 Paola Bonaventura1,2
Paola Bonaventura1,2 Tala Shekarian1,2Vincent Alcazer1,2Jenny Valladeau-Guilemond2Sandrine Valsesia-Wittmann1,2
Tala Shekarian1,2Vincent Alcazer1,2Jenny Valladeau-Guilemond2Sandrine Valsesia-Wittmann1,2 Sebastian Amigorena3
Sebastian Amigorena3 Christophe Caux1,2
Christophe Caux1,2 Stéphane Depil1,2,4*
Stéphane Depil1,2,4*- 1Centre Léon Bérard, Lyon, France
- 2INSERM U1052, Centre de Recherche en Cancérologie de Lyon, Lyon, France
- 3Institut Curie, PSL Research University, INSERM, U932, Paris, France
- 4Université Claude Bernard Lyon 1, Lyon, France
Therapeutic monoclonal antibodies targeting immune checkpoints (ICPs) have changed the treatment landscape of many tumors. However, response rate remains relatively low in most cases. A major factor involved in initial resistance to ICP inhibitors is the lack or paucity of tumor T cell infiltration, characterizing the so-called “cold tumors.” In this review, we describe the main mechanisms involved in the absence of T cell infiltration, including lack of tumor antigens, defect in antigen presentation, absence of T cell activation and deficit of homing into the tumor bed. We discuss then the different therapeutic approaches that could turn cold into hot tumors. In this way, specific therapies are proposed according to their mechanism of action. In addition, ‘‘supra-physiological’’ therapies, such as T cell recruiting bispecific antibodies and Chimeric Antigen Receptor (CAR) T cells, may be active regardless of the mechanism involved, especially in MHC class I negative tumors. The determination of the main factors implicated in the lack of preexisting tumor T cell infiltration is crucial for the development of adapted algorithms of treatments for cold tumors.
Immune checkpoint inhibitors (ICIs) have changed the treatment landscape of many tumors, inducing durable responses in some cases, Tumor mutational load, CD8+ T cell density and Programmed cell Death Ligand−1 (PD-L1) expression have each been proposed as distinct biomarkers of response to PD-1/-L1 antagonists. The lymphocyte infiltration and IFN-γ status may be key factors for effective anti-PD-1/-L1 therapy by defining a “T cell inflamed” phenotype (“hot tumors”). In contrast, lack of T cells infiltrating the tumor characterizes “non-inflamed” or “cold tumors” (in which other immune populations or myeloid cells can however be observed). Immunological treatment of cold tumors is a great challenge as no adaptive immune response has been set up or maintained. In this review, we discuss the possible issues that the immune system could encounter at different steps of the anti-tumor immune cycle (1), leading to the absence of T cell infiltration: lack of tumor antigens, defect in Antigen Presenting Cells (APCs), absence of T cell activation and deficit of homing into the tumor bed (Figure 1). The potential therapeutic strategies to overcome these problems will be described in the second part of this review. We will not discuss here the mechanisms of immune escape developed by inflamed tumors, reviewed elsewhere (2).
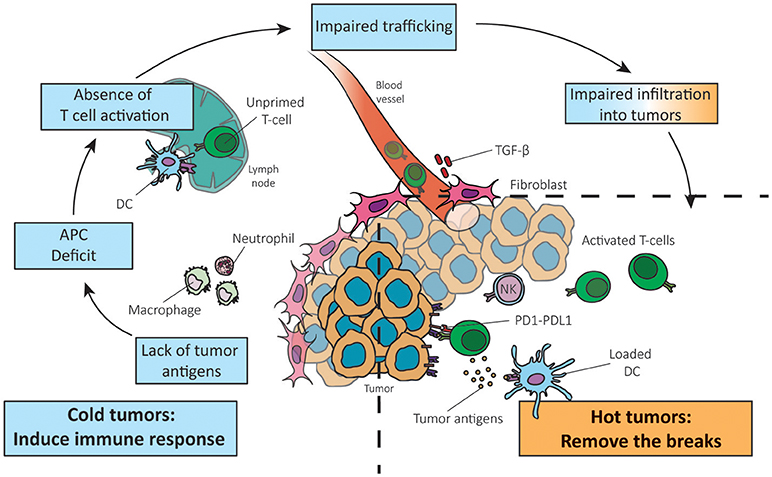
Figure 1. Reversing a cold into a hot tumor. Adapted from Chen and Mellman (1). The absence of T cells in the tumor can be due to the lack of tumor antigens, APC deficit, absence of T cell priming/activation and impaired trafficking of T cells to the tumor mass (left panel). Understanding which step of the anti-cancer immune response is not functional in cancers is crucial to adapt therapies to the cancer phenotype.
Lack of Tumor Antigens
Tumor antigens can be divided into three main classes: tumor specific antigens (TSA), cancer-germline antigens (CGA), and tumor associated antigens (TAA) (3). TSA are expressed only by cancer cells and not in healthy tissues (4). TSA include mutation-associated neoantigens (MANA) and viral antigens. MANAs arise from DNA mutation/rearrangement in a gene coding sequence and play a crucial role in the recognition of tumor cells by CD8+ T cells after immune checkpoint treatment (5). Viral antigens may also represent the target for immune recognition of virus-associated tumors (6). CGA are expressed in tumor cells of different histological origins, but they are silent in normal adult tissues, except in the male germ line and trophoblastic cells (7). Their expression is associated to the demethylation of their promoter. TAA correspond to antigens with low expression in normal tissues and overexpressed in tumor cells, like HER2 (8), or melanocyte differentiation proteins (9).
Tumor mutation burden (TMB) is a quantitative measure of the total number of mutations per coding area of a tumor genome that has been shown to predict responses to ICIs in a range of advanced cancers (10). Tumors with a high TMB are believed to express more MANA. Interestingly, a correlation between MANA and CGA has been observed (11). However, even if some tumors are characterized by low expression of both MANA and CGA, the quantity of tumor neoantigens does not seem to be the main limiting factor for the induction of a T cell response. A recent study by Spranger et al. analyzed the impact of the presence of differentiation antigens, CGA and MANA on T cell infiltration in malignant melanoma. They reported that non-T-cell-inflamed melanomas do not lack antigens for T-cell recognition, arguing for other mechanisms causing the lack of T cell priming and recruitment. Moreover, the number of neoantigens and the mutational load was still comparable between non-T cell-inflamed and T cell-inflamed subtypes in other solid tumors (12). Finally, the kinetics of tumor antigens release, associated with different modalities of spontaneous or induced cancer cell death, may also influence the quality of the T cell response (13).
Absence of T Cell Priming/Activation
Defective Recruitment of APCs
The second step of the anti-tumor immune response consists in the presentation of tumor antigens by dendritic cells (DCs), resulting in the priming and activation of specific effector T cells. Several DCs subsets such as classical DCs (type 1 cDC1s and type 2 cDC2s), Langerhans cells, inflammatory DCs and plasmacytoid DCs (pDCs) exist and are specialized in different functions to shape the immune response and cope with the threat of diversity (14). In particular some evidences exist showing that a higher ratio of cDC1s over monocytes/macrophages in the tumor bed favors protective anti-tumoral adaptive immune responses (15). Spranger et al. have also shown in melanoma that MHC expression and DCs infiltration is associated with T cell infiltration (12).
Among DCs, cDC1s excel at inducing anti-tumoral CD8 T cell responses through cross-presentation of exogenous antigens on MHC-I (16–18). A strong correlation between CD8 gene transcript and cDC1s markers was observed, suggesting that lack of T cell activation and infiltration in the non-T-cell-inflamed tumor microenvironment is mainly associated with a defective recruitment and activation of cDC1 (19–21). Moreover, recent papers published by M. Krummel and C. Reis e Sousa teams recently demonstrated a critical role of the cross-talk between cDC1s and NK cells for the CTL infiltration in melanoma (22, 23). In mice, several reports have shown that cDC1s are necessary for the natural rejection of transplanted tumors and for the efficiency of anti-tumoral immunotherapies including ICIs or adoptive transfer of anti-tumoral CD8 T cells (24).
Lack of T Cell Co-stimulation and Activation After Antigen Presentation
The maturation and activation of antigen-presenting DCs is a critical step for activating an efficient T cell-response. In this context, the DC activation marker DC-LAMP is a good prognostic marker in solid tumors (25). Naive T cells require contact with activated APCs to be primed in an appropriate context of “danger signal” (26). APCs expressing Pattern Recognition Receptors (PRRs) can be directly activated by Pathogen-Associated Molecular Patterns (PAMPs) or Danger Associated Molecular Patterns (DAMPs) to become competent to prime T cell responses (27). Engagement of PRRs on DCs induces NF-κB activation, up-regulation of co-stimulatory molecules, production of cytokines and promotion of cross-priming (28, 29). Various DAMPs are produced by tumor cells undergoing immunological cell death [e.g., calreticulin, HighMobility Group Box 1 protein (HMGB1) or Sin3A Associated Protein 130 (SAP130)] (30). The absence or low production of DAMPs could induce a lack of DCs maturation as well as production of immunosuppressive factors such as transforming growth factor beta (TGF-β) leading to the absence of CD4+ T cell help (30, 31). Recent works demonstrate the importance of the protein Formyl Peptide Receptor 1 (FPR1) expressed by tumoral DCs in the anthracycline-induced immunogenic cell death. DCs lacking or presenting a variant of FRP1, failed in antigen presentation and activation of T cells, resulting in poor anticancer immune responses and reduced overall survival in breast and colon cancer (32).
Stimulation of CD40 on APCs through CD40L expressed on helper CD4+ T cells is another crucial step for the activation of APCs to prime CD8+ T cells. Moreover, the stimulation of CD40 on DCs regulates the expression of the co-stimulatory molecules CD80 and CD86, enhances the production of cytokines (most notably IL-12 and IFN-I) and promotes the cross-priming to exogenous antigens (33). As a consequence, reduced CD8+ T cell responses are largely due to impaired activation of APCs or to the absence of co-stimulation.
Deficit of Homing to the Tumor Bed
CD8+ T Cell Exclusion by the Immunosuppressive Peritumoral Stroma and Tumor Cell Alterations
When DCs are mature and T-cells correctly primed and activated, the access of T cell to the tumor bed could be compromised by the stromal compartment (34). The exclusion of CD8+ T cells from the vicinity of cancer cells was shown to correlate with a poor long-term clinical outcome in colorectal cancer, ovarian cancer and pancreatic ductal adenocarcinoma (35, 36). Interestingly, Spranger et al. reported an inverse relationship between intrinsic β-catenin signaling of tumor cells and intra-tumoral T cells in melanoma. Using a genetically engineered mouse model they showed that melanomas arising from mice with active β-catenin were characterized by an almost complete absence of both CD8+ T cells and cDC1 subsets (37). A second pathway identified to play a role in T cell exclusion is PI3K pathway activation/PTEN loss. Loss of PTEN in tumor cells in preclinical models of melanoma was shown to increase the expression of immunosuppressive cytokines, inhibit T cell-mediated tumor killing and decrease T cell trafficking into tumors. Furthermore, in patients PTEN loss correlated with decreased T cell infiltration at tumor sites and inferior outcome after PD-1 inhibitor therapy (13). PTEN-deficient prostate tumors similarly induce an immunosuppressive tumor microenvironment by upregulating PTPN11/SHP2 and inducing activity of the Jak2-Stat3 pathway (38). Loss of PTEN was recently associated with resistance to anti-PD1 therapy in metastatic uterine leiomyosarcoma (39) and the blockade of this pathway in vivo contributed to an improved tumor control (13).
Tauriello et al. investigated how genetic alterations and the tumor microenvironment (TME) interact in a metastatic colorectal carcinoma (CRC) model. A Tumor Growth Factor (TGF)-β activity correlating with T cell exclusion and a low TMB was described (40). Recently, a study associated a TGF-β signature of stromal cells with lack of response to anti PD-L1 in the excluded tumor–immune phenotype (41). Blockade of TGF-β in a pancreatic ductal adenocarcinoma model improved the cure rate of mice by decreasing the presence of immune suppressive cells in the TME and enhancing CD8+ T cell infiltration within the tumor (42).
Modified Production of Chemokines and Cytokines Affecting Cell Trafficking and Activation
Cytokines and chemokines may influence cell trafficking to the tumor bed. Besides the steady-state influx of immature dendritic cells (iDCs) within tissues, chemokines, abundantly secreted under inflammatory conditions, can provoke influx of iDCs in the tumor bed (43). Lack of those chemokines and the consequent reduced influx of iDCs in the tumor bed can be the cause of the reduced activation and migration of T cells at the tumor site. Chemokines acting on iDCs are the Monocyte Chemoattractant Proteins (CCL2, CCL7, CCL8) as well as CCL3/MIP-1alpha, CCL5/RANTES, and CCL4/MIP-1beta (44). Cytokines are also necessary to generate active DCs: as an example type I interferon (IFN-I) produced by DCs can act in an autocrine manner to generate fully active DC1s (45). Moreover, DC1s are a source of CXCL-9/10 and their absence lead to a reduced production of these chemokines (20). The chemokine CXCL16, produced by DCs, and its receptor CXCR6 for example have been associated with an increased CD4+ and CD8+ T cell recruitment and a good prognosis in CRC (46). The disruption of the CXCL16/CXCR6 pathway could lead to a reduced tumor T cell infiltration.
The deregulation of trafficking can directly involve T cells: DCs-activated T cells against tumor antigens have to reach the tumor bed to perform their anti-cancer activity. Tumors can disrupt chemokine expression to deregulate the immune response and chemokines involved in effector T-cell recruitment is significantly reduced in tumors lacking a CD8+ T-cell infiltrate. CXCL9 and CXCL10 (CXCL11 in humans) are key chemokines in the recruitment of CD8+ T cells engaging the CXCR3 on their surface and their production is generally deregulated in “non-inflamed” tumors (47). CXCL9/10 can be produced by the tumor cell itself where a methylation of chemokine genetic loci results in a reduced CD8+ T cell infiltration. The use of demethylating agents restores chemokine production and T-cell recruitment, showing that epigenetic modification is a mechanism of tumor escape which could lead to the lack of immune cells infiltration (48). Tumors can also alter the chemistry of certain chemokines to preferentially recruit myeloid cells: as an example the nitrosylated CCL2 eliminates the ability to recruit CTLs and Th1 effector cells (49), while selectively recruiting myeloid dendritic stem cells (MDSCs) to tumor sites.
Therapeutic Approaches
Different therapeutic approaches can theoretically be used to overcome the absence of T cell infiltration in tumors. These strategies are summarized in Figure 2. The demonstration that these therapies can effectively transform a cold into hot tumor remains to be done in the clinic in most instances.
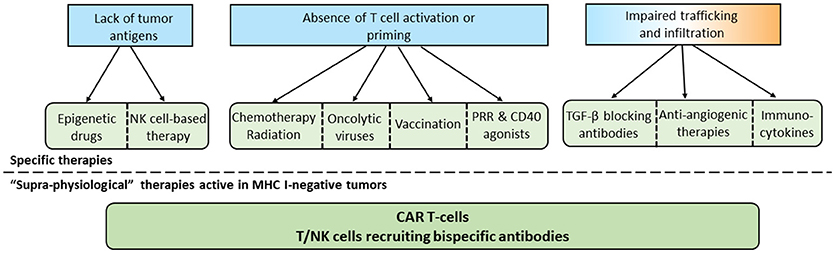
Figure 2. Specific and common approaches to overcome the absence of T cells in tumors. According to the mechanism involved in the lack of T cell infiltration in tumors, specific therapies can be selected. In the case of MHC-I negative tumors or if specific therapies are not sufficient, “supra-physiological therapies” can be used.
Specific Therapies for Tumors Expressing Few Antigens
Demethylating Agents
It has been shown that DNA methyltransferase inhibitors (DNMTi) and histone deacetylase inhibitors can enhance the expression of tumor antigens and components of antigen processing and presenting machinery pathways, as well as other immune related genes (50, 51). These agents can also induce the expression of retroelements such as endogenous retroviruses (ERVs), usually silent and able to induce a type I IFN response (52). Epigenetic drugs have been reported to induce transcription from normally repressed ERV LTR, that may cause ectopic expression of transcripts with canonical or novel open reading frames, leading to the production of immunogenic peptides (53, 54). DNMTi and Histone-lysine N-methyltransferase EZH2 inhibitors have also been shown to reverse epigenetic silencing of Th1-type chemokines in tumor cells, which is negatively associated with CD8+ T cells in tumors and patient outcome (48). There is thus a strong rationale to combine epigenetic therapy and immunotherapy and many clinical trials are currently ongoing (55).
NK Cell-Based Approaches
Natural killer (NK) cells are lymphocytes of the innate immune system able to recognize and kill tumors lacking self-MHC class I molecules, by recognizing stressed cells. For this reason NK approaches could be suitable in the absence of tumor antigens or in case of deficient antigen presentation machinery (e.g., lack of MHC class I). While a large portion of cancer immunotherapies focus on targeting T cells, NK cell system for therapeutic intervention stays relatively underexplored. Nevertheless, different NK cell-based approaches have been described, such as ex vivo activated NK cells or NK cells transduced with a chimeric antigen receptor (CAR) to target specific cancer cell surface antigen (56, 57). Antibodies-mediated targeting of NK activating receptors such as NKG2D and NKP46, or inhibitory receptors such as KIR and NKG2a, is under deep investigation (58). Lirilumab is a fully human antibody directed against KIR2DL-1,-2,-3 inhibitory receptors expressed predominantly on NK cells and is being tested in combination with ipilimumab or nivolumab for the treatment of patients bearing advanced solid malignancies (59).
Specific Therapies for Tumors With Defective Priming or T Cell Activation
Chemo/Radiotherapy Inducing Immunogenic Cell Death (ICD)
Several chemotherapies were found to work mainly in immunocompetent subjects, accumulating evidences that tumor inhibition partially relies on the immune system competence and not only on the direct anti-tumor toxicity of chemotherapy (60). In this regard ICD inducing chemotherapeutic agents can be classified within cancer immunotherapy strategies. Radiotherapy was initially designed to selectively kill tumor cells within the irradiated field. However, emerging evidence indicates that radiotherapy, by inducing ICD, harnesses the host's immune system to attack the tumor cells outside the irradiation field, explaining the “abscopal” effect (regression of tumor lesions outside the irradiation field) (61). Based on this rationale, many trials are ongoing to combine chemotherapy and radiotherapy with PD-1/PD-L1 antibodies. Of note, the same rationale also applies to antibody-drug conjugates (ADCs) with cytotoxic payloads capable of inducing ICD, justifying the initiation of clinical trials combining ADCs and immunotherapy (62).
Oncolytic Viruses
Oncolytic viruses are common viruses that can selectively target, replicate in and destroy cancer cells (63). Most oncolytic viruses can induce cancer cell death and directly eliminate tumor cells, but they also initiate systemic immune responses through different mechanisms such as induction of ICD and release of danger signals (DAMPs) and tumor antigens from virus-infected cells. They also release viral PAMPs contributing to APCs maturation that conduct to activation of antigen-specific CD4+ and CD8+ T cell responses (64). Moreover, the infected cells are directly recognized by the innate immune system such as NK cells or macrophages (65). It has been recently shown in a melanoma phase I trial that use of oncolytic viro-therapy was able to convert a cold into hot tumor, as patients with a low level of immune cell infiltrate and a negative IFNγ signature before treatment responded well to the combination of talimogene laherparepvec with the PD-1 antagonist pembrolizumab (66).
PRR Agonists
PRRs consist of five families including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and cytoplasmic DNA sensors. PRRs agonists have notably the ability to activate PRR pathways inducing antigen presentation by myeloid cells residing in the tumor micro-environment (67–69).
TLR agonists showed controversial results in pre-clinical studies by either promoting or inhibiting tumor progression depending on the TLR and the tumor type (70). Multiple TLR agonists are currently in clinical development. As an example, the intra-tumoral injections of a TLR-9 agonist, the CpG-rich oligonucleotide PF-3512676, showed significant activity in both injected and non-injected lesions of B- and T-cell lymphomas when used concomitantly with low-dose (2 × 2 Gy) local irradiation (71).
STING has been shown to play an important role in the innate immune response against cancer. In the TME, tumor cell DNA detected by APCs is correlated with activation of STING pathway that leads to IFN-β production (25), enhancing CD8+ T cell priming and trafficking of effector T cells. One STING agonist, ADU-S100 (Aduro Biotech/Novartis), is currently evaluated in phase I clinical trial by intra-tumoral administration in cutaneously accessible tumors (72).
CD 40 Agonistic Antibodies
CD40 is broadly expressed on immune cells, predominantly on DCs, B cells and macrophages. A major role of CD40 signaling is to activate and “license” DCs to prime effective cytotoxic CD8+ T cell responses. CD40 signaling can also be effectively triggered using agonistic antibodies or CD40L, thus bypassing the need for CD4+ helper T cells (73). In preclinical studies, agonistic CD40 antibodies have demonstrated T cell-dependent anti-tumor activity, in particular in combination with conventional chemotherapy and immune checkpoint inhibitors (ICIs). Many CD40 antibodies are under clinical development (74). Toxicity profile is acceptable in monotherapy and combination trials are ongoing (75).
Tumor Vaccines
The therapeutic breakthrough provided by ICIs and the demonstration of the role of MANA in T-cell mediated antitumor response have paved the way for a next generation of personalized cancer vaccines based on the use of MANA specific of the tumor. Preclinical results showed induction of efficient antitumor response and clinical trials providing a clinical proof of concept in melanoma have been published (76–78). A specific CD8+ and CD4+ T cell immune response characterized by the induction or the amplification of a preexisting response against MANA has been shown. Encouraging clinical activity seems to be associated but larger trials are awaited to firmly demonstrate the clinical activity of this therapeutic approach. Theoretically, cancer vaccines may either reinforce the activity and therapeutic margin of ICIs by increasing the number of specific effector T cells, or convert cold into inflamed tumors. The potential limitations are the availability of T cell repertoire in cancer patients and the risk of specific loss of heterozygosity (LOH) of the HLA presenting MANA in advanced metastatic tumors (79).
Specific Therapies for Tumors With Impaired T Cell Trafficking to the Tumor
TGF-β Blocking Antibodies and TGF-β-Receptor Antagonists
TGF-β has been involved in cell proliferation, angiogenesis, epithelial-to-mesenchymal transition, immune infiltration, metastases dissemination, and drug resistance (80). Of note TGF-β produced by the tumor cells mediates alterations in tumor-associated pDCs functions, e.g., impaired capacity to produce IFN type I, leading to a lacking/unbalanced T cell recruitment (81, 82). Recent reports have shown that TGF- β in the peritumoral area was a major factor involved in T cell exclusion from the tumor. Mariathasan et al. used a preclinical model recapitulating T cell exclusion and showed that combination of a TGFβ blocking antibody with a PD-L1 antibody induced T cell penetration into the center of tumors, allowing anti-tumor immunity and tumor regression (41). Several TGF-β antibodies or small molecules TGF-β-receptor antagonists are in clinical development and could be tested in this setting.
Anti-angiogenic Therapies
The clinical activity of anti-angiogenic drugs is modest when used as single agent. However, it has been shown that anti-angiogenic drugs normalize the tumor vasculature and induce the upregulation of the leukocyte adhesion molecules ICAM-1 and VCAM-1 on tumor endothelial cells (83), leading to increased T cell infiltration (84). These therapies may thus represent a treatment of choice for tumors characterized by T cells blocked in the periphery of the tumor in order to enhance intratumoral penetration of T cells. It has been shown in the clinic that anti-angiogenic therapies could synergize with ICIs in metastatic melanoma (85).
Immunocytokines
Cytokines such as IL-2, TNF, IL-12 mediate the influx and expansion of leukocytes at the tumor site. However, these cytokines, and especially IL-12, induce significant toxicity when administrated systemically in clinical trials (86). Next generation immunocytokines combining a fragment from a specific tumor antigen antibody with a modified cytokine are being developed with the aim of activating specifically the immune system inside the tumor to reduce systemic side effects. For example, cergutuzumab amunaleukin (CEA-IL2v), is a novel monomeric CEA-targeted immunocytokine that comprises a single IL-2 variant (IL2v) moiety with abolished CD25 binding (to avoid activation of regulatory T cells) fused to the C-terminus of a high affinity bivalent carcinoembryonic antigen (CEA)-specific antibody devoid of Fc-mediated effector functions. A superior efficacy over the respecting monotherapies was observed with CEA-IL-2v in combination with PD-L1 antibody and ADCC competent antibodies in CEA-positive solid tumor models (87).
Therapeutic Approaches Active in Different Immune Contexts
Adoptive T Cell Therapy and Chimeric Antigen Receptor (CAR) T Cells
Historically, cell-based therapies have focused on cytotoxic T cells targeting MHC-restricted antigens. This approach remains promising, in particular with the development of T cell receptor (TCR)-engineered T cells (88) and improvement of tumor infiltrating lymphocytes (TILs) infusion (89). However, their efficacy may be limited by the tendency of tumors to downregulate MHC molecules. CARs are engineered receptors made of the combination of an antigen binding domain of a monoclonal antibody specific for a cancer antigen (not MHC restricted) together with an intracellular domain of the CD3-zeta chain (90). CAR T cells responses can be further enhanced by addition of costimulatory domains, such as CD28 and CD137 (4-IBB) to support the expansion and persistence of genetically engineered cells in vivo (91). The reinfusion of CAR T-cells is preceded by a “lymphodepleting” chemotherapy regimen used to physically create enough space for the expansion and persistence of CAR T cell clones. Over the past decade multiple tumor antigens have been targeted by CARs. Outstanding activity of CAR T cells targeting CD19 has been observed in hematological malignancies, in particular in acute lymphoblastic leukemia and Diffuse Large B cell lymphoma with two CAR T-cell therapies approved by the Food and Drug Administration (FDA) in 2017 (92). The clinical activity of CAR in solid tumors is still to demonstrate and will probably require optimization of CAR functions [for review see (93)].
T-Cell Recruiting Bi-specific Antibodies
Bi-specific antibodies (bsAbs) are engineered antibodies that can bind two different antigens. One of the main strategies in the development of bsAbs is the recruitment and activation of immune effector T cells by targeting CD3 domain of the TCR complex (T-cell recruiting bsAbs) together with another antigen abnormally expressed on the tumor cell surface. The approval of catumaxomab (anti-epitelial cell adhesion molecule EpCAM and anti-CD3) and blinatumomab (anti-CD19 and anti-CD3) has become a major milestone in the development of bsAbs. BsAbs can be divided into two categories: immunoglobulin G (IgG)-like molecules and non-IgG-like molecules. Non-IgG-like bsAbs are smaller in size, leading to enhanced tissue penetration, but shorter half-life (94). Currently, more than 60 different bsAbs formats exist, some of them making their way into clinical trials. As for CAR T-cells, activity of T cell-recruiting bsAbs is not dependent on MHC class I expression on tumor cells and both approaches represent thus very promising treatments for MHC I-negative cold tumors. It is however not know whether a minimum threshold of T cells inside the tumor is required for the activity of bsAb, alone or in combination with other therapies. A comparison between CAR T cells and T-cell recruiting bsAbs is proposed in Table 1.
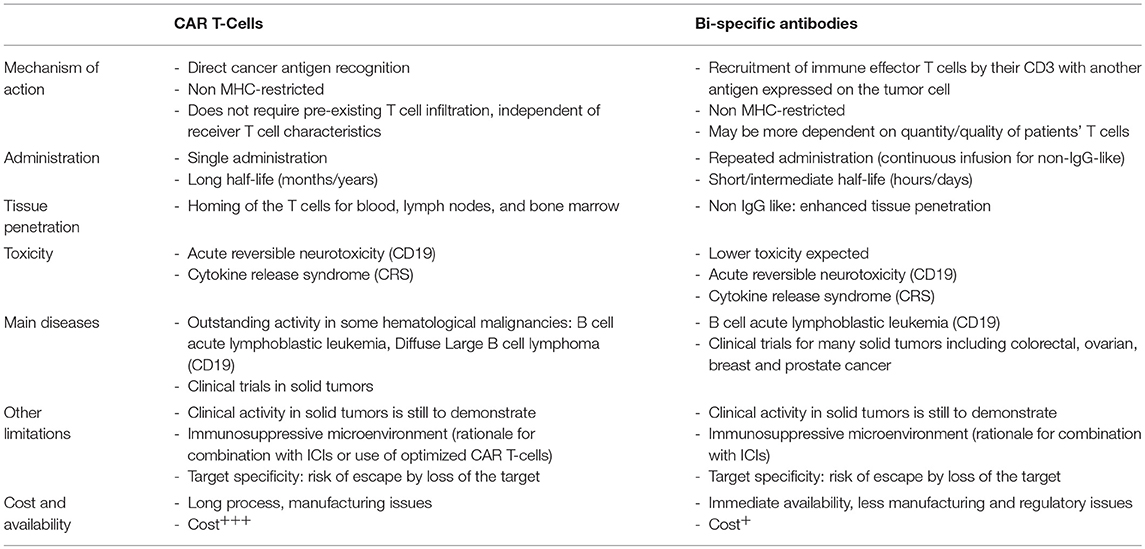
Table 1. Comparison between CAR T-cells and T-cell recruiting bi-specific antibodies.
Perspectives
Changing the natural history of a tumor characterized by the absence of T cells remains a great therapeutic challenge. However, as discussed above, many therapeutic approaches can be evaluated in this context. A major issue will be to determine the origin of this lack of T cell response to adapt the therapy to the physiology of the tumor. Figure 2 summarizes the possibilities of treatment according to this tumor context.
Conventional anticancer approaches like chemotherapy and radiotherapy have still a room in the therapeutic armamentarium, not only to potentially induce ICD but also to reduce the tumor burden and thus potentially decrease the selection of immune-resistant clones. In other cases, “in situ” vaccination using PRR or CD40 agonists may be used to induce a specific immune antitumor response against naturally presented tumor antigens. Personalized cancer vaccines also represent a very promising strategy especially in highly mutated tumors. Finally, “supra-physiological” approaches like CAR T cells or T-cell recruiting bsAbs could be efficient in tumors characterized by the absence of MHC expression or even LOH of the HLA alleles presenting tumor antigens. It is likely that in the near future, algorithms of treatment will be developed to adapt the therapeutic strategy to the immune context of the tumor, considering also space and time evolution for adequate sequential strategies. At the end, while the efficacy of the therapy in inflamed tumors depends principally in the remove of the breaks induced by the immune activation itself, the conversion of a cold into an inflamed tumor will require a prior combination of therapies to induce immune infiltration and then different immune checkpoint modulators to remove the breaks.
Author Contributions
PB, TS, VA, JV-G, and SD contributed to the writing. SD and CC contributed to the conception. SV-W, SA, CC, and SD participated in the lecture and corrections.
Funding
This work was supported by grants from Programme de Recherche Translationnelle en Cancérologie INCa-DGOS (INCa PRT-K2017-072), the Rhône comity of the Ligue Contre le Cancer, the SIRC project (LYRICAN, INCa-DGOS-Inserm_12563), and the LABEX DEVweCAN (ANR-10-LABX-0061) of the University of Lyon, within the program Investissements d'Avenir organized by the French National Agency (ANR).
Conflict of Interest Statement
SD is also an employee for Cellectis and reports personal fees from AstraZeneca, Elsalys, Erytech Pharma, and Netris Pharma.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
2. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
3. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 Inhibition. N Engl J Med. (2017) 377:2500–1. doi: 10.1056/NEJMc1713444
4. Kelderman S, Kvistborg P. Tumor antigens in human cancer control. Biochim Biophys Acta BBA Rev Cancer (2016) 1865:83–9. doi: 10.1016/j.bbcan.2015.10.004
5. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
6. Mitch L. Mutation Burden Predicts Anti-PD-1 Response. Cancer Discov. (2018) 8:258. doi: 10.1158/2159-8290.CD-NB2018-005
7. Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. (2001) 61:5544–51.
8. Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. (1995) 181:2109–17.
9. Coulie PG, Brichard V, Van Pel A, Wölfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med. (1994) 180:35–42.
10. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer (2017) 17:209–222. doi: 10.1038/nrc.2016.154
11. Ilyas S, Yang JC. Landscape of tumor antigens in T-cell immunotherapy. J Immunol. (2015) 195:5117–22. doi: 10.4049/jimmunol.1501657
12. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell–inflamed tumor microenvironment in melanoma. Proc Natl Acad Sci USA. (2016) 113:E7759–68. doi: 10.1073/pnas.1609376113
13. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. (2016) 6:202–216. doi: 10.1158/2159-8290.CD-15-0283
14. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
15. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
16. Jongbloed SL, Kassianos AJ, McDonald KJ, Clark GJ, Ju X, Angel CE, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. (2010) 207:1247–60. doi: 10.1084/jem.20092140
17. Bachem A, Güttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. (2010) 207:1273–81. doi: 10.1084/jem.20100348
18. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
19. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi: 10.1126/science.1164206
20. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector t cell trafficking and adoptive t cell therapy. Cancer Cell (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
21. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
22. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
23. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
24. Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. (2016) 6:71–9. doi: 10.1158/2159-8290.CD-15-0510
25. Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
26. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
27. Akira S, Uematsu STO. Pathogen recognition and innate immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
28. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
29. Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. (2006) 6:476–83. doi: 10.1038/nri1845
30. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
31. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. (2009) 9:353–63. doi: 10.1038/nri2545
32. Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science (2015) 350:972–8. doi: 10.1126/science.aad0779
33. Ridge JP, Di Rosa FMP. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature (1998) 393:474–8. doi: 10.1038/30989
34. Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. (2005) 175:6169–76. doi: 10.4049/jimmunol.175.9.6169
35. Lohneis P, Sinn M, Bischoff S, Jühling A, Pelzer U, Wislocka L, et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur J Cancer (2017) 83:290–301. doi: 10.1016/j.ejca.2017.06.016
36. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti–PD-1 treatment. Proc Natl Acad Sci USA. (2018) 115:E4041–50. doi: 10.1073/pnas.1720948115
37. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
38. Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. (2014) 9:75–89. doi: 10.1016/j.celrep.2014.08.044
39. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with resistance to Anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity (2017) 46:197–204. doi: 10.1016/j.immuni.2017.02.001
40. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature (2018) 554:538–43. doi: 10.1038/nature25492
41. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554:544–8. doi: 10.1038/nature25501
42. Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, et al. TGF-β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget (2015) 6:43005–15. doi: 10.18632/oncotarget.5656
43. Randolph GJ, Ochando J, Partida-Sánchez S. Migration of dendritic cell subsets and their precursors. Annu Rev Immunol. (2008) 26:293–316. doi: 10.1146/annurev.immunol.26.021607.090254
44. Luther SA, Cyster JG. Chemokines as regulators of T cell differentiation. Nat Immunol. (2001) 2:102–7. doi: 10.1038/84205
45. Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood (2002) 99:3263–3271. doi: 10.1182/blood.V99.9.3263
46. Hojo S, Koizumi K, Tsuneyama K, Arita Y, Cui Z, Shinohara K, et al. High-level expression of chemokine CXCL16 by tumor cells correlates with a good prognosis and increased tumor-infiltrating lymphocytes in colorectal cancer. Cancer Res. (2007) 67:4725–31. doi: 10.1158/0008-5472.CAN-06-3424
47. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
48. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527:249–53. doi: 10.1038/nature15520
49. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med. (2011) 208:1949–62. doi: 10.1084/jem.20101956
50. Srivastava P, Paluch BE, Matsuzaki J, James SR, Collamat-Lai G, Karbach J, et al. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells. Leuk Res. (2014) 38:1332. doi: 10.1016/j.leukres.2014.09.001
51. Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. (2017) 87:227–39. doi: 10.1016/j.molimm.2017.04.012
52. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA including endogenous retroviruses. Cell (2015) 162:974–86. doi: 10.1016/j.cell.2015.07.011
53. Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. (2017) 49:1052–60. doi: 10.1038/ng.3889
54. Licht JD. DNA Methylation inhibitors in cancer therapy: the immunity dimension. Cell (2015) 162:938–9. doi: 10.1016/j.cell.2015.08.005
55. Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigenetic and immune therapy to combat cancer. Cancer Res. (2016) 76:1683. doi: 10.1158/0008-5472.CAN-15-2125
56. Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. (2017) 8:533. doi: 10.3389/fimmu.2017.00533
57. Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo anti-tumor activity against human multiple myeloma. Leukemia (2014) 28:917–27. doi: 10.1038/leu.2013.279
58. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. (2015) 14:561–84. doi: 10.1038/nrd4591
59. Aranda F, Vacchelli E, Eggermont A, Galon J, Fridman WH, Zitvogel L, et al. Trial watch: immunostimulatory monoclonal antibodies in cancer therapy. Oncoimmunology (2014) 3:e27297. doi: 10.4161/onci.27297
60. DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. (2011) doi: 10.1158/2159-8274.CD-10-0028
61. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
62. Parslow AC, Parakh S, Lee FT, Gan HK, Scott AM. Antibody-drug conjugates for cancer therapy. Biomedicines (2016) 4:14 doi: 10.3390/biomedicines4030014
63. Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. (2012) 30:658–70. doi: 10.1038/nbt.2287
64. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
65. Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. (2006) 16:141–7. doi: 10.1038/sj.cr.7310019
66. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell (2017) 170:1109–19.e10. doi: 10.1016/j.cell.2017.08.027
67. Bevers RFM, Kurth K-H, Schamhart DHJ. Role of urothelial cells in BCG immunotherapy for superficial bladder cancer. Br J Cancer (2004) 91:607–12. doi: 10.1038/sj.bjc.6602026
68. Jahrsdörfer B, Hartmann G, Racila E, Jackson W, Mühlenhoff L, Meinhardt G, et al. CpG DNA increases primary malignant B cell expression of costimulatory molecules and target antigens. J Leukoc Biol. (2001) 69:81–8. doi: 10.1189/jlb.69.1.81
69. Smits ELJM, Cools N, Lion E, Van Camp K, Ponsaerts P, Berneman ZN, et al. The Toll-like receptor 7/8 agonist resiquimod greatly increases the immunostimulatory capacity of human acute myeloid leukemia cells. Cancer Immunol Immunother. (2010) 59:35–46. doi: 10.1007/s00262-009-0721-8
70. Pradere JP, Dapito DH, Schwabe RF. The Yin and Yang of toll-like receptors in cancer. Oncogene (2014) 33:3485–95. doi: 10.1038/onc.2013.302
71. Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. (2010) 28:4324–32. doi: 10.1200/JCO.2010.28.9793
72. Safety, and Efficacy of MIW815 (ADU-S100) +/- Ipilimumab in Patients With Advanced/Metastatic Solid Tumors or Lymphomas - Full Text View - ClinicalTrials,.gov. Available online at: https://clinicaltrials.gov/ct2/show/NCT02675439 (Accessed May 25, 2018).
73. Khong A, Nelson DJ, Nowak AK, Lake RA, Robinson BWS. The use of agonistic anti-CD40 therapy in treatments for cancer. Int Rev Immunol. (2012) 31:246–66. doi: 10.3109/08830185.2012.698338
74. Vonderheide RH, Burg JM, Mick R, Trosko JA, Li D, Shaik MN, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology (2013) 2:e23033. doi: 10.4161/onci.23033
75. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell (2018) 33:563–9. doi: 10.1016/j.ccell.2018.03.008
76. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature (2017) 547:217–221. doi: 10.1038/nature22991
77. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature (2017) 547:222–6. doi: 10.1038/nature23003
78. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348:803–8. doi: 10.1126/science.aaa3828
79. McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell (2017) 171:1259–71.e11. doi: 10.1016/j.cell.2017.10.001
80. Gramont A de, Faivre S, Raymond E. Novel TGF-β inhibitors ready for prime time in onco-immunology. Oncoimmunology (2017) 6:e1257453. doi: 10.1080/2162402X.2016.1257453
81. Labidi-Galy SI, Sisirak V, Meeus P, Gobert M, Treilleux I, Bajard A, et al. Quantitative and functional alterations of plasmacytoid dendritic cells contribute to immune tolerance in ovarian cancer. Cancer Res. (2011) 71:5423–34. doi: 10.1158/0008-5472.CAN-11-0367
82. Sisirak V, Faget J, Vey N, Blay JY, Ménétrier-Caux C, Caux C, et al. Plasmacytoid dendritic cells deficient in IFNα production promote the amplification of FOXP3+ regulatory T cells and are associated with poor prognosis in breast cancer patients. Oncoimmunology (2013) 2:e22338: doi: 10.4161/onci.22338
83. Kandalaft LE, Motz GT, Busch J, Coukos G. Angiogenesis and the tumor vasculature as antitumor immune modulators: the role of vascular endothelial growth factor and endothelin. Curr Top Microbiol Immunol. (2011) 344:129–48. doi: 10.1007/82_2010_95
84. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res. (2010) 70:6171–80. doi: 10.1158/0008-5472.CAN-10-0153
85. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Front Oncol (2015) 5:202. doi: 10.3389/fonc.2015.00202
86. Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother. (2014) 63:419–35. doi: 10.1007/s00262-014-1523-1
87. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology (2017) 6:e1277306. doi: 10.1080/2162402X.2016.1277306
88. Matsuda T, Leisegang M, Park JH, Ren L, Kato T, Ikeda Y, et al. Induction of neoantigen-specific cytotoxic t cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin Cancer Res. (2018) 24:5357–67. doi: 10.1158/1078-0432.CCR-18-0142
89. Doherty M, Leighl NB, Feld R, Bradbury PA, Wang L, Nie J, et al. Phase I/II study of tumor-infiltrating lymphocyte (TIL) infusion and low-dose interleukin-2 (IL-2) in patients with advanced malignant pleural mesothelioma (MPM). J Clin Oncol. (2015) 33:TPS7586. doi: 10.1200/jco.2015.33.15_suppl.tps7586
90. Irving BA, Weiss A. The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell (1991) 64:891–901.
91. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
92. Research AA for C. CAR T-cell therapies produce durable remissions. Cancer Discov. (2018) 8:379. doi: 10.1158/2159-8290.CD-NB2018-017
93. Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer (2016) 16:566–81. doi: 10.1038/nrc.2016.97
Keywords: cold tumors, T cells, tumor antigen, presentation, priming, trafficking, immunotherapy
Citation: Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C and Depil S (2019) Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 10:168. doi: 10.3389/fimmu.2019.00168
Received: 04 December 2018; Accepted: 21 January 2019;
Published: 08 February 2019.
Edited by:
Salem Chouaib, Institut Gustave Roussy, FranceReviewed by:
Viktor Umansky, German Cancer Research Center (DKFZ), GermanyDaniel Olive, Aix Marseille Université, France
Copyright © 2019 Bonaventura, Shekarian, Alcazer, Valladeau-Guilemond, Valsesia-Wittmann, Amigorena, Caux and Depil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stéphane Depil, c3RlcGhhbmUuZGVwaWxAbHlvbi51bmljYW5jZXIuZnI=