 Martin Christopher James Maiden
Martin Christopher James Maiden- Department of Zoology, University of Oxford, Oxford, United Kingdom
Since it became available as a routine tool in biology, the determination and analysis of nucleotide sequences has been applied to the design of vaccines and the investigation of their effectiveness. As vaccination is primarily concerned with the interaction of biological molecules with the immune system, the utility of sequence data is not immediately obvious and, indeed, nucleotide sequence data are most effective when used to complement more conventional immunological approaches. Here, the impact of sequencing on the field of vaccinology will be illustrated with reference to the development and implementation of vaccines against Neisseria meningitidis (the meningococcus) over the 30-year period from the late-1980s to the late-2010s. Nucleotide sequence-based studies have been important in the fight against this aggressive pathogen largely because of its high genetic and antigenic diversity, properties that were only fully appreciated because of sequence-based studies. Five aspects will be considered, the use of sequence data to: (i) discover vaccine antigens; (ii) assess the diversity and distribution of vaccine antigens; (iii) determine the evolutionary and population biology of the organism and their implications for immunization; and (iv) develop molecular approaches to investigate pre- and post-vaccine pathogen populations to assess vaccine impact. One of the great advantages of nucleotide sequence data has been its scalability, which has meant that increasingly large data sets have been available, which has proved invaluable in the investigation of an organism as diverse and enigmatic as the meningococcus.
Introduction
The 40 years following the introduction of the Sanger dideoxy method in 1977 (1) saw a revolution in biology, which was driven by the improvement of nucleotide sequencing technologies. At the start of this period, determining a DNA or RNA sequence was a highly specialized task, which was undertaken in a very few laboratories most often using their own home-made equipment and reagents. Only individual genes or viruses could be sequenced, and then at great expense. By 2018, nucleotide sequencing was conducted on an industrial scale, employing mass-produced reagents and highly automated equipment, often in large factory-like installations. Complete genome sequences were assembled routinely for tens of thousands, even hundreds of thousands of organisms, including those with the largest genomes. Major advances had also been made in the computer equipment and software available to interpret the data produced, although it is fair to say that while issues of data generation were for all practical purposes resolved, data interpretation remained a major hurdle. In common with most areas of biology, vaccine development, and evaluation were transformed by these advances and this transformation will be illustrated here using meningococcal vaccines as an example.
Many of the most successful bacterial and viral vaccines were developed in the mid- to late- twentieth century, without recourse to the detailed genetic information that nucleotide sequencing provides; however, most of these conventionally-developed vaccines targeted antigenically stable pathogens, such as the smallpox virus, or those that rely on a single, stable, molecule for their pathogenicity, such as Corynebacterium diphtheriae (diphtheria) and Clostridium tetani (tetanus). The bacterium Neisseria meningitidis, the meningococcus, very nearly falls in to this category, as almost all invasive meningococcal disease (IMD) is caused by bacteria that express one of only six capsular polysaccharide antigens, referred to as serogroups A, B, C, W, X, and Y. Polysaccharide and, especially, protein-conjugate polysaccharide, vaccines are effective in protecting against disease for five of these (serogroups A, C, X, W, X, and Y) and provide a means of eliminating most IMD worldwide (2). Unfortunately, however, vaccines directed against meningococcal serogroup B polysaccharide have not been developed. This is a consequence of poor immunogenicity and fear of inducing host autoimmune disease, due to similarities of the serogroup B polysaccharide with human antigens (3, 4). The search for alternative antigens that target serogroup B meningococci has been complicated by the high variability of virtually all other possible meningococcal vaccine components. Ever-increasing volumes of nucleotide sequence data have been used in this search.
As successful immunization is almost always a population process, population studies based on nucleotide sequences have important applications in the development, deployment, and evaluation of vaccines. Here I shall describe how nucleotide sequence technologies have contributed to vaccine development and the assessment of vaccines, outlining the development, and implementation of meningococcal vaccines from the late 1970s to the time of writing.
Structure and Variation in Individual Antigens
Determining the nucleotide sequence of a gene encoding a vaccine antigen permits the deduction of a wealth of information concerning the protein and enables a wide range of follow-up studies. For example, the PorA protein was established as a potential vaccine component for “serogroup B substitute” vaccines (i.e., vaccines developed as an alternative to those including serogroup B capsular polysaccharide as an antigen) in the late 1970s and early 1980s, on the basis of immunological and biochemical studies (5, 6). The cloning and sequencing of the porA gene in 1991 provided much additional information, for example confirming that it was a typical porin related to those found in the gonococcus (7). The meningococcal and gonococcal sequences were used to design oligonucleotide primers for the then new PCR technique, enabling the amplification, and sequencing of the porA gene from multiple meningococcal isolates (8), illustrating how sequence technologies lead to cumulative gains in knowledge. These comparative studies enabled structural models to be proposed and established that the antigenic variability identified by subtype-specific monoclonal antibodies was mostly determined by the peptide sequences of two major variable regions (VR1 and VR2) and one less variable region (VR3 or sVR) of the PorA protein. Combining sequencing and immunological studies enabled these antigens and their interaction with immune molecules to be defined (8–10).
The combination of PCR amplification and Sanger sequencing enables multiple variants of a given gene to be characterized accurately and rapidly at high volume. This permits the antigenic variability of a given protein vaccine component to be established. In the case of meningococcal PorA, the variants of which were initially identified with monoclonal antibodies (11), an ever-increasing diversity of variants have been identified by sequencing in the past 30 years. This required that the original nomenclature, which was based on monoclonal antibody reactivity, needed to be replaced with an updated nomenclature scheme that was based on the peptide sequences themselves, rather than the antibodies that reacted with them (12). It was important that this scheme was backwards compatible with the designations of the established antibody-based system. The scheme needed to be infinitely expandable, and to this end variant numbers were used, which could be added to as each new variant was discovered. At the time of writing at total of 324 peptide variants had be defined or PorA VR1 and 906 for PorA VR2. An advantage of sequence-based nomenclature is that the sequences can be recorded on open-access web based databases (e.g., https://pubmlst.org/neisseria/PorA/), enabling easy access to the nomenclature (13). Similar approaches have since been used to catalog the variation of a number of meningococcal vaccine candidates including: factor H binding protein (fHbp) (14); the ferric enterochelin receptor, FetA (15); Neisseria adhesin A (NadA) (16); and the heamoglobin receptor (HmbR) (17).
In addition to describing the nature and extent of variation of genes, including those encoding vaccine antigens, sequence analyses help to reveal the mechanisms whereby this variation occurs. In the case of PorA, the immunogenic VRs are relatively short continuous sequences, encoding surface-exposed parts of the porin structure (18), which vary by point mutation, insertion, and deletion. Each of these processes can have an impact on the binding of immune molecules such as antibodies to the expressed protein. These impacts have been assessed by a combination of sequence comparison, biochemical, structural, and immunological analyses (19, 20) (Figure 1). Sequencing studies have also established that protein antigen expression can be influenced by the sequence of control regions, with polynucleotide tracts playing an important role in the expression of a number of meningococcal antigens (21). Another mechanism of variation that sequencing studies identified is the exchange of genetic material via horizontal genetic transfer (HGT), and the PorA protein was one of the first bacterial genes in which this was extensively documented (22). The recognition of the importance of HGT in bacterial evolution came from studies of antibiotic resistance and vaccine antigens in the gonococcus (23), meningococcus (22), and pneumococcus (24), with a major impact on our understanding on bacterial population biology and evolution (25).
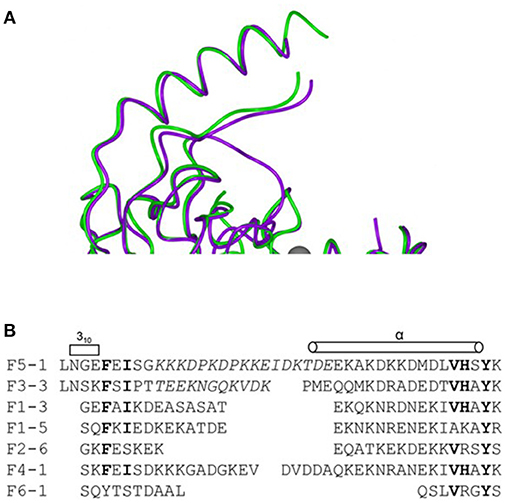
Figure 1. Combining sequence data with and structural information to define variation in a meningococcal antigen. The structure and alignment of the FrpB (FetA) hypervariable sequence regions. (A) Superposition of two hypervariable regions F5-1 purple, F3-3 (A chain) green. (B) Sequence alignment of the HR regions from the major FrpB variants. Reproduced without modification from Saleem et al. (20) under CC BY.
Bacterial Population Biology and Evolutionary Studies
Knowledge of the extent of variation in vaccine antigens inevitably poses questions of how this variation comes about and how it moves through the population. Both topics have important implications for vaccine design, as it is essential to know how effective a given vaccine will be once it has been introduced and how easily vaccine escape variants might arise and spread. Population studies of the pathogenic Neisseria, both the meningococcus and Neisseria gonorrheae (the gonococcus), have played a major role in developing bacterial population genetics. In the case of the meningococcus, these studies have also been central in the design and implementation of new vaccines.
Bacterial population studies predate the sequencing era, with seminal investigations of the meningococcus using a combination of multi-locus enzyme electrophoresis (MLEE) (26) and immunological typing (27–29), establishing fundamental concepts that have been built on subsequently by sequence-based investigations. MLEE investigations conducted on isolates from cases of invasive disease, showed that a limited number of groups of closely related organisms, known as “clones” or “genetic lineages,” each of which was associated with particular antigenic characteristics, were predominant causes of disease (27, 29–32). By contrast, meningococci isolated from asymptomatic carriers were much more diverse (33, 34). This led to the concept of “hypervirulent” or “hyperinvasive” meningococci: persistent genotypes that were especially likely to cause IMD (35). One such lineage, associated with electrophoretic type 5 (ET-5), caused an international outbreak of serogroup B IMD (30), resulting in the development and implementation of specific outer membrane vesicle (OMV) vaccines generated against the local outbreak strain in Norway (36) and Cuba (37). This approach was repeated in New Zealand 15 years later (38).
The high levels of HGT observed in genes encoding antigens (22, 39) and antibiotic resistance (23), sometimes involving inter-species transfer (23, 40), are also a observed in “housekeeping” genes: those expected to be subject to stabilizing selection for the conservation of metabolic function (41). This demonstrates the important of HGT in bacterial evolution generally, and especially in the Neisseria. One consequence of this is the a range of population structures observed in the meningococcus (25). MLEE technology was difficult to implement and compare across studies and, as sequencing technology developed, it was replaced by a nucleotide sequence-based approach, multilocus sequence typing (MLST), which indexed the variation in seven ~400 bp fragments of housekeeping genes: abcZ; adk; aroE; fumC; gdh; pdhC; and pgm. MLST was much easier to implement at scale and had the advantage of being easily portable among laboratories (42). Originally developed for Neisseria meningitis, MLST has had wide application to many bacterial species and, in combination with web-accessible databases, has become a widely used method, with applications in, for example: evolutionary studies; epidemiology; population biology; and taxonomy (42). MLST studies enabled genetic lineages to be defined in terms of groups of related STs called clonal complexes (ccs), which were named after the ST that was used to define that complex. Thus, ST-11 is the defining sequence type for clonal complex 11 (cc11), also known as the “ST-11 complex.” These molecular designations have been incorporated with other typing schemes such as serogrouping (for capsule) (43) (Figure 2), subtyping (for PorA) (12) and typing of the FetA antigen (Figure 1) (15) into a standardized typing nomenclature with the form: B: P1.19,15: F5-1: ST-33 (cc32), referring to: group (in this case B); PorA VR1 and VR2 type (P1.19,15); FetA VR type (F5-1); sequence type (ST-33); and clonal complex (cc32), respectively (44).
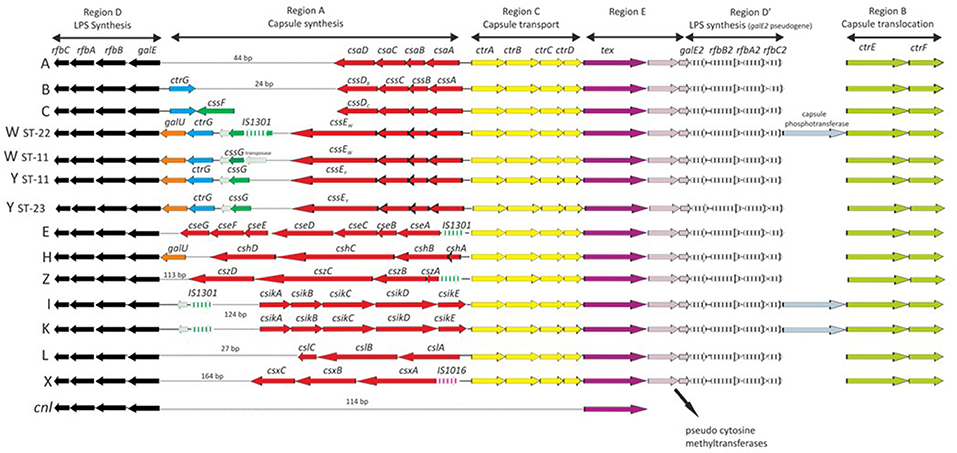
Figure 2. Genetic organization of the cps locus among Neisseria meningitidis serogroups revealed by nucleotide sequencing. Serogroups A (N. meningitidis isolate Z2491); B (H44/76); C (FAM18, 053442, and 29013); W (α275); W (WUE171); Y (α162); Y (WUE172); E (α707); H (29031); I (29043); K (29046); L (WUE3608); X (α388); and Z (WUE173); and a capsule null (cnl) isolate. Letters on left represent serogroups. Arrows depict gene orientation. Reproduced without modification from Harrison et al. (43) under CC BY.
The Population Approach to Vaccine Assessment
High-throughput sequencing approaches, which permit the characterization of hundreds or thousands of bacteria isolates at multiple loci, enable a population genetic approach to be taken to the assessment of vaccination programs. In the case of N. meningitidis, these approaches played a central role in understanding the impact of conjugate polysaccharide vaccines, which targeted the meningococcal capsular antigen (2). In late 1999, just after the publication of the MLST approach in 1998, the United Kingdom (UK) Department of Health implemented a novel monovalent serogroup C conjugate polysaccharide vaccine, in response to a dramatic increase in serogroup C meningococcal disease in the UK (45). The protein-polysaccharide vaccines had significant advantages over the older “plain” polysaccharide vaccines, in that the conjugate vaccines generated more effective immune responses. This included eliciting protective responses in younger individuals, affinity maturation, and a memory response, none of which were generated by the “plain” polysaccharide vaccines (46). A combination of the urgency of the public health response and the epidemiology of IMD meant that the new meningococcal C conjugate (MCC) vaccines were implemented without the benefit of a phase III efficacy trial (2). This led to several uncertainties, not least that it was unknown whether the vaccine would affect asymptomatic carriage. Although the prospect of directly preventing disease was wholly positive, there was a concern that population effects might lead to the emergence of vaccine escape variants—variants of the epidemic clone that had acquired a different capsule by HGT (47). Investigation of this required monitoring, not only disease isolates to establish impact on disease, but also carried meningococci, during vaccine implementation period.
The United Kingdom Meningococcal Carriage (UKMenCar) study was established to measure the impact of MCC on carriage and surveyed 47,765 teenagers from 1999 to 2001, immediately before and for 2 years after vaccine implementation (48–50). Oropharyngeal swab samples and conventional culturing were used to isolate meningococci from the throats of the subjects. In combination with phenotyping, high-throughput single-locus sequencing of the capsular region combined with MLST analysis enabled the nature and status of the serogroups to be determined, together with the clonal complex of each isolate. These data showed that, before vaccine implementation, the point prevalence of carriage of the epidemic-causing meningococcus (C:cc11) was low (0.31% of individuals), which is characteristic of a highly hyperinvasive meningococcus. In 2001, 2 years after implementation, this carriage rate decreased by 80% (to 0.04% of individuals). As a proportion of the meningococci isolated, the C:cc11 epidemic strain dropped by nearly 90%, from 1.83 to 0.21%, with a coincident reduction in the proportion expressing the capsular antigen of around 50% (48). This resulted in a significant herd immunity (protection) of the unvaccinated (51), which was fortunate as the initial vaccination schedule used for infants (2, 3, and 4 months) provided no direct protection more than a year after immunization (52). The vaccination program had no significant effect on the carriage of other genotypes and serogroups, although there was some evidence for secular changes over the period of the surveys (50).
The success of the monovalent MCC vaccines, demonstrated to be in large degree due to their ability to induce herd immunity, catalyzed interest in developing a similar vaccine to target the periodic very large epidemics seen in the African Meningitis Belt. These represent one of the most important manifestations of IMD globally and were first described by Lapeyssonnie (53). Since that time, repeated serogroup A epidemics had been observed, with especially large outbreaks in the late 1990s (54). The meningitis vaccine project (MVP) was established in 2001 as a partnership between PATH (formerly the Program for Appropriate Technology in Health) and the World Health Organization (WHO), funded by the Bill and Melinda Gates foundation. The aim of the MVP was the elimination of meningococcal epidemics in Africa by means of an affordable serogroup A protein-polysaccharide conjugate vaccine. Employing an innovative public-private approach with northern and southern partners, this goal was achieved, with the vaccine PsA-TT, “MenAfriVac®,” introduced in December 2010, with a plan to immunize all African Meningitis Belt countries by 2013 (55, 56).
The Meningococcal Carriage in Africa (MenAfriCar) consortium was established in 2008 to measure the impact of the PsA-TT vaccine on carriage and the impact of herd immunity (57). Up until that time, knowledge of the carriage of meningococci in this region was incomplete, with a number of studies undertaken at various times employing a variety of techniques. This limited the collation of consistent information and a wide range of carriage prevalence rates (3–30%) had been reported (58). The MenAfriCar consortium aimed to conduct large-scale carriage surveys across the meningitis belt before and after the introduction of the PsA-TT vaccine using consistent methods. Risk factor data for carriage were simultaneously collected, as in the UKMenCar surveys, although the risk factors included were somewhat different from those seen in the UK (59, 60). One unknown was the impact of the carriage of other Neisseria species, especially Neisseria lactamica, which is commonly isolated from individuals in Africa (61). This was challenging as members of the genus are very similar: they are poorly distinguishable by 16SrRNA sequencing (62), for example. The large number of isolates that would have to be processed by laboratories in resource-limiting environments presented further challenges that were met by the exploitation of nucleotide sequence-based approaches, made possible by the availability of genomic technologies.
The MenAfriCar surveys employed a combination of conventional culture, biochemical, and sequence-based methodologies. As in UKMenCar surveys, oropharyngeal swab samples were collected and cultured on selective media with putative Neisseria identified using biochemical tests in local laboratories (60). From these cultures, boiled cell preparations were made, which were shipped to Oxford where the molecular analyses were performed (61). To solve the speciation problem a novel assay was designed. This took advantage of an extended MLST scheme, ribosomal MLST (rMLST), which indexes the 53 genes encoding ribosomal proteins, and enables bacterial isolate characterization “from domain to strain” (63). From the rMLST sequences extracted from WGS data from 44 isolates of diverse species, a 413 bp fragment of the rplF gene was identified, the sequence of which was diagnostic for each Neisseria species. This fragment could be amplified and sequenced at high-throughput, enabling Neisseria species identification which was rapid, accurate, and cost-effective (64). The capsule genes were identified with a real time PCR assay and fine typing performed by sequencing the porA and fetA loci (60).
The MenAfriCar studies showed great diversity of meningococcal carriage across the belt and over time, which differed from the patterns of carriage typically observed in high-income countries with temperate climates, where carriage is more consistent (65). The age profiles of carriers was also different, with meningococcal carriage highest in individuals aged 5–14 (60), rather than in adults and adolescents as typically seen elsewhere (65). Transmission among children within households was shown to be important (66), again different from other settings, where social interactions outside the family are important (59). There was also much more diversity in the non-meningococcal Neisseria species isolated, the carriage of which was also age-related (61). This different dynamic was consistent with the unique epidemiology observed in African meningitis belt countries, and with the idea that the occurrence of seasonal epidemics in the meningitis belt were dependent on the transmission of epidemic clones.
Most of the counties in which PsA-TT was introduced including the first, Burkina Fasso, were not experiencing an epidemic of serogroup A IMD at the time of introduction, making assessment of the herd effects of the vaccine difficult, although effects on disease and carriage rates were consistent with such effects (67). An epidemic in Chad during the introduction there, however, enabled a direct demonstration of efficacy against IMD and carriage of the epidemic strain (68) (Figure 3). The rollout of the vaccines in different districts in over two epidemic years, combined with the MenAfriCar sampling and sequence-based isolate characterization protocols, demonstrated high efficacy of the vaccination against both IMD and carriage (68). Samples collected in this study also demonstrated that even at genomic levels of isolate characterization, there were no consistent differences between carried and invasive meningococci (69).
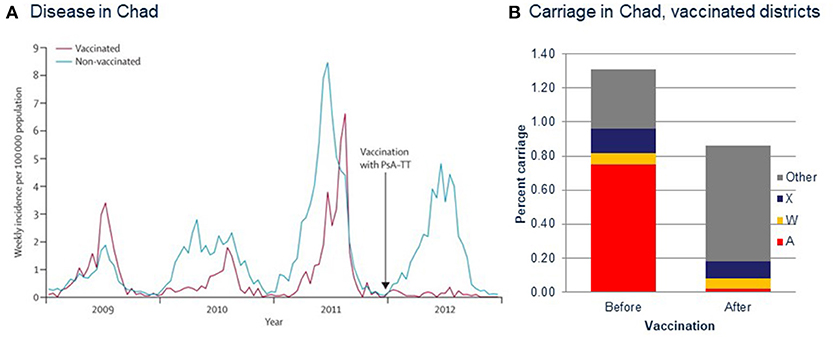
Figure 3. Measuring vaccine efficacy in the meningitis belt during PsA-TT (MenAfriVac) introduction in Chad using genomic sequencing techniques. (A) Incidence of reported cases of meningitis in Chad, 2009–2012. Vaccination with PsA–TT was undertaken in patients aged 1–29 years at the end of 2011 (arrow). PsA–TT=serogroup A meningococcal polysaccharide–tetanus toxoid conjugate vaccine. (B) Carriage rates of meningococci of different serogroups before and after vaccination. (A) Reproduced from Daugla et al. (68) without modification under CC BY.
The marked successes of the conjugate vaccines in different settings provided the prospect of a “meningitis free world,” so long as an effective group B vaccine could be developed (70); however, no vaccine against the meningococcal serogroup B polysaccharide, either plain or protein-conjugate, has been developed to the time of writing and none was under development (3). This was due to a combination of the poor immunogenicity of the antigen combined with fears of the induction of auto-immunity, as a consequence of its similarity of the antigen to human neural polysaccharides (4). Consequently, even with the prospect of conjugate polysaccharide vaccines that target serogroups A, C, W, X, and Y (71), there is a need for alternative or “substitute serogroup B” vaccines, if IMD is to be comprehensively prevented (3).
Genomic Disease Surveillance: Understanding and Combatting Virulence and Vaccine Escape
The development and validation of next generation sequencing approaches for the determination of high-quality draft WGSs of meningococci (72), led to the establishment of the Meningitis Research Foundation Meningococcus Genome Library (MRF-MGL) (73) (Figure 4). This repository contained the WGS data for all meningococci isolated from cases of IMD in the UK. The MRF-MGL provides the opportunity to identify and react to IMD outbreaks in real time or near real time. An example of this is the reaction of the UK public health authorities to a serogroup W IMD outbreak. From the early 2010s onwards, coinciding with the establishment of the MRF-MGL, there was a year-on-year increase in cases of serogroup W meningococcal disease, which data from the MRF-MGL showed to be W:cc11 (74). A similar increase had been observed a decade before (75), just after the successful introduction of the MCC vaccines, causing some alarm (76). The former increase had been associated with the global spread of a particular W:cc11 strain after the Hajj pilgrimage and had fortunately been transitory (77); however, the epidemiology of the cases in the 2010s was somewhat different, leading to the concern that this might be a different circumstance and that a larger epidemic might occur (74). A WGS study of a global collection of 750 diverse cc11 meningococci demonstrated that, although the cases in the UK were indeed related to the Hajj-associated isolates, they were much more closely related at the WGS level to W:cc11 meningococci that had caused large-scale epidemic outbreaks in South America (78). In combination with other epidemiological information, these data formed the basis of a decision to implement tetravalent A, C, W, Y conjugate vaccines into the UK immunization program for teenagers (79). Certainly, this was the first use of genomics to change national vaccination policy for meningococci and perhaps for any organism.
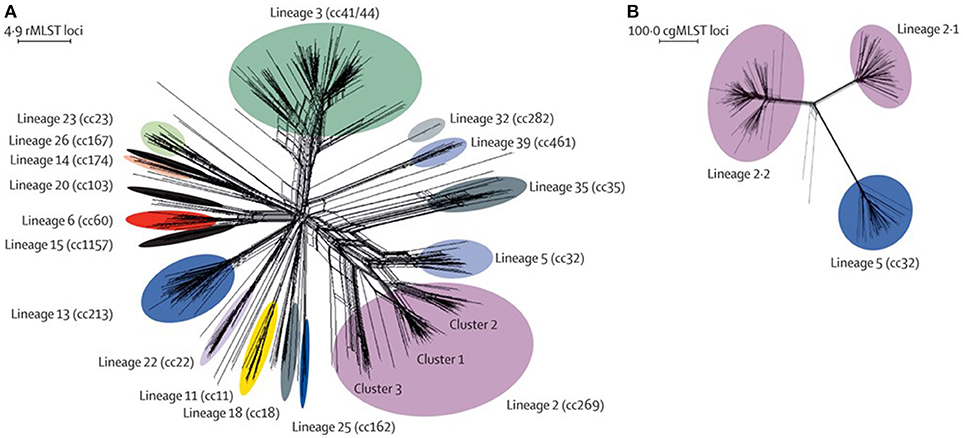
Figure 4. Genetic diversity of disease-associated meningococcal isolates in the Meningitis Research Foundation Meningococcus Genome Library for England and Wales (A) Neighbor-Net graph showing the relationships of all 498 rMLST profiles (ribosomal sequence types) within the 899 isolates available for epidemiological years 2010–2011 and 2011–2012. (B) Relationships among isolates belonging to lineage 2 (cc269; n = 171) and lineage 5 (cc32) isolates (n = 42) assessed with 1605 cgMLST loci. This analysis illustrates the improved resolution achieved with cgMLST for the substructures within and between lineages, compared with rMLST. rMLST=ribosomal multilocus sequence typing. cc=clonal complex. cgMLST=core-genome multilocus sequence typing. Reproduced without modification from Hill et al. (73) Under CC BY.
Genome Sequencing Vaccine Design and Assessment
The availability of whole genome sequences (WGS) of bacterial pathogens also provided novel opportunities in the development of vaccines. In the case of the meningococcus, the first meningococcal WGSs, from bacterial isolates MC58 (80) and Z2491 (81) both published 2000 played a role in the development of two “serogroup B substitute” vaccines: Bexsero® (4CMenB, developed in Siena, Italy) (82); and Trumenba® (LP2086, developed in Pearl River, USA) (83). In the case of the Bexsero® vaccine a “reverse vaccinology” (84) approach was adopted using the MC58 genome sequences as its starting point. In contrast to more conventional approaches, which took the interaction of a bacterial isolate as a starting point, reverse vaccinology started by predicting potential vaccine antigens (i.e., surface-exposed proteins) from a genome sequence, and then assessing these in animals. This identified three potential targets, fHbp, NadA, and NHBA that were eventually included in the final Bexsero® formulation, along with the MenNZB OMV vaccine, made against the New Zealand outbreak strain (38). Interestingly, a more conventional vaccine development approach, but which did use sequence information from the Z2498 meningococcal isolate to find the gene sequence from protein sequences, identified the fHbp protein (also known as LP2086) as an important vaccine candidate (83).
Given the known diversity of meningococcal protein antigens, assessment of the levels of diversity of these new vaccine components, especially the leading candidate fHbp (LP2086) formed a major part of the preclinical studies. Based on the analysis of deduced peptide sequences, the Pearl River group (studying “LP086”) proposed two subfamilies of the protein, A and B, whereas the Siena group (studying “GNA1870” later called fHbp), using a similar analysis of a different meningococcal collection, proposed three subvariants (1, 2, and 3, with subvariants 2 and 3 more closely related to each other) (85). As further sequencing was performed of this antigen, it became necessary to cross-reference and unify the sequence nomenclatures between the two typing schemes and a single nomenclature was proposed (14) (Figure 5), along with a web accessible database enabling the cross-referencing of the various nomenclature schemes (https://pubmlst.org/neisseria/fHbp/). At the time of writing (September 2018), a total of 1,157 peptide sequence variants of this protein were described on the database.
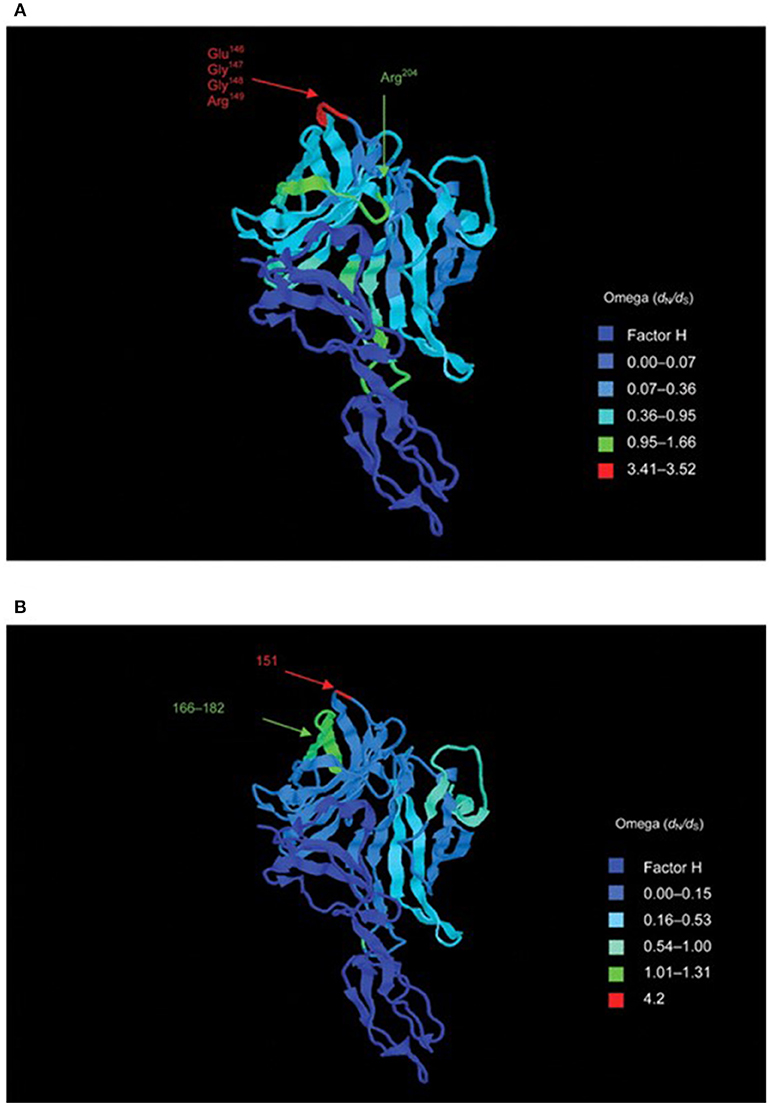
Figure 5. Structure of the fH–fHbp complex, with temperature coloring using per-site point estimates of selection (ω) for (A) subfamily B/variant 1 sequences and (B) subfamily A/variant 2 sequences. Peptides indicated in (A) are putative bactericidal epitopes. In (B) positively selected sites are indicated. Note: subfamily A/variant 2 differs in length from subfamily B/variant 1 by +4 bp (e.g., Glu151 is equivalent to Glu147 in variant 1). Reproduced from Brehony et al. (14) without modification under CC BY.
As with the capsular polysaccharide-conjugate vaccines, it was impractical to conduct phase III efficacy studies on these novel “serogroup B substitute” vaccines. Further, the correlates of protection were less well-established than those for the capsular polysaccharide vaccines, where bactericidal assays using blood samples from vaccinees were employed (3). This was a particular problem in assessing the breadth of coverage of these vaccines, given the diversity of meningococcal protein antigens. This prompted the development of indirect assessments of coverage: the Meningococcal Antigen Typing System (MATS) assay for Bexsero®, which incorporates sequence data from the porA gene (86); and the Meningococcal Antigen Surface Expression (MEASURE) Assay for Trumenba® (87). Both vaccines were licensed and used based on phase II clinical studies and studies using these assays (88, 89).
Pre- and post-introduction assessment of the coverage of these vaccines is an important component of assessing their likely and continued efficacy, especially for an organism as diverse and dynamic as the meningococcus. An efficient way to achieve this is by the extraction of antigen gene sequences from genome data collected as part of routine surveillance. The PubMLST website, which hosts data for the MRF-MGL and a number of other important global isolate collections, indexes all known genes and these can be flexibly grouped into “schemes,” which are groups of genetic loci that are analyzed together for typing or functional purposes (13, 90). It was straightforward to combine the typing schemes for the various antigens in Bexsero® into a Bexsero® Vaccine Antigen Typing Scheme (BAST) (91), which enabled the assessment of the changing prevalence of the vaccine antigen variants in the UK and Ireland (91, 92) (Figure 6). Approaches such as this have great potential for supporting vaccine development implementation and monitoring into the future.
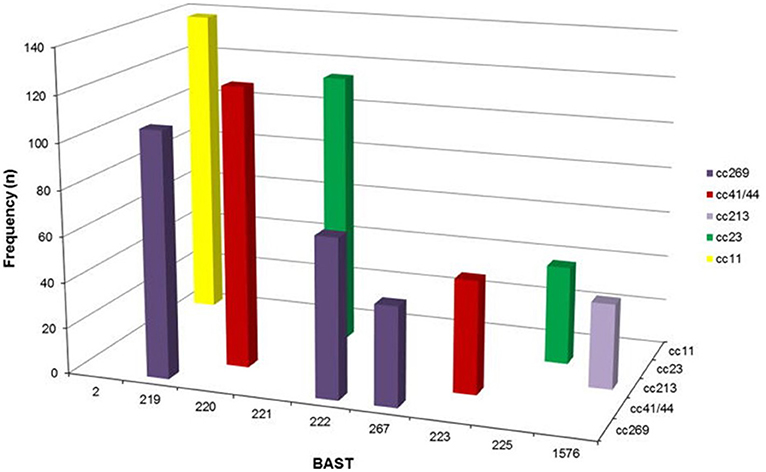
Figure 6. Frequency distribution of Bexsero Antigen Sequence Typing (BAST) by clonal complex for the nine most frequently occurring BASTs (n = 775/2,016, 39.4%) in invasive meningococcal disease isolates in the epidemiological years 2010/2011 to 2013/2014. BASTs shown on the x axis are structured by clonal complex (z axis) for a proportion of frequently occurring dominant clones circulating in Great Britain and Ireland from 2010/2014, for example BAST-2 is only found in isolates of clonal complex 11. Clonal complex 269 has three predominant BASTs, 219, 222, and 267 not found in other clonal complexes. Reproduced without modification from Brehony et al. (91) under CC BY.
Future Prospects
As demonstrated by the examples outlined above, nucleotide sequence data, ranging from single gene fragments from individual bacterial specimens to whole completed genomes that are representative of populations, have many applications in vaccinology. These data are particularly useful in the rapid and cost-effective characterization of bacterial isolates. Combining such data with population and evolutionary analyses generates many informative inferences; however, whilst this complements data on the interaction of bacterial components with the host immune system, sequence analyses cannot wholly replace immunological studies. In an era where nucleotide sequences are low-cost commodities, the important advances of the future will depend on the interpretation and open-access dissemination of these data. In addition to novel statistical genetic techniques and integration with phenotypic data, the implementation of visualization tools is likely to be important in the further exploitation of this rich source of biological information.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
During the earlier parts of this work (1988–1997) MM was employed by the UK National Health Service at the National Institute for Biological Standards and Control (NIBSC), he was funded for a sabbatical in the laboratory of Mark Achtman by the Alexander von Humboldt Stiftung (1996–7) from 1997 to 2015 he was Wellcome Trust Senior Fellow and from 2004-present Professor of Molecular Epidemiology and a Fellow of Hertford College at the University of Oxford. He is grateful to the European Union (especially grants QLK2-CT-2001-01436 and FP7-278864-2), the Wellcome Trust (especially grants: 047072/Z/96/B, 047072/Z/96/C, 062057/Z/00/Z, 081494/Z/06/Z, 087622/Z/08/A, 086546/Z/08/Z, 091634/Z/10/Z, and 104992/Z/14/Z), the Meningitis Research Foundaton, the UK Department of Health (especially contract PR-ST-0915-10015) and the Oxford Martin School for funding.
Conflict of Interest Statement
As an employee of the University of Oxford, MM has, over the past 20 years, undertaken contract research and consultancy for, and has been paid expenses and honoraria by, companies involved in vaccine development including GSK, Chiron, Novartis, Wyeth, and Pfizer.
Acknowledgments
I would like to express my thanks to the members of past and present MaidenLab and the many collaborators who have contributed to my work over the years. Special thanks go to Ian Feavers of NIBSC, whom was the crucial collaborator in establishing this program of work.
References
1. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA (1977) 74:5463–7. doi: 10.1073/pnas.74.12.5463
2. Maiden MCJ. The impact of protein-conjugate polysaccharide vaccines: an endgame for meningitis? Philos T R Soc B (2013) 368:20120147. doi: 10.1098/rstb.2012.0147
3. Feavers IM, Maiden MC. Recent progress in the prevention of serogroup B meningococcal disease. Clin Vaccine Immunol. (2017) 24:e00566–16. doi: 10.1128/CVI.00566-16
4. Finne J, Leinonen M, Makela PH. Antigenic similarities between brain components and bacteria causing meningitis. Implications for vaccine development and pathogenesis. Lancet (1983) 2:355–7. doi: 10.1016/S0140-6736(83)90340-9
5. Poolman JT, Hopman CTP, Zanen HC. Immunogenicity of meningococcal antigens as detected in patient sera. Infect Immun. (1983) 40:398–406.
6. Poolman JT, Beuvery EC, Hopman CTP, Witvliet MH, Timmermans HAM, Teerlink T, et al. Class 1/3 outer membrane protein vaccine against Group B, Type 15, Subtype 16 Meningococci. Dev Biol Stand. (1986) 63:147–52.
7. Barlow AK, Heckels JE, Clarke IN. The class 1 outer membrane protein of Neisseria meningitidis: gene sequence and structural and immunological similarities to gonococcal porins. Mol Microbiol. (1989) 3:131–9. doi: 10.1111/j.1365-2958.1989.tb01802.x
8. Maiden MCJ, Suker J, McKenna AJ, Bygraves JA, Feavers IM. Comparison of the class 1 outer membrane proteins of eight serological reference strains of Neisseria meningitidis. Mol Microbiol. (1991) 5:727–36. doi: 10.1111/j.1365-2958.1991.tb00743.x
9. McGuinness B, Barlow AK, Clarke IN, Farley JE, Anilionis A, Poolman JT, et al. Deduced amino acid sequences of class 1 protein (PorA) from three strains of Neisseria meningitidis.Synthetic peptides define the epitopes responsible for serosubtype specificity. J Exp Med. (1990) 171:1871–82. doi: 10.1084/jem.171.6.1871
10. van der Ley P, Heckels JE, Virji M, Hoogerhout P, Poolman JT. Topology of outer membrane porins in pathogenic Neisseria species. Infect Immun. (1991) 59:2963–71.
11. Frasch CE, Zollinger WD, Poolman JT. Serotype antigens of Neisseria meningitidis and a proposed scheme for designation of serotypes. Rev Infect Dis. (1985) 7:504–10. doi: 10.1093/clinids/7.4.504
12. Russell JE, Jolley KA, Feavers IM, Maiden MC, Suker J. PorA variable regions of Neisseria meningitidis. Emerg Infect Dis. (2004) 10:674–8. doi: 10.3201/eid1004.030247
13. Jolley KA, Maiden MC. Automated extraction of typing information for bacterial pathogens from whole genome sequence data: Neisseria meningitidis as an exemplar. Eurosurveillance (2013) 18:20379. doi: 10.2807/ese.18.04.20379-en
14. Brehony C, Wilson DJ, Maiden MC. Variation of the factor H-binding protein of Neisseria meningitidis. Microbiology (2009) 155:4155–69. doi: 10.1099/mic.0.027995-0
15. Thompson EAL, Feavers IM, Maiden MCJ. Antigenic diversity of meningococcal enterobactin receptor FetA, a vaccine component. Microbiology (2003) 149(Pt 7):1849–58. doi: 10.1099/mic.0.26131-0
16. Bambini S, De Chiara M, Muzzi A, Mora M, Lucidarme J, Brehony C, et al. Neisseria adhesin A variation and revised nomenclature scheme. Clin Vaccine Immunol. (2014) 21:966–971. doi: 10.1128/CVI.00825-13
17. Harrison OB, Evans NJ, Blair JM, Grimes HS, Tinsley CR, Nassif X, et al. Epidemiological evidence for the role of the hemoglobin receptor, HmbR, in meningococcal virulence. J Infect Dis. (2009) 200:94–8. doi: 10.1086/599377
18. van den Elsen JM, Herron JN, Hoogerhout P, Poolman JT, Boel E, Logtenberg T, et al. Bactericidal antibody recognition of a PorA epitope of Neisseria meningitidis: crystal structure of a Fab fragment in complex with a fluorescein-conjugated peptide. Proteins (1997) 29:113–25.
19. Tzitzilonis C, Prince SM, Collins RF, Achtman M, Feavers IM, Maiden MC, et al. Structural variation and immune recognition of the P1.2 subtype meningococcal antigen. Proteins (2006) 62:947–55. doi: 10.1002/prot.20800
20. Saleem M, Prince SM, Rigby SE, Imran M, Patel H, Chan H, et al. Use of a molecular decoy to segregate transport from antigenicity in the FrpB iron transporter from Neisseria meningitidis. PLoS ONE (2013) 8:e56746. doi: 10.1371/journal.pone.0056746
21. Moxon ER, Rainey PB, Nowak MA, Lenski RE. Adaptive evolution of highly mutable loci in pathogenic bacteria. Curr Biol. (1994) 4:24–33. doi: 10.1016/S0960-9822(00)00005-1
22. Feavers IM, Heath AB, Bygraves JA, Maiden MCJ. Role of horizontal genetic exchange in the antigenic variation of the class 1 outer membrane protein of Neisseria meningitidis. Mol Microbiol. (1992) 6:489–95. doi: 10.1111/j.1365-2958.1992.tb01493.x
23. Spratt BG. Hybrid penicillin-binding proteins in penicillin-resistant strains of Neisseria gonorrhoeae. Nature (1988) 332:173–6. doi: 10.1038/332173a0
24. Dowson CG, Hutchison A, Brannigan JA, George RC, Hansman D, Liñares J, et al. Horizontal transfer of penicillin-binding protein genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Proc Natl Acad Sci USA (1989) 86:8842–6. doi: 10.1073/pnas.86.22.8842
25. Maynard Smith J, Dowson CG, Spratt BG. Localized sex in bacteria. Nature (1991) 349:29–31. doi: 10.1038/349029a0
26. Selander RK, Caugant DA, Ochman H, Musser JM, Gilmour MN, Whittam TS. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl Environ Microbiol. (1986) 51:837–84.
27. Wang JF, Caugant DA, Morelli G, Koumaré B, Achtman M. Antigenic and epidemiological properties of the ET-37 complex of Neisseria meningitidis. J Infect Dis. (1993) 167:1320–9. doi: 10.1093/infdis/167.6.1320
28. Olyhoek T, Crowe BA, Achtman M. Clonal population structure of Neisseria meningitidis serogroup A isolated from epidemics and pandemics between 1915 and 1983. Rev Infect Dis. (1987) 9:665–82. doi: 10.1093/clinids/9.4.665
29. Caugant DA, Mocca LF, Frasch CE, Frøholm LO, Zollinger WD, Selander RK. Genetic structure of Neisseria meningitidis populations in relation to serogroup, serotype, and outer membrane protein pattern. J Bacteriol. (1987) 169:2781–92. doi: 10.1128/jb.169.6.2781-2792.1987
30. Caugant DA, Frøholm LO, Bovre K, Holten E, Frasch CE, Mocca LF, et al. Intercontinental spread of a genetically distinctive complex of clones of Neisseria meningitidis causing epidemic disease. Proc Natl Acad Sci USA (1986) 83:4927–31. doi: 10.1073/pnas.83.13.4927
31. Caugant DA, Zollinger WD, Mocca LF, Frasch CE, Whittam TS, Frøholm LO, et al. Genetic relationships and clonal population structure of serotype 2 strains of Neisseria meningitidis. Infect Immun. (1987) 55:1503–13.
32. Wang JF, Caugant DA, Li X, Hu X, Poolman JT, Crowe BA, et al. Clonal and antigenic analysis of serogroup A Neisseria meningitidis with particular reference to epidemiological features of epidemic meningitis in China. Infect Immun. (1992) 60:5267–5282.
33. Caugant DA, Kristiansen BE, Frøholm LO, Bovre K, Selander RK. Clonal diversity of Neisseria meningitidis from a population of asymptomatic carriers. Infect Immun. (1988) 56:2060–8.
34. Caugant DA, Høiby EA, Rosenqvist E, Frøholm LO, Selander RK. Transmission of Neisseria meningitidis among asymptomatic military recruits and antibody analysis. Epidemiol Infect. (1992) 109:241–53. doi: 10.1017/S0950268800050196
35. Maiden MCJ, Caugant DA. The population biology of Neisseria meningitidis: implications for meningococcal disease, epidemiology and control. In: Frosch M, Maiden MCJ, editors. Handbook of Meningococcal Disease. Weinheim: Wiley-VCH Verlag GmbH & Co KGaA (2006). p. 17–35. doi: 10.1002/3527608508.ch2
36. Bjune G, Høiby EA, Grønnesby JK, Arnesen O, Fredriksen JH, Halstensen A, et al. Effect of outer membrane vesicle vaccine against group B meningococcal disease in Norway. Lancet (1991) 338:1093–6. doi: 10.1016/0140-6736(91)91961-S
37. Sierra GVG, Campa HC, Varcacel NM, Garcia IL, Izquierdo PL, Sotolongo PF, et al. Vaccine against group B Neisseria meningitidis: protection trial and mass vaccination results in Cuba. NIPH Ann. (1991) 14:195–207.
38. Oster P, Lennon D, O'Hallahan J, Mulholland K, Reid S, Martin D. MeNZB: a safe and highly immunogenic tailor-made vaccine against the New Zealand Neisseria meningitidis serogroup B disease epidemic strain. Vaccine (2005) 23:2191–6. doi: 10.1016/j.vaccine.2005.01.063
39. Halter R, Pohlner J, Meyer TF. Mosaic-like organization of IgA protease genes in Neisseria gonorrhoeae generated by horizontal genetic exchange in vivo. EMBO J. (1989) 8:2737–44. doi: 10.1002/j.1460-2075.1989.tb08415.x
40. Spratt BG, Zhang Q-Y, Jones DM, Hutchison A, Brannigan JA, Dowson CG. Recruitment of a penicillin-binding protien gene from Neisseria flavescens during the emergence of penicillin resistance in Neisseria meningitidis. Proc Natl Acad Sci USA (1989) 86:8988–92. doi: 10.1073/pnas.86.22.8988
41. Feil E, Zhou J, Maynard Smith J, Spratt BG. A comparison of the nucleotide sequences of the adk and recA genes of pathogenic and commensal Neisseria species: evidence for extensive interspecies recombination within adk. J Mol Evol. (1996) 43:631–40. doi: 10.1007/BF02202111
42. Maiden MC. Multilocus sequence typing of bacteria. Annu Rev Microbiol. (2006) 60:561–88. doi: 10.1146/annurev.micro.59.030804.121325
43. Harrison OB, Claus H, Jiang Y, Bennett JS, Bratcher HB, Jolley KA, et al. Description and nomenclature of Neisseria meningitidis capsule locus. Emerg Infect Dis. (2013) 19:566–73. doi: 10.3201/eid1904.111799
44. Jolley KA, Brehony C, Maiden MC. Molecular typing of meningococci: recommendations for target choice and nomenclature. FEMS Microbiol Rev. (2007) 31:89–96. doi: 10.1111/j.1574-6976.2006.00057.x
45. Miller E, Salisbury D, Ramsay M. Planning, registration, and implementation of an immunisation campaign against meningococcal serogroup C disease in the UK: a success story. Vaccine (2001) 20(Suppl. 1):S58–67. doi: 10.1016/S0264-410X(01)00299-7
46. Pollard AJ, Perrett KP, Beverley PC. Maintaining protection against invasive bacteria with protein-polysaccharide conjugate vaccines. Nat Rev Immunol. (2009) 9:212–20. doi: 10.1038/nri2494
47. Maiden MCJ, Spratt BG. Meningococcal conjugate vaccines: new opportunities and new challenges. Lancet (1999) 354:615–6. doi: 10.1016/S0140-6736(99)00252-4
48. Maiden MC, Ibarz-Pavon AB, Urwin R, Gray SJ, Andrews NJ, Clarke SC, et al. Impact of meningococcal serogroup c conjugate vaccines on carriage and herd immunity. J Infect Dis. (2008) 197:737–43. doi: 10.1086/527401
49. Maiden MC, Stuart JM UK_Meningococcal_Carriage_Group. Carriage of serogroup C meningococci 1 year after meningococcal C conjugate polysaccharide vaccination. Lancet (2002) 359:1829–31. doi: 10.1016/S0140-6736(02)08679-8
50. Ibarz-Pavon AB, Maclennan J, Andrews NJ, Gray SJ, Urwin R, Clarke SC, et al. Changes in serogroup and genotype prevalence among carried meningococci in the United Kingdom during vaccine implementation. J Infect Dis. (2011) 204:1046–53. doi: 10.1093/infdis/jir466
51. Trotter CL, Maiden MC. Meningococcal vaccines and herd immunity: lessons learned from serogroup C conjugate vaccination programs. Exp Rev Vaccines (2009) 8:851–61. doi: 10.1586/erv.09.48
52. Trotter CL, Andrews NJ, Kaczmarski EB, Miller E, Ramsay ME. Effectiveness of meningococcal serogroup C conjugate vaccine 4 years after introduction. Lancet (2004) 364:365–7. doi: 10.1016/S0140-6736(04)16725-1
53. Lapeyssonnie L. La méningite cérébrospinale en Afrique. Bull World Health Organ (1963) 28:53–114.
54. Greenwood B. Manson lecture. Meningococcal meningitis in Africa. Trans R Soc Trop Med Hyg. (1999) 93:341–53. doi: 10.1016/S0035-9203(99)90106-2
55. Tiffay K, Jodar L, Kieny MP, Socquet M, LaForce FM. The evolution of the meningitis vaccine project. Clin Infect Dis. (2015) 61:S396–403. doi: 10.1093/cid/civ594
56. Sow SO, Okoko BJ, Diallo A, Viviani S, Borrow R, Carlone G, et al. Immunogenicity and safety of a meningococcal a conjugate vaccine in Africans. N Engl J Med. (2011) 364:2293–304. doi: 10.1056/NEJMoa1003812
57. Ali O, Aseffa A, Bedru A, Lemma T, Moti T, Worku A, et al. Meningococcal carriage in the African meningitis belt. Trop Med Int Health (2013) 18:968–78. doi: 10.1111/tmi.12125
58. Trotter CL, Greenwood BM. Meningococcal carriage in the African meningitis belt. Lancet Infect Dis. (2007) 7:797–803. doi: 10.1016/S1473-3099(07)70288-8
59. MacLennan J, Kafatos G, Neal K, Andrews N, Cameron JC, Roberts R, et al. Social behavior and meningococcal carriage in British teenagers. Emerg Infect Dis. (2006) 12:950–7. doi: 10.3201/eid1206.051297
60. Ali O, Aseffa A, Bedru A, Lema T, Moti T, Tekletsion Y, et al. The diversity of meningococcal carriage across the african meningitis belt and the impact of vaccination with a group a meningococcal conjugate vaccine. J Infect Dis. (2015) 212:1298–307. doi: 10.1093/infdis/jiv211
61. Diallo K, Trotter C, Timbine Y, Tamboura B, Sow SO, Issaka B, et al. Pharyngeal carriage of Neisseria species in the African meningitis belt. J Infect. (2016) 72:667–77. doi: 10.1016/j.jinf.2016.03.010
62. Harmsen D, Singer C, Rothganger J, Tonjum T, de Hoog GS, Shah H, et al. Diagnostics of neisseriaceae and moraxellaceae by ribosomal DNA sequencing: ribosomal differentiation of medical microorganisms. J Clin Microbiol. (2001) 39:936–42. doi: 10.1128/JCM.39.3.936-942.2001
63. Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony CM, Colles FM, et al. Ribosomal multi-locus sequence typing: universal characterization of bacteria from domain to strain. Microbiology (2012) 158:1005–15. doi: 10.1099/mic.0.055459-0
64. Bennett JS, Watkins ER, Jolley KA, Harrison OB, Maiden MC. Identifying Neisseria species using the 50S ribosomal protein L6 (rplF) gene. J Clin Microbiol. (2014) 52:1375–81. doi: 10.1128/JCM.03529-13
65. Christensen H, May M, Bowen L, Hickman M, Trotter CL. Meningococcal carriage by age: a systematic review and meta-analysis. Lancet Infect Dis. (2010) 10:853–61. doi: 10.1016/S1473-3099(10)70251-6
66. Ali O, Aseffa A, Omer AB, Lema T, Demissie TM, Tekletsion Y, et al. Household transmission of Neisseria meningitidis in the African meningitis belt: a longitudinal cohort study. Lancet Glob Health (2016) 4:E989–95. doi: 10.1016/S2214-109X(16)30244-3
67. Kristiansen PA, Diomande F, Ba AK, Sanou I, Ouedraogo AS, Ouedraogo R, et al. Impact of the serogroup a meningococcal conjugate vaccine, MenAfriVac, on carriage and herd immunity. Clin Infect Dis. (2013) 56:354–63. doi: 10.1093/cid/cis892
68. Daugla DM, Gami JP, Gamougam K, Naibei N, Mbainadji L, Narbe M, et al. Effect of a serogroup A meningococcal conjugate vaccine (PsA-TT) on serogroup A meningococcal meningitis and carriage in Chad: a community study [corrected]. Lancet (2014) 383:40–7. doi: 10.1016/S0140-6736(13)61612-8
69. Diallo K, Gamougam K, Daugla DM, Harrison OB, Bray JE, Caugant DA, et al. Hierarchical genomic analysis of carried and invasive serogroup A Neisseria meningitidis during the 2011 epidemic in Chad. BMC Genom. (2017) 18:398. doi: 10.1186/s12864-017-3789-0
70. Black S, Pizza M, Nissum M, Rappuoli R. Toward a meningitis-free world. Sci Transl Med. (2012) 4:123ps5. doi: 10.1126/scitranslmed.3003859
71. Chen WH, Neuzil KM, Boyce CR, Pasetti MF, Reymann MK, Martellet L, et al. Safety and immunogenicity of a pentavalent meningococcal conjugate vaccine containing serogroups A, C, Y, W, and X in healthy adults: a phase 1, single-centre, double-blind, randomised, controlled study. Lancet Infect Dis. (2018) 18:1088–96. doi: 10.1016/S1473-3099(18)30400-6
72. Bratcher HB, Corton C, Jolley KA, Parkhill J, Maiden MC. A gene-by-gene population genomics platform: de novo assembly, annotation and genealogical analysis of 108 representative Neisseria meningitidis genomes. BMC Genome. (2014) 15:1138. doi: 10.1186/1471-2164-15-1138
73. Hill DMC, Lucidarme J, Gray SJ, Newbold LS, Ure R, Brehony C, et al. Genomic epidemiology of age-associated meningococcal lineages in national surveillance: an observational cohort study. Lancet Infect Dis. (2015) 15:1420–8. doi: 10.1016/S1473-3099(15)00267-4
74. Ladhani SN, Beebeejaun K, Lucidarme J, Campbell H, Gray S, Kaczmarski E, et al. Increase in endemic Neisseria meningitidis capsular group W sequence type 11 complex associated with severe invasive disease in england and wales. Clin Infect Dis. (2015) 60:578–85. doi: 10.1093/cid/ciu881
75. Taha MK, Achtman M, Alonso JM, Greenwood B, Ramsay M, Fox A, et al. Serogroup W135 meningococcal disease in Hajj pilgrims. Lancet (2000) 356:2159. doi: 10.1016/S0140-6736(00)03502-9
76. Mayer LW, Reeves MW, Al-Hamdan N, Sacchi CT, Taha MK, Ajello GW, et al. Outbreak of W135 meningococcal disease in 2000: not emergence of a new W135 strain but clonal expansion within the electophoretic type-37 complex. J Infect Dis. (2002) 185:1596–605. doi: 10.1086/340414
77. Aguilera JF, Perrocheau A, Meffre C, Hahne S. Outbreak of serogroup W135 meningococcal disease after the Hajj pilgrimage, Europe, 2000. Emerg Infect Dis. (2002) 8:761–7. doi: 10.3201/eid0808.010422
78. Lucidarme J, Hill DM, Bratcher HB, Gray SJ, du Plessis M, Tsang RS, et al. Genomic resolution of an aggressive, widespread, diverse and expanding meningococcal serogroup B, C and W lineage. J Infect. (2015) 71:544–52. doi: 10.1016/j.jinf.2015.07.007
79. Ladhani SN, Ramsay M, Borrow R, Riordan A, Watson JM, Pollard AJ. Enter B and W: two new meningococcal vaccine programmes launched. Arch Dis Child. (2016) 101:91–5. doi: 10.1136/archdischild-2015-308928
80. Tettelin H, Saunders NJ, Heidelberg J, Jeffries AC, Nelson KE, Eisen JA, et al. Complete genome sequence of Neisseria meningitidis serogroup B strain MC58. Science (2000) 287:1809–15. doi: 10.1126/science.287.5459.1809
81. Parkhill J, Achtman M, James KD, Bentley SD, Churcher C, Klee SR, et al. Complete DNA sequence of a serogroup A strain of Neisseria meningitidis Z2491. Nature (2000) 404:502–6. doi: 10.1038/35006655
82. Vernikos G, Medini D. Bexsero® chronicle. Pathog Glob Health (2014) 108:305–16. doi: 10.1179/2047773214Y.0000000162
83. Zlotnick GW, Jones TR, Liberator P, Hao L, Harris S, McNeil LK, et al. The discovery and development of a novel vaccine to protect against Neisseria meningitidis Serogroup B Disease. Hum Vaccines Immunother. (2015) 11:5–13. doi: 10.4161/hv.34293
84. Pizza M, Scarlato V, Masignani V, Giuliani MM, Arico B, Comanducci M, et al. Identification of vaccine candidates against serogroup B meningococcus by whole-genome sequencing. Science (2000) 287:1816–20. doi: 10.1126/science.287.5459.1816
85. Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med. (2003) 197:789–99. doi: 10.1084/jem.20021911
86. Donnelly J, Medini D, Boccadifuoco G, Biolchi A, Ward J, Frasch C, et al. Qualitative and quantitative assessment of meningococcal antigens to evaluate the potential strain coverage of protein-based vaccines. Proc Natl Acad Sci USA (2010) 107:19490–5. doi: 10.1073/pnas.1013758107
87. Donald RG, Hawkins JC, Hao L, Liberator P, Jones TR, Harris SL, et al. Meningococcal serogroup B vaccines: estimating breadth of coverage. Hum Vaccines Immunother. (2017) 13:255–65. doi: 10.1080/21645515.2017.1264750
88. Vogel U, Taha MK, Vazquez JA, Findlow J, Claus H, Stefanelli P, et al. Predicted strain coverage of a meningococcal multicomponent vaccine (4CMenB) in Europe: a qualitative and quantitative assessment. Lancet Infect Dis. (2013) 13:416–25. doi: 10.1016/S1473-3099(13)70006-9
89. McNeil LK, Donald RGK, Gribenko A, French R, Lambert N, Harris SL, et al. Predicting the susceptibility of meningococcal serogroup b isolates to bactericidal antibodies elicited by Bivalent rLP2086, a novel prophylactic vaccine. Mbio (2018) 9:e00036–18. doi: 10.1128/mBio.00036-18
90. Jolley KA, Maiden MC. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform. (2010) 11:595. doi: 10.1186/1471-2105-11-595
91. Brehony C, Rodrigues CM, Borrow R, Smith A, Cunney R, Moxon ER, et al. Distribution of Bexsero® Antigen Sequence Types (BASTs) in invasive meningococcal disease isolates: Implications for immunisation. Vaccine (2016) 34:4690–7. doi: 10.1016/j.vaccine.2016.08.015
Keywords: Neisseria meningitidis, conjugate polysaccharide vaccines, outer membrane vesicle vaccines, population biology, herd immunity, efficacy
Citation: Maiden MCJ (2019) The Impact of Nucleotide Sequence Analysis on Meningococcal Vaccine Development and Assessment. Front. Immunol. 9:3151. doi: 10.3389/fimmu.2018.03151
Received: 19 October 2018; Accepted: 20 December 2018;
Published: 15 January 2019.
Edited by:
Pedro A. Reche, Complutense University of Madrid, SpainReviewed by:
Scott D. Gray-Owen, University of Toronto, CanadaWilliam William Shafer, Emory University School of Medicine, United States
Copyright © 2019 Maiden. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Christopher James Maiden, bWFydGluLm1haWRlbkB6b28ub3guYWMudWs=