 Djuro Karanovic1
Djuro Karanovic1 Ian C. Michelow2
Ian C. Michelow2 Anthony R. Hayward3Suk See DeRavin1
Anthony R. Hayward3Suk See DeRavin1 Ottavia M. Delmonte1
Ottavia M. Delmonte1 Michael E. Grigg4
Michael E. Grigg4 Adam Kerry Dobbs1
Adam Kerry Dobbs1 Julie E. Niemela5Jennifer Stoddard5
Julie E. Niemela5Jennifer Stoddard5 Zaid Alhinai2Natasha Rybak6Nancy Hernandez7Stefania Pittaluga8
Zaid Alhinai2Natasha Rybak6Nancy Hernandez7Stefania Pittaluga8 Sergio D. Rosenzweig5Gulbu Uzel1
Sergio D. Rosenzweig5Gulbu Uzel1 Luigi D. Notarangelo1*
Luigi D. Notarangelo1*- 1Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 2Division of Infectious Diseases, Department of Pediatrics, Brown University and Rhode Island Hospital, Providence, RI, United States
- 3Division of Allergy and Immunology, Department of Pediatrics, Brown University and Rhode Island Hospital, Providence, RI, United States
- 4Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
- 5Immunology Service, Department of Laboratory Medicine, NIH Clinical Center, Bethesda, MD, United States
- 6Division of Infectious Diseases, Department of Medicine, Brown University and The Miriam Hospital, Providence, RI, United States
- 7Department of Medicine and Pediatrics, Brown University and Rhode Island Hospital, Providence, RI, United States
- 8Center for Cancer Research, National Cancer Institute (NCI), National Institutes of Health (NIH), Bethesda, MD, United States
Phosphoinositide 3-kinase (PI3K) plays an integral role in lymphocyte function. Mutations in PIK3CD and PIK3R1, encoding the PI3K p110δ and p85α subunits, respectively, cause increased PI3K activity and result in immunodeficiency with immune dysregulation. We describe here the first cases of disseminated and congenital toxoplasmosis in a mother and child who share a pathogenic mutation in PIK3R1 and review the mechanisms underlying susceptibility to severe Toxoplasma gondii infection in activated PI3Kδ syndrome (APDS) and in other forms of primary immunodeficiency.
Introduction
Phosphoinositide 3-kinases (PI3Ks) control essential functions in cellular activation, development and differentiation through generation of phosphatidylinositol (3,4,5)-trisphosphate (PIP3) (1). In lymphocytes, the PI3K complex is made up of a heterodimer consisting of a catalytic subunit (p110δ) and a regulatory subunit (p85α) that are encoded by the PIK3CD and PIK3R1 genes, respectively (1). Heterozygous gain-of-function mutations in PIK3CD lead to constitutive activation of the PI3K pathway and cause activated PI3K delta syndrome type 1 (APDS1); similarly, heterozygous PIK3R1 mutations that affect interaction of p85α with p110δ also lead to constitutive activation of PI3K and cause APDS2 (2–7). The clinical presentation of these primary immunodeficiencies (PIDs) may include recurrent upper respiratory tract infections often leading to bronchiectasis, humoral immunodeficiency with elevated IgM, diffuse lymphadenopathy, Epstein Barr virus, and/or cytomegalovirus viremia, and an increased risk of lymphoma (8, 9). In addition, mutations in the donor splice site in intron 11 of PIK3R1Δ434 _475, resulting in exclusion of exon 11 from the cDNA, cause APDS2, short stature-hyperextensibility of joints-ocular depression-Rieger anomaly-teething delay (SHORT) syndrome, or a combination of the two (10).
We describe a female with APDS2 manifesting with short stature, diffuse lymphadenopathy, recurrent upper respiratory tract infections, elevated IgM and disseminated toxoplasmosis who gave birth to a genetically affected daughter with severe, congenital toxoplasmosis.
Case Report
The patient was born to non-consanguineous healthy parents of Hispanic and East Asian descent. In early childhood, she was diagnosed with recurrent sinopulmonary and otitis infections. Due to persistent diffuse lymphadenopathy since age 5, she underwent multiple lymph node and bone marrow biopsies that were negative for malignancy. She also suffered from multiple episodes of Herpes Simplex oral infection and genital warts, which required electrosurgical excision of precancerous lesions. At the age of 40, she complained of painful cervical lymphadenopathy and was diagnosed with toxoplasmosis based on positive IgM serology results. An ophthalmological and MRI exam were normal. She was treated for 2 weeks with an unknown agent(s) with the presumption that her infection was cured. Then, she immigrated to Rhode Island (USA), where she gave birth to a female baby at 37 weeks gestation. There were concerns for craniosyntosis, occasional episodes of staring and myoclonic jerks that led to a brain CT scan at 3 months of age that showed marked hydrocephalus, enlarged lateral and third ventricles, and extensive cerebral atrophy (Figure 1A). There were widespread calcifications at the gray-white matter interface and in the basal ganglia. The clinical, laboratory and radiologic features were consistent with a congenitally acquired infection. Tests for CMV, HIV, HSV and syphilis were negative while serologic tests for Toxoplasma gondii were positive (IgG was 1:512, IgM was positive by the ISAGA method, IgA antibodies were negative; Palo Alto Toxoplasma Serology Laboratory). T. gondii PCR assays of the blood and CSF were negative. A dried blood spot from the newborn screen was positive when retrospectively tested for T. gondii IgM antibodies at Massachusetts Department of Health.
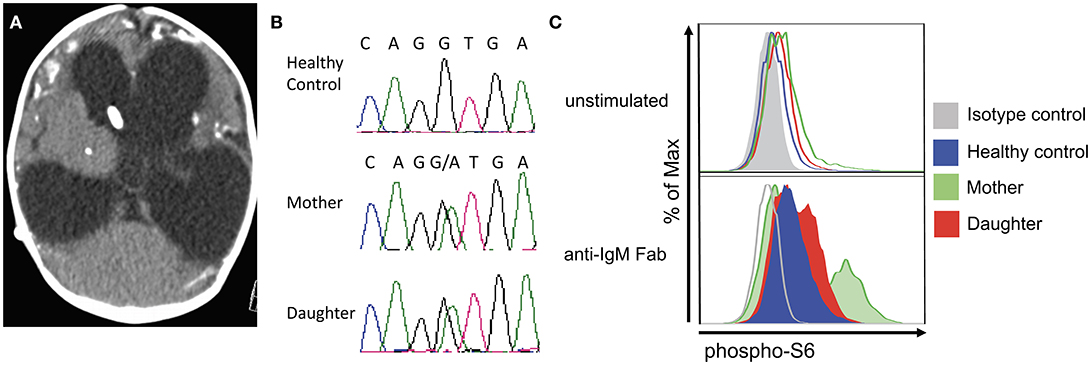
Figure 1. Clinical and laboratory features in mother and daughter with Toxoplasmosis and APDS2. (A) Brain CT in the patient's daughter at 3 months of age, showing marked hydrocephalus with enlarged lateral and third ventricles, profound brain atrophy and basal ganglia calcifications. (B) Chromatogram demonstrating heterozygosity for the c.1425+1g > a at the PIK3R1 locus in the patient and her daughter. (C) Analysis of phospho-S6 in CD20+ cells from a healthy control, the mother, and the daughter at resting conditions (top) and upon in vitro activation with anti-IgM (bottom).
The child met criteria for congenital toxoplasmosis (11) and was treated with oral pyrimethamine, sulfadiazine and leucovorin. During the following year, the child had refractory seizures despite treatment with topiramate, levetiracetam and clonazepam, her microcephaly progressed to <1st percentile, and static encephalopathy with poor feeding necessitated a gastrostomy tube. The anti-toxoplasma IgG titer decreased while on antimicrobial therapy and was undetectable by 36 weeks of treatment. Two months after completion of a 1-year course of anti-parasitic therapy, repeat anti-Toxoplasma IgG testing showed a rebound to a titer of 1:8,000. At 2 years of age, repeat anti-Toxoplasma IgG (1:3,072) and IgM (7.6, normal < 2.0) levels remained elevated. She has elevated serum IgG (1,399 mg/dL) and IgM (215 mg/dL) and undetectable IgA. Her length has consistently remained below the 3rd percentile.
When the child was hospitalized at age 4 months, the mother was not acutely ill, but she had chronic non-tender bilateral cervical lymphadenopathy. Her laboratory tests were significant for strongly positive toxoplasmosis serology thought to be secondary to ongoing chronic infection (IgG was 1:16,000; IgG avidity was high, IgM ELISA was 4.1 (normal < 2.0), and AC/HS ratio of 1,600/3,200). A cervical lymph node biopsy was positive for toxoplasma PCR and she was started on oral pyrimethamine, sulfadiazine, and leucovorin. After 7 months of treatment and moderate improvement in lymphadenopathy, she was switched to suppressive therapy with trimethoprim-sulfamethoxazole (TMP/SMX). When this suppressive regimen was discontinued, the lymphadenopathy worsened. To evaluate for a potential underlying immunodeficiency, both the mother and her daughter were enrolled in NIH protocol 05-I-0213 upon informed consent.
At age 42, the mother was noted to be short (148 cm, <3rd percentile), and to have generalized lymphadenopathy. A mild persistent EBV viremia (up to 2.58log10) and an intermittent CMV viremia (< 3.08log10) was observed.
Immunological investigations revealed normal IgG (986 mg/dL) and IgA (69 mg/dL), with elevated IgM (571 mg/dL). The total lymphocyte count was 1,950 cells/μL. Analysis of lymphocyte subsets by flow cytometry demonstrated decreased CD20+ CD27+ memory B cells (6 cells/μL), increased proportion of CD19+ CD10+ transitional B cells (36.4% of total B cells), and lack of CD20+ CD27+ IgM− switched memory B cells. Specific antibody responses to Streptococcus pneumoniae were not protective to all serotypes. T-cell studies were significant for markedly reduced number of naïve CD4+ CD62L+ CD45RA+ cells (10 cells/μL) and increased number of central (CD62L+ CD45RA−, 265 cells/μL) and effector memory (CD62L− CD45RA−, 456 cells/μL) CD8+ cells.
Whole exome gene sequencing with targeted analysis of 362 PID genes (Table 1) identified a heterozygous mutation at an essential donor splice site of PIK3R1 (NM_181523.2:c.1425+1g> a), which was confirmed with Sanger sequencing (Figure 1B). The mutation results in the skipping of exon 11, which encodes a part of the inter-SH2 domain of the regulatory p85α subunit, and results in hyperactivation of the PI3K pathway (6). DNA analysis of the patient's daughter demonstrated the same PIK3R1 c.1425+1g>a mutation.
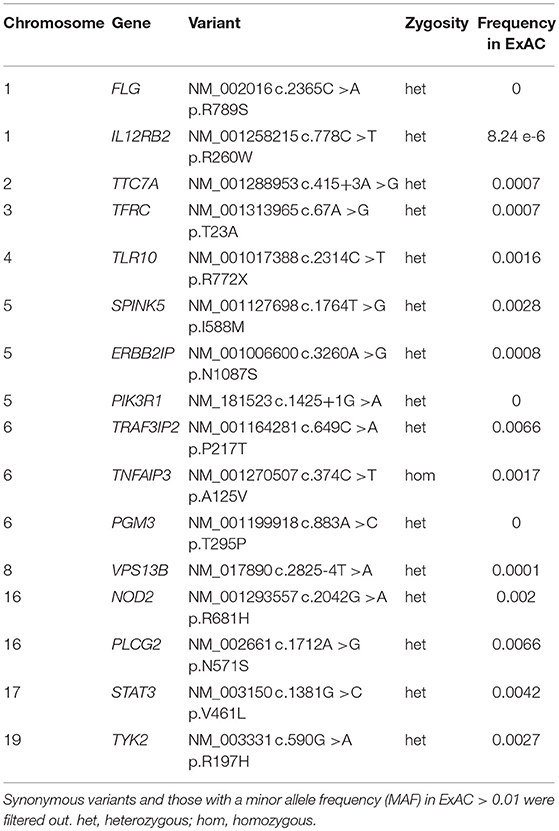
Table 1. Rare genomic variants identified by whole exome sequencing (WES) and targeted analysis of Primary Immune Deficiency genes in the mother with disseminated Toxoplasmosis.
The phosphorylation of Akt is an important downstream event in the activation of the PIK3 pathway. In turn, phosphorylated Akt activates mTOR which phosphorylates S6. Functional studies on the patient's CD8+ and her daughter's CD20+ cells confirmed an increased S6 phosphorylation after IL-2 and anti-IgM stimulation (Figure 1C), which suggests hyperactivation of the PI3K-mTOR signaling pathway.
Finally, during admission at the NIH, a PET-scan was performed on the mother, confirming hypermetabolic generalized lymphadenopathy. In order to better define the nature of the lymphoproliferative process, an excisional biopsy of a left cervical lymph node was performed, which revealed an abundance of PD-1+ T follicular helper (TFH) cells, a striking predominance of IgM+ plasma cells (with few IgG+ plasma cells), and abnormal germinal centers (Figure 2). The proportion of CD4+ CXCR5+ CD45RA− CD25− TFH cells was also markedly increased in circulation in the patient as compared to two healthy controls (41.7 vs. 13.7% and 16.6%).
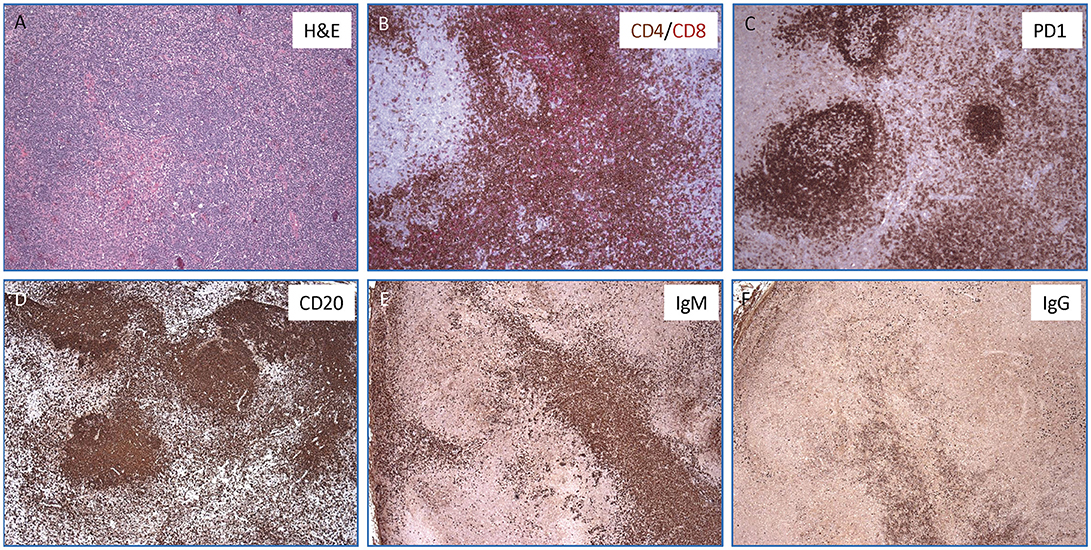
Figure 2. Lymph node histopathology in the mother with APDS2. (A) Hematoxylin & Eosin stained section show an ill-defined secondary follicle with “naked”germinal center; bottom left shows a cluster of monocytoid cells with pale cytoplasm (magnification, 10x). In the inlet, CMV positive cells are identified by immunohistochemistry (magnification, 40x). (B) Double immunohistochemisrty staining for CD4+ (in brown) and CD8+ (in red) T cells (magnification, 10x). (C) Immunohistochemistry staining for PD-1 highlights numerous T-follicular helper cells within the germinal centers (stronger expression) as well as in the interfollicular areas (magnification, 10X). (D–F) Immunohistochemical stains for CD20+ (D), IgM+ (E), and IgG+ cells (F) showing a marked increase of IgM positive plasma cells over IgG (magnification, 4x).
Upon functional and genetic confirmation of APDS2, we initiated treatment with sirolimus in the mother to inhibit mTOR activation, and added IgG replacement therapy and Toxoplasma suppressive therapy with TMP-SMX. This treatment has resulted in improvement of the lymphadenopathy. She remains negative for CMV and EBV viremia by quantitative PCR. Her daughter has been started on TMP-SMX to prevent reactivation of T. gondii. At the time of her last follow-up visit (3 years and 3 months of age), she presented with typical immunological hallmarks of the disease, including an increased proportion of central memory CD4+ cells (44.1% of total CD4+ cells) and of TEMRA CD8+ cells (36.1%), along with an elevated proportion of transitional B cells (31.6% of total CD19+ cells).
Discussion
Immune Deficiencies Reveal Critical Mechanisms Involved in Protection Against T. Gondii Infection
T. gondii is an obligate intracellular parasite that establishes a relatively benign, life-long infection with only immunocompromised hosts showing signs of clinical disease (12). Encephalitis and ocular infections are reported in secondary immunodeficiencies due to HIV, chemotherapy and post solid organ or hematopoietic stem cell transplant. Humans and mice act as intermediate hosts and are infected either by ingesting undercooked meat laden with T. gondii tissue cysts or by drinking oocyst contaminated water. Tachyzoites released from oocysts or tissue cysts then replicate in intestinal epithelium and myeloid cells can disseminate the infection (13).
Murine models suggest that different lymphocyte subsets are involved in the acute and chronic phases of parasite control. The acute phase of infection is mediated principally by macrophages, dendritic cells, NK and CD4+ cells (14–16). Upon engagement of the chemokine receptor 5 (CCR5), Toll-like receptors (TLR) 2 and 11, and the inflammasome sensors NLRP1 and NLRP3 by Toxoplasma molecules, antigen presenting cells are activated to produce IL-12 and IL-18 (17–19), triggering IFN-γ production by CD4+ and NK cells. In turn, IFN-γ upregulates production of reactive oxygen species (ROS) in macrophages thereby inhibiting parasite replication (20, 21). On the other hand, the memory immune response during chronic infection or upon antigenic challenge is driven by IFN-γ producing CD8+ T cells (22).
Identifying human primary immunodeficiencies that are associated with Toxoplasma infection is critical for developing an understanding of host defenses against this parasite. A review of the literature yields 24 cases with a diverse set of primary immunodeficiencies that predispose human hosts to severe toxoplasmosis (Table 2). The heterogeneity of these diseases suggests that multiple immune system defects may contribute to toxoplasmosis pathogenesis.
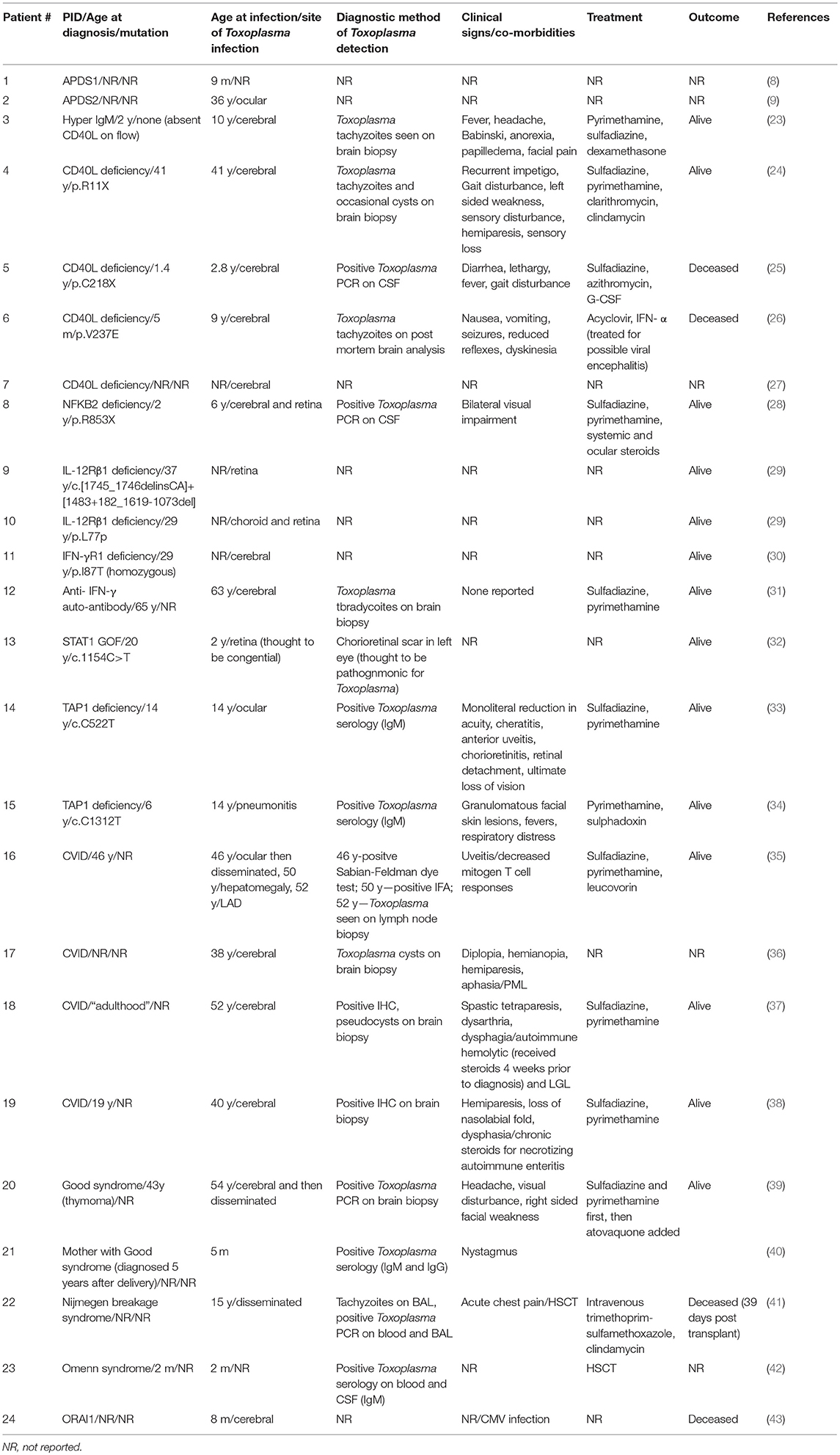
Table 2. Cases of toxoplasmosis in patients with primary immune deficiencies.
Defects of IL-12/IFN-γ Loop
The IL-12/IFN-γ loop facilitates T cell activation and proliferation and intracellular macrophage-mediated parasite killing. Upon T cell receptor (TCR) recognition of a MHC:Toxoplasma peptide complex, T lymphocytes are activated and express the CD40 ligand (CD40L) molecule on the cell surface, enabling T cell interaction with macrophages and dendritic cells that constitutively express CD40. CD40L:CD40 signaling activates the non-canonical NF-κB pathway (NFKB2), which increases the cell surface expression of the co-stimulatory molecules CD80/86 (44). In addition, NFKB2 upregulates IL-12 production in activated macrophages (45). The ligation of the IL-12R on T lymphocytes by IL-12 leads to IFN-γ release. In turn, ligation of the IFN-γR on macrophages leads to activation of the JAK/STAT pathway, with phosphorylation, dimerization and nuclear translocation of Signal Transducer and Activator of Transcription 1 (STAT1), which helps produce anti-Toxoplasma effectors like intracellular ROS.
Review of the literature identified a total of 10 patients with severe Toxoplasma infection involving five distinct gene defects along the IL-12/ IFN-γ loop: four cases of CD40L deficiency, one case of heterozygous mutation in NFKB2, two cases of IL-12Rβ1 deficiency, one case of STAT1 gain-of-function mutation, and two cases of autosomal recessive IFN-γR1 deficiency (23–30). Furthermore, anti-IFN-γ autoantibodies were demonstrated in one patient with severe toxoplasmosis (31). Interestingly, disruption of IFN-γ signaling (receptor deficiency or auto-antibody production) and CD40L deficiency result in more severe disease, as these defects were associated with Toxoplasma encephalitis.
There is evidence that TNF-α may also provide protection against T. gondii. In particular, early work by Jack Remington established that regulation of TNF expression determines susceptibility to toxoplasmic encephalitis in mice (46). Furthermore, Jannsen et al. demonstrated in patients with partial IFN-γR1 deficiency that the addition of TNF-α can limit T. gondii replication in vitro (47). The important role of TNF-α in parasite control was also confirmed by the observation that treatment with infliximab may lead to reactivation of cerebral toxoplasmosis (48).
As mentioned previously, CD8+ cells mediate the memory response by recognizing Toxoplasma antigens in the context of MHC class I molecules. Transporter associated with antigen processing (TAP) is critical in transporting peptides to the lumen of the endoplasmic reticulum and facilitating peptide loading onto MHC class I molecules. Ocular and pulmonary toxoplasmosis has been reported in two patients with TAP1 deficiency (33, 34). On the other hand, Tap1−/− mice succumb to infection faster than Cd8−/− mice, and CD4+ T cells and NK lymphocytes from Tap1−/− mice produce reduced amounts of IFN-γ (49), indicating that TAP deficiency affects both acute and chronic phases of Toxoplasma infection.
Common Variable Immunodeficiency
Humoral responses are not thought to play an essential role in limiting Toxoplasma replication. Three cerebral and one case of disseminated toxoplasmosis in common variable immunodeficiency are published (35–38). However, the clinical history and laboratory findings of these cases, characterized by chronic steroid administration for necrotizing autoimmune enteritis, development of large granular leukemia, progressive multifocal leukoencephalopathy and significant reduction in mitogen-induced T cell proliferation, are suggestive of co-existing cellular defects.
Good Syndrome
Patients with Good Syndrome have thymomas with combined cellular and humoral immunodeficiency and autoimmunity. Cerebral and disseminated Toxoplasma infection was reported in one patient with Good syndrome (39), and Toxoplasma reactivation in pregnancy resulting in congenital toxoplasmosis has been described in another patient with the same disease (40). Further studies are needed to characterize the molecular and cellular mechanisms predisposing to Toxoplasma infection in this patient population.
Other T Cell Immunodeficiencies
Fulminant and fatal reactivation of T. gondii infection has been reported early after allogeneic hematopoietic cell transplantation (HCT) in a patient with Nijmegen breakage syndrome (41). In addition to the underlying immunodeficiency, both the Toxoplasma seronegative status of the donor, and use of immunosuppressive therapy pre- and post-transplantation may have contributed to the fatal outcome in this patient. Invasive toxoplasmosis after allogeneic HCT has bene reported in 4–6% of seropositive recipients, with an estimated mortality rate as high as 60–90% (50, 51).
In spite of the severity of T cell immunodeficiency, few cases of toxoplasmosis have bene reported in patients with severe combined immune deficiency (SCID) and related disorders. The rarity of this association is probably related to the fact that infants with SCID die early in life unless immune reconstitution is achieved with HCT. However, cerebral toxoplasmosis has been reported in an infant with Omenn syndrome (42) and in another infant with severe T cell immunodeficiency due to calcium flux defect (43).
APDS
Toxoplasma infection has been previously reported in a 9-month-old infant with APDS1 (8), and ocular involvement has been described in a 36-year-old patient with APDS2 (9). T. gondii may evade host defense by inducing the activation of the PI3K/AKT signaling pathway which reduces intracellular reactive oxygen species (ROS) through NOX4 suppression (52) (Figure 3). Active AKT phosphorylates and thereby inactivates the transcription factor FOXO1, thereby preventing p22phox transcription. The resultant decrease of ROS in macrophages leads to reduced activation of AP-1, MAPK, and NF-κB and decreased production of macrophage migration inhibitory factor (MIF), a pro-inflammatory cytokine that plays an important role in the immune response to Toxoplasma (53).
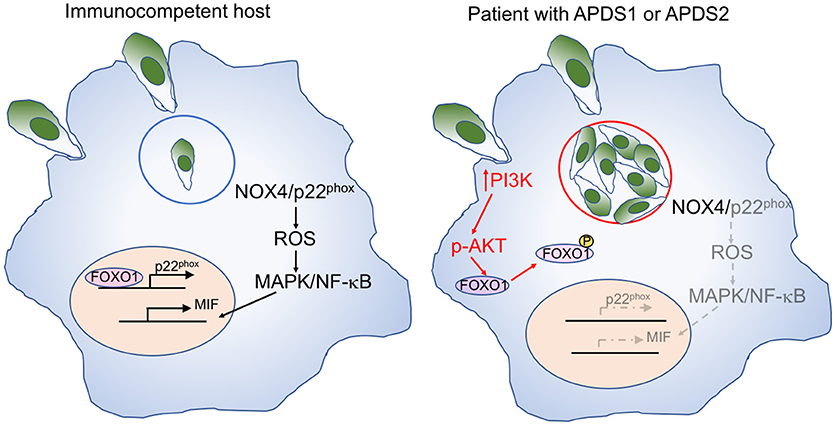
Figure 3. Mechanisms of macrophage-mediated response against Toxoplasma, and effects of increase PI3K signaling. Left: In immunocompetent hosts, intracellular Toxoplasma infection with tachyzoites within parasitophorous vesicles (blue circle) elicits a macrophage response mediated by NOX4 and p22phox. Expression of the latter is controlled by the FOXO1 transcription factor. Activation of the NOX4/p22phox complex allows generation of reactive oxygen species (ROS), activation of MAP kinase (MAPK), and NF-κB signaling, and production of the pro-inflammatory macrophage inhibitory factor (MIF). Right: In patients with APDS1/2, increased PI3K signaling induces AKT phosphorylation, which in turn mediates phosphorylation of FOXO1, impairing p22phox gene expression (in gray). This causes reduced production of ROS, defective activation of MAPK and NF-κB, and impaired production of MIF (all in gray) in response to Toxoplasma infection. Furthermore, the favorable metabolic environment supported by enhanced PI3K activity promotes intracellular replication of Toxoplasma tachyzoites.
Moreover, AKT phosphorylation promotes glycolysis and activation of the amino acid sensor mechanistic target of rapamycin complex 1 (mTORC1), thereby creating an intracellular metabolic environment that is favorable to the intracellular proliferation of Toxoplasma parasites, that can utilize micronutrients for their own replication cycle.
Concluding Remarks
We have described two patients, a mother and daughter, with an essential donor splice site mutation of PIK3R1 and disseminated and congenital toxoplasmosis, respectively. Few cases of parasitic infections have been previously reported in patients with APDS, including diarrhea due to Cryptosporidium parvum or to Giardia lamblia (8–10). This is the first time that systemic and severe congenital toxoplasmosis are reported in a mother and child with APDS2, an immune defect that may favor Toxoplasma replication. For patients with APDS who are considered for HCT, both donor and recipient Toxoplasma serology must be included in the screening work-up. Finally, the identification of severe Toxoplasma infection in patients with inborn errors of immunity has helped identify cellular and molecular mechanisms that are critically involved in the immune response against this parasite.
Ethics Statement
This study was carried out in accordance with the recommendations of the Declaration of Helsinki and consistent with Good Clinical Practice and the applicable regulatory requirements, according to protocol 05-I-0213 which was approved by the IRB of the National Institutes of Health and is registered at ClinicalTrials.Gov as NCT00128973. Written informed consent has been obtained from the patient for the publication of this case report.
Author Contributions
DK, IM, ZA, LN, and MG wrote the manuscript. IM, AH, SD, OD, ZA, NR, and NH identified the patient's condition and provided clinical care. AD, JN, JS, SR, and GU performed experiments. SP performed tissue pathology studies. LN supervised and contributed to the whole work.
Funding
This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol. (2016) 16:702–14. doi: 10.1038/nri.2016.93
2. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science (2013) 342:866–71. doi: 10.1126/science.1243292
3. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol. (2014) 15:88–97. doi: 10.1038/ni.2771
4. Chudasama KK, Winnay J, Johansson S, Claudi T, König R, Haldorsen I, et al. SHORT syndrome with a partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. (2013) 93:150–7. doi: 10.1016/j.ajhg.2013.05.023
5. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Bagel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. (2013) 93:158–66. doi: 10.1016/j.ajhg.2013.06.005
6. Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, et al. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest. (2014) 124:3923–8. doi: 10.1172/JCI75746
7. Lougaris V, Faletra F, Lanzi G, Vozzi D, Marcuzzi A, Valencic E, et al. Altered germinal center reaction and abnormal B cell peripheral maturation in PI3KR1-mutated patients presenting with HIGM-like phenotype. Clin Immunol. (2015) 159:33–6. doi: 10.1016/j.clim.2015.04.014
8. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: a large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606. doi: 10.1016/j.jaci.2016.06.021
9. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: a cohort study. J Allergy Clin Immunol. (2016) 138:210–8. doi: 10.1016/j.jaci.2016.03.022
10. Bravo Garcia-Morato M, Garcia-Minaur S, Molina Garciano J, Santos Simarro F, Del Pino Molina L, López-Granados E, et al. Mutations in PIK3R1 can lead to APDS2, SHORT syndrome or a combination of the two. Clin Immunol. (2017) 179:77–80. doi: 10.1016/j.clim.2017.03.004
11. Kimberlin DW, Long SS, Brady MT, Jackson MAeditors. Red Journal 2018: Report of the Committee on Infectious Diseases. 31st ed. Itasca, IL: American Academy of Pediatrics. (2018).
12. Sturge CR, Yarovinsky F. Complex immune cell interplay in the gamma interferon response during Toxoplasma gondii infection. Infect Immun. (2014) 82:3090–7. doi: 10.1128/IAI.01722-14
13. Peterson E, Dubey JP. Biology of toxoplasmosis. In: Joynson DHM, Wreghitt TG, editors. Toxoplasmosis. Cambridge: Cambridge University Press (2001) p. 1–42.
14. Gazzinelli RT, Heiny S, Wynn TA, Wolf S, Sher A. Interleukin 12 is required for the T-lymphocyte-independent induction of interferon γ by an intracellular parasite and induces reisistance in T-cell-deficient hosts. Proc Natl Acad Sci USA. (1993) 90:6115–9. doi: 10.1073/pnas.90.13.6115
15. Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, et al. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J Immunol. (2006) 177:31–5. doi: 10.4049/jimmunol.177.1.31
16. Reis E, Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, et al. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J Exp Med. (1997) 186:1819–29.
17. Aliberti J, Reis e Sousa C, Schito M, Hieny S, Wells T, Huffnagle GB, et al. CCR5 provides a signal for microbial induced production of IL-12 for CD8α+ dendritic cells. Nat Immunol. (2000) 1:83–7. doi: 10.1038/76957
18. Mun HS, Aosai F, Norose K, Chen M, Piao LX, Takeuchi O, et al. TLR2 as an essential molecule for protective immunity against Toxoplasma gondii infection. Int Immunol. (2003) 15:1081–7. doi: 10.1093/intimm/dxg108
19. Yarovinsky F, Zhang D, Anderson JF, Bannenberg GL, Serhan CN, Hayden MS, et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science (2005) 308:1626–9. doi: 10.1126/science.1109893
20. Khan IA, Matsuura T, Kasper LH. Interleukin-12 enhances murine survival against acute toxoplasmosis. Infect Immun. (1994) 62:1639–42.
21. Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-γ as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. (1983) 158:670–89. doi: 10.1084/jem.158.3.670
22. Gazzinelli RT, Hakim FT, Hieny S, Shearer GM, Sher A. Synergistic role of CD4+ and CD8+ T lymphocytes in IFN-γ production and protective immunity induced by an attenuated Toxoplasma gondii vaccine. J Immunol. (1991) 146:286–92.
23. Leiva LE, Junprasert J, Hollenbaugh D, Sorensen RU. Central nervous system toxoplasmosis with an increased proportion of circulating γδ t cells in a patient with hyper-IgM syndrome. J Clin Immunol. (1998) 18:283–9. doi: 10.1023/A:1027337923709
24. Yong PFK, Post FA, Gilmour KC, Grosse-Kreul D, King A, Easterbrook P, et al. Cerebral toxoplasmosis in a middle-aged man as first presentation of primary immunodeficiency due to a hypomorphic mutation in the CD40 ligand gene. J Clin Pathol. (2008) 61:1220–2. doi: 10.1136/jcp.2008.058362
25. Liu X, Zhou K, Yu D, Cai X, Hua Y, Zhou H, et al. A delayed diagnosis of X-linked hyper IgM syndrome complicated with toxoplasmic encephalitis in a child. Medicine (2017) 96:e8989. doi: 10.1097/MD.0000000000008989
26. Tsuge I, Matsuoka H, Nakagawa A, Kamachi Y, Aso K, Negoro T, et al. Necrotizing toxoplasmic encephalitis in a child with the X-linked hyper-IgM syndrome. Eur J Pediatr. (1998) 157:735–57. doi: 10.1007/s004310050925
27. Levy J, Espanol-Boren T, Thomas C, Fischer A, Tovo P, Bordigoni P, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. (1997) 131:47–54. doi: 10.1016/S0022-3476(97)70123-9
28. Maccari ME, Scarselli A, Di Cesare S, Floris M, Angius A, Deodati A, et al. Severe Toxoplasma gondii infection in a member of a NFKB2-deficient family with T and B cell dysfunction. Clin Immunol. (2017) 183:273–7. doi: 10.1016/j.clim.2017.09.011
29. De Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (2010) 89:381–402. doi: 10.1097/MD.0b013e3181fdd832
30. Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernández-Pérez L, Chapgier A, et al. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients with 11 kindreds. Human Mol Genet. (2011) 20:1509–23. doi: 10.1093/hmg/ddr029
31. Zerbe CS, Marciano BE, Katial RK, Santos CB, Adamo N, Hsu AP, et al. Progressive multifocal leukoencephalopathy in primary immune deficiencies: STAT1 gain of function and review of the literature. Clin Infect Dis. (2016) 62:986–94. doi: 10.1093/cid/civ1220
32. Hanitsh LG, Löbel M, Müller-Redetzky H, Schürmann M, Suttorp N, Unterwalder N, et al. Late-onset disseminated Mycobacterium avium intracellulare complex infection (MAC), cerebral toxoplasmosis and salmonella sepsis in a German caucasian patient with unusual anti-interferon-gamma IgG1 autoantibodies. J Clin Immunol. (2015) 35:361–5. doi: 10.1007/s10875-015-0161-5
33. Parissiadis A, Dormoy A, Fricker D, Hanau D, de la Salle H, Cazenave JP, et al. Unilateral necrotizing toxoplasmic retinochoroiditis as the main clinical manifestation of a peptide transporter (TAP) deficiency. Br J Ophthalmol. (2005) 89:1661–2. doi: 10.1136/bjo.2005.078097
34. Dogu F, Ikincigullari A, Fricker D, Bozdogan G, Aytekin C, Ileri M, et al. A novel mutation for TAP deficiency and its possible association with Toxoplasmosis. Parasitol Int. (2006) 55:219–22. doi: 10.1016/j.parint.2006.02.003
35. Shachor J, Shneyour A, Radnay J, Steiner ZP, Bruderman I. Toxoplasmosis in a patient with common variable immunodeficiency. Am J Med Sci. (1984) 287:36–8.
36. Scotton PG, Vaglia A, Carniato A, Marchiori GC. Progressive multifocal leukoenceophalopathy in a patient with common variable immunodeficiency. Clin Infect Dis. (1998) 26:215–6.
37. Holtkamp M, Okuducu AF, Klingebiel R, Ploner CJ. Cerebral toxoplasmosis in a patient with common variable immunodeficiency. Neurology (2004) 63:2192–3. doi: 10.1212/01.WNL.0000145833.69409.2B
38. Hofmann A, Zaharatos G, Miller M. Case report and review of the literature: Toxoplasma gondii encephalitis in a 40-year-old woman with common variable immunodeficiency and a new diagnosis of large granular lymphocytic leukemia. Can J Infect Dis Med Microbiol. (2008) 19:309–10. doi: 10.1155/2008/614279
39. Sasson S, Davies S, Chan R, Davies L, Garsia R. Cerebral toxoplasmosis in a patient with myasthenia gravis and thymoma with immunodeficiency/Good's syndrome: a case report. BMC Infect Dis. (2016) 16:457. doi: 10.1186/s12879-016-1801-y
40. Tahiria J, Fouyssac F, Morel O, Maatouk A. Good's syndrome and congential toxoplasmosis due to maternal reactivation during pregnancy. Rev Med Interne (2017) 38:337–9. doi: 10.1016/j.revmed.2016.08.005
41. Stajner T, Vasiljević Z, Vujić D, Marković M, Ristić G, Mićić D, et al. Atypical strain of Toxoplasma gondii causing fatal reactivation after hematopoietic stem cell transplantation in a patient with an underlying immunological deficiency. J Clin Microbiol. (2013) 51:2686–90. doi: 10.1128/JCM.01077-13
42. Cansever M, Pinarbasi AS, Baydilli N, Karakukcu M, Patiroglu T. A case of Omenn syndrome accompanied by bilateral cystine stones: case report. Urol Nephrol Open Access J. (2018) 6:35–8. doi: 10.15406/unoaj.2018.06.00200
43. Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, et al. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood (1995) 85:1053–62.
44. Munroe ME, Bishop GA. A costimulatory function for T cell CD40. J Immunol. (2007) 178:671–82. doi: 10.4049/jimmunol.178.2.671
45. Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. (1995) 15:5258–67. doi: 10.1128/MCB.15.10.5258
46. Freund YR, Sgarlato G, Jacob CO, Suzuki Y, Remington JS. Polymorphisms in the tumor necrosis factor α (TNF-α) gene correlate with murine resistance of development of toxoplasmic encephalitis and with levels of TNF-α mRNA in infected brain tissue. J Exp Med. (1992) 175:683–8. doi: 10.1084/jem.175.3.683
47. Jannsen R, Van Wengen A, Verhard E, De Boer T, Zomerdijk T, Ottenhoff TH, et al. Divergent role for TNF-α in IFN-γ-induced killing of Toxoplasma gondii and Salmonella typhimurium contributes to selective suscceptibility of patients with partial IFN-γ receptor 1 deficiency. J Immunol. (2002) 169:3900–7. doi: 10.4049/jimmunol.169.7.3900
48. Young JD, McGwire BS. Infliximab and reactivation of cerebral toxoplasmosis. N Eng J Med. (2005) 353:1530–1. doi: 10.1056/NEJMc051556
49. Goldszmid RS, Bafica A, Jankovic D, Feng CG, Caspar P, Winkler-Pickett R, et al. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J Exp Med. (2007) 204:2591–602. doi: 10.1084/jem.20070634
50. Martino R, Maertens J, Bretagne S, Rovira M, Deconinck E, Ullmann AJ, et al. Toxoplasmosis after hematopoietic stem cell transplantation. Clin Infect Dis. (2000) 31:1188–94. doi: 10.1086/317471
51. Martino R, Bretagne S, Einsele H, Maertens J, Ullmann AJ, Parody R, et al. Early detection of Toxoplasma gondii infectiuon by molecular monitoring of Toxoplasma gondii in peripheral blood samples after allogeneic stem cell transplantation. Clin Infect Dis. (2005) 40:67–78. doi: 10.1086/426447
52. Zhou W, Quan JH, Lee YH, Shin DW, Cha GH. Toxoplasma gondii proliferation require down-regulation of host Nox-4 expression via activation of PI3 kinase/Akt signaling pathway. PLoS ONE (2013) 8:e66306. doi: 10.1371/journal.pone.0066306
Keywords: immunodeficiency, toxoplasmosis, APDS2, PI3K3R1, PI3K
Citation: Karanovic D, Michelow IC, Hayward AR, DeRavin SS, Delmonte OM, Grigg ME, Dobbs AK, Niemela JE, Stoddard J, Alhinai Z, Rybak N, Hernandez N, Pittaluga S, Rosenzweig SD, Uzel G and Notarangelo LD (2019) Disseminated and Congenital Toxoplasmosis in a Mother and Child With Activated PI3-Kinase δ Syndrome Type 2 (APDS2): Case Report and a Literature Review of Toxoplasma Infections in Primary Immunodeficiencies. Front. Immunol. 10:77. doi: 10.3389/fimmu.2019.00077
Received: 18 September 2018; Accepted: 11 January 2019;
Published: 14 February 2019.
Edited by:
Sergey Nejentsev, University of Cambridge, United KingdomReviewed by:
Michel J. Massaad, American University of Beirut Medical Center, LebanonPérsio Roxo-Junior, University of São Paulo, Brazil
Copyright © 2019 Karanovic, Michelow, Hayward, DeRavin, Delmonte, Grigg, Dobbs, Niemela, Stoddard, Alhinai, Rybak, Hernandez, Pittaluga, Rosenzweig, Uzel and Notarangelo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luigi D. Notarangelo, bHVpZ2kubm90YXJhbmdlbG8yQG5paC5nb3Y=