. Autophagy generally precedes apoptosis under cellular stresses and the apoptotic program is activated when such stresses exceed a critical duration or intensity threshold (98). Here, we describe the important roles of CHOP in ER stress induced apoptosis. Recent studies have identified CHOP as a direct regulator of numerous genes involved in the pro-survival autophagic process (99, 100). CHOP has been implicated in autophagy induced by amino acid starvation, ER stress, and infection with microorganisms (101, 102). Research has shown that knockdown of CHOP not only enhances tunicamycin-induced autophagy, but also significantly attenuates ER stress-induced apoptosis in human colon cancer cells. Moreover, CHOP modulates the induction of autophagosomes during ER stress, as evidenced by the inhibition of LC3-II expression and GFP-LC3B dots (103). Another study also showed that UPR-activated CHOP elicited complete autolysosome maturation during hepatitis C virus-induced autophagy via an LC3B-II-dependent mechanism (104). During amino acid starvation and ER stress, CHOP can up-regulate the expression of autophagy-related genes at the onset, while in the later stages of starvation, CHOP can inhibit the occurrence of autophagy and gradually initiate apoptosis (105). Therefore, CHOP plays a pivotal role in switching between apoptosis and autophagy.
The Functions of CHOP-induced Apoptosis During Microbial Infection
Many pathogenic microorganisms infect host cells and can cause ER stress (106). When ER stress occurs at a high intensity or is prolonged, homeostasis is not restored and apoptosis will occur. Many microbes infect host cells to induce apoptosis through regulating the expression of CHOP. CHOP-induced apoptosis can be beneficial to the microorganisms or be detrimental to their growth and reproduction (Table 1). Here, we summarize recent findings in the functions of CHOP-induced apoptosis during microbial infection.
Table 1
| Microbial type | Key proteins | Involved mechanisms and phenotype | Functions of CHOP | Research models | References |
|---|---|---|---|---|---|
| Porcine circovirus type 2 | CHOP, ATF4 Caspase 3, |
PERK-eIF2α-ATF4 -CHOP-BCL2 | Apoptosis and beneficial to PCV2 replication | PK-15 cells | (107, 108) |
| Infectious bronchitis virus | CHOP, TRIB3, BCL2 | PERK-eIF2α-ATF4 PKR-eIF2α-ATF4 | Apoptosis and facilitates virus release | H1299 cells | (109, 110) |
| Newcastle disease virus | CHOP, ATF4, cyclin D1 | PERK-ATF4-CHOP | Apoptosis and beneficial to NDV replication | Asynchronously growing cells | (111) |
| West Nile virus | CHOP, PARP, Caspase 3, |
PERK-ATF4-CHOP | Apoptosis and limit WNV replication | Human neuroblastoma cells, | (112) |
| Coxsackie virus B3 | CHOP, BAX, Caspase 3, |
CHOP-BAX- Caspase 3 |
Apoptosis and beneficial to CVB3 replication | Mice | (113) |
| Japanese encephalitis virus | CHOP, p38, BCL2 | p38-CHOP-BCL2 | Apoptosis | BHK-21 cells | (114, 115) |
| HIV | CHOP, BCL2, BAX | XBP-1-CHOP- Caspase 3/9 |
Apoptosis | Human brain endothelial cells | (116, 117) |
| M. tuberculosis | CHOP, Caspase3/9 |
p-eIF2α-CHOP | limits M. tuberculosis replication | RAW 264.7 cells | (26, 118) |
| Helicobacter pylori | CHOP, BIM, BAX, TRIB3 | PERK-CHOP PKR-eIF2α-ATF4 | Damage the cell and tissue (gastric injury) | AZ-521 cells | (119, 120) |
| Escherichia coli | CHOP, DR5, TRAIL | CHOP-DR5- Caspase 3/8 |
Damage the cell and tissue (renal failure) | THP-1 cells | (121, 122) |
| Shigella dysenteriae | CHOP, DR5 | p38-CHOP-DR5 | Damage the cell and tissue (neurological abnormalities) | RPE cells | (123) |
The functions of CHOP in ER stress-induced apoptosis in microbial models.
The Functions of CHOP in Viral Infection
After viruses invade the host they must adapt to the intracellular environment, and use host cell factors to self-replicate. The ER is an essential organelle for virus assembly and replication, and the replication and proliferation of the virus can affect the function of the ER and cause ER stress. Under prolonged ER stress, cell apoptosis is activated. Under certain conditions, apoptosis is beneficial or essential for virus replication and the release of virus particles. Under other conditions, apoptosis can inhibit the proliferation and spread of the virus in host cells to protect uninfected cells.
After DNA viruses infect host cells, CHOP plays a critical role in apoptosis induced by ER stress. Porcine circovirus type 2 (PCV2) has been reported to elicit the UPR and mediate apoptosis following ER stress (107). PCV2 triggers the UPR in PK-15 cells by activating the PERK-eIF2α-ATF4-CHOP pathway, without the concomitant activation of IRE1 or ATF6. Over-expression of GRP78 can enhance viral capsid expression and/or viral titers. Thus, PCV2 deploys the UPR to enhance its replication (107). Replicase (Rep) and capsid (Cap) proteins of PCV2 activate the eIF2α-ATF4-CHOP pathway. Cap expression significantly reduced the expression of anti-apoptotic BCL2 and increased caspase-3 cleavage by increasing the expression of CHOP. Knockdown of PERK by RNA interference significantly reduces Cap-induced CHOP expression and caspase-3 cleavage. Cap induces UPR and apoptosis via the PERK-eIF2α-ATF4-CHOP-BCL2 pathway (108). When PERK is inhibited by GSK2606414 or eIF2α dephosphorylation is suppressed by salubrinal, viral replication is limited, suggesting that CHOP is involved in apoptosis induced by PCV2 and may be beneficial to viral replication (107).
CHOP also plays an important role in RNA virus-infected host cells. H1299 cells infected with Infectious Bronchitis Virus (IBV) induced apoptosis via ER stress. Post-IBV infection, the IRE1α-XBP1 pathway of the UPR was activated. IRE1α protects infected cells from IBV-induced apoptosis, which requires both its kinase and RNase activities. IRE1α antagonizes IBV-induced apoptosis by modulating the phosphorylation of the proapoptotic c-Jun N-terminal kinase (JNK) and the pro-survival RAC-alpha serine/threonine-protein kinase (Akt) (124). At the same time, IBV infection also activates two other pathways, including PERK-eIF2α-ATF4 and PKR-eIF2α-ATF4. Following activation, ATF4, ATF3 and CHOP are upregulated. CHOP affects GADD34 and the dephosphorylation of eIF2α. This leads to the recovery of protein translation and increasing ER stress and cell apoptosis. CHOP can up-regulate the expression of the apoptotic precursor protein, pseudokinase tribbles homolog 3 (TRIB3), which inhibits the ERK pro-survival pathway, thereby promoting apoptosis. During apoptosis induced by IBV, CHOP also decreases the expression of BCL2, which contributes to CHOP-mediated apoptosis. Thus, IBV induces apoptosis through the IRE/JNK and PERK/PKR pathways. More importantly, in CHOP-deficient cells, IBV-induced apoptosis is attenuated and virus replication is inhibited. Thus, all such results suggest that CHOP-induced apoptosis is beneficial to IBV replication (109, 110).
Coxsackie virus B3 (CVB3) can trigger the UPR and induce apoptosis by mediating the production of CHOP (125, 126) and reducing the cardiac Bcl-2/Bax ratio. That finding supports that CHOP-mediated apoptosis plays a role in acute viral myocarditis (AVMC), which occurs primarily through a mitochondria-dependent pathway. However, the precise mechanism leading to CHOP-mediated apoptosis remains unclear and requires further investigation. Researches show that CHOP deficiency reduces CVB3 replication, cardiac damage, and promotes survival in CVB3-mediated acute viral myocarditis, in vivo (113, 125).
CHOP-mediated apoptosis in premature cells may function as a host defense response by limiting virus replication and pathogenesis. West Nile Virus (WNV) can induce CHOP expression through the PERK-ATF4-CHOP pathway, and CHOP induces the expression of the downstream target gene, GADD34. eIF2α dephosphorylation also occurs, leading to the restoration of protein translation, which increases ER stress and cell apoptosis. Simultaneously, CHOP induces caspase-3 activation, leading to apoptosis. In CHOP-deficient mouse embryonic fibroblasts (MEFs), WNV grows to significantly higher viral titers than that in wild-type MEFs, suggesting that CHOP-mediated apoptosis functions to control WNV replication in vitro (112, 127).
The Functions of CHOP in Bacterial Infection
ER stress-induced apoptosis plays an important role in bacteria-infected host cells. The prevention of apoptosis provides a survival advantage because it facilitates bacterial replication inside host cells (128).
Mycobacterium tuberculosis (Mtb) infected host cells activate three signaling pathways of ER stress (IRE1, PERK, and ATF6) (129). Recent evidence suggests that Mtb and its 38 kDa antigen can activate the PERK/eIF2α/CHOP pathway (25, 26). Mycobacterium tuberculosis and its antigens have been shown to be associated with IRE1α/TRAF2/ASK1/JNK/p38MAPK activation and to result in apoptosis (130). Lim et al. reported that CHOP production induced by the 38 kDa antigen decreased when the JNK pathway was inhibited (25). In A549 cells, JNK phosphorylated Bcl-2 to inhibit its anti-apoptotic activity, and also phosphorylated Bax. Phosphorylated Bax then translocated to the mitochondria and activated Bak to promote apoptosis (130). Thus, CHOP can cooperate with IRE1α/TRAF2/ASK1/JNK to regulate the occurrence of apoptosis.
During M. tuberculosis infection, excessive expression of CHOP can promote cell apoptosis by at least two ways: (i) activation of ERO1α. The 38 kDa antigen induces the production of ROS and the subsequent ERS via ERO1α, which leads to apoptosis through high concentrations of peroxide in the ER environment (25). (ii) Inhibition of Bcl-2. Researchers have reported that ESAT-6 induces CHOP to form dimers with CREB, leading to decreased Bcl-2 expression and increased Bax expression (130, 131). More importantly, when CHOP is interfered with siRNA, it can significantly increase the survival of M. tuberculosis in host cells (26). Collectively, CHOP-induced apoptosis is beneficial to combat Mtb infection.
Vacuolating cytotoxin (VacA) is a critical virulence factor of Helicobacter pylori. VacA can upregulate the expression of CHOP after gastric epithelial cells are stimulated with cytotoxin A, and can also upregulate the expression of BIM and activate BAX, and TRIB3, leading to apoptosis. CHOP is transcriptionally activated by PERK via the phosphorylation of eIF2-α, which is also augmented by NH4Cl. Knockdown of CHOP or TRIB3 could also decrease VacA-induced mitochondrial dysfunction and apoptosis. CHOP is not only involved in apoptosis, but also in autophagy induced by VacA. Knockdown of the ER stress effectors, CHOP or TRIB3, could drastically decrease the formation of autolysosomes and cell death in VacA-treated gastric cancer cells. Therefore, VacA induces autophagy and cell death in the AGS cells by triggering ER stress, which involves the upregulation of CHOP and TRIB3 (119, 120).
Type I Shiga toxin produced by Escherichia coli can also cause apoptosis through endogenous and exogenous pathways, in which CHOP and DR5 play important roles. Silencing the expression of CHOP selectively blocks the activation of caspase and attenuates cell apoptosis (121, 132). Shiga toxins produced by Shigella dysenteriae serotype I activate both apoptotic cell death signaling and the ER stress response. Treatment of human retinal pigment epithelial cells (RPE cells) with Stxs results in the activation of JNK and p38MAPK, and up-regulation of CHOP and DR5 expression (123, 132). Collectively, characterization of CHOP functions during microbial infection will help us to understand the pathogenesis of microorganisms and provide a better theoretical basis to control and prevent diseases.
CHOP plays important functions during microbial infections, and therefore, may be an important potential target for new therapeutic approaches. Research has shown that the regulation of CHOP expression plays an important role in metabolic diseases and in some cancers (83, 133). CHOP deficiencies attenuate oxidative stress and renal ischemia-reperfusion-induced acute renal injury, in vitro and in vivo (83). The regulation of CHOP expression has been accepted as an approach to remove cancer cells through the induction of apoptosis (134). As mentioned above, small molecule inhibitors that inhibit ER stress (UPR) and the expression of CHOP may act as therapeutic options to prevent ER stress and microbial infections. Using the small molecule inhibitors, GSK2606414 or salubrinal, to inhibit the PERK-eIF2α pathway and the expression of CHOP can limit PCV2 replication (107). Thus, targeting CHOP may be a good therapeutic approach for the treatment of PCV2 infection. The chemical chaperone TUDCA is a classic ER stress inhibitor that improves ER folding capacity, which has been protective in various diseases, including diabetes mellitus, hypertension, calcification, and even cardiac dysfunction by preventing ER stress (135–137). TUDCA administration markedly suppresses cardiac ER stress and CHOP induction; thus, preventing cardiomyocyte apoptosis, cardiac inflammation and injury, cardiac dysfunction, reducing CVB3 replication in vivo, and increasing survival rates in CVB3 inoculation-induced AVMC models (113). Therefore, small molecule inhibitors that prevent ER stress and CHOP expression are potential therapeutic approaches for CVB3 infections. Additionally, in CHOP-deficient cells, the apoptosis caused by IBV was attenuated and IBV replication was limited and CVB3 replication was also limited (110, 113). Thus, overexpression or knock-out of the CHOP gene may be a therapeutic approach to treat related diseases. Therefore, understanding how CHOP functions during microbial infections will provide better therapeutic approaches to control and prevent diseases.
Conclusions and Future Perspectives
ER stress induced by pathogenic microorganism infection and subsequent apoptosis play pivotal roles in the regulation of infection (106, 138). CHOP is an important molecule in the ER stress-induced apoptosis pathway. CHOP-induced apoptosis also plays a pivotal role during virus or bacterium infection. Therefore, it is necessity to illustrate CHOP induced apoptosis pathway clearly. As mentioned above, there are extensive in-depth studies on the upstream regulatory genes of CHOP and its downstream target genes in the context of ER stress (a diagram summarizing the regulation of CHOP is shown in Figure 4).
Figure 4
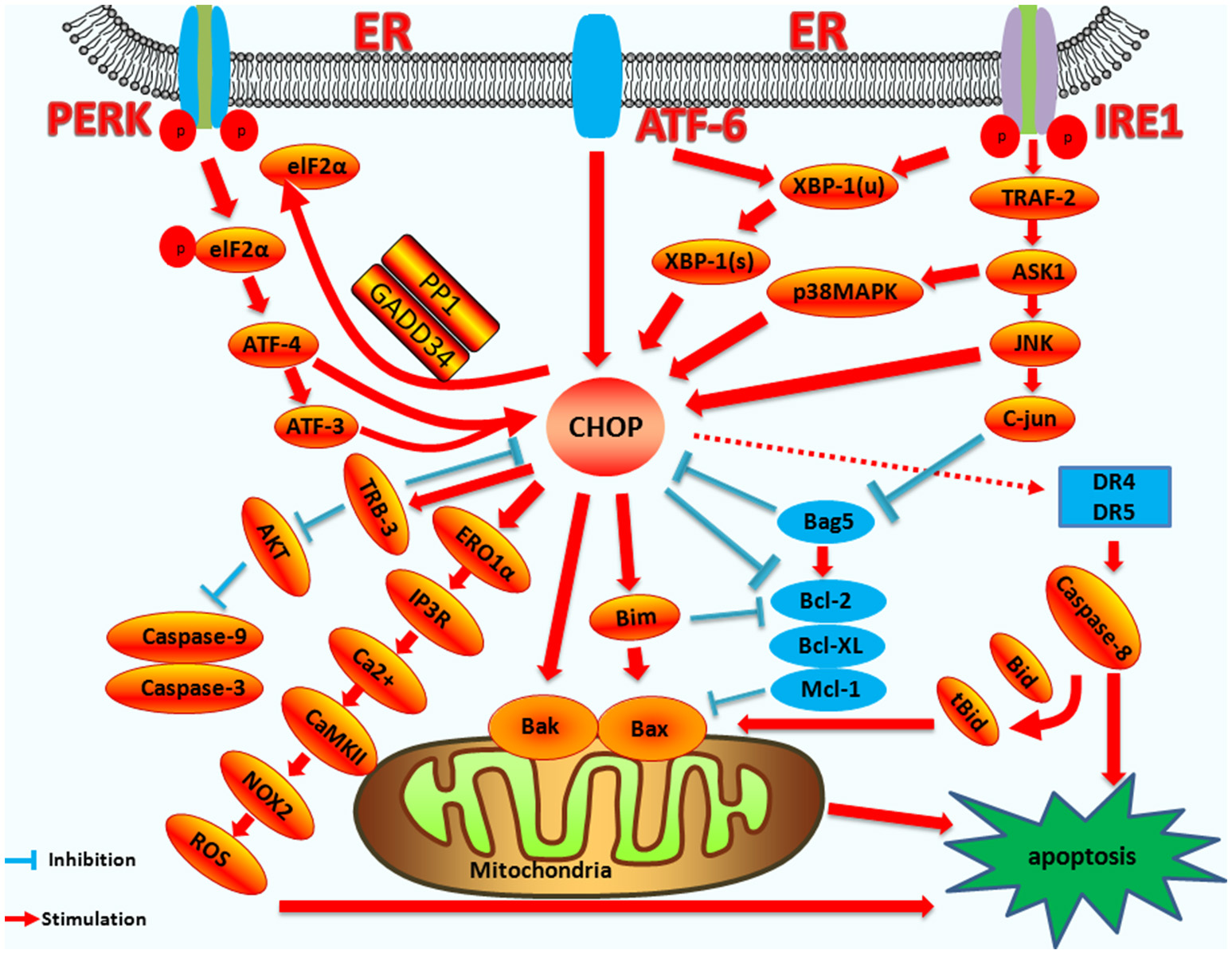
The mechanism of CHOP regulation in ER stress induced apoptosis. Upstream regulatory pathway: Activated PERK phosphorylates eIF2α, which results in the translation of ATF4. ATF4 translocates to the nucleus, which increases the expression of CHOP and ATF3. CHOP and ATF3 then bind to the promoters of the target genes, upregulating their expression. ATF6 translocates to Golgi apparatus where it is activated by proteolysis. Activated ATF6 transcriptionally upregulates CHOP expression. Additionally, ATF6 can regulate XBP-1 to activate CHOP. Activation of IRE1α processes unspliced XBP-1 mRNA into activated XBP1(s), which enters the nucleus and controls the expression of CHOP. IRE1α activates the apoptotic signaling kinase 1 (ASK1), which in turn phosphorylates p38 MAPK/JNK, and activates CHOP. In addition, Bag5 can reduce CHOP expression and increase Bcl-2 gene expression. Downstream regulatory pathway: CHOP triggers the intrinsic apoptotic pathway through the inhibition of BCL-2, BCL-XL, MCL-1, and the upregulation of BIM, which regulates BAX-BAK-mediated mitochondrial outer membrane permeabilization. This leads to cytochrome c release and the caspase cascade. CHOP can also regulate apoptosis by upregulating the expression of the TRB3 gene and preventing Akt phosphorylation, which inhibits the activity of caspases-3/9. CHOP triggers the extrinsic apoptotic pathway through the upregulation of DR4 and DR5, which regulate the caspase-8-mediated cascade. This leads to Bid cleavage into tBid, which regulates the BAX-BAK-mediated mitochondrial apoptosis pathways. CHOP can also trigger the ERO1α-IP3R-Ca2+-CaMKII pathway. ROS can also trigger Ca2+-dependent mitochondrial apoptosis. CHOP can directly activate GADD34 (DNA damage protein), which, combined with phosphatase 1 protein (PP1), dephosphorylates eIF2α, and results in protein translation recovery, increased ER stress, and cell apoptosis.
Increasing evidence has suggested that infections are relevant to the abnormal expression of CHOP during ER stress and most of the time, the expression of CHOP can induce apoptosis. However, the mechanisms and pathways triggered by different pathogens vary and current researches indicate that CHOP plays an important role in apoptosis induced by pathogenic microorganisms. Some of those roles are beneficial to the microorganisms, while others are detrimental to the microorganisms. Most researches aim to show the apoptosis pathways induced by CHOP, and the functions of apoptosis to microorganisms. However, it is unknown under which conditions apoptosis induced by CHOP is beneficial to the release and spread of pathogenic microorganisms, or if such processes trigger a more intense immune response. Thus, how microorganisms use CHOP-induced apoptosis to regulate growth and reproduction, or how the host cells use CHOP-induced apoptosis to restrict a microorganism's replication and transmission are important aspects that need to be investigated further. The intricate balance between the two effects is worth further study, and the detailing of such mechanisms may help in the development of therapeutic approaches for the ER stress and microbial infection.
Statements
Author contributions
HH, MT, and CD drafted the review. SY critically revised the review. All authors read and approved the final manuscript.
Funding
This work was supported by the Scientific and Technical Innovation Project of the Chinese Academy of Agricultural Science (Grant No. SHVRI-ASTIP-2014-8).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- CHOP
C/EBP-homologous protein
- ER
endoplasmic reticulum
- ATF4
activating transcription factor 4
- ATF6
activating transcription factor 6
- DISC
death-inducing signaling complex
- DR
death receptor
- eIF2α
eukaryotic translation initiation factor 2α
- IRE1
inositol-requiring enzyme 1
- JNK
c-Jun N-terminal kinase 1
- PERK
protein kinase RNA-like endoplasmic reticulum kinase
- ROS
reactive oxygen species
- TRAIL
TNF-related apoptosis-inducing ligand
- UPR
unfolded protein response
- XBP1
X-box-binding protein 1
- XIAP
X-linked inhibitor of apoptosis protein
- TRIB3
pseudokinase tribbles homolog 3
- p38MAPK
p38 mitogen-activated protein kinase
- CaMKII
calcium-dependent protein kinase
- PDI
protein disulfide isomerase
- FADD
Fas-associated death domain protein.
Abbreviations
References
1.
Sano R Reed JC . ER stress-induced cell death mechanisms. Biochim Biophys Acta (2013) 1833:3460–70. 10.1016/j.bbamcr.2013.06.028
2.
Han J Kaufman RJ . The role of ER stress in lipid metabolism and lipotoxicity. J Lipid Res. (2016) 57:1329–38. 10.1194/jlr.R067595
3.
Sovolyova N Healy S Samali A Logue SE . Stressed to death - mechanisms of ER stress-induced cell death. Biol Chem. (2014) 395:1–13. 10.1515/hsz-2013-0174
4.
Luethy JD Holbrook NJ . Activation of the gadd153 promoter by genotoxic agents: a rapid and specific response to DNA damage. Cancer Res. (1992) 52:5–10.
5.
Pahl HL . Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. (1999) 79:683–701. 10.1152/physrev.1999.79.3.683
6.
Ariyasu D Yoshida H Hasegawa Y . Endoplasmic reticulum (ER) stress and endocrine disorders. Int J Mol Sci. (2017) 18:E382. 10.3390/ijms18020382
7.
Yoshida H . ER stress and diseases. Febs J. (2007) 274:630–58. 10.1111/j.1742-4658.2007.05639.x
8.
Ayaub EA Kolb PS Mohammed-Ali Z Tat V Murphy J Bellaye PS et al . GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J Pathol. (2016) 239:411–25. 10.1002/path.4738
9.
Lucke-Wold BP Turner RC Logsdon AF Nguyen L Bailes JE Lee JM et al . Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg. (2016) 124:687–702. 10.3171/2015.3.JNS141802
10.
Kropski JA Blackwell TS . Endoplasmic reticulum stress in the pathogenesis of fibrotic disease. J Clin Invest. (2018) 128:64–73. 10.1172/JCI93560
11.
Oyadomari S Mori M . Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. (2004) 11:381–9. 10.1038/sj.cdd.4401373
12.
Hetz C . The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. (2012) 13:89–102. 10.1038/nrm3270
13.
Chen S Wang Y Yang Y Xiang T Liu J Zhou H et al . psoralen inhibited apoptosis of osteoporotic osteoblasts by modulating IRE1-ASK1-JNK pathway. BioMed Res Int. (2017) 2017:3524307. 10.1155/2017/3524307
14.
Zeng T Peng L Chao H Xi H Fu B Wang Y et al . IRE1alpha-TRAF2-ASK1 complex-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to CXC195-induced apoptosis in human bladder carcinoma T24 cells. Biochem Biophys Res Commun. (2015) 460:530–6. 10.1016/j.bbrc.2015.03.064
15.
Zhang Q Liu J Chen S Liu J Liu L Liu G et al . Caspase-12 is involved in stretch-induced apoptosis mediated endoplasmic reticulum stress. Apoptosis (2016) 21:432–42. 10.1007/s10495-016-1217-6
16.
Liu L Liu C Lu Y Liu L Jiang Y . ER stress related factor ATF6 and caspase-12 trigger apoptosis in neonatal hypoxic-ischemic encephalopathy. Int J Clin Exp Pathol. (2015) 8:6960–6.
17.
Tong Q Wu L Jiang T Ou Z Zhang Y Zhu D . Inhibition of endoplasmic reticulum stress-activated IRE1alpha-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson's disease. Eur J Pharmacol. (2016) 776:106–15. 10.1016/j.ejphar.2016.02.042
18.
Fornace AJ Jr Alamo I Jr Hollander MC . DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci USA. (1988) 85:8800–4. 10.1073/pnas.85.23.8800
19.
Ubeda M Wang XZ Zinszner H Wu I Habener JF Ron D . Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol Cell Biol. (1996) 16:1479–89. 10.1128/MCB.16.4.1479
20.
Matsumoto M Minami M Takeda K Sakao Y Akira S . Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett. (1996) 395:143–7. 10.1016/0014-5793(96)01016-2
21.
Nam DH Han JH Lee TJ Shishido T Lim JH Kim GY et al . CHOP deficiency prevents methylglyoxal-induced myocyte apoptosis and cardiac dysfunction. J Mol Cell Cardiol. (2015) 85:168–77. 10.1016/j.yjmcc.2015.05.016
22.
Rao J Zhang C Wang P Lu L Qian X Qin J et al . C/EBP homologous protein (CHOP) contributes to hepatocyte death via the promotion of ERO1alpha signalling in acute liver failure. Biochem J. (2015) 466:369–78. 10.1042/BJ20140412
23.
Tabas I Ron D . Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. (2011) 13:184–90. 10.1038/ncb0311-184
24.
Sanchez-Lopez E Zimmerman T Gomez del Pulgar T Moyer MP Lacal Sanjuan JC Cebrian A . Choline kinase inhibition induces exacerbated endoplasmic reticulum stress and triggers apoptosis via CHOP in cancer cells. Cell Death Dis. (2013) 4:e933. 10.1038/cddis.2013.453
25.
Lim YJ Choi JA Lee JH Choi CH Kim HJ Song CH . Mycobacterium tuberculosis 38-kDa antigen induces endoplasmic reticulum stress-mediated apoptosis via toll-like receptor 2/4. Apoptosis (2015) 20:358–70. 10.1007/s10495-014-1080-2
26.
Lim YJ Choi JA Choi HH Cho SN Kim HJ Jo EK et al . Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS ONE (2011) 6:e28531. 10.1371/journal.pone.0028531
27.
Ron D Habener JF . CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. (1992) 6:439–53. 10.1101/gad.6.3.439
28.
Yao Y Lu Q Hu Z Yu Y Chen Q Wang QK . A non-canonical pathway regulates ER stress signaling and blocks ER stress-induced apoptosis and heart failure. Nat Commun. (2017) 8:133. 10.1038/s41467-017-00171-w
29.
Korennykh A Walter P . Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. (2012) 28:251–77. 10.1146/annurev-cellbio-101011-155826
30.
Michel S Canonne M Arnould T Renard P . Inhibition of mitochondrial genome expression triggers the activation of CHOP-10 by a cell signaling dependent on the integrated stress response but not the mitochondrial unfolded protein response. Mitochondrion (2015) 21:58–68. 10.1016/j.mito.2015.01.005
31.
Han J Back SH Hur J Lin YH Gildersleeve R Shan J et al . ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. (2013) 15:481–90. 10.1038/ncb2738
32.
Harding HP Zhang Y Zeng H Novoa I Lu PD Calfon M et al . An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell (2003) 11:619–33. 10.1016/S1097-2765(03)00105-9
33.
Liu ZX Shi Q Song XF Wang YX Wang YW Song EQ et al . Activating transcription factor 4 (ATF4)-ATF3-C/EBP homologous protein (CHOP) cascade shows an essential role in the ER stress-induced sensitization of tetrachlorobenzoquinone-challenged PC12 cells to ROS-mediated apoptosis via death receptor 5 (DR5) signaling. Chem Res Toxicol. (2016) 29:1510–8. 10.1021/acs.chemrestox.6b00181
34.
Chen Y Gui D Chen J He D Luo Y Wang N . Down-regulation of PERK-ATF4-CHOP pathway by Astragaloside IV is associated with the inhibition of endoplasmic reticulum stress-induced podocyte apoptosis in diabetic rats. Cell Physiol Biochem. (2014) 33:1975–87. 10.1159/000362974
35.
Cao J Dai DL Yao L Yu HH Ning B Zhang Q et al . Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. (2012) 364:115–29. 10.1007/s11010-011-1211-9
36.
Ye J Rawson RB Komuro R Chen X Dave UP Prywes R et al . ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell (2000) 6:1355–64. 10.1016/S1097-2765(00)00133-7
37.
Senkal CE Ponnusamy S Bielawski J Hannun YA Ogretmen B . Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. (2010) 24:296–308. 10.1096/fj.09-135087
38.
Xu W Gao L Li T Zheng J Shao A Zhang J . Apelin-13 Alleviates early brain injury after subarachnoid hemorrhage via suppression of endoplasmic reticulum stress-mediated apoptosis and blood-brain barrier disruption: possible involvement of ATF6/CHOP pathway. Neuroscience (2018) 388:284–96. 10.1016/j.neuroscience.2018.07.023
39.
Yoshida H Okada T Haze K Yanagi H Yura T Negishi M et al . ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. (2000) 20:6755–67. 10.1128/MCB.20.18.6755-6767.2000
40.
Ron D Hubbard SR . How IRE1 reacts to ER stress. Cell (2008) 132:24–6. 10.1016/j.cell.2007.12.017
41.
Han D Lerner A Vande Walle L Upton J Xu W Hagen A et al . IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell (2009) 138:562–75. 10.1016/j.cell.2009.07.017
42.
Yoshida H Matsui T Yamamoto A Okada T Mori K . XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell (2001) 107:881–91. 10.1016/S0092-8674(01)00611-0
43.
Hetz C Martinon F Rodriguez D Glimcher LH . The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol Rev. (2011) 91:1219–43. 10.1152/physrev.00001.2011
44.
Madhusudhan T Wang H Dong W Ghosh S Bock F Thangapandi VR et al . Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. (2015) 6:6496. 10.1038/ncomms7496
45.
Yang Y Liu L Naik I Braunstein Z Zhong J Ren B . Transcription factor C/EBP homologous protein in health and diseases. Front Immunol. (2017). 8:1612. 10.3389/fimmu.2017.01612
46.
Kim I Xu W Reed JC . Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. (2008) 7:1013–30. 10.1038/nrd2755
47.
Sari FR Widyantoro B Thandavarayan RA Harima M Lakshmanan AP Zhang S et al . Attenuation of CHOP-mediated myocardial apoptosis in pressure-overloaded dominant negative p38alpha mitogen-activated protein kinase mice. Cell Physiol Biochem. (2011) 27:487–96. 10.1159/000329970
48.
Wang XZ Ron D . Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science (1996) 272:1347–9. 10.1126/science.272.5266.1347
49.
Guo X Meng Y Sheng X Guan Y Zhang F Han Z et al . Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anticancer Drugs (2017) 28:66–74. 10.1097/CAD.0000000000000431
50.
Kim BJ Ryu SW Song BJ . JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. (2006) 281:21256–65. 10.1074/jbc.M510644200
51.
Estaquier J Vallette F Vayssiere JL Mignotte B . The mitochondrial pathways of apoptosis. Adv Exp Med Biol. (2012) 942:157–83. 10.1007/978-94-007-2869-1_7
52.
Bromati CR Lellis-Santos C Yamanaka TS Nogueira TC Leonelli M Caperuto LC et al . UPR induces transient burst of apoptosis in islets of early lactating rats through reduced AKT phosphorylation via ATF4/CHOP stimulation of TRB3 expression. Am J Physiol Regul Integr Comp Physiol. (2011) 300:R92–100. 10.1152/ajpregu.00169.2010
53.
Campos G Schmidt-Heck W Ghallab A Rochlitz K Putter L Medinas DB et al . The transcription factor CHOP, a central component of the transcriptional regulatory network induced upon CCl4 intoxication in mouse liver, is not a critical mediator of hepatotoxicity. Arch Toxicol. (2014) 88:1267–80. 10.1007/s00204-014-1240-8
54.
Singh K Briggs JM . Functional Implications of the spectrum of BCL2 mutations in Lymphoma. Mutation Res Rev. (2016) 769:1–18. 10.1016/j.mrrev.2016.06.001
55.
Hata AN Engelman JA Faber AC . The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov. (2015) 5:475–87. 10.1158/2159-8290.CD-15-0011
56.
Kvansakul M Caria S Hinds MG . The Bcl-2 family in host-virus interactions. Viruses (2017) 9:290. 10.3390/v9100290
57.
Maes ME Schlamp CL Nickells RW . BAX to basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. (2017) 57:1–25. 10.1016/j.preteyeres.2017.01.002
58.
Ghosh AP Klocke BJ Ballestas ME Roth KA . CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PLoS ONE (2012) 7:e39586. 10.1371/journal.pone.0039586
59.
Galehdar Z Swan P Fuerth B Callaghan SM Park DS Cregan SP . Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. (2010) 30:16938–48. 10.1523/JNEUROSCI.1598-10.2010
60.
Iurlaro R Munoz-Pinedo C . Cell death induced by endoplasmic reticulum stress. Febs J. (2016) 283:2640–52. 10.1111/febs.13598
61.
Tsukano H Gotoh T Endo M Miyata K Tazume H Kadomatsu T et al . The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2010) 30:1925–32. 10.1161/ATVBAHA.110.206094
62.
Tuzlak S Kaufmann T Villunger A . Interrogating the relevance of mitochondrial apoptosis for vertebrate development and postnatal tissue homeostasis. Genes Dev. (2016) 30:2133–51. 10.1101/gad.289298.116
63.
Wang W Cheng J Sun A Lv S Liu H Liu X et al . TRB3 mediates renal tubular cell apoptosis associated with proteinuria. Clin Exp Med. (2015) 15:167–77. 10.1007/s10238-014-0287-4
64.
Izrailit J Jaiswal A Zheng W Moran MF Reedijk M . Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene (2017) 36:1048–57. 10.1038/onc.2016.276
65.
Avery J Etzion S DeBosch BJ Jin X Lupu TS Beitinjaneh B et al . TRB3 function in cardiac endoplasmic reticulum stress. Circ Res. (2010) 106:1516–23. 10.1161/CIRCRESAHA.109.211920
66.
Morse E Schroth J You YH Pizzo DP Okada S Ramachandrarao S et al . TRB3 is stimulated in diabetic kidneys, regulated by the ER stress marker CHOP, and is a suppressor of podocyte MCP-1. Am J Physiol Renal Physiol. (2010) 299:F965–72. 10.1152/ajprenal.00236.2010
67.
Kopecka J Salaroglio IC Righi L Libener R Orecchia S Grosso F et al . Loss of C/EBP-beta LIP drives cisplatin resistance in malignant pleural mesothelioma. Lung Cancer (2018) 120:34–45. 10.1016/j.lungcan.2018.03.022
68.
Zhang P Sun Q Zhao C Ling S Li Q Chang YZ et al . HDAC4 protects cells from ER stress induced apoptosis through interaction with ATF4. Cell Signal. (2014) 26:556–63. 10.1016/j.cellsig.2013.11.026
69.
Ohoka N Yoshii S Hattori T Onozaki K Hayashi H . TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. (2005) 24:1243–55. 10.1038/sj.emboj.7600596
70.
Du K Herzig S Kulkarni RN Montminy M . TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science (2003) 300:1574–7. 10.1126/science.1079817
71.
Li Y Zhu D Hou L Hu B Xu M Meng X . TRB3 reverses chemotherapy resistance and mediates crosstalk between endoplasmic reticulum stress and AKT signaling pathways in MHCC97H human hepatocellular carcinoma cells. Oncol Lett. (2018) 15:1343–9. 10.3892/ol.2017.7361
72.
Datta SR Dudek H Tao X Masters S Fu H Gotoh Y et al . Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91:231–41. 10.1016/S0092-8674(00)80405-5
73.
Cravero JD Carlson CS Im HJ Yammani RR Long D Loeser RF . Increased expression of the Akt/PKB inhibitor TRB3 in osteoarthritic chondrocytes inhibits insulin-like growth factor 1-mediated cell survival and proteoglycan synthesis. Arthritis Rheum. (2009) 60:492–500. 10.1002/art.24225
74.
Shimizu K Takahama S Endo Y Sawasaki T . Stress-inducible caspase substrate TRB3 promotes nuclear translocation of procaspase-3. PLoS ONE (2012) 7:e42721. 10.1371/journal.pone.0042721
75.
Shang YY Wang ZH Zhang LP Zhong M Zhang Y Deng JT et al . TRB3, upregulated by ox-LDL, mediates human monocyte-derived macrophage apoptosis. Febs J. (2009) 276:2752–61. 10.1111/j.1742-4658.2009.06998.x
76.
Li T Su L Lei Y Liu X Zhang Y Liu X . DDIT3 and KAT2A Proteins regulate TNFRSF10A and TNFRSF10B expression in endoplasmic reticulum stress-mediated apoptosis in human lung cancer cells. J Biol Chem. (2015) 290:11108–18. 10.1074/jbc.M115.645333
77.
Chen P Hu T Liang Y Li P Chen X Zhang J et al . Neddylation inhibition activates the extrinsic apoptosis pathway through ATF4-CHOP-DR5 axis in human esophageal cancer cells. Clin Cancer Res. (2016) 22:4145–57. 10.1158/1078-0432.CCR-15-2254
78.
Edagawa M Kawauchi J Hirata M Goshima H Inoue M Okamoto T et al . Role of activating transcription factor 3 (ATF3) in endoplasmic reticulum (ER) stress-induced sensitization of p53-deficient human colon cancer cells to tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated apoptosis through up-regulation of death receptor 5 (DR5) by zerumbone and celecoxib. J Biol Chem. (2014) 289:21544–61. 10.1074/jbc.M114.558890
79.
Chang CC Kuan CP Lin JY Lai JS Ho TF . Tanshinone IIA facilitates TRAIL sensitization by up-regulating DR5 through the ROS-JNK-CHOP signaling axis in human ovarian carcinoma cell lines. Chem Res Toxicol. (2015) 28:1574–83. 10.1021/acs.chemrestox.5b00150
80.
Jung KJ Min KJ Bae JH Kwon TK . Carnosic acid sensitized TRAIL-mediated apoptosis through down-regulation of c-FLIP and Bcl-2 expression at the post translational levels and CHOP-dependent up-regulation of DR5, Bim, and PUMA expression in human carcinoma caki cells. Oncotarget (2015) 6:1556–68. 10.18632/oncotarget.2727
81.
Lu M Lawrence DA Marsters S Acosta-Alvear D Kimmig P Mendez AS et al . Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science (2014) 345:98–101. 10.1126/science.1254312
82.
Elmore S . Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. 10.1080/01926230701320337
83.
Chen BL Sheu ML Tsai KS Lan KC Guan SS Wu CT et al . CCAAT-Enhancer-binding protein homologous protein deficiency attenuates oxidative stress and renal ischemia-reperfusion injury. Antioxid Redox signal. (2015) 23:1233–45. 10.1089/ars.2013.5768
84.
Simmen T Lynes EM Gesson K Thomas G . Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta (2010) 1798:1465–73. 10.1016/j.bbamem.2010.04.009
85.
Gross E Sevier CS Heldman N Vitu E Bentzur M Kaiser CA et al . Generating disulfides enzymatically: reaction products and electron acceptors of the endoplasmic reticulum thiol oxidase Ero1p. Proc Natl Acad Sci USA. (2006) 103:299–304. 10.1073/pnas.0506448103
86.
Ramming T Okumura M Kanemura S Baday S Birk J Moes S et al . A PDI-catalyzed thiol-disulfide switch regulates the production of hydrogen peroxide by human Ero1. Free Radic Biol Med. (2015) 83:361–72. 10.1016/j.freeradbiomed.2015.02.011
87.
Li G Mongillo M Chin KT Harding H Ron D Marks AR et al . Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol. (2009) 186:783–92. 10.1083/jcb.200904060
88.
Li G Scull C Ozcan L Tabas I . NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. (2010) 191:1113–25. 10.1083/jcb.201006121
89.
Timmins JM Ozcan L Seimon TA Li G Malagelada C Backs J et al . Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. (2009) 119:2925–41. 10.1172/JCI38857
90.
Song B Scheuner D Ron D Pennathur S Kaufman RJ . Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest. (2008) 118:3378–89. 10.1172/JCI34587
91.
Yin L Dai Y Cui Z Jiang X Liu W Han F et al . The regulation of cellular apoptosis by the ROS-triggered PERK/EIF2alpha/chop pathway plays a vital role in bisphenol A-induced male reproductive toxicity. Toxicol Appl Pharmacol. (2017) 314:98–108. 10.1016/j.taap.2016.11.013
92.
Marciniak SJ Yun CY Oyadomari S Novoa I Zhang Y Jungreis R et al . CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. (2004) 18:3066–77. 10.1101/gad.1250704
93.
Liu CL He YY Li X Li RJ He KL Wang LL . Inhibition of serine/threonine protein phosphatase PP1 protects cardiomyocytes from tunicamycin-induced apoptosis and I/R through the upregulation of p-eIF2alpha. Int J Mol Med. (2014) 33:499–506. 10.3892/ijmm.2013.1603
94.
Mkrtchian S . Targeting unfolded protein response in cancer and diabetes. Endocr Relat Cancer (2015) 22:C1–4. 10.1530/ERC-15-0106
95.
Mihailidou C Chatzistamou I Papavassiliou AG Kiaris H . Improvement of chemotherapeutic drug efficacy by endoplasmic reticulum stress. Endocr Relat Cancer (2015) 22:229–38. 10.1530/ERC-15-0019
96.
Gupta MK Tahrir FG Knezevic T White MK Gordon J Cheung JY et al . GRP78 interacting partner Bag5 responds to ER stress and protects cardiomyocytes from ER stress-induced apoptosis. J Cell Biochem. (2016) 117:1813–21. 10.1002/jcb.25481
97.
Bruchmann A Roller C Walther TV Schafer G Lehmusvaara S Visakorpi T et al . Bcl-2 associated athanogene 5 (Bag5) is overexpressed in prostate cancer and inhibits ER-stress induced apoptosis. BMC Cancer (2013) 13:96. 10.1186/1471-2407-13-96
98.
Maiuri MC Zalckvar E Kimchi A Kroemer G . Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. (2007) 8:741–52. 10.1038/nrm2239
99.
Rouschop KM van den Beucken T Dubois L Niessen H Bussink J Savelkouls K et al . The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. (2010) 120:127–41. 10.1172/JCI40027
100.
B'Chir W Maurin AC Carraro V Averous J Jousse C Muranishi Y et al . The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. (2013) 41:7683–99. 10.1093/nar/gkt563
101.
Kong FJ Ma LL Guo JJ Xu LH Li Y Qu S . Endoplasmic reticulum stress/autophagy pathway is involved in diabetes-induced neuronal apoptosis and cognitive decline in mice. Clin Sci (Lond). (2018) 132:111–25. 10.1042/CS20171432
102.
Ogbechi J Hall BS Sbarrato T Taunton J Willis AE Wek RC et al . Inhibition of Sec61-dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis. (2018) 9:397. 10.1038/s41419-018-0427-y
103.
Shimodaira Y Takahashi S Kinouchi Y Endo K Shiga H Kakuta Y et al . Modulation of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1alpha (IRE1alpha) in human colon cancer cells. Biochem Biophys Res Commun. (2014) 445:524–33. 10.1016/j.bbrc.2014.02.054
104.
Ke PY Chen SSL . Autophagy: a novel guardian of HCV against innate immune response. Autophagy (2011) 7:533–5. 10.4161/auto.7.5.14732
105.
B'Chir W Chaveroux C Carraro V Averous J Maurin AC Jousse C et al . Dual role for CHOP in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell Signal. (2014) 26:1385–91. 10.1016/j.cellsig.2014.03.009
106.
Li S Kong L Yu X . The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit Rev Microbiol. (2015) 41:150–64. 10.3109/1040841X.2013.813899
107.
Zhou Y Qi B Gu Y Xu F Du H Li X et al . Porcine circovirus 2 deploys PERK pathway and GRP78 for its enhanced replication in PK-15 cells. Viruses (2016) 8:56. 10.3390/v8020056
108.
Zhou YS Gu YX Qi BZ Zhang YK Li XL Fang WH . Porcine circovirus type 2 capsid protein induces unfolded protein response with subsequent activation of apoptosis. J Zhejiang Univ Sci B (2017) 18:316–23. 10.1631/jzus.B1600208
109.
Liao Y Fung TS Huang M Fang SG Zhong Y Liu DX . Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol. (2013) 87:8124–34. 10.1128/JVI.00626-13
110.
Zhong Y Liao Y Fang S Tam JP Liu DX . Up-regulation of Mcl-1 and Bak by coronavirus infection of human, avian and animal cells modulates apoptosis and viral replication. PLoS ONE (2012) 7:e30191. 10.1371/journal.pone.0030191
111.
Wang Y Wang R Li Y Sun Y Song C Zhan Y et al . Newcastle disease virus induces G0/G1 cell cycle arrest in asynchronously growing cells. Virology (2018) 520:67–74. 10.1016/j.virol.2018.05.005
112.
Medigeshi GR Lancaster AM Hirsch AJ Briese T Lipkin WI Defilippis V et al . West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol. (2007) 81:10849–60. 10.1128/JVI.01151-07
113.
Cai Z Shen L Ma H Yang J Yang D Chen H et al . Involvement of endoplasmic reticulum stress-mediated C/EBP homologous protein activation in coxsackievirus B3-induced acute viral myocarditis. Circ Heart Fail. (2015) 8:809–18. 10.1161/CIRCHEARTFAILURE.114.001244
114.
Su HL Liao CL Lin YL . Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J Virol. (2002) 76:4162–71. 10.1128/JVI.76.9.4162-4171.2002
115.
Huang M Xu A Wu X Zhang Y Guo Y Guo F et al . Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Arch Virol. (2016) 161:699–703. 10.1007/s00705-015-2715-5
116.
Ma R Yang L Niu F Buch S . HIV Tat-mediated induction of human brain microvascular endothelial cell apoptosis involves endoplasmic reticulum stress and mitochondrial dysfunction. Mol Neurobiol. (2016) 53:132–42. 10.1007/s12035-014-8991-3
117.
Shah A Vaidya NK Bhat HK Kumar A . HIV-1 gp120 induces type-1 programmed cell death through ER stress employing IRE1alpha, JNK and AP-1 pathway. Sci Rep. (2016) 6:18929. 10.1038/srep18929
118.
Seimon TA Kim MJ Blumenthal A Koo J Ehrt S Wainwright H et al . Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE (2010) 5:e12772. 10.1371/journal.pone.0012772
119.
Akazawa Y Isomoto H Matsushima K Kanda T Minami H Yamaghchi N et al . Endoplasmic reticulum stress contributes to Helicobacter pylori VacA-induced apoptosis. PLoS ONE (2013) 8:e82322. 10.1371/journal.pone.0082322
120.
Zhu P Xue J Zhang ZJ Jia YP Tong YN Han D et al . Helicobacter pylori VacA induces autophagic cell death in gastric epithelial cells via the endoplasmic reticulum stress pathway. Cell Death Dis. (2017) 8:3207. 10.1038/s41419-017-0011-x
121.
Lee SY Lee MS Cherla RP Tesh VL . Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. (2008) 10:770–80. 10.1111/j.1462-5822.2007.01083.x
122.
Lee MS Cherla RP Leyva-Illades D Tesh VL . Bcl-2 regulates the onset of shiga toxin 1-induced apoptosis in THP-1 cells. Infect Immun. (2009) 77:5233–44. 10.1128/IAI.00665-09
123.
Park JY Jeong YJ Park SK Yoon SJ Choi S Jeong DG et al . Shiga toxins induce apoptosis and ER stress in human retinal pigment epithelial cells. Toxins (2017) 9:E319. 10.3390/toxins9100319
124.
Fung TS Liao Y Liu DX . The endoplasmic reticulum stress sensor IRE1alpha protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J Virol. (2014) 88:12752–64. 10.1128/JVI.02138-14
125.
Zhang HM Ye X Su Y Yuan J Liu Z Stein DA et al . Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol. (2010) 84:8446–59. 10.1128/JVI.01416-09
126.
Liu Z Zhang HM Yuan J Ye X Taylor GA Yang D . The immunity-related GTPase Irgm3 relieves endoplasmic reticulum stress response during coxsackievirus B3 infection via a PI3K/Akt dependent pathway. Cell Microbiol. (2012) 14:133–46. 10.1111/j.1462-5822.2011.01708.x
127.
Ambrose RL Mackenzie JM . West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. (2011) 85:2723–32. 10.1128/JVI.02050-10
128.
Faherty CS Maurelli AT . Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol. (2008) 16:173–80. 10.1016/j.tim.2008.02.001
129.
Cui Y Zhao D Barrow PA Zhou X . The endoplasmic reticulum stress response: a link with tuberculosis?Tuberculosis (Edinburgh, Scotland) (2016) 97:52–6. 10.1016/j.tube.2015.12.009
130.
Choi HH Shin DM Kang G Kim KH Park JB Hur GM et al . Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. (2010) 584:2445–54. 10.1016/j.febslet.2010.04.050
131.
Derrick SC Morris SL . The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. (2007) 9:1547–55. 10.1111/j.1462-5822.2007.00892.x
132.
Lee MS Cherla RP Lentz EK Leyva-Illades D Tesh VL . Signaling through C/EBP homologous protein and death receptor 5 and calpain activation differentially regulate THP-1 cell maturation-dependent apoptosis induced by Shiga toxin type 1. Infect Immun. (2010) 78:3378–91. 10.1128/IAI.00342-10
133.
Cubillos-Ruiz JR Bettigole SE Glimcher LH . Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell (2017) 168:692–706. 10.1016/j.cell.2016.12.004
134.
Wang HQ Du ZX Zhang HY Gao DX . Different induction of GRP78 and CHOP as a predictor of sensitivity to proteasome inhibitors in thyroid cancer cells. Endocrinology (2007) 148:3258–70. 10.1210/en.2006-1564
135.
Battson ML Lee DM Jarrell DK Hou S Ecton KE Phan AB et al . Tauroursodeoxycholic acid reduces arterial stiffness and improves endothelial dysfunction in type 2 diabetic mice. J Vasc Res. (2017) 54:280–7. 10.1159/000479967
136.
Ding W Wang B Zhang M Gu Y . Involvement of endoplasmic reticulum stress in uremic cardiomyopathy: protective effects of tauroursodeoxycholic acid. Cell Physiol Biochem. (2016) 38:141–52. 10.1159/000438616
137.
Dromparis P Paulin R Stenson TH Haromy A Sutendra G Michelakis ED . Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation (2013) 127:115–25. 10.1161/CIRCULATIONAHA.112.133413
138.
Okamoto T Suzuki T Kusakabe S Tokunaga M Hirano J Miyata Y et al . Regulation of apoptosis during flavivirus infection. Viruses (2017) 9:E243. 10.3390/v9090243
Summary
Keywords
C/EBP homologous protein, endoplasmic reticulum stress, apoptosis, microorganisms, virus, bacteria
Citation
Hu H, Tian M, Ding C and Yu S (2019) The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 9:3083. doi: 10.3389/fimmu.2018.03083
Received
04 September 2018
Accepted
13 December 2018
Published
04 January 2019
Volume
9 - 2018
Edited by
Marina De Bernard, Università degli Studi di Padova, Italy
Reviewed by
Matthew J. Robson, University of Cincinnati, United States; Frank Kruyt, University Medical Center Groningen, Netherlands
Updates
Copyright
© 2019 Hu, Tian, Ding and Yu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengqing Yu yus@shvri.ac.cn
This article was submitted to Microbial Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.