 Matteo Vecellio1,2,3*†
Matteo Vecellio1,2,3*† Carla J. Cohen1,2,3
Carla J. Cohen1,2,3 Amity R. Roberts4Paul B. Wordsworth1,2,3†
Amity R. Roberts4Paul B. Wordsworth1,2,3† Tony J. Kenna5,6*†
Tony J. Kenna5,6*†- 1Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, United Kingdom
- 2Oxford Musculoskeletal Biomedical Research Unit, National Institute for Health Research, Oxford, United Kingdom
- 3Oxford Comprehensive Biomedical Research Centre, Botnar Research Centre, National Institute for Health Research, Nuffield Orthopaedic Centre, Oxford, United Kingdom
- 4Dunn School of Pathology, University of Oxford, Oxford, United Kingdom
- 5Translational Research Institute, Princess Alexandra Hospital, Brisbane, QLD, Australia
- 6Faculty of Health, Institute of Health and Biomedical Innovation, Queensland University of Technology, Brisbane, QLD, Australia
Susceptibility to ankylosing spondylitis (AS) is polygenic with more than 100 genes identified to date. These include HLA-B27 and the aminopeptidases (ERAP1, ERAP2, and LNPEPS), which are involved in antigen processing and presentation to T-cells, and several genes (IL23R, IL6R, STAT3, JAK2, IL1R1/2, IL12B, and IL7R) involved in IL23 driven pathways of inflammation. AS is also strongly associated with polymorphisms in two transcription factors, RUNX3 and T-bet (encoded by TBX21), which are important in T-cell development and function. The influence of these genes on the pathogenesis of AS and their potential for identifying drug targets is discussed here.
Introduction: AS Genetics
Ankylosing Spondylitis (AS) is a chronic inflammatory enthesitis predominantly affecting the axial skeleton (1). It is often associated with other conditions, such as uveitis, psoriasis, and inflammatory bowel disease (IBD), with which it has significant genetic overlap (2–5). The strongest genetic association of AS is with the HLA-B27 immune response gene (6). About 85% of British AS cases carry HLA-B27 (odds ratio ~60) but probably no more than ~5% of those who are HLA-B27 positive actually develop the condition, suggesting that other genes are also involved. Twin studies also support polygenic susceptibility (7) and this has been amply confirmed by genome-wide association studies (GWAS) (8–10). More than 100 loci have already been implicated in AS but for most the mechanisms underlying the associations are unknown. One exception appears to be the multiple “hits” in genes involved in interleukin (IL)-23 driven pro-inflammatory pathways (e.g., IL23R, IL12B, IL6R, TYK2, IL27R, IL1R2, IL1R1, and STAT3) (9–11). In murine models of the disease, resident IL23R-expressing cells can be demonstrated at the entheses (stress-bearing fibrous or fibro cartilaginous attachment of connective tissues, such as tendons or ligaments to bone). Further, the main features of the disease can be recapitulated simply by inducing hepatic overexpression of IL-23 without any evidence of recruitment of additional cell types to the entheses (12).
HLA-B27 and Aminopeptidases
Because HLA-B27 plays a key role in antigen presentation to CD8+ cytotoxic T-cells (13), it is plausible that the inflammation in AS might be mediated by HLA-B27-restricted CD8+ cytotoxic T-cells, perhaps generated in response to microbial infection (14). However, it is also possible that depressed CD8+ T-cell function might be responsible, which could account for certain animal models of spondyloarthropathy (SpA) that develop in the absence of CD8+ T-cells (15). Further, in some models of autoimmune demyelinating disease, such as multiple sclerosis, CD8+ T-cells can act as either effector or suppressor cells (16). Specifically, the absence of CD8+ T-cells can be associated with more severe chronic disease or with increased susceptibility to relapses (17).
One of the earliest and most exciting GWAS findings indicated strong association between AS and certain aminopeptidases, initially with ERAP1 (endoplasmic reticulum aminopeptidase 1) but subsequently also with ERAP2 and the related LNPEP (leucyl/cystinyl aminopeptidase) (9). Further, some of these associations were replicated in related conditions, including psoriasis and IBD (18, 19). ERAP1 has the second strongest association with AS and displays a synergistic interaction with HLA-B27 (18–21); the ERAP1 association is lost in HLA-B27 negative cases but a weaker association with ERAP2 can be seen both in HLA-B27 negative and positive cases (22). ERAP1 and other aminopeptidases appear to play a significant role in trimming peptides transported from the cytosol to the endoplasmic reticulum to optimal length (8 or 9 amino acids) for loading on HLA class I molecules (23, 24). We and others have shown that AS-associated ERAP1 variants are involved in dysregulated peptide trimming that profoundly affects the range of peptide antigens presented to the immune system (25–27).
The protective allele of the ERAP1 rs30187 polymorphism is associated with reduced peptidase activity resulting in alterations in the HLA-interacting peptidome (24, 28) of both pathogen-derived and host-derived antigens compared to the high-risk allele. Theoretically small molecule inhibitors of ERAP1 might therefore be of interest in the treatment of AS.
The Contribution of IL23R
IL23R (encoding the specific portion of the heterodimeric interleukin 23 receptor) was the first non-MHC gene to be associated with AS (29, 30). The primary IL23R association with AS (also psoriasis and IBD) is with rs11209026, a coding SNP in the cytoplasmic tail, which alters IL-23R signaling (31, 32). In addition, a second independent association signal has been identified in the intergenic region downstream of IL23R and upstream of IL12RB2 (encoding the 130kd β2 chain of the IL-12 receptor) (10). Our group recently identified a putative enhancer in this region of independent association, where the rs11209032 SNP it is likely to be the candidate causal SNP in this region. Allelic variation of rs11209032 may influence Th1-cell numbers (33). Further work is necessary to explain the mechanisms for these important observations.
IL-23 has critical roles in the pathogenesis of autoimmunity: it induces the Th17 cell population with a unique inflammatory gene signature (IL17A, IL17F, IL6, CSF2, TNF, CCL20, CCL22, IL1R1, and IL23R) (34).
The multiple associations (IL23R, IL6R, STAT3, JAK2, IL1R1/2, IL12B, and IL7R) with AS in the IL-23-driven pathways played a prominent role in promoting the successful targeting of the pro-inflammatory cytokine IL17A with the therapeutic monoclonal antibody secukinumab (35). Recently it has also been demonstrated that it is possible to use epigenetic approaches to inhibit Th17 cell maturation (integral to the pathogenesis of SpA) by targeting the bromodomains of transcriptional co-activators (such as CBP and p300) that recognize acetylated lysine residues in the regulatory elements associated with pro-inflammatory genes (36). However, the precise mechanisms involved in these therapeutic approaches are not entirely straightforward. Thus, although secukinumab is highly effective in AS (37), recent clinical trials of risankizumab, which targets the p19 subunit of IL-23 have proved ineffective (38). It is possible that there is a pharmacokinetic reason for this failure but this seems relatively unlikely given that the drug seems to be effective in similar doses in psoriasis and IBD (39, 40). It would seem more likely that there are alternative pathways through which IL17 production is sustained other than purely through IL23-driven mechanisms. Analogous therapeutic differences have been noted previously in the failure of secukinumab to benefit those with IBD despite its obvious immunogenetic similarities to AS (41). Further studies, both basic and clinical, are needed to unpick the mechanistic differences between the outcomes of the secukinumab and risankizumab trials.
GM-CSF: a Novel Target in AS
GM-CSF, a cytokine involved in hematopoiesis and in immune responses, has been recently identified as a promising target in AS therapy. GM-CSF and its receptor are overexpressed in the synovial joints of patients with immune-mediated inflammatory SpA (42–44). A unique subset of GM-CSF+ CD4 T-cells has been identified by transcriptomics that represents an effector population distinct from Th1 and Th17 cells (45). GM-CSF promotes joint damage by recruiting granulocyte and macrophage precursors from activated bone marrow adjacent to synovial joints and causes the release of pro-inflammatory chemokines such as CCL17 (46). These processes, which are expanded in the synovial fluid of patients with axial SpA, led by GM-CSF, also induce the destruction of the cartilage and the resorption of the bone, inducing several matrix metalloproteinases and the osteoclast activating factor—RANKL (Receptor Activator of Nuclear Factor Kappa-B Ligand) (47, 48).
The use of monoclonal antibodies to neutralize GM-CSF have been very effective in preventing the progression of joint damage and inflammation in arthritic mice (49) and long-term phase II studies in RA are very encouraging (50). Furthermore, based on the evidences discussed above, a clinical trial with antibody targeting GM-CSF has recently started in AS patients.
Transcription Factors Associated With AS
Most DNA single nucleotide polymorphisms (SNP) associated with complex diseases like AS do not cause protein coding changes; the R381N polymorphism in the cytoplasmic tail of the IL23 receptor associated with susceptibility to AS and IBD is the exception rather than the rule in this regard (32). On the contrary, most genetic associations in such multifactorial conditions are in non-coding regions of the genome where they exert epigenetic regulatory effects (51). Such associations in intergenic regions are well described in AS, and in at least some cases, probably alter susceptibility to the disease by influencing gene expression levels. Disease associated polymorphisms might influence regulatory elements (enhancers, silencers, promoters) through changes in chromatin remodeling or transcription factor binding. SNPs in regulatory regions could affect gene expression through changes in transcription factors (TFs) recruitment, DNA methylation, histone modification or miRNA expression or binding.
RUNX3
The AS association at the RUNX3 locus (Runt-related transcription factor 3) provides a good example of this type of genetic regulatory influence (10). There is convincing evidence that RUNX3 is associated with AS and other forms of SpA, including psoriatic arthritis (52). RUNX3 plays a prominent role in the development and differentiation of CD8+ T-cells, which have been implicated in the pathogenesis of AS (53, 54). Three other AS–associated genes (EOMES, IL7R, and ZMIZ1) impacting on variation in CD8+ lymphocyte counts were also identified by the International Genetics of Ankylosing Spondylitis (IGAS) consortium (10). The robust genetic associations with RUNX3, IL7R, and EOMES (eomesodermin), all of which impact on CD8+ T-cells differentiation, support the involvement of CD8 T-cell pathology in AS. However, the precise mechanisms involved are likely to be more complex than simple effects on T-cell numbers. Thus, although the risk haplotype at RUNX3 (rs6600247) is associated with lower CD8+ T cell counts the opposite is seen with the risk haplotype at IL7R (rs991570), suggesting involvement of an unknown mechanism related to the IL7R/RUNX3 pathway.
RUNX3 also has other fundamental roles in many other cell types. Its deletion leads to the dysregulation of cells including neurons, chondrocytes, Th1 helper cells, dendritic cells and NK cells (55).
In particular, RUNX3 is downstream of the TGF-β signaling pathway and may play a key role in CD4+ T-cell differentiation, potentially driving the imbalance of Th17/Treg cell in AS (56). RUNX3 is also a pivotal TF for the function of Innate Lymphoid Cells 3 (ILC3) via driving the expression of RORγt and its downstream target aryl hydrocarbon receptor (AHR), in ILC3 cells. RUNX3 deletion increases the susceptibility of ILCs to infection with Citrobacter rodentium (57).
Previously we have shown that an AS-associated SNP (rs4648889), located upstream of the RUNX3 promoter affects RUNX3 gene expression in CD8+ T-cells through changes in transcription factor binding (58). These findings implicate CD8+ T-cells in the pathophysiology of AS, and raise the possibility that reduced CD8+ T-cell numbers and/or altered function may be contribute to its pathogenesis (Figure 1). Subsequently, we described another SNP (rs4265380) also upstream of RUNX3 and only 500 bp from rs4648889, which has potential regulatory functions in monocytes rather than T-cells (59). The role of RUNX3 in the myeloid compartment has not been extensively studied in immune biology (60, 61), but clearly could be very important in our understanding of the pathophysiology of AS. Similar genetic associations at the RUNX3 locus have been also described in psoriatic arthritis, thereby revealing an unsurprising degree of genetic overlap between these two related forms of SpA (52).
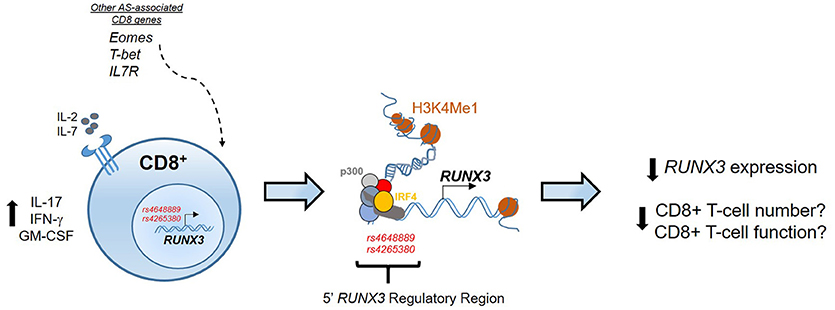
Figure 1. Epigenetic regulation at the RUNX3 AS-associated locus. In CD8+ T-cells the regulatory region upstream the promoter of RUNX3 is characterized by binding of several TFs, included p300 and IRF4, and enhancer histone marks (H3K4Me1). RUNX3 AS-risk allele has an epigenetic effect to reduce RUNX3 expression that might affect CD8+ T-cell numbers and function. The contribution of other genes associated with AS, like T-bet, Eomes and IL7R strengthen the involvement of CD8+ T-cells development pathway in AS pathogenesis.
Further evidence from Al-Mossawi and colleagues supports the plausible role of monocytes in AS pathogenesis. Their study showed that monocytes upregulate IL7R expression and soluble IL7R secretion after LPS treatment in a functional, genotype- and TNFα-dependant manner. These data draw attention to an unappreciated key myeloid role for AS risk variants at IL7R (62).
These findings suggest that the IL7R/RUNX3 axis might have a plausible role in monocyte biology and in the pathogenic process of AS, which up to now has been poorly investigated.
TBX21
AS is associated with rs11657479, a SNP in the 3′ untranslated region (UTR) of TBX21 (encoding the transcription factor T-bet) (10). Although the causal variant has not yet been defined since the locus has not been fine mapped. Expression of TBX21 and T-bet is increased in patients with AS and homozygosity for the rs11657479 risk allele increases T-bet expression. In the SKG mouse model of SpA, loss of T-bet expression protects against disease (63) suggesting that T-bet is an important regulator of immune responses in SpA. Genetic associations have also been reported at TBX21 in AS and IBD (64), suggesting that T-bet may play an important role in these diseases, most likely involving control of mucosal barrier defenses.
T-bet was first described in CD4 T-cells as the key Th1 lineage-defining transcription factor controlling expression of IFN-γ and CXCR3 (65). It is now clear, however, that T-bet is widely expressed in immune cell subsets and controls functional differentiation of many cell types including CD8 T cells, NK cells, B cells.
Enhanced T-bet expression in AS is predominantly seen in CD8 T-cells and NK cells where it influences function of these cell types (63), both of which are strongly implicated in AS pathogenesis (66). The development and terminal maturation of immature NK cells, characterized by the expression of TNF-related apoptosis-inducing ligand (TRAIL) (67, 68), are dependent on T-bet (69). However, the importance of T-bet in defining NK cell biology may be tissue or context specific. For example in mice, liver resident NK cells (or ILC1 cells) depend on T-bet while conventional NK cells are strictly dependent on eomesodermin (67, 68).
T-bet regulation of CD8 T-cells responses is similarly context dependent. T-bet promotes differentiation of short-lived effector CD8 T-cells but does not influence long-lived memory CD8 T-cells (70). Importantly, T-bet cooperates with eomesodermin and other transcription factors to fine tune CD8 T-cell functions. For example, cooperative effects of T-bet and eomesodermin induce IFN-γ production by CD8 T cells (71). Indeed, in mice much is known about cooperation between T-bet and several transcription factors such as ZEB2 (72) and Blimp-1 (73). Yet our knowledge of cooperation between T-bet and other transcription factors in humans and in disease settings is limited. Given the genetic associations with TBX21, EOMES, and RUNX3 in AS (10) further investigation of transcription factor biology in AS is warranted. More broadly, a better understanding of T-bet regulation of CD8 T cell responses in humans would be beneficial.
While studies in Tbx21−/− mice have highlighted some important T-bet functions the regulation of immune cell responses by T-bet is nuanced. For example, graded expression of T-bet in CD4 T-cells responding to Listeria monocytogenes infections determines which cells produce IFN-γ (T-bethi) and which ones do not mount a protective IFN- γ response (T-betlo) (74). Of direct relevance to AS, IL-12 induces graded expression of T-bet in mouse CD8 T-cells which in turn controls functional potential of those CD8 T-cells (70) while T-bet expression CD4 Th17 cells stabilizes pathogenic Th17 cells (75). In mice at least, T-bet plays a role in controlling transcription of Il23r (76) and expression of Cxcr3 in multiple immune cells (77), thereby shaping their tissue migratory potential. It is therefore clear that T-bet, alone and in concert with other transcription factors, fine tunes functions of multiple immune cell types but the relevance of this to chronic immune-mediated diseases in humans is yet to be investigated. Since T-bet also play important roles in gut barrier function (78, 79) dissecting the role T-bet plays in host-microbiome responses in chronic immune-mediated diseases is also likely to be of value to understanding disease- and tissue-specific immunopathogenesis processes (Figure 2).
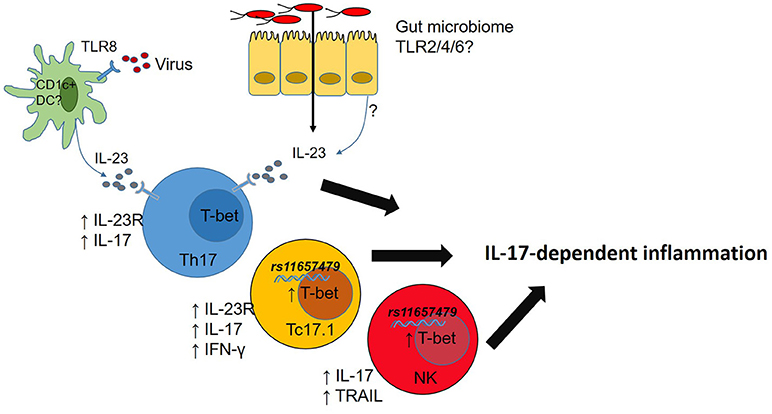
Figure 2. Microbial dysbiosis or viral infection drives IL-23 production. T-bet expression in T cells promotes IL-23R expression licensing IL-23-mediated inflammation. Carriage of rs11657479 enhances T-bet expression in CD8 T cells and NK cells and enhances pro-inflammatory functions in those cell types.
Coordinated Effects of AS-Associated Transcription Factors
There is still much to do to define all the functional consequences of AS-associated genetic variations in transcription factors like T-bet, eomesodermin and RUNX3. This is made more complicated by the fact that their effects are likely to be cell type specific.
It has been demonstrated that together with T-bet, RUNX3 regulates interferon-γ and interleukin 4 expression in T-helper Th1 cells. Moreover, in Th1 cells the Runx3 expression is induced in a T-bet-dependent manner (80). This fact implies a plausible synergy between these two factors and a plausible role in AS pathogenesis.
Although AS is highly heritable, the contribution of individual genes (or biological pathways) has been relatively poorly explored, with the notable exception of IL23-driven inflammation and a few other examples. The RUNX3/T-bet/eomesodermin axis could represent a key pathway for exploring the differentiation of many key immune cells relevant to AS (66).
The different examples we have here reported clearly illustrated how genetic associations can provide useful information about the finding and the development of potential drug targets.
Of note, the interplay between chromatin state, TF occupancy and tissue-specific gene expression is critical (81), and an integrative approach is needed in order to increase the power of the analysis and fully understand the AS-associated regulatory mechanism, of which the RUNX3/T-bet axis is not exempt.
Next Steps in RUNX3 and T-Bet Biology in AS
Current data support the importance of RUNX3+ CD8 T-cells, T-bet+ CD8 T-cells, and T-bet+ NK cells in the pathogenesis of AS. These data in turn validate the need to study these immune cell populations in further detail in AS cases. The functional properties of RUNX3+ CD8 T-cells and T-bet+ CD8 T-cells and NK cells are still largely unresolved. The effects of manipulating these cell populations in mice and humans requires further investigation. A detailed functional genomic approach, such as cell-type specific ChIP-Seq and RNA-seq, will help to identify the downstream targets of RUNX3 and T-bet in these cells. Only through these more comprehensive studies will we be able to define useful therapeutic targets more accurately.
Conclusions and Perspectives
GWAS have the potential to provide key insights into the pathogenesis of complex polygenic disorders even where the strength of individual associations seem weak. Even modest genetic associations may reveal therapeutic targets with profound biological effects. Thus, the relatively modest but robust association of the R381N SNP in the cytoplasmic tail of IL-23R, which has functional consequences, clearly implicates IL-23 pathways in AS; targeting the main effector cytokine IL-17A in this pathway with the therapeutic antibody secukinumab has proved highly effective (35).
Nevertheless, considerable work will be required to finely dissect the biological mechanisms at play to elucidate why, for example, anti-IL-23 antibodies work in IBD and psoriasis but not in AS (38–40). It will be also vital to see the results of the ongoing trials targeting GPR65, a G-protein coupled receptor overexpressed in AS patients, with an antibody against GM-CSF: a positive outcome will open new ways for future investigations.
In general, such coding polymorphisms are unusual in the context of polygenic diseases and more subtle genetic influences, arising from polymorphisms in regulatory elements influencing gene expression, are more likely to be implicated in complex diseases like AS (82). For example, additive influences have been described between the various genes in the IL-23 signaling pathway on the effector functions of Th1 and Th17 cells in patients with SpA (83), but the full range of factors involved in the regulation of Th17 cells is only just beginning to be appreciated (84). This is the goal also for the RUNX3/T-bet/eomesodermin axis. Targeting these transcription factors, such as RUNX3, directly will not only be technically challenging but may prove harmful due to their broad effects on cell biology and human immunity. However, determining the interplay between them and defining their targets, both individual and collective, in the context of AS will advance our understanding of the pathogenesis of AS. Ultimately this may identify novel cell-specific therapeutic targets with acceptable risk benefit profiles.
Author Contributions
MV, AR, PW, and TK conceived the manuscript. MV, PW, and TK drafted the manuscript and all the authors revised the final version prior submission.
Funding
MV was funded by Arthritis Research UK (grant 21428). AR and CC were funded by Arthritis Research UK (grant 20402). TK was funded by a Marion A. Simpson Grant from Arthritis Australia. Additional funding was provided by Arthritis Research UK (18797, 19536, 20402, 20796), the NIHR Thames Valley Collaborative Research Network and National Ankylosing Spondylitis Society (UK).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors apologize to those authors whose work is not cited due to space constraints.
References
1. Dougados M, van der Linden S, Juhlin R, Huitfeldt B, Amor B, Calin A, et al. The European spondylarthropathy study group preliminary criteria for the classification of spondylarthropathy. Arthritis Rheum. (1991) 34:1218–27. doi: 10.1002/art.1780341003
2. Robinson PC, Claushuis TA, Cortes A, Martin TM, Evans DM, Leo P, et al. Genetic dissection of acute anterior uveitis reveals similarities and differences in associations observed with ankylosing spondylitis. Arthritis Rheum. (2015) 67:140–51. doi: 10.1002/art.38873
3. Danoy P, Pryce K, Hadler J, Bradbury LA, Farrar C, Pointon J, et al. Association of variants at 1q32 and STAT3 with Ankylosing spondylitis suggests genetic overlap with Crohn's disease. PLoS Genet. (2010) 6:e1001195. doi: 10.1371/journal.pgen.1001195
4. Robinson PC, Brown MA. Genetics of ankylosing spondylitis. Mol Immunol. (2014) 57:2–11. doi: 10.1016/j.molimm.2013.06.013
5. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. (2013) 14:661–73. doi: 10.1038/nrg3502
6. Brewerton DA, Hart FD, Nicholls A, Caffrey M, James DC, Sturrock RD. Ankylosing spondylitis and HL-A 27. Lancet (1973) 1:904–7. doi: 10.1016/S0140-6736(73)91360-3
7. Brown MA, Kennedy LG, MacGregor AJ, Darke C, Duncan E, Shatford JL, et al. Susceptibility to ankylosing spondylitis in twins: the role of genes, HLA, and the environment. Arthritis Rheum. (1997) 40:1823–8. doi: 10.1002/art.1780401015
8. Australo-Anglo-American Spondyloarthritis Consortium (TASC) Reveille JD, Sims AM, Danoy P, Evans DM, Leo P, et al. Genome-wide association study of ankylosing spondylitis identifies non-MHC susceptibility loci. Nat Genet. (2010) 42:123–7. doi: 10.1038/ng.513
9. Evans DM, Spencer CC, Pointon JJ, Su Z, Harvey D, Kochan G, et al. Interaction between ERAP1 and HLA-B27 in ankylosing spondylitis implicates peptide handling in the mechanism for HLA-B27 in disease susceptibility. Nat Genet. (2011) 43:761–7. doi: 10.1038/ng.873
10. International Genetics of Ankylosing Spondylitis Consortium (IGAS), Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, et al. (2013) Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat Genet. 45:730–40. doi: 10.1038/ng.2667
11. Davidson SI, Liu Y, Danoy PA, Wu X, Thomas GP, Jiang L, et al. Association of STAT3 and TNFRSF1A with ankylosing spondylitis in Han Chinese. Ann Rheum Dis. (2011) 70:289–92. doi: 10.1136/ard.2010.133322
12. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
13. Fiorillo MT, Maragno M, Butler R, Dupuis ML, Sorrentino R. CD8+ T-cell autoreactivity to an HLA-B27–restricted self-epitope correlates with ankylosing spondylitis. J Clin Invest. (2000) 106:47–53. doi: 10.1172/JCI9295
14. Schirmer M, Goldberger C, Würzner R, Duftner C, Pfeiffer KP, Clausen J, et al. Circulating cytotoxic CD8(+) CD28(-) T cells in ankylosing spondylitis. Arthritis Res. (2002) 4:71–6. doi: 10.1186/ar386
15. Taurog JD, Dorris ML, Satumtira N, Tran TM, Sharma R, Dressel R, et al. Spondylarthritis in HLA-B27/human beta2-microglobulin-transgenic rats is not prevented by lack of CD8. Arthritis Rheum. (2009) 60:1977–84. doi: 10.1002/art.24599
16. Najafian N, Chitnis T, Salama AD, Zhu B, Benou C, Yuan X, et al. Regulatory functions of CD8+CD28–T cells in an autoimmune disease model. J Clin Invest. (2003) 112:1037–48. doi: 10.1172/JCI17935
17. Jiang H, Zhang SI, Pernis B. Role of CD8+ T cells in murine experimental allergic encephalomyelitis. Science (1992) 256:1213–5. doi: 10.1126/science.256.5060.1213
18. Genetic Analysis of Psoriasis Consortium and the Wellcome Trust Case Control Consortium 2, Strange A, Capon F, Spencer CC, Knight J, Weale ME, et al. A Genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet. (2010) 42:985–90. doi: 10.1038/ng.694
19. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
20. Agrawal N, Brown MA. Genetic associations and functional characterization of M1 aminopeptidases and immune-mediated diseases. Genes Immun. (2014) 15:521–7. doi: 10.1038/gene.2014.46
21. Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. (2010) 42:1118–25. doi: 10.1038/ng.717
22. Robinson PC, Costello ME, Leo P, Bradbury LA, Hollis K, Cortes A, et al. ERAP2 is associated with ankylosing spondylitis in HLA-B27-positive and HLA-B27-negative patients. Ann Rheum Dis. (2015) 74:1627–9. doi: 10.1136/annrheumdis-2015-207416
23. Chen L, Fischer R, Peng Y, Reeves E, McHugh K, Ternette N, et al. Critical role of endoplasmic reticulum aminopeptidase 1 in determining the length and sequence of peptides bound and presented by HLA-B27. Arthritis Rheumatol. (2014) 66:284–94. doi: 10.1002/art.38249
24. Kochan G, Krojer T, Harvey D, Fischer R, Chen L, Vollmar M, et al. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc Natl Acad Sci USA. (2011) 108:7745–50. doi: 10.1073/pnas.1101262108
25. Martin-Esteban A, Gomez-Molina P, Sanz-Bravo A, López de Castro JA. Combined effects of ankylosing spondylitis –associated ERAP1 polymorphisms outside the catalytic and peptide-binding sites on the processing of natural HLA-B27 ligands. J Biol Chem. (2014) 289:3978–90. doi: 10.1074/jbc.M113.529610
26. Chen L, Ridley A, Hammitzsch A, Al-Mossawi MH, Bunting H, Georgiadis D, et al. Silencing or inhibition of endoplasmic reticulum aminopeptidase 1 (ERAP1) suppresses free heavy chain expression and Th17 responses in ankylosing spondylitis. Ann Rheum Dis. (2016) 75:916–23. doi: 10.1136/annrheumdis-2014-206996
27. Nagarajan NA, de Verteuil DA, Sriranganadane D, Yahyaoui W, Thibault P, Perreault C, et al. ERAAP shapes the peptidome associated with classical and non-classical MHC class I molecules. J Immunol. (2016) 197:1035–43. doi: 10.4049/jimmunol.1500654
28. Cortes A, Pulit SL, Leo PJ, Pointon JJ, Robinson PC, Weisman MH, et al. Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat Commun. (2015) 6:7146. doi: 10.1038/ncomms8146
29. Wellcome Trust Case Control Consortium; Australo-Anglo-American Spondylitis Consortium (TASC), Burton PR, Clayton DG, Cardon LR, Craddock N, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. (2007) 39:1329–37. doi: 10.1038/ng.2007.17
30. Karaderi T, Harvey D, Farrar C, Appleton LH, Stone MA, Sturrock RD, et al. Association between the interleukin 23 receptor and ankylosing spondylitis is confirmed by a new UK case–control study and meta-analysis of published series. Rheumatology (2009) 48:386–9. doi: 10.1093/rheumatology/ken501
31. Di Meglio P, Villanova F, Napolitano L, Tosi I, Terranova Barberio M, Mak RK, et al. The IL23R A/Gln381 allele promotes IL-23 unresponsiveness in human memory T-helper 17 cells and impairs Th17 responses in psoriasis patients. J Invest Dermatol. (2013) 133:2381–9. doi: 10.1038/jid.2013.170
32. Di Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, Villanova F, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE (2011) 6:e17160. doi: 10.1371/journal.pone.0017160
33. Roberts AR, Vecellio M, Chen L, Ridley A, Cortes A, Knight JC, et al. An ankylosing spondylitis-associated genetic variant in the IL23R-IL12RB2 intergenic region modulates enhancer activity and is associated with increased Th1-cell differentiation. Ann Rheum Dis. (2016) 75:2150–6. doi: 10.1136/annrheumdis-2015-208640
34. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
35. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet (2013) 382:1705–13. doi: 10.1016/S0140-6736(13)61134-4
36. Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci USA. (2015) 112:10768–73. doi: 10.1073/pnas.1501956112
37. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N Engl J Med. (2015) 373:2534–48. doi: 10.1056/NEJMoa1505066
38. Baeten D, Østergaard M, Wei JC, Sieper J, Järvinen P, Tam LS, et al. (2018). Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann Rheum Dis. 77:1295–302. doi: 10.1136/annrheumdis-2018-213328
39. Feagan BG, Sandborn WJ, D'Haens G, Panés J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin 23 inhibitor risankizumab in patients with moderate-to-severe Crohn's disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet (2017) 389:1699–709. doi: 10.1016/S0140-6736(17)30570-6
40. Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. (2017). Risankizumab versus ustekinumab for moderate to severe plaque psoriasis. N Eng J Med. 376:1551–1560. doi: 10.1056/NEJMoa1607017
41. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. (2012). Sekukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's Disease: unexpected results of a randomized, double-blind placebo-controlled trial. Gut 61:1693–700. doi: 10.1136/gutjnl-2011-301668
42. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. (2015) 75:899–907. doi: 10.1136/annrheumdis-2014-206578
43. Zhang L, Fu J, Sheng K, Li Y, Song S, Li P, et al. Bone marrow CD11b(+)F4/80(+) dendritic cells ameliorate collagen-induced arthritis through modulating the balance between Treg and Th17. Int Immunopharmacol. (2015) 25:96–105. doi: 10.1016/j.intimp.2015.01.014
44. Al-Mossawi MH, Ridley A, Chen L, de Wit J, Bowness P. Role of lymphocytes producing GM-CSF in human spondyloarthritis. Lancet (2017) 389:S21. doi: 10.1016/S0140-6736(17)30417-8
45. Al-Mossawi MH, Chen L, Fang H, Ridley A, de Wit J, Yager N, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. (2017) 8:1510. doi: 10.1038/s41467-017-01771-2
46. Achuthan A, Cook AD, Lee MC, Saleh R, Khiew HW, Chang MWN, et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest. (2016) 126:3453–66. doi: 10.1172/JCI87828
47. Udagawa N, Kotake S, Kamatani N, Takahashi N, Suda T. The molecular mechanism of osteoclastogenesis in rheumatoid arthritis. Arthritis Res. (2002) 4:281–9. doi: 10.1186/ar431
48. Balani D, Dolderb S, Aeberli D, Kamgang R, Hofstetter W, Seitz M. IL-17A inhibits osteoclast development by inducing the release of GM-CSF in osteoblast lineage cells. Bone (2012) 50:S88. doi: 10.1016/j.bone.2012.02.260
49. Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol. (2014) 193:849–59. doi: 10.4049/jimmunol.1303255
50. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovský J, Godwood A, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor α monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:679–89. doi: 10.1002/art.40420
51. Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. (2009) 106:9362–7. doi: 10.1073/pnas.0903103106
52. Apel M, Uebe S, Bowes J, Giardina E, Korendowych E, Juneblad K, et al. Variants in RUNX3 contribute to susceptibility to psoriatic arthritis, exhibiting further common ground with ankylosing spondylitis. Arthritis Rheum. (2013) 65:1224–31. doi: 10.1002/art.37885
53. Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, et al. Differential requirements for runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell (2002) 111:621–33. doi: 10.1016/S0092-8674(02)01111-X
54. Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. (2009) 206:51–9. doi: 10.1084/jem.20081242
55. Lotem J, Levanon D, Negreanu V, Bauer O, Hantisteanu S, Dicken J, et al. Runx3 at the interface of immunity, inflammation and cancer. Biochim Biophys Acta (2015) 1855:131–43. doi: 10.1016/j.bbcan.2015.01.004
56. Fainaru O, Woolf E, Lotem J, Yarmus M, Brenner O, Goldenberg D, et al. Runx3 regulates mouse TGF-beta-mediated dendritic cell function and its absence results in airway inflammation. EMBO J. (2004) 23:969–79. doi: 10.1038/sj.emboj.7600085
57. Asquith M, Rosenbaum JT. The interaction between host genetics and the microbiome in the pathogenesis of spondyloarthropathies. Curr Opin Rheumatol. (2016) 28:405–12. doi: 10.1097/BOR.0000000000000299
58. Vecellio M, Roberts AR, Cohen CJ, Cortes A, Knight JC, Bowness P, et al. The genetic association of RUNX3 with ankylosing spondylitis can be explained by allele-specific effects on IRF4 recruitment that alter gene expression. Ann Rheum Dis. (2016) 75:1534–40. doi: 10.1136/annrheumdis-2015-207490
59. Vecellio M, Cortes A, Roberts AR, Ellis J, Cohen CJ, Knight JC, et al. Evidence for a second ankylosing spondylitis-associated RUNX3 regulatory polymorphism. RMD Open. (2018) 4:e000628. doi: 10.1136/rmdopen-2017-000628
60. Puig-Kröger A, Aguilera-Montilla N, Martínez-Nuñez R, Domínguez-Soto A, Sánchez-Cabo F, Martín-Gayo E, et al. The novel RUNX3/p33 isoform is induced upon monocyte-derived dendritic cell maturation and downregulates IL-8 expression. Immunobiology (2010) 215:812–20. doi: 10.1016/j.imbio.2010.05.018
61. Sánchez-Martín L, Estecha A, Samaniego R, Sánchez-Ramón S, Vega MÁ, Sánchez-Mateos P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood (2011) 117:88–97. doi: 10.1182/blood-2009-12-258186
62. Al-Mossawi H, Lau L, Danielli S, de Wit J, Makino S, Yager N, et al. The Autoimmune Disease Risk Allele rs6897932 Modulates Monocyte IL7R Surface and Soluble Receptor Levels in a context-Specific Manner. bioRxiv: 262410 (2018). doi: 10.1101/262410
63. Lau MC, Keith P, Costello ME, Bradbury LA, Hollis KA, Thomas R, et al. Genetic association of ankylosing spondylitis with TBX21 influences T-bet and pro-inflammatory cytokine expression in humans and SKG mice as a model of spondyloarthritis. Ann Rheum Dis. (2017) 76:261–9. doi: 10.1136/annrheumdis-2015-208677
64. Soderquest K, Hertweck A, Giambartolomei C, Henderson S, Mohamed R, Goldberg R, et al. Genetic variants alter T-bet binding and gene expression in mucosal inflammatory disease. PLoS Genet. (2017) 13:e1006587. doi: 10.1371/journal.pgen.1006587
65. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell (2000) 100:655–669. doi: 10.1016/S0092-8674(00)80702-3
66. Li Z, Haynes K, Pennisi DJ, Anderson LK, Song X, Thomas GP, et al. Epigenetic and gene expression analysis of ankylosing spondylitis-associated loci implicate immune cells and the gut in the disease pathogenesis. Genes Immun. (2017) 18:135–43. doi: 10.1038/gene.2017.11
67. Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity (2012) 36:55–67. doi: 10.1016/j.immuni.2011.11.016
68. Daussy C, Faure F, Mayol K, Viel S, Gasteiger G, Charrier E, et al. T-bet and Eomes instruct the development of two distinct natural killer cell lineages in the liver and in the bone marrow. J Exp Med. (2014) 211:563–77. doi: 10.1084/jem.20131560
69. Townsend MJ, Weinmann AS, Matsuda JL, Salomon R, Farnham PJ, Biron CA, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity (2004) 20:477–94. doi: 10.1016/S1074-7613(04)00076-7
70. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
71. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. (2005) 6:1236–44. doi: 10.1038/ni1268
72. Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, et al. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J Exp Med. (2015) 212:2041–56. doi: 10.1084/jem.20150186
73. Xin A, Masson F, Liao Y, Preston S, Guan T, Gloury R, et al. A molecular threshold for effector CD8(+) T cell differentiation controlled by transcription factors Blimp-1 and T-bet. Nat Immunol. (2016) 17:422–32. doi: 10.1038/ni.3410
74. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. (2011) 12:255–63. doi: 10.1038/ni.1993
75. Wang Y, Godec J, Ben-Aissa K, Cui K, Zhao K, Pucsek AB, et al. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-γ-producing T helper 17 cells. Immunity (2014) 40:355–66. doi: 10.1016/j.immuni.2014.01.002
76. Gocke AR, Cravens PD, Ben LH, Hussain RZ, Northrop SC, Racke MK, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. (2007) 178:1341–8. doi: 10.4049/jimmunol.178.3.1341
77. Matsuda JL, George TC, Hagman J, Gapin L. Temporal dissection of T-bet functions. J Immunol. (2007) 178:3457–65. doi: 10.4049/jimmunol.178.6.3457
78. Sciumé G, Hirahara K, Takahashi H, Laurence A, Villarino AV, Singleton KL, et al. Distinct requirements for T-bet in gut innate lymphoid cells. J Exp Med. (2012) 209:2331–8. doi: 10.1084/jem.20122097
79. Ermann J, Staton T, Glickman JN, de Waal Malefyt R, Glimcher LH. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice. Proc Natl Acad Sci USA. (2014) 111:E2559–66. doi: 10.1073/pnas.1408540111
80. Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells Nat Immunol. (2007) 8:145–53. doi: 10.1038/ni1424
81. Wong ES, Schmitt BM, Kazachenka A, Thybert D, Redmond A, Connor F, et al. Interplay of cis and trans mechanisms driving transcription factor binding and gene expression evolution. Nat Commun. (2017) 8:1092. doi: 10.1038/s41467-017-01037-x
82. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet. (2016) 48:510–8. doi: 10.1038/ng.3528
83. Coffre M, Roumier M, Rybczynska M, Sechet E, Law HK, Gossec L, et al. Combinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin-23 signaling pathway in spondyloarthritis. Arthritis Rheum. (2013) 65:1510–21. doi: 10.1002/art.37936
Keywords: ankylosing spondylitis, inflammation, functional genomics, autoimmunity, therapy
Citation: Vecellio M, Cohen CJ, Roberts AR, Wordsworth PB and Kenna TJ (2019) RUNX3 and T-Bet in Immunopathogenesis of Ankylosing Spondylitis—Novel Targets for Therapy? Front. Immunol. 9:3132. doi: 10.3389/fimmu.2018.03132
Received: 14 September 2018; Accepted: 18 December 2018;
Published: 10 January 2019.
Edited by:
Francesco Ciccia, University of Palermo, ItalyReviewed by:
James Frederick Burrows, Queen's University Belfast, United KingdomYong-Gil Kim, University of Ulsan College of Medicine, South Korea
Copyright © 2019 Vecellio, Cohen, Roberts, Wordsworth and Kenna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Vecellio, bWF0dGVvLnZlY2VsbGlvQG5kb3Jtcy5veC5hYy51aw==
Tony J. Kenna, dG9ueS5rZW5uYUBxdXQuZWR1LmF1
†These authors have contributed equally to this work