 Vasil V. Vasilev1†
Vasil V. Vasilev1† Maria Radanova2†
Maria Radanova2† Valentin J. Lazarov1
Valentin J. Lazarov1 Marie-Agnes Dragon-Durey3,4,5,6
Marie-Agnes Dragon-Durey3,4,5,6 Veronique Fremeaux-Bacchi3,4,5,6
Veronique Fremeaux-Bacchi3,4,5,6 Lubka T. Roumenina4,5,6*
Lubka T. Roumenina4,5,6*- 1Nephrology Clinic, University Hospital “Tsaritsa Yoanna-ISUL,” Medical University–Sofia, Sofia, Bulgaria
- 2Department of Biochemistry, Molecular Medicine and Nutrigenomics, Medical University Varna, Varna, Bulgaria
- 3Assistance Publique–Hôpitaux de Paris, Service d'Immunologie Biologique, Hôpital Européen Georges Pompidou, Paris, France
- 4INSERM, UMR_S 1138, Centre de Recherche des Cordeliers, Paris, France
- 5Sorbonne Universités, UPMC Univ Paris 06, Paris, France
- 6Université Paris Descartes, Sorbonne Paris Cité, Paris, France
The complement component C3 is at the heart of the complement cascade. It is a complex protein, which generates different functional activated fragments (C3a, C3b, iC3b, C3c, C3d). C3b is a constituent of the alternative pathway C3 convertase (C3bBb), binds multiple regulators, and receptors, affecting thus the functioning of the immune system. The activated forms of C3 are a target for autoantibodies. This review focuses on the discovery, disease relevance, and functional consequences of the anti-C3b autoantibodies. They were discovered about 70 years ago and named immunoconglutinins. They were found after infections and considered convalescent factors. At the end of the twentieth century IgG against C3b were found in systemic lupus erythematosus and recently in lupus nephritis, correlating with the disease severity and flare. Cases of C3 glomerulopathy and immune complex glomerulonephritis were also reported. These antibodies recognize epitopes, shared between C3(H2O)/C3b/iC3b/C3c and have overt functional activity. They correlate with low plasmatic C3 levels in patients. In vitro, they increase the activity of the alternative pathway C3 convertase, without being C3 nephritic factors. They perturb the binding of the negative regulators Complement Receptor 1 and Factor H. The clear functional consequences and association with disease severity warrant further studies to establish the link between the anti-C3b autoantibodies and tissue injury. Comparative studies with such antibodies, found in patients with infections, may help to uncover their origin and epitopes specificity. Patients with complement overactivation due to presence of anti-C3b antibodies may benefit from therapeutic targeting of C3.
Introduction
The complement system is apart of the innate immune defense (1, 2). Autoantibodies against complement components and regulators have proven pathogenic effect. These antibodies (Ab) may cause acquired functional deficiencies of the complement cascade or induce amplification of an already activated complement (3–6). This review is focused on the auto-antibodies, recognizing the activation products of the central complement component C3. The history of their discovery, their prevalence in different diseases as well as their functional and clinical relevance are discussed. Multiple unanswered questions open avenues for future studies.
The Complement Component C3—Structure and Function
C3 is the central component of the complement system (7). It is the convergent point of the three pathways of the cascade (1, 2). C3 is a 185-kDa glycoprotein, which belongs to the α2-macroglobulin family, part of the thioester-containing protein superfamily. C3 consists of two polypeptide chains—α-chain (110 kDa) and β-chain (75 kDa), linked by one disulfide bond and by non-covalent forces (8). It contains a globular thioester domain (TED) with an intrachain thioester bond, capable to attach covalently to surfaces; eight macroglobulin (MG) domains, and CUB domain, which frames and holds the TED domain (Figure 1A).
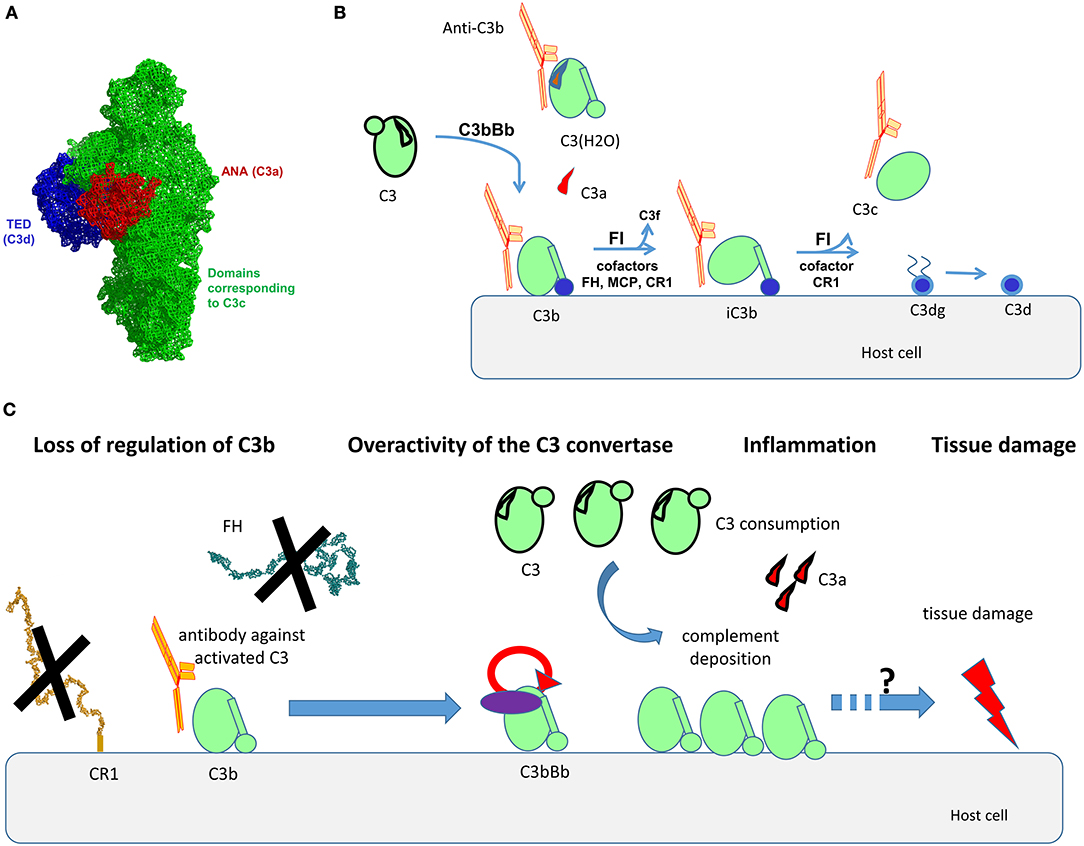
Figure 1. Structural organization of C3, its activation forms, localization of the binding epitopes, and proposed mode of action of the anti-C3b auto-antibodies. (A) Structural organization of C3, based on the crystal structure published in Janssen et al. (8). The different domains, corresponding to the different fragments of the molecule are depicted with different color. Of note are the ANA domain, corresponding to C3a (red), the domains corresponding to C3c (green), and the TED domain, corresponding to C3d (blue). (B) Steps of cleavage of C3 by the C3 convertase of the alternative pathway C3bBb and generation of its activation forms. The cofactors needed for the cleavage of C3b by Factor I (FI) are indicated. The activation forms of C3, recognized by the autoantibodies (C3(H2O), C3b, iC3b, C3c) are indicated as complexed with an antibody in orange. (C) Proposed mode of action of the anti-C3b autoantibodies. The Ab will bind to C3b, preventing its interaction with CR1, and in the context of lupus nephritis, with Factor H (FH). This loss of regulation, together with the direct stabilization/enhancement of formation of the alternative pathway C3 convertase C3bBb will result in overactivation of complement, generating inflammation (via C3a), massive deposits of C3 activation fragments and finally, tissue damage. The exact mechanism linking complement overactivation to tissue injury and its contribution to organ injury need further studies.
C3 undergoes low-rate spontaneous conformational change, leading to hydrolysis of its thioester bond (9). This generates an activated form of the protein, called C3(H2O). It does not bind to surfaces (as it no longer possesses a thioester group), but it resembles C3b in many of its functional and structural features, despite its retention of the ANA domain. C3(H2O) is the initiator of the alternative pathway, since it binds Factor B (FB) and Factor D (FD) and generates a fluid phase initiating convertase C3(H2O)Bb, capable to cleave C3. C3 is cleaved also by the classical or alternative pathway surface C3 convertases to generate C3a (9 kDa), a small potent pro-inflammatory molecule, and C3b (177 kDa) (Figure 1B). Generated C3b changes conformation, uncovering the binding sites for its ligands, including FB, Factor H (FH), complement receptor 1 (CR1, CD35), properdin, etc. (10). C3b interacts with Factor B to form the C3 convertase of the alternative pathway C3bBb (11, 12). Properdin is the only positive regulator of complement, stabilizing the convertase (13). The activity of C3b is tightly regulated to avoid accidental host tissue damage by negative complement regulators, like membrane-bound CR1, membrane cofactor protein (MCP; CD46), decay acceleration factor (DAF; CD55) or circulating regulators like FH, Factor-H-like protein 1 (FHL-1), and Factor I (FI) (14, 15). FI cleaves C3b in presence of cofactors (like the regulators MCP, FH, CR1) to generate iC3b (inactivated C3b, unable to form convertases). In presence of CR1 the cleavage by FI proceeds to C3dg and finally C3d fragments, remaining attached to the surface. When iC3b is cleaved, C3c is released (Figure 1B).
C3 activation fragments not only participate in cleaning of pathogens, apoptotic cells, and cellular debris but also in a number of homeostatic processes. This includes tissue regeneration, synapse pruning, controlling tumor cell progression, etc. (7). Therefore, alteration of the function of the activation fragments of C3 by genetic abnormalities results in pathological conditions (7). Indeed, mutations in the C3 gene result in an abnormal protein, promoting complement overactivation and predisposing to renal injury (atypical hemolytic uremic syndrome) due to loss of regulation or direct overactivation of the C3 convertase (16–18). On the other hand, complete C3 deficiency shows increased susceptibility to bacterial infections in early childhood (19). Critical role of intracellular C3 activation for T cells function was recently described (20). This intracellular C3 activation, as well as the C3-based recycling pathway and C3 being a driver and programmer of cell metabolism suggest that the complement system utilizes C3 to guard not only extracellular but also the intracellular environment (21).
The activation products of C3 are also a target for autoantibodies. The following parts if this review describe the functional consequences and clinical relevance of the autoantibodies targeting C3b.
From Immunoconglutinins to Anti-C3b Ab
The notion of Ab, recognizing activated forms of C3, dates from the mid-twentieth century, when they were named immunoconglutinins (22). Like rheumatoid factors are Ab binding to IgG, immunoconglutinins are Ab, binding components of complement. By definition, the immunoconglutinins are a group of Ab, formed in response to antigenic stimulation by components of an animal's own fixed complement components C3, but sometimes C4. They react against newly-formed epitopes, created after activation of C3 and C4, when the proteins change their conformations. Immunoconglutinins appear after viral or bacterial infections, the titers peak about 2 weeks following infection and usually drop rapidly afterwards (23–26). The Ab are frequently from IgM class. In chronic infections in animal models, high titres immunoconglutinins persisted over a prolonged period of time (27). At this period, it was concluded that the immunoconglutinins are convalescence factor, helping the healing process (25). Indeed, pretreatment of mice with immunoconglutinins prior to challenge with virulent strains of bacteria resulted in prolonged survival and decreased mortality (28). It was hypothesized that immunoconglutinins could enhance the clearance of bacteria by phagocytes.
This view was challenged, when immunoconglutinins/anti-C3 activated forms Ab were established in patients with autoimmune diseases. Such Ab were detected in systemic lupus erythematosus (SLE) (29–32), lupus nephritis (LN) (33, 34), in Crohn disease (35), in some nephrotic kidney diseases (36–38), in dense deposit disease (DDD) (39), in C3 glomerulopathy (C3G), and Immune Complex glomerulonephritis (IC-GN) (40) as well as in autoimmune-prone mice (32). However, these Ab has not been detected in primary biliary cirrhosis or rheumatoid arthritis (30, 32). A single patient with atypical hemolytic uremic syndrome, positive for anti-C3b Ab was also reported (41). These Ab were IgG and were measured as anti-C3 or anti-C3b Ab by ELISA (29, 31–34, 39–41) or as immunoconglutinins (36–38). In SLE they are predominantly belonging to IgG1 and IgG3 subclasses (30).
Clinical Relevance of the Anti-C3b Ab
SLE and LN
Systemic lupus erythematosus is a heterogeneous, multisystem autoimmune disease (42). Kidney involvement in SLE, also known as LN, is a common and serious organ complication that determines the quality of life and prognosis in patients with SLE and is characterized by specific clinical (nephritic or nephrotic syndrome), laboratory (proteinuria, hematuria), immune, and morphological (proliferative or non-proliferative glomerulopathy with mesangial, subendothelial, and subepithelial deposition of immune complexes, tubulointerstitial, and vascular lesions) manifestations. Different disorders of the regulation of the immune response with production of a wide range of Ab directed to various self-antigens (DNA, nuclear proteins, ribosomal proteins, and complement component C1q), are among the main characteristics of SLE and LN. The complement system plays a critical role in inflammatory and immune responses, in clearance of immune complexes and apoptotic cells, and autoreactivity to complement may have considerable pathological consequences (1, 2). The classical pathway has a predominant role in the initiation of the complement activation in SLE and LN, but the complement-mediated damage is often caused by the alternative pathway amplification loop (43).
Most of the reports in the literature related to anti-C3b concern cohorts of SLE or LN. The levels of anti-C3b Ab (measured as immunoconglutinins) were higher in active SLE compared to patients with inactive disease (29). We reported for the first time that more than 30% of LN patients (12/39) were positive for anti-C3 Ab (measured against C3 immobilized on an ELISA plate and confirmed against C3b) (33). Here again the levels of anti-C3b Ab were higher in the LN patients with severe disease, compared to a milder one. In the anti-C3b Ab-positive LN patients, the plasma levels of C4 and C3 were also lower compared to the negative ones, showing thus association of these Ab with complement activation. An additional case of LN revealed positivity against C3b, but also against FI, FB, C3, and properdin (44). The patient carried also a heterozygous mutation in the gene of C3.
In the same time Birmingham et al. showed in a bigger cohort an association of Ab against C3b with LN activity and their diagnostic and prognostic value (34). In this study, among 74 LN patients, 36% were positive for anti-C3b Ab, while only 1 out of 41 cases (2%) with non-renal SLE showed such positivity. Here again, the presence of anti-C3b Ab correlated with plasmatic C3 consumption. In the cross-sectional assessment, compared with anti-C1q IgG, anti-C3b IgG was less sensitive but more specific for lupus nephritis. In a longitudinal analysis, the rise of the levels of anti-C1q Ab were prognostic markers for LN flare only in anti-C3b Ab positive patients, thus defining anti-C3b Ab as a useful marker for to identify at risk patients for disease flare (34).
The presence of ant-C3b Ab correlated with the presence of anti-dsDNA (33). It was hypothesized that the dual presence of anti-dsDNA and anti-C1q Ab may coincide with, and possibly drive, a level of complement activation that leads to the onset of anti-C3b Ab (45).
C3G and IC-GN
C3G is a renal disorder characterized by the presence of glomerular C3 staining in the absence of significant immunoglobulins staining (46). Subsets of C3G include DDD and C3 glomerulonephritis (C3GN). C3G is marked by the presence of glomerular deposits. They are electron dense (by electron microscopy) and localized in the lamina densa of the glomerular basement membrane in case of DDD or are subepithelial and subendothelial in patients with C3GN. The deposits in C3G contain complement components of the alternative pathway—C3b, iC3b, C3dg, C3c (47). The dysregulation of the alternative complement pathway, often caused by the presence of C3NeF, maintains a permanent activation of the complement (48).
In a cohort of 141 patients with C3G and IC-GN, only eight patients were positive for anti-C3b Ab, among which 5 were also positive for anti-FB Ab (40). These eight patients showed increased Bb fragment in plasma. Interestingly, the patients positive for anti-C3b Ab and anti-FB Ab in this cohort had higher rates of infections. Another study described two DDD patients with combined anti-C3b and anti-FB Ab (39). There is still not sufficient data that could determine the diagnostic value of anti-C3b Ab in patient with C3G and IC-GN.
Methods of Detection and Binding Epitopes
Nowadays the presence of anti-C3 activation fragments Ab can be detected routinely by ELISA (31–34, 40). More in depth characterization of the binding is done by surface plasmon resonance (SPR), allowing evaluation of the antigen-antibody interaction in real time as well as running functional tests for the formation, and regulation of the C3 convertase (33, 40, 49).
From the early studies it was known that the immunoconglutinins do not recognize native C3, but rather interact with its activated forms (22). Later studies revealed binding to immobilized C3 (hence C3(H2O)-like); C3b, iC3b, C3c with variable intensity but rarely to C3d and C3a (30, 33, 40). Therefore, the screening for diagnostic purpose should be done using coating with C3b, since it contains all recognized epitopes. C3 was also used as an antigen for the ELISA of detection (33), since it changes its conformation upon immobilization to the plastic, adopting most likely C3b(H2O)-like appearance, revealing the binding epitopes (8, 10). In some patients, positive for anti-C3b Ab, reactivity against immobilized C4 was detected as well (31, 33), but it is difficult to conclude whether these are separate Ab or a cross-reactivity of the anti-C3b ones. Nevertheless, all LN patients, positive for anti-C3b were negative for anti-C4b Ab in one of the published cohorts (34), suggesting that anti-C4/C4b Ab are most likely a separate entity occurring in some patients.
The anti-C3b Ab are different than the C3 Nephritic factor (C3Nef), since by definition C3Nef binds to the convertase C3bBb but not to its isolated components C3b and Bb (50). For one patient with very high titers of anti-C3b Ab, the C3Nef functional test remained negative (33). Moreover, anti-C3b Ab, together with Ab against FB, were detected in two unrelated patients with DDD without C3NeF activity (39).
The complexity of the C3 structure and the dramatic conformational change that the protein undergoes during its cleavage steps expose neoepitopes, which may drive pathological immune response. The ensemble of the results for the epitope mapping suggests that the immunodominant epitope is located within the domains, corresponding to the portion of the protein, which will become C3c after cleavage. This is supported by the fact that in the majority of the cases the reactivity is revealed toward C3b, immobilized C3, and C3c but rarely C3a and C3d (30, 33, 39–41). The domains corresponding to the C3c portion within C3b represent the central part of the protein, where are located the binding sites for FB (12) and numerous negative regulators, as FH, CR1, MCP, and DAF (14), Figure 1B. These data are relevant only to the anti-C3b Ab, found in autoimmune disease. Unfortunately, there is no epitope data about these Ab, occurring after infection. It is still unclear how the anti-C3b Ab originate and what is the immunization mechanism, but it could be explained by immune response to C3b neoepitopes. It was found that C3 could be phosphorylated at various sites, which influence its functional activity (51). It could be a reason for immunogenic properties of molecule, because increased levels of phosphorylated C3 are detected in SLE patients (52).
The activation products of C3 have diverse binding partners and functions, therefore it may not be surprising that acting on one epitope, the antibody will facilitate the pathogen clearance and on another—the complement overactivation, loss of regulation, and tissue damage. Another hypothesis could be that the binding epitopes are similar in both cases. In physiology, complement-overactivating anti-C3b IgM may help to clear the infection and disappear afterwards. Nevertheless, if they persist over time, switch to IgG, undergo epitope spreading, and reach high titers, the anti-C3b Ab could be of harm. This will be particularly relevant in autoimmune-prone background. Alternatively, C3 activation within the kidney may result in a C3b deposited in a way that presents neoeptiopes that drive anti-C3b IgG production.
Functional Consequences
It is critical to determine whether the anti-C3b Ab are protective, a disease-relevant factor or a simple epiphenomenon, one among many, arising from the dysregulated immune response in the autoimmune diseases (53, 54). Although limited, the experimental studies clearly describe functional consequences for these Ab (Table 1). It was found that anti-C3b positive IgG, purified from plasma of patients with LN, SLE or C3G, and IC-GN, trigger overactivation of the complement cascade by the alternative pathway (30, 33, 39, 40). The mechanism of the complement activation, however, vary depending on the particular disease. In LN, C3G, and IC-GN, IgG from the patients enhanced C3 cleavage and the formation of new convertases. Moreover, they induced C3 activation fragments deposition on the surface of endothelial cells (33, 40), which could contribute to the disease process (55). These Ab, as well as the ones from SLE patients, inhibited also the interaction of C3b with its negative regulator CR1 and the Factor I-mediated cleavage (30, 33, 40). For two SLE samples, the anti-C3b Ab also perturbed the factor I-mediated release of immune complexes from CR1 (30). Nevertheless, only the tested Ab from LN perturbed the interaction of C3b with factor H (33, 40). Interestingly, these Ab differed also in terms of the stability of the formed complexes with C3b. The ones from C3G/IC-GN showed a rapid association but fast dissociation, while the C3-Ab from LN had a slow association rate but formed more stable complexes (49). This may explain in part the apparent functional differences. Based on the available functional data, the anti-C3b Ab perturb the binding of the negative regulators FH and CR1 to C3b, causing mostly loss of regulation and to a milder degree—direct overactivation of the C3 convertase.
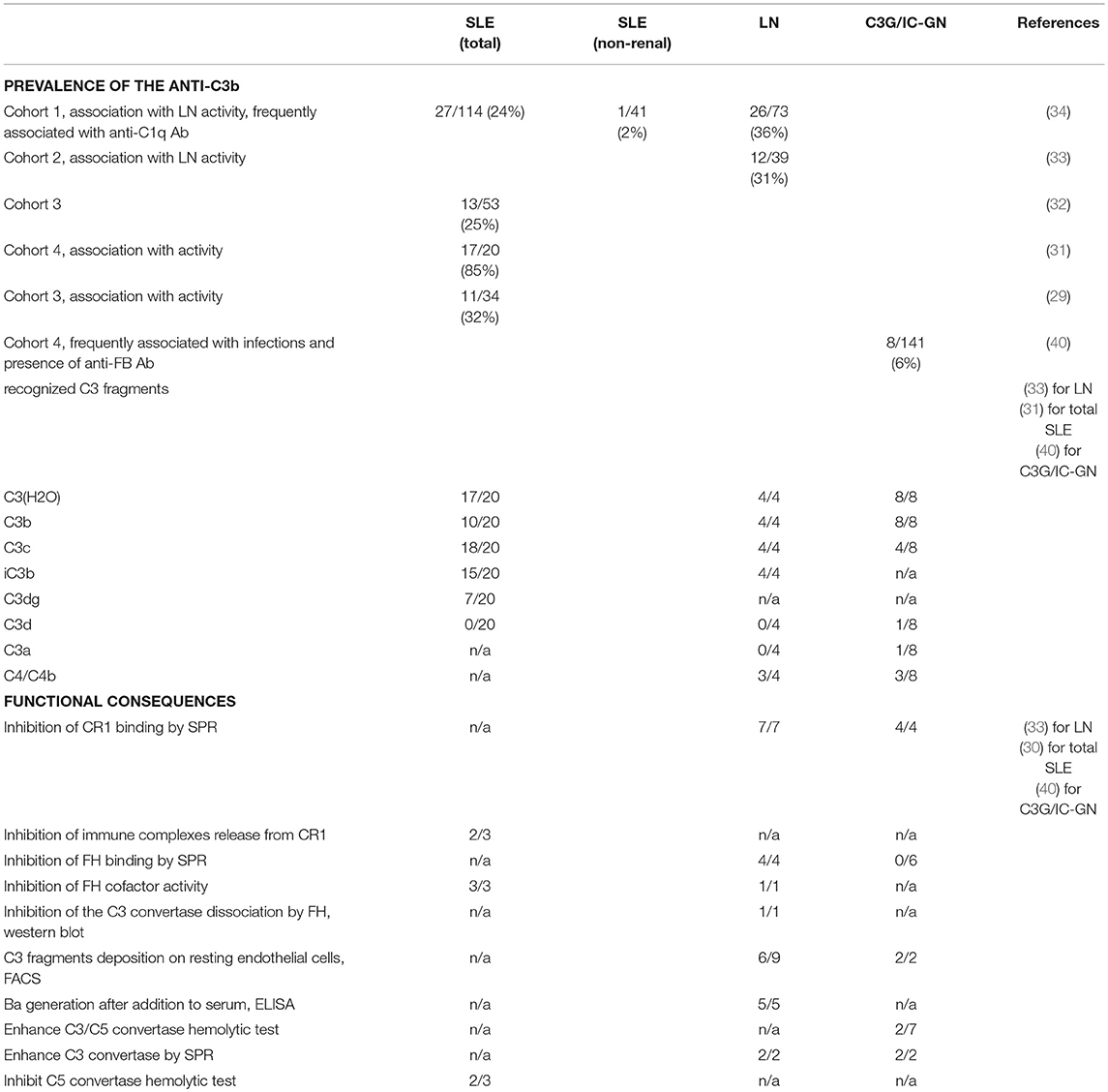
Table 1. Prevalence and functional consequences of the anti-C3b Ab in autoimmune diseases (cohort studies).
Although the LN is clearly disease of overactivation of the classical pathway, it is important to note that the alternative pathway amplification loop is the main source of the terminal pathway complex C5b-9 (43). It is the one that generates a large number of C3b molecules that make both classical and alternative pathway C5 convertases. The capacity of anti-C3b Ab to perturb the regulation at the level of C3b would, therefore, result in an acceleration in the deposition of the C3 activation fragments, and might also result in enhanced C5b-9 deposits. The putative mechanism of action of the anti-C3b Ab is presented at Figure 1C.
Limited data is available for the action of anti-C3b Ab at the level of the C5 convertase. This process is not studied in LN. Nevertheless, anti-C3b/C3c Ab from two SLE patients inhibited the alternative pathway C3 convertase in an in vitro model, deprived of action of the complement regulators (30). On the contrary, in a relatively similar model, 2 out of 3 tested anti-C3b Ab from C3G/IC-GN patients enhanced C3/C5 convertase formation (40). Such data is not available for LN. Further studies are needed to elucidate the action of the anti-C3b Ab on the C5 convertase in different disease. It is tempting to speculate that in different pathological contexts the anti-C3b Ab will affect differently the convertases. In C3G, C3Nef, and C5Nef are described, acting differentially on the two convertases and having distinct disease associations (56).
Another aspect, which remains unexplored is the capacity of the anti-C3b Ab to bind to the C3b deposits in the kidney and to amplify the complement activation. This is possible in LN and IC-GN, where renal immunoglobulin deposits are clearly present and may, in part, be related to immune complexes, containing C3b as an antigen. In C3G, by definition, the patients do not have or have very limited, IgG deposited in the kidney. It is interesting to note that in C3G the presence of anti-C3b Ab is in fact, extremely rare, contrary to the high frequency in LN.
Apart from the action of the anti-C3b Ab at the level of the complement cascade, additional functions were described for these Ab. Suppression of apoptotic cell disposal by Ab against deposited C3 may contribute to increasing severity and/or exacerbations in different autoimmune diseases (32). Indeed, anti-C3b Ab blocked the recognition of C3b-opsinized cells by macrophages in a mouse model.
Further functional studies are needed in larger cohorts to determine to what extend the anti-C3b Ab have a pathogenic potential and whether they can transfer the disease, for example in a mouse model. Comparative studies are lacking for the anti-C3b Ab from patients/mice recovering from infections and the ones with autoimmune diseases. It is tempting to speculate that the functional consequences in the two contexts will be clearly different.
Conclusion
Growing body of experimental data revealed that the anti-C3b Ab have overt functional consequences (30, 32, 33, 40) and correlate with disease severity at least in LN (33, 34). Additional clinical and experimental studies are needed to confirm their role in the disease pathogenesis and their relevance as a biomarker for the clinical practice. Nevertheless, the current state of the art shows arguments that identify the anti-C3b Ab as a potential diagnostic and prognostic marker in LN patients. The presence of anti-C3b Ab indicates a level of complement activation sufficient to initiate and accelerate the kidney damage. Therefore, these Ab should be taken into consideration in the management of LN. We hypothesize that patients with complement overactivation due to presence of anti-C3b Ab may benefit from therapeutic targeting of C3.
Author Contributions
LR, VV, and MR reviewed the literature and wrote the first draft. VF-B, M-AD-D, and VL revised the manuscript. All authors validated the submission.
Funding
This work was supported by a grant for French-Bulgarian collaboration by the program RILA No DNTS/France 01/11 National Science Fund, Bulgaria (to MR) and Campus France, 38666QH (to M-AD-D) as well as by INSERM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
2. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
3. Dragon-Durey MA, Blanc C, Marinozzi MC, Van Schaarenburg RA, Trouw LA. Autoantibodies against complement components and functional consequences. Mol Immunol. (2013) 56:213–21. doi: 10.1016/j.molimm.2013.05.009
4. Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol. (2016) 7:36. doi: 10.3389/fimmu.2016.00036
5. Hristova MH, Stoyanova VS. Autoantibodies against complement components in systemic lupus erythematosus - role in the pathogenesis and clinical manifestations. Lupus (2017) 26:1550–5. doi: 10.1177/0961203317709347
6. Dumestre-Perard C, Clavarino G, Colliard S, Cesbron JY, Thielens NM. Antibodies targeting circulating protective molecules in lupus nephritis: Interest as serological biomarkers. Autoimmun Rev. (2018) 17:890–9. doi: 10.1016/j.autrev.2018.03.013
7. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
8. Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, et al. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature (2005) 437:505–11. doi: 10.1038/nature04005
9. Chen ZA, Pellarin R, Fischer L, Sali A, Nilges M, Barlow PN, et al. Structure of complement C3(H2O) revealed by quantitative cross-linking/mass spectrometry and modeling. Mol Cell Proteomics (2016) 15:2730–43. doi: 10.1074/mcp.M115.056473
10. Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature (2006) 444:213–6. doi: 10.1038/nature05172
11. Rooijakkers SH, Wu J, Ruyken M, Van Domselaar R, Planken KL, Tzekou A, et al. Structural and functional implications of the alternative complement pathway C3 convertase stabilized by a staphylococcal inhibitor. Nat Immunol. (2009) 10:721–7. doi: 10.1038/ni.1756
12. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science (2010) 330:1816–20. doi: 10.1126/science.1195821
13. Pedersen DV, Roumenina L, Jensen RK, Gadeberg TA, Marinozzi C, Picard C, et al. Functional and structural insight into properdin control of complement alternative pathway amplification. EMBO J. (2017) 36:1084–99. doi: 10.15252/embj.201696173
14. Forneris F, Wu J, Xue X, Ricklin D, Lin Z, Sfyroera G, et al. Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. (2016) 35:1133–49. doi: 10.15252/embj.201593673
15. Xue X, Wu J, Ricklin D, Forneris F, Di Crescenzio P, Schmidt CQ, et al. Regulator-dependent mechanisms of C3b processing by factor I allow differentiation of immune responses. Nat Struct Mol Biol. (2017) 24:643–51. doi: 10.1038/nsmb.3427
16. Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood (2008) 112:4948–52. doi: 10.1182/blood-2008-01-133702
17. Roumenina LT, Frimat M, Miller EC, Provot F, Dragon-Durey MA, Bordereau P, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood (2012) 119:4182–91. doi: 10.1182/blood-2011-10-383281
18. Schramm EC, Roumenina LT, Rybkine T, Chauvet S, Vieira-Martins P, Hue C, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood (2015) 125:2359–69. doi: 10.1182/blood-2014-10-609073
19. Reis SE, Falcao DA, Isaac L. Clinical aspects and molecular basis of primary deficiencies of complement component C3 and its regulatory proteins factor I and factor H. Scand J Immunol. (2006) 63:155–68. doi: 10.1111/j.1365-3083.2006.01729.x
20. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
21. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev. (2016) 274:9–15. doi: 10.1111/imr.12474
22. Lachmann PJ. Conglutinin and immunoconglutinins. Adv Immunol. (1967) 6:479–527. doi: 10.1016/S0065-2776(08)60527-1
23. Coombs AM, Coombs RR. The conglutination phenomenon. IX the production of immuno-conglutinin in rabbits. J Hyg. 51:509–31. doi: 10.1017/S0022172400036792
24. Ingram DG. The production of immuno-conglutinin. IX in acute bacterial infections. Can J Microbiol. (1965) 11:151–9. doi: 10.1139/m65-021
25. Rytel MW, Lytle RI. Immunoconglutinin response in patients with acute viral respiratory disease. Clin Exp Immunol. (1973) 14:227–35.
26. Makhdoomi GM, Tiku ML, Beutner KR, Ogra PL. Serum immunoconglutinin titers during acute and chronic hepatitis B virus infection. Infect Immun. (1980) 28:842–5.
27. Ingram DG. The production of immuno-conglutinin. X in chronic bacterial infections. Can J Microbiol. (1965) 11:161–5. doi: 10.1139/m65-022
28. Ingram DG. The conglutination phenomenon. XIV the resistance enhancing effect of conglutinin and immuno-conglutinin in experimental bacterial infections. Immunology (1959) 2:334–45.
29. Durand CG, Burge JJ. A new enzyme-linked immunosorbent assay (ELISA) for measuring immunoconglutinins directed against the third component of human complement. Findings in systemic lupus erythematosus. J Immunol Methods (1984) 73:57–66. doi: 10.1016/0022-1759(84)90031-0
30. Nilsson B, Ekdahl KN, Svarvare M, Bjelle A, Nilsson UR. Purification and characterization of IgG immunoconglutinins from patients with systemic lupus erythematosus: implications for a regulatory function. Clin Exp Immunol. (1990) 82:262–7. doi: 10.1111/j.1365-2249.1990.tb05437.x
31. Nilsson B, Ekdahl KN, Sjoholm A, Nilsson UR, Sturfelt G. Detection and characterization of immunoconglutinins in patients with systemic lupus erythematosus (SLE): serial analysis in relation to disease course. Clin Exp Immunol. (1992) 90:251–5. doi: 10.1111/j.1365-2249.1992.tb07937.x
32. Kenyon KD, Cole C, Crawford F, Kappler JW, Thurman JM, Bratton DL, et al. IgG autoantibodies against deposited C3 inhibit macrophage-mediated apoptotic cell engulfment in systemic autoimmunity. J Immunol. (2011) 187:2101–11. doi: 10.4049/jimmunol.1003468
33. Vasilev VV, Noe R, Dragon-Durey MA, Chauvet S, Lazarov VJ, Deliyska BP, et al. Functional characterization of autoantibodies against complement component C3 in patients with lupus nephritis. J Biol Chem. (2015) 290:25343–55. doi: 10.1074/jbc.M115.647008
34. Birmingham DJ, Bitter JE, Ndukwe EG, Dials S, Gullo TR, Conroy S, et al. Relationship of circulating anti-C3b and anti-C1q IgG to lupus nephritis and its flare. Clin J Am Soc Nephrol. (2016) 11:47–53. doi: 10.2215/CJN.03990415
35. Potter BJ, Brown DJ, Watson A, Jewell DP. Complement inhibitors and immunoconglutinins in ulcerative colitis and Crohn's disease. Gut (1980) 21:1030–4. doi: 10.1136/gut.21.12.1030
36. Ngu JL, Soothill JF. Immunoconglutinin and complement changes in children with acute nephritis. Clin Exp Immunol. (1969) 5:557–66.
37. Ngu JL, Barratt TM, Soothill JF. Immunoconglutinin and complement changes in steroid sensitive relapsing nephrotic syndrome of children. Clin Exp Immunol. (1970) 6:109–16.
38. Ngu JL, Blackett K. Complement and immunoconglutinin changes in the nephrotic syndrome of adult Africans. J Trop Med Hyg. (1970) 73:250–4.
39. Chen Q, Muller D, Rudolph B, Hartmann A, Kuwertz-Broking E, Wu K, et al. Combined C3b and factor B autoantibodies and MPGN type II. N Engl J Med. (2011) 365:2340–2. doi: 10.1056/NEJMc1107484
40. Marinozzi MC, Roumenina LT, Chauvet S, Hertig A, Bertrand D, Olagne J, et al. Anti-Factor B and Anti-C3b autoantibodies in C3 glomerulopathy and Ig-Associated membranoproliferative GN. J Am Soc Nephrol. (2017) 28:1603–13. doi: 10.1681/ASN.2016030343
41. Jozsi M, Reuter S, Nozal P, Lopez-Trascasa M, Sanchez-Corral P, Prohaszka Z, et al. Autoantibodies to complement components in C3 glomerulopathy and atypical hemolytic uremic syndrome. Immunol Lett. (2014) 160:163–71. doi: 10.1016/j.imlet.2014.01.014
42. Tsokos GC. Systemic lupus erythematosus. N Engl J Med. (2011) 365:2110–21. doi: 10.1056/NEJMra1100359
43. Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. (2009) 104:115–49. doi: 10.1016/S0065-2776(08)04004-2
44. Nozal P, Garrido S, Martinez-Ara J, Picazo ML, Yebenes L, Alvarez-Doforno R, et al. Case report: lupus nephritis with autoantibodies to complement alternative pathway proteins and C3 gene mutation. BMC Nephrol. (2015) 16:40. doi: 10.1186/s12882-015-0032-6
45. Birmingham DJ, Merchant M, Waikar SS, Nagaraja H, Klein JB, Rovin BH. Biomarkers of lupus nephritis histology and flare: deciphering the relevant amidst the noise. Nephrol Dial Transplant. (2017) 32:i71–9. doi: 10.1093/ndt/gfw300
46. Fakhouri F, Fremeaux-Bacchi V, Noel LH, Cook HT, Pickering MC. C3 glomerulopathy: a new classification. Nat Rev Nephrol. (2010) 6:494–9. doi: 10.1038/nrneph.2010.85
47. Sethi S, Vrana JA, Fervenza FC, Theis JD, Sethi A, Kurtin PJ, et al. Characterization of C3 in C3 glomerulopathy. Nephrol Dial Transplant. (2017) 32:459–65. doi: 10.1093/ndt/gfw290
48. Servais A, Noel LH, Roumenina LT, Le Quintrec M, Ngo S, Dragon-Durey MA, et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. (2012) 82:454–64. doi: 10.1038/ki.2012.63
49. Noe R, Chauvet S, Togarsimalemath SK, Marinozzi MC, Radanova M, Vasilev VV, et al. Detection of autoantibodies to complement components by surface plasmon resonance-based technology. Methods Mol Biol. (2019) 1901:271–80. doi: 10.1007/978-1-4939-8949-2_24
50. Daha MR, Fearon DT, Austen KF. C3 nephritic factor (C3NeF): stabilization of fluid phase and cell-bound alternative pathway convertase. J Immunol. (1976) 116:1–7.
51. Nilsson Ekdahl K, Nilsson B. Phosphorylation of plasma proteins with emphasis on complement component C3. Mol Immunol. (1999) 36:233–9. doi: 10.1016/S0161-5890(99)00037-1
52. Ekdahl KN, Ronnblom L, Sturfelt G, Nilsson B. Increased phosphate content in complement component C3, fibrinogen, vitronectin, and other plasma proteins in systemic lupus erythematosus: covariation with platelet activation and possible association with thrombosis. Arthritis Rheum. (1997) 40:2178–86. doi: 10.1002/art.1780401212
53. Rekvig OP, Putterman C, Casu C, Gao HX, Ghirardello A, Mortensen ES, et al. Autoantibodies in lupus: culprits or passive bystanders? Autoimmun Rev. (2012) 11:596–603. doi: 10.1016/j.autrev.2011.10.021
54. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. (2015) 14:75–9. doi: 10.1016/j.autrev.2014.10.003
55. Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al. Endothelium structure and function in kidney health and disease. Nat Rev Nephrol. (2019) 15:87–108. doi: 10.1038/s41581-018-0098-z
Keywords: anti-C3 autoantibodies, anti-C3b autoantibodies, complement, immunoconglutinins, autoimmunity, systemic lupus erythematosus, lupus nephritis, C3 glomerulopathy
Citation: Vasilev VV, Radanova M, Lazarov VJ, Dragon-Durey M-A, Fremeaux-Bacchi V and Roumenina LT (2019) Autoantibodies Against C3b—Functional Consequences and Disease Relevance. Front. Immunol. 10:64. doi: 10.3389/fimmu.2019.00064
Received: 02 November 2018; Accepted: 11 January 2019;
Published: 29 January 2019.
Edited by:
Alexandre Belot, Claude Bernard University Lyon 1, FranceReviewed by:
Josh Thurman, University of Colorado, United StatesMarie Nathalie Sarda, Université de Lyon, France
Copyright © 2019 Vasilev, Radanova, Lazarov, Dragon-Durey, Fremeaux-Bacchi and Roumenina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lubka T. Roumenina, bHVia2Eucm91bWVuaW5hQGNyYy5qdXNzaWV1LmZy
†These authors have contributed equally to this work