 Anja ten Brinke1,2*
Anja ten Brinke1,2* Marc Martinez-Llordella3
Marc Martinez-Llordella3 Nathalie Cools4
Nathalie Cools4 Catharien M. U. Hilkens5
Catharien M. U. Hilkens5 S. Marieke van Ham1,2,6
S. Marieke van Ham1,2,6 Birgit Sawitzki7
Birgit Sawitzki7 Edward K. Geissler8
Edward K. Geissler8 Giovanna Lombardi9
Giovanna Lombardi9 Piotr Trzonkowski10
Piotr Trzonkowski10 Eva Martinez-Caceres11,12
Eva Martinez-Caceres11,12- 1Department of Immunopathology, Sanquin Research, Amsterdam, Netherlands
- 2Landsteiner Laboratory, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
- 3Department of Inflammation Biology, MRC Centre for Transplantation, School of Immunology and Microbial Sciences, Institute of Liver Studies, King's College London, London, United Kingdom
- 4Laboratory of Experimental Hematology, Faculty of Medicine and Health Sciences, Vaccine and Infectious Disease Institute (Vaxinfectio), University of Antwerp, Antwerp, Belgium
- 5Institute of Cellular Medicine, Newcastle University, Newcastle upon Tyne, United Kingdom
- 6Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, Netherlands
- 7Charité-Universitaetsmedizin Berlin, Berlin Institute of Health, Institute for Medical Immunology, Humboldt-Universitaet zu Berlin, Berlin, Germany
- 8Section of Experimental Surgery, Department of Surgery, University Hospital Regensburg, University of Regensburg, Regensburg, Germany
- 9Division of Transplantation Immunology and Mucosal Biology, MRC Centre for Transplantation, Guy's Hospital, King's College London, London, United Kingdom
- 10Department of Clinical Immunology and Transplantology, Medical University of Gdansk, Gdansk, Poland
- 11Division of Immunology, Germans Trias i Pujol University Hospital, LCMN, IGTP, Badalona, Spain
- 12Department of Cellular Biology, Physiology and Immunology, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, Spain
Clinical studies with cellular therapies using tolerance-inducing cells, such as tolerogenic antigen-presenting cells (tolAPC) and regulatory T cells (Treg) for the prevention of transplant rejection and the treatment of autoimmune diseases have been expanding the last decade. In this perspective, we will summarize the current perspectives of the clinical application of both tolAPC and Treg, and will address future directions and the importance of immunomonitoring in clinical studies that will result in progress in the field.
Introduction
The number of patients with autoimmune diseases, severe allergies and recipients of organ or stem cell transplants is increasing worldwide. Currently, many of these patients require lifelong administration of immunomodulatory drugs, which cause generalized immunosuppression and hereby only partially alleviate the symptoms but do not cure the disease. Besides these drugs are inevitably associated with a risk of immediate or late-occurring severe adverse effects (e.g., life-threatening infections, cancer). Targeting the fundamental cause of autoimmunity, i.e., loss of tolerance to self-antigens, or inhibiting induction or execution of undesired immunity in transplantation and allergy will provide the next steps forward to avoid general immunosuppression. Accumulating knowledge on mechanisms of immune activation and tolerance has led to the development of tolerance-inducing cellular therapies with regulatory T cells (Treg) and tolerogenic antigen presenting cells (tolAPC), such as tolerogenic dendritic cells (tolDC) and regulatory macrophages (Mreg) [as reviewed in (1, 2)], with the specific objective to restrain unwanted immune reactions long-term.
The development of cell-based therapies is clinically attractive for many reasons, not in the least through their potential of being of low-toxicity, to simultaneously control many different inflammatory cells and induction of antigen-specific immunity. Since immunological tolerance is a self-reinforcing state (3), the therapeutic effects of cell therapy are expected to outlast the lifespan of the therapeutic cells themselves, opening the possibility of curative treatments. Production costs for these tolerogenic cell products range from 10,000 to 40,000 Euro depending on the therapeutic cell product and production site, which is relatively low considering that few injections of cells may be sufficient to induce long-lasting tolerance.
In recognition of the potential of tolerance inducing cell-based therapies and to join forces in the ongoing efforts in the field, A FACTT (Action to Focus and Accelerate Cell Based Tolerance-inducing Therapies (CTT) was initiated through EU COST Action Funding. Our goal was to initiate a network that would coordinate European CTT efforts to minimize overlap and maximize comparison of the diverse approaches across Europe. Now, looking back at 4 years of very active network interactions, we have evaluated our combined current stance in the field and defined avenues to support future directions.
Tolerogenic Therapy With Antigen Presenting Cells in Clinical Practice
Over the past 20 years extensive experimental research has been invested in the generation and characterization of tolAPC, including tolDC and Mreg, with the aim to restore tolerance in autoimmune diseases (4–10) and transplant rejection (5, 11–14). To date, clinical trials exploring the safety, feasibility and efficacy of different types of tolAPC are a reality [reviewed in (1) and Table 1], and have confirmed so far that tolAPC therapy is safe, with no relevant side effects, and is well-tolerated by patients. Hence, to advance tolerogenic therapy with antigen-presenting cells (APC), we should stand on the shoulders of these pioneers and address remaining challenges, such as the optimal dose, injection route, frequency of administration, antigen-specificity, and the related issue of suitable biomarkers of cell therapy-induced reduction of general inflammatory state and induction of tolerance, in the design of the next-generation clinical trials.
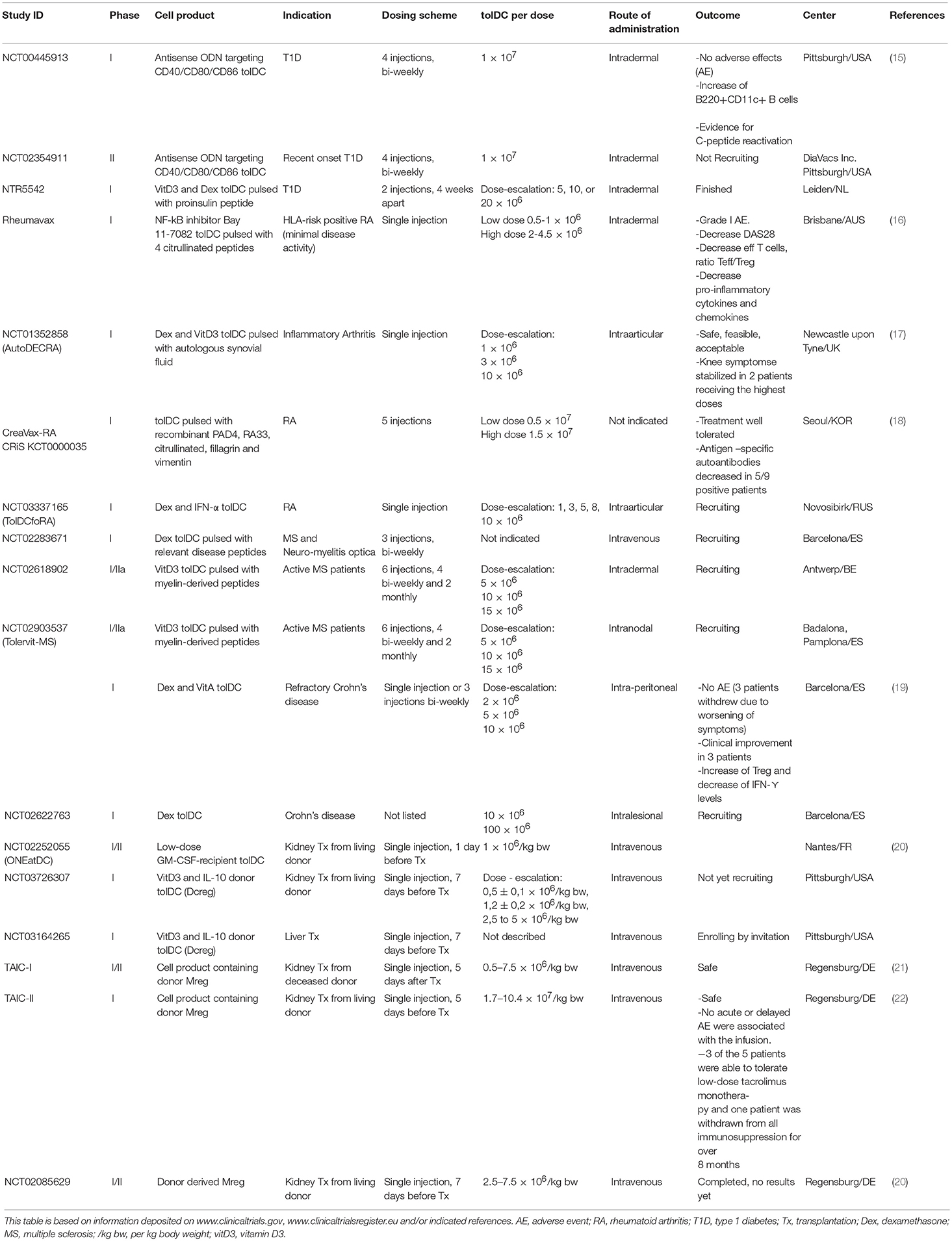
Table 1. Completed or ongoing trials with tolAPC in autoimmunity and transplantation.
Phenotypic and Functional Identification of in vitro Generated TolAPC
Both tolDC and Mreg can be generated in vitro starting from CD14+ monocytes and share some phenotypic and functional characteristics. Indeed, both tolAPC types express low to intermediate levels of T-cell costimulatory molecules, and secrete low amounts of pro-inflammatory cytokines, indicative of a partially matured APC. Similarly, immature DC (iDC) display minimal expression of costimulatory molecules and little secretion of inflammatory cytokines, demonstrating potential optimal requirements for tolerance induction in vivo (23, 24). However, iDC are unstable and may differentiate into immunogenic DC under inflammatory conditions (25, 26). This invalidates their putative use as therapeutic products for tolerance induction. Therefore, different strategies to generate stable tolAPC have been explored, including treatment with pharmacological agents or cocktails of immunomodulatory cytokines, genetic engineering, and exposure to apoptotic cells (9, 27, 28). Most of these in vitro conditioning regimens aim at stabilizing a semi-mature state of tolDC, maintaining the capacity to induce immune hyporesponsiveness of T cells, even in presence of powerful pro-inflammatory signals.
Importantly, tolAPC inhibit T cell proliferation, albeit through different immunosuppressive mechanisms depending on the approach used to generate tolAPC in vitro. Induction of peripheral T cell anergy and apoptosis (29), attenuation of effector and memory T cell responses and the generation and activation of Treg populations (30, 31) result in part from presentation of low levels of antigen in the absence of costimulation; these are typical mechanisms attributed to a variety of tolerogenic subtypes (10, 32, 33). Additionally, tolerogenic DC may express various inhibitory receptors such as programmed death-ligand (PD-L)1, PD-L2 (34), immunoglobulin-like transcripts (ILT) (35), FasL (36, 37), and TRAIL (38). Secretion of anti-inflammatory cytokines such as IL-10 (39, 40) and TGF-β (41, 42), as well as reduced expression of pro-inflammatory cytokines, also may contribute to tolerance induction. A study comparing tolDC generated in presence of dexamethasone and rapamycin demonstrated that while both tolDC subsets were able to impair T cell proliferation, rapamycin-treated tolDC have a mature phenotype and are not able to produce IL-10 upon stimulation with LPS, as opposed to vitamin D3- or dexamethasone-treated tolDC (27). Whereas, it was demonstrated that rapamycin-treated tolDC induce Treg (27), vitD3-treated tolDC induce T-cell hyporesponsiveness and antigen-specific Treg (7, 43). Moreover, DC-10 induce Tr1 cells (44), while autologous tolDC have a weak capacity to stimulate allogeneic T cells and suppress T-cell proliferation and IFN-γ production (45). Mreg have been shown to convert allogeneic CD4+ T cells to IL-10-producing, TIGIT+, FoxP3+-induced Treg (46). Variations in the process to generate tolAPC may initiate regulation through distinctive mechanisms, making it difficult to compare these different types of tolAPC. Therefore, efforts have been made to find common features unique for tolerance-inducing cells (47). For example, since tolDC conditioned using vitamin D3 and dexamethasone exhibit high cell surface expression of TLR2 (48) or CD52 (49), such markers might be considered to assess the quality and stability of tolDC in future cell-based clinical trial protocols. In addition, it was demonstrated that the expression of single immunoglobulin IL-1-related receptor has a role in maintaining low levels of costimulatory molecules and in regulation of Treg expansion (50). Others demonstrated that C-lectin receptor CLEC-2 upregulation by DC is associated with Treg induction (51). So far, however, gene expression studies comparing different tolDC and Mreg protocols have not been able to identify common biomarkers of tolerance induction [reviewed by (52, 53) and (54)].
The difficulty in comparing characteristics of different clinical tolAPC suggests the need for a uniform set of metrics for their description, including full characterization of (at least) the immune phenotype and their functional activity (potency). Hence, a better identification of the characteristics that identify the tolerance-inducing properties of tolAPC, irrespective of the conditioning regimen, would be valuable for safe cell therapy delivery into patients. Joint efforts in translating tolAPC into the clinic by harmonizing protocols and defining functional quality parameters have been initiated (1, 55). A Minimum Information Model on Antigen-presenting cells (MITAP) has been defined to harmonize reporting on tolAPC therapy to ultimately allow the uncovering of commonalities between tolAPC and to define common quality control biomarkers and potency assays for the various tolAPC products for clinical use (55). Likewise, using similar immunomonitoring protocols in different clinical trials could help to better understand the in vivo mechanism of action of these cells (56).
Antigen Specificity of TolAPC-Based Immunomodulation
Targeted regulation of antigen-specific T cell responses would avoid generalized immunosuppression as observed with immunosuppressive drugs and monoclonal antibodies currently in use in the clinics and may thus overcome occurrence of impaired immune-surveillance leading to infections or development of malignancies. Ex vivo generated tolAPC have the potential to therapeutically induce, enhance, or restore antigen-specific tolerance. Indeed, after loading these cells with exogenous or endogenous antigens, one major advantage is their capability to act in an antigen-specific manner.
A number of in vivo studies demonstrate that antigen loading of tolAPC is indispensable to reach efficient clinical responsiveness following tolAPC therapy. For instance, a beneficial effect of vitamin D3-tolDC loaded with MOG40−55 peptide was demonstrated in experimental autoimmune encephalomyelitis (EAE), whereas no clear beneficial effect on the clinical score of EAE mice was found when mice were treated with vitamin D3- tolDC not loaded with myelin peptides (57, 58). Similar findings have been demonstrated in other animal models of autoimmune diseases, including collagen-induced arthritis and autoimmune thyroiditis (59–61). Altogether, these findings suggest that selection of the target self-antigen is critical for disease-specific tolerance induction in vivo. Suitable disease-associated self-antigens responsible for T cell priming have been identified for T1D and multiple sclerosis (MS). However, this is not the case for other autoimmune diseases, such as rheumatoid arthritis or Crohn's disease, for which specific disease-associated antigens are unknown or not tissue specific. Moreover, not all patients uniformly display the same set of self-antigens for a given disease. MS, for example, is associated with a range of self-antigens and auto-antibodies that are differentially expressed among patients and at different points during the disease (62). While the targetable autoreactive T cell populations may be limited to a select number of antigens at the onset of clinical disease, other “late antigen” or spread epitope autoreactive T cell populations may drive autoimmunity during progression of the disease. To overcome this hurdle, some groups have chosen to load the tolerance-inducing therapeutic cells with a broad pool of distinct, candidate disease-related peptides (63–65) (NCT02283671, NCT02618902, and NCT02903537).
In contrast to autoimmunity, transplant rejection is mediated by an undesired immune response against epitopes that differ between the transplanted donor graft and the recipient host, so-called allorecognition (66). Specifically, recipient T cells may initiate a strong immune response leading to transplant rejection in the absence of adequate immunosuppression. To avoid transplant rejection, the induction of tolerance to donor-specific antigens has been coined as a therapeutic target for decades. For this, both donor tolAPC and recipient tolAPC loaded with donor-specific antigens are being considered for development of cell-based immunotherapeutic protocols in the transplantation setting (67). However, whereas the clinical use of donor-derived tolAPC is only feasible in the context of living donor transplantation and entails a risk of sensitization (including development of allo-antibodies), the use of autologous, i.e., recipient-derived, tolAPC is a less risky approach to begin with. Indeed, the use of recipient autologous tolAPC is likely to be more feasible than that of donor-derived tolAPC, since cell products can be prepared from the peripheral blood of the recipient before transplantation, stored while the patient is on the waiting list, and loaded with donor-derived antigens (such as HLA peptides or donor cell lysates) at the time of transplantation. In the context of the ONE study, two trials using tolDC and Mreg are being performed in living-donor kidney transplant recipients (Table 1).
Route of Administration of TolAPC
Although it is generally accepted that the route of administration is important for optimal tolAPC effector function, the best route of tolAPC administration is not known. To date, a variety of routes of administration have been used (see Table 1), including intradermal, intraperitoneal (19), intravenous and intra-articular (17). Different routes of administration may be required to allow tolDC to reach the relevant draining lymphoid tissue for T cell encounter or to end up at the site of inflammation. Especially since tolDC demonstrate a reduced expression of CCR7 and consequently a reduced (but not absent) ability to migrate in response to the CCR7 ligand CCL19 (68), the capacity of tolDC to reach the lymph nodes is a critical concern. While the migration of DC toward the lymph nodes increases following intradermal as compared to subcutaneous administration, only 2–4% of DC migrate to the draining lymph nodes after intradermal administration, but the situation may be different in patients with autoimmune diseases where monocyte-derived DC from MS patients have shown a significantly higher proportion of CCR7-expressing cells compared to healthy controls (69). Given these observations, and that in the setting of cancer vaccine development, DC injected into a lymphatic vessel showed a prolonged half-life as compared to DC injected intravenously (70, 71), direct intranodal injection of tolDC is being evaluated in a clinical setting (see Table 1).
As an alternative to lymph node targeting of tolAPC, tolAPC may also be introduced directly into the site of inflammation. Indeed, injection into the affected disease site (i.e., an inflamed joint) where the tolAPC could suppress auto-reactive effector T cell responses is logical. In this context, intra-articular injection of tolDC differentiated using dexamethasone and vitamin D3 and loaded with autologous synovial fluid in patients with rheumatoid arthritis was demonstrated to be safe and feasible. Hence, despite the fact that tolDC were directly injected at the site of inflammation, no adverse events were observed in most patients and hypertrophy, vascularity and synovitis were stable in all treated cohorts. Moreover, two patients receiving 10 million cells showed a decrease in synovitis score (17). Similarly, a phase I randomized clinical study currently evaluates the safety and efficacy of tolDC injected into the intestinal lesions in patients with refractory Crohn's disease (Table 1). In some conditions such as T1D, direct injection of cells in the inflammatory site, e.g., pancreas, might not be possible and require tolDC administration adjacent to the inflammation site. For the treatment of inflammatory diseases of the brain, the blood–brain barrier (BBB) may represent a major hurdle. Considering this potential problem, it was demonstrated that enhancing CCR5 expression in tolDC using mRNA electroporation endowed these cells with CCR5-driven migratory capacity. This enabled the cells to migrate to inflammatory sites, even when it required crossing of functional barriers such as the BBB (72). Similarly, introducing CCR7 expression in tolDC using the proposed approach of chemokine receptor mRNA electroporation could overcome the limited lymphoid homing capacity of tolDC. Indeed, DC transduced with lentiviral vectors coding for CCR7 and IL-10 genes were able to migrate to the lymph nodes and spleen, prolonging cardiac allograft survival in mice (73). However, there are still many unknowns and there is a clear clinical need to characterize the pharmacodynamics of tolDC in humans and relate this to clinical efficacy. Advances in cell imaging techniques, for example magnetic resonance imaging of 19F-labeled cells, have made it possible to address this question in future studies.
TolAPC Therapy: What Does the Future Hold?
In vivo Targeting
While our knowledge of tolAPC biology has expanded greatly, and in vitro generated tolDC and Mreg are currently being used in various clinical trials (Table 1), clinical-grade manufacturing of tolAPC is still a time-consuming and expensive process. It requires cell precursors that need to be isolated from the patient's blood, modulated ex vivo and reintroduced into the patient. Direct antigen delivery to tolAPC in vivo may limit the workload and costs. Indeed, specific antigen-targeting of DC-restricted endocytic receptors (DEC-205) with monoclonal antibodies has been shown to induce antigen-specific T cell hyporesponsiveness in experimental models (74). Interestingly, a phase I clinical trial demonstrated that in vivo targeting of human DC could be achieved by antibodies against DEC205 with subsequent antigen presentation and robust humoral and cellular responses (75). In vivo targeting of DC with biomaterials such as liposomes, microparticles and nanoparticles is also a promising approach [as reviewed in (76–78)]. This is exemplified by the fact that liposomes loaded with NFkB inhibitors targeting APC in situ, suppress the cellular responsiveness to NF-kB and induce antigen-specific FoxP3+ regulatory T cells in an animal model of arthritis (79) and that administration of phosphatidylserine-rich liposomes loaded with disease-specific autoantigens lead to a beneficial effect in experimental models of T1D and MS (80, 81). Nevertheless, DC represent a heterogeneous cell population arising from bone marrow-restricted precursors identified in humans. While multiple subsets of DC have been found in the peripheral blood, lymphoid organs and tissues, most of the hallmark DC markers are promiscuously expressed making it difficult to unambiguously discriminate between DC subpopulations and specifically target those DC subpopulations that induce tolerance. Extensive phenotypic screening combined with gene expression profiling allows the identification of tolerance-inducing DC counterparts present in vivo. For instance, Gregori and co-workers identified a DC subset expressing HLA-DR+CD14+CD16+ that exhibits potent tolerogenic activity (44). In targeting only such tolerance-inducing DC cell type-specific targeting may emerge as another promising approach in DC-based immunotherapy.
Combination Therapy
Since a variety of often complementary mechanisms are involved in the maintenance of immune tolerance, a more complex therapeutic approach using combinations that target different pathways that contribute to induction and maintenance of tolerance may be required to fully control autoimmunity. For instance, combinations of tolAPC with disease-modifying treatments that reduce the frequency of disease-causing cells, e.g., alemtuzumab, should be explored as the latter therapy reduces the disease-causing cells to a number that may be more effectively controlled by antigen-specific tolerance-inducing strategies, such as tolDCs. Alternatively, therapies like fingolimod, an antagonist of sphingosine−1-phosphate receptor which retains naïve and central memory T cells in the lymph nodes, are promising as co-medication with tolAPC as it could increase the number of tolDC-T cell interactions in the lymph node thereby facilitating Treg priming and consequently the efficacy of tolDC-based strategies. Also in the context of solid organ transplantation, tolDC therapy could be improved by the addition of a complementary treatment. For instance, the combination of adoptive transfer of tolDC and CTLA-4 Ig, a fusion protein that blocks B7-CD28 costimulation, resulted in extended survival of MHC-mismatched heart allografts in mice (82, 83), while the pancreatic islet allograft survival improved by combination of autologous tolDC and CD3 targeting antibodies (84). With the recent data on the important role of other coinhibitory molecules for T cell-mediated inflammation such as CD96 the portfolio of combination therapies might increase in the next years (85). However, since antigens can easily trigger immunity instead of tolerance, a primary concern remains the safety of combining two immune-modulatory vaccination strategies in autoimmune diseases and in the prevention of transplant rejection. Although one can envisage that concomitant use of immunosuppressive therapies might synergistically reduce the risk to unexpectedly worsen antigen-specific reactions (86), any novel manipulation of the immune system may involve an unpredictable risk. Furthermore, combination therapy may introduce confounding factors inducing additive, synergistic or antagonistic effects complicating the evaluation of the precise mode of action.
Conclusion
Several protocols to generate and administer tolAPC have been tested in phase I clinical trials with highly encouraging results from a safety point of view and in terms of adverse effects (Table 1). Further phase I/II studies are under way in Crohn's disease, T1D, rheumatoid arthritis, MS and kidney transplantation (Table 1). However, for the success of future tolAPC trials, there is great need to define the optimal vaccination protocol; to ensure optimal in vivo-acting of the tolAPC, future trials may require changes in administration route, dose or could demand repeated tolAPC administration. Furthermore, the identification of a common set of tolerogenic markers would enable optimized comparison of tolAPC products and their tolerance-inducing potential and provide an improved understanding of how these cells modulate the T cell response both locally and systemically. It would be of great help to analyze critical pathways contributing to programming and function of tolAPC. Ultimately, this may set the stage for new approaches improving the therapeutic potential of tolAPC for the future.
CD4+ Regulatory T Cells (Treg)
CD4+ Treg are recognized as a dominant cell population responsible for induction and maintenance of immune tolerance. They may be generated either in the thymus as natural regulatory T cells (nTreg or tTreg) or in the periphery as induced regulatory T cells (iTreg). Both subsets can induce tolerance toward auto- and alloantigens utilizing a variety of mechanisms including cell-to-cell contacts, secretion of immunosuppressive cytokines and inhibitory molecules (e.g., adenosine or prostaglandin E), local depletion of IL-2, or through killing of other cells (87). Treg actively traffic to inflammatory sites and the suppressive activity is usually localized without a significant impact on the general immunity. Since their more precise identification 2–3 decades ago in the mouse, and more recently in the human, steady advances in understanding Treg biology have eventually provided sufficient knowledge to culture, manipulate and expand the cells in vitro under Good Manufacturing Practice (GMP) conditions for therapeutic purposes. Indeed, Treg have become a promising cellular drug that can potentially be used to control disease-causing immune responses.
Treg in Clinical Practice
While the application of Treg for the treatment of autoimmune diseases is currently under intense investigation, Treg were first used in the clinic to treat patients with graft vs. host disease (GvHD) after hematopoietic stem cell transplantation (HSCT) (88) (Table 2). Results from the clinical trials in GvHD with polyclonal expanded Treg have suggested that altogether these cells are safe, but there is some concern about the occurrence of mild to moderate infections, and it still is unclear whether Treg treatment could promote cancer (92, 94). The latter problem has been reported in only one trial to date, however it was concluded that the tumor was present before the therapy with Treg was applied (94). The safety and feasibility of adoptive transfer of ex vivo expanded Treg was further confirmed in T1D patients (2), which has driven the application of Treg therapy to clinical trials in other autoimmune conditions such as MS, autoimmune hepatitis, systemic lupus erythematosus, Crohn's disease, and autoimmune uveitis (102) (Table 2). Another clinical trial was recently published where polyclonal Treg were injected into T1D patients; results from this trial confirm the safety of this type of therapy and also show for the first time, by deuterium labeling of the Treg, that some of the injected Treg can be detected for up to 1 year after infusion (103).
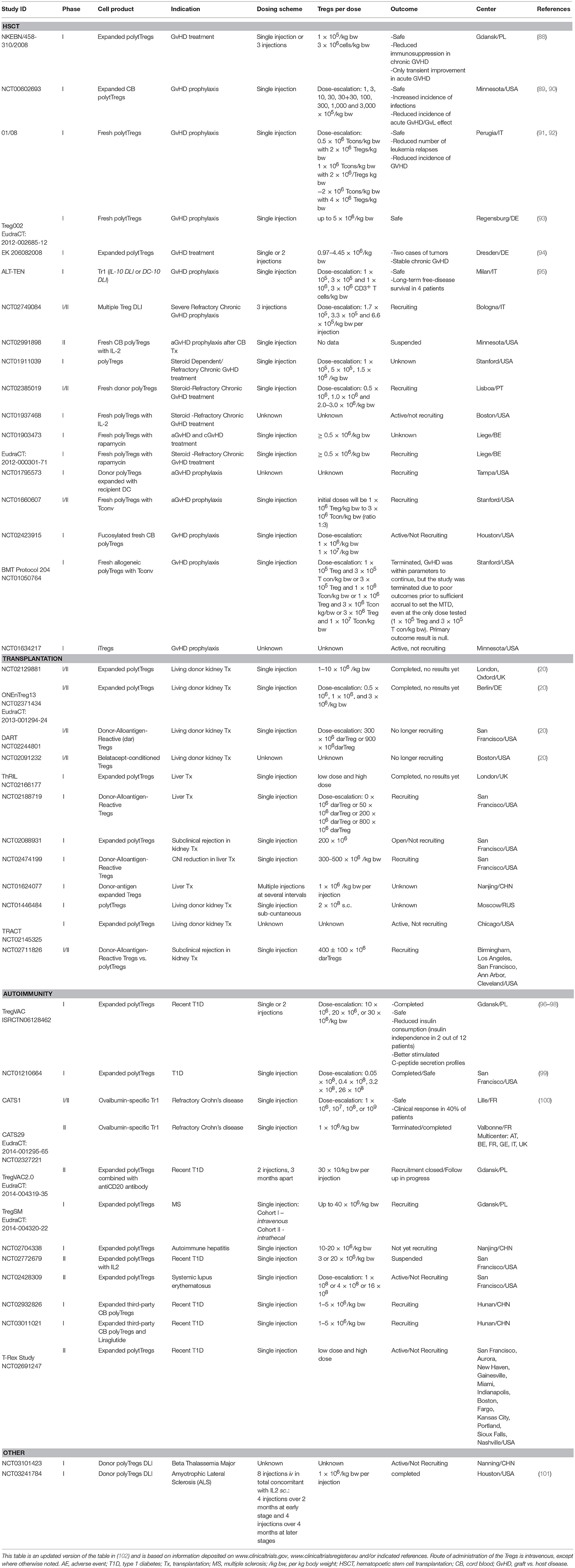
Table 2. Completed or ongoing trials with Treg in autoimmunity and transplantation.
Treg therapy is now being applied as a “tolerogenic” therapy to reduce dependency on immunosuppressant drugs in patients receiving solid organ transplants. The idea behind this strategy is very similar to the application of Treg in autoimmune diseases, namely to tilt the balance toward Treg dominance over rejection-causing Teff cells. The first reports using adoptive transfer of Treg in kidney transplant patients have been recently published demonstrating the safety of this strategy in the context of solid organ transplantation (103, 104). Recently, clinical trials are being completed using different variations of Treg products (The ONE Study and ThRIL) (Table 2). The ONE Study includes Phase I clinical trials comparing the safety of different types of regulatory cells, including polyclonal and donor-reactive Treg in patients receiving kidney transplants (www.onestudy.org) (20). The ThRIL trial is a Phase I/IIa dose-escalation clinical trial in the setting of liver transplantation. Results from the ThRIL and the various ONE Study trials are currently being prepared for publication. The impact of Treg on the recipient immune system will be revealed only when the very detailed immunomonitoring is completed, which is a major objective of the described clinical trials (105–107).
Altogether, from the outcomes of the completed clinical trials so far, it can be concluded that adoptive transfer of Treg is safe and technically feasible (Table 2). Therefore, increasing efforts are currently focusing on clinical trials to test their therapeutic efficacy. Importantly, several lessons have been learned from recent experiences with Treg to improve future trial designs. For example, the clinical state of the patients has been shown to influence the function and properties of Treg, and therefore can condition the success of ex vivo cell product expansion (108). Furthermore, the specific expansion protocol can affect Treg function and specificity, and can improve tissue targeting and suppression capacity (108). Finally, the immune modulatory therapies received by patients at the time of Treg adoptive transfer can positively or negatively impact therapeutic outcome (109, 110).
Treg Therapy: What Does the Future Hold?
Antigen-Specificity of Treg Therapy
Studies in preclinical models using murine Treg have demonstrated that specificity for either the auto or allo (transplantation) -antigens may offer an advantage for Treg function compared to polyclonal Treg (111). Adoptively transferred allospecific murine Treg generated by using either donor-derived APC or TCR transduction promote indefinite heart allograft survival, even in completely mismatched mouse strains (111). This strategy was subsequently applied to human Treg in a humanized mouse model, where Treg were generated in the presence of donor APC /DCs or B cells) and shown to be superior to polyclonal Treg in protecting from human skin graft rejection (112). More recently, by conferring specificity using a chimeric antigen receptor (CAR), human Treg transduced with a lentivirus encoding for HLA-A2-CAR were superior to polyclonal Treg in protecting HLA-A2+ human skin grafts (113–115). CAR constructs are now being developed to increase Treg stability and function.
Based on promising results with antigen-specific Treg in pre-clinical models, the use of alloantigen-specific Treg generated by culturing recipient Treg with donor-specific cells, either using activated donor-derived B cells (112) or donor-derived DCs is being tested in clinical trials (Table 2). Compared to the transplantation field where the antigens are known, generation of antigen-specific Treg in autoimmunity is more challenging because the inciting antigens are often not known (see also paragraph Antigen Specificity of tolAPC-Based Immunomodulation). New data suggest that regulatory and effector cell subsets are driven by different epitopes (116, 117). In addition, the auto-antigen triggering the autoimmune condition can change during disease progression due to epitope spreading and antigen-specific Treg may thus need to be tuned toward specific stages of disease.
Combination Therapy
Since adoptive transfer with Treg alone, particularly with a one-time infusion, may not be sufficient to control the immune response, combined or successive therapies are being tested. One approach is based on evidence that low doses of IL-2 can preferentially increase the endogenous pool of Treg; so far, low-dose IL-2 treatment has been safe in inflammatory conditions such as GvHD after HSCT (118). In a preclinical model it was demonstrated that IL-2/anti-IL-2 complexes not only promote Treg proliferation, they increase Treg survival and function while synergizing with calcineurin inhibitors to prolong graft survival (119). Recently, in a murine model of transplantation it was shown that by combining donor-specific Treg with the IL-2/anti-IL-2 complexes, a synergistic effect in extending skin transplant survival is observed (Ratnasothy et al., unpublished data). These results pave the way for the first clinical trial in liver transplant patients to combine Treg and low-dose IL-2 therapy (NCT02949492). More caution is being exercised regarding similar trials using low-dose IL-2 in autoimmune diseases, since in contrast to HSCT or solid organ transplantation, autoimmune patients are not lymphopenic and are likely to produce IL-2 themselves (98). However, in some conditions such as systemic lupus erythematosus an intrinsic defect in Tregs contributes to disease progression and here low-dose IL-2 therapy was shown to correct the defect (120, 121). Furthermore, in T1D settings low dose IL-2 treatment is currently being investigated (NCT02411253).
The positive effects of rapamycin on Treg in vitro or in a transplant setting (122–124) could not be effectively translated to the in vivo treatment of autoimmune syndromes. For example, while rapamycin administered to T1D patients preferentially increased Treg levels, pancreas function deteriorated due to islet toxicity (125). Nonetheless, the complex and specific pathogenesis of autoimmune syndromes may provide hints toward the design of new combined therapies. Tandemly targeting different effector mechanisms involved in particular syndromes with Treg therapy may improve outcomes. For example, an ongoing trial in early phase of T1D (EudraCT: 2014-004319-35) supports the idea of a synergistic approach by combining Treg administration with B cell depletion.
New Technical Advancements
It is now accepted that the optimal way to manufacture Treg for clinical use requires an efficient ex vivo expansion rate while maintaining purity and suppression potency before GMP product release. Two main Treg isolation strategies are currently being used in clinical trials to purify the starting Treg, immunomagnetic selection and flow cytometry cell sorting. The magnetic platform (CliniMACS CD4+CD25+ selection) provides a highly automated GMP-grade approach which is easy to standardize across centers. However, the resultant Treg product does generally contain a minor population of CD127+ cells that could jeopardize product purity after expansion. Addition of rapamycin to the culture media has helped to maintain Treg purity and function, without reducing the expansion rate too much (126). The second method, which uses a flow cytometry approach, results in a highly pure Treg population due to CD127+ cell depletion ability. However, this cell-sorting strategy entails more complex protocols and challenges to maintain GMP grade. With the appearance of new GMP-compatible cell sorters (Tyto MacQuant, Miltenyi or Influx, BD Biosciences), this sorting approach is likely to become the preferential Treg isolation method.
Although cell-sorting of Treg increases their initial purity, a search for an optimally stable final Treg products is critical therapeutically. It is well known that prolonged in vitro culture results in epigenetic changes in Treg—methylation of TSDR region of foxp3 gene—which in-turn reduces suppressive ability (127). The use of anti-methylation agents in cultures to prevent epigenetic changes has failed due to culture viability issues. The addition of rapamycin to the expansion culture, as used in both The ONE Study and ThRIL, was proposed as a remedy to such changes as the drug preferentially expands Treg both in vitro and in vivo (122). However, proper choice of media, addition of autologous serum, limited time frame for the expansion and temperature decreases during in vitro culture to ≈33°C, all impact the suppressive capacity of final Treg products (128–130).
Conclusion
Treg therapies are currently undergoing intense testing. An interest in therapeutic Treg preparations has resulted in several ongoing clinical trials in the transplantation setting, in autoimmune diseases, but now also in conditions such as beta thalassemia major and amyotrophic lateral sclerosis (101) (Table 2). We await new testing in the setting of hypersensitivity and cardiovascular disease (102), and anticipate that Treg therapy will be considered for any condition where there is evidence for an immune regulation imbalance.
Timing of Tolerance-Inducing Cell Therapy
Timing of tolerance-inducing cell therapy in relation to the transplant or in the course of autoimmune disease development needs specific consideration. Clinical trials using Treg for GvHD indicate that Treg should be injected as early as possible, preferentially before disease onset (89, 91, 131). Early treatment is particularly important to achieve a high ratio between Treg and detrimental effector T cells, and thus prevent development of acute rejection or GvHD. In both HSCT and solid organ transplantation, tolerance-inducing cells are therefore mainly given around, just before or shortly after, the transplantation (see Tables 1, 2). Although early treatment is likely more effective, this approach encounters limitations such as increased immunosuppression doses and anti-CD25 treatment, which can interfere with the activity of infused cells. Thus, Tregs have also been infused at later time points e.g., 6 months after kidney transplantation in patients with biopsies showing evidence of inflammatory infiltrates to treat ongoing chronic rejection (103). It must also be considered that disease diagnosis timing is a factor in this respect. Although it is likely best to give the tolerance-inducing cell treatment as early as possible in disease development, it is currently not feasible in treating autoimmunity since a majority of autoimmune diseases start long before clinical symptoms and diagnosis, this brings in the added factor that the functional capacity of the attacked organ may already be irreparably damaged by the ongoing autoimmune process. Future insight into autoimmune disease development and early biomarkers will hopefully allow for earlier treatment with tolerance-inducing cell products.
Regulations
The perspectives of tolerance-inducing cellular therapy depend also on recently introduced regulations. The majority of tested preparations in Europe are now classified as drugs under the 1394/2007 EU directive on advanced therapy medicinal products (ATMP). This significantly changes the legal path for their registration requirements, for manufacturing license and marketing authorization. While the idea of an ATMP in Europe is relatively nascent, and it is continuously evolving through public consultations with interested parties, cellular drugs have a distinct central paneuropean path for registration. While the Committee for Advanced Therapies (CAT) steers this process in Europe to optimize safety for patients, it would be useful to introduce wider rules allowing for introduction of the cells as a routine treatment. To accelerate the whole process, acknowledgment of flexible new types of manufacturing cGMP equipment and reagents could open the way to more widespread ATMP use. Furthermore, measures to reduce manufacturing costs would lessen this major limitation to new trials. When considering cell therapy, scientists, physicians and regulators must keep in mind that ATMP must be affordable for patients and society. Interestingly, scientists are largely responsible for current guidelines, and should revisit those recommendations based on factors such as cost (55, 132).
Immunomonitoring of Tolerance-Inducing Cellular Therapies
Tackling Immunomonitoring in Tolerogenic Therapies
Since cell-based therapies are becoming more common, it is important to reliably monitor the immune system for both desired and undesired immunological effects. Rigorous immunomonitoring will therefore provide information about the safety of these treatments, ideally at early time points after CTT administration. In addition it will give insight in the therapy-related mechanisms of tolerance-induction and maintenance and may aid in patient-tailoring of therapy. To accurately measure the effects, especially across different trials, it will require introduction of harmonized and validated immunomonitoring assays.
There are a number of possible assays that can determine cell therapy effects in humans. For instance, measurement of circulating cytokines, C-reactive protein or changes in antibody titer can determine immune status. Similarly, measuring CD4+ T cell responses after viral-antigen stimulation is possible by flow cytometry via CD40L expression or cytokine production in a functional assay; these assays could potentially identify non-specific immunosuppressive effects (133). Although the completed clinical trials have shown so far that cell-based tolerogenic therapies are safe and do not cause serious undesirable immune responses (96, 99, 103, 104), the extent to which these treatments achieve therapeutic efficacy remains largely undetermined. Current cell-based tolerogenic trials in autoimmunity and transplantation include clinical outcome measures such as C-peptide response, insulin consumption or reduction of immunosuppressive doses. However, clinical endpoints may not necessary reflect the efficacy of cell-based therapies, since the tolerogenic effect of the transferred cells may not directly lead to immediate changes in systemic parameters such as inflammation, potentially underestimating a longer term effect. It is therefore important that future clinical trials incorporate suitable monitoring methods to assess the immunomodulatory effects of cellular therapies.
General vs. Specific Monitoring Assays
To assess therapeutic effectiveness, different methods have been proposed to identify tolerogenic responses. The assessment of in vitro autoreactive or donor-specific T cells responses prior to and after treatment could provide a precise evaluation of therapeutic efficacy. Antigen-specific assays allow discrimination between targeted tolerance to the induction of general immune suppression and loss of responses to pathogens. In addition, these methods provide an efficacy readout for antigen-specific therapies such as tolerogenic APC loaded with antigens or the generation of donor-specific Treg. The identification of antigen-specific immune responses by ELISPOT (134) or through flow cytometry detection of CD40L upregulation (135) have shown promising results in predicting kidney and liver allograft rejection, suggesting potential applicability for the evaluation of tolerogenic therapies. Monitoring targets will be different depending on the main immune population involved in the disease (e.g., CD4, CD8 T cells or antibodies). Unfortunately, the antigens mediating the immune responses in autoimmunity are not always available or identified (as discussed in paragraph Antigen Specificity of tolAPC-Based Immunomodulation); HLA antigens in the case of transplantation are known and can be used, or stimulation with donor or donor-matched cells is possible. Though less specific, identification of phenotypic or functional changes by flow cytometry on the total pool of cells targeted (e.g., Treg) by the tolerogenic treatment may reveal therapeutic effectiveness. Indeed, the acquisition of tolerance in animal models and in the clinic is associated with an increased number of regulatory cells and decreased pro-inflammatory function of innate and adaptive immune cells (5, 136, 137). Therefore, flow cytometry analysis to delineate the distribution and activation status of different cell types, or in vitro assays to evaluate the suppressive and inflammatory function of circulating cells can provide a non-specific approach to assess the development of tolerogenic properties (138). Other non-specific strategies such as gene expression profiling of circulating immune cells or tissue biopsies can add to the functional assessment of immune responses. Furthermore, there is a growing body of work focusing on the validation of transcriptional signatures to predict transplantation tolerance in liver and kidney transplant recipients (139–141), which may prove to be a valuable tool in assessing the efficacy of tolerogenic therapies.
Tracking of Cellular Product
The evaluation of homeostatic characteristics of infused cells such as survival, stability or tissue migration, constitutes another monitoring objective to assess therapeutic efficacy. Being able to track infused cells will help to determine the best site for administration/application of tolerogenic cell products. While simple phenotypic detection (flow cytometry) of the transferred cell subset after treatment suggests the presence of infused cells, it does not distinguish transferred from endogenous cells. Therefore, current techniques for tracking infused cells depend on direct cell labeling strategies, such as indium labeling or deuterium introduction during ex vivo expansion, with subsequent isotope detection in the different tissues or compartments (99, 103, 142). Unfortunately, this method is only semi-quantitative, since individual cells are not detected. Individual cells can be labeled with rare earth metals and detected with precision in vivo using laser ablation-inductively coupled plasma-mass spectrometry, but so far this has only been tested in mouse models (143, 144). Other emerging therapeutic approaches such as CAR-Treg could take advantage of genetic modifications to adapt reported gene imaging strategies to detect the transferred cells by non-invasive methods (e.g., MRI, PET, SPECT) (145, 146). Additionally, the use of T cell receptor (TCR) engineered Treg or ex vivo expanded antigen-specific Treg create the opportunity to track infused cells by predefining the TCR clones and identifying them among the T cell compartment repertoire through TCR sequencing analysis (147).
Harmonization to Allow Comparison
Current tolerogenic cell-based trials are highly heterogeneous, comprising different cell types with variable manufacturing approaches, and targeting various autoimmune diseases and transplantation settings. Furthermore, by their very nature such trials tend to involve low numbers of patients, often participating in different centers or countries. It is therefore essential to establish common immunomonitoring strategies in the research community to achieve robust and reliable data which can be compared and combined between trials. First, not all the immunomonitoring methods are similarly standardisable, and second, not all the centers have the technical expertise or infrastructure to perform certain assays. Sample collection and storage of whole blood or tissue biopsies for transcriptional analysis does not involve extensive sample manipulation, making standardization of this method achievable. On the contrary, while flow cytometry analysis of circulating blood is an accessible technology, instrumentation must be calibrated appropriately to accurately define cell subsets, and analysis of the data needs to be strictly regulated. Although centralized phenotypic analysis in multicenter trials is feasible (105), this involves significant logistical challenges, including the decision to either test fresh or frozen samples; frozen samples sacrifice accuracy due to loss of certain cell populations during separation and freezing procedures. In general, flow cytometry standardization requires extensive cooperation between centers and precise planning. To further harmonize flow cytometry data implementation of automated gating approaches will be of outmost importance in the future (148–150). Finally, functional in vitro assays also represent a challenging method to standardize, involving different approaches depending of the cell type and function. Nevertheless, several efforts have been made to establish a minimal harmonization of antigen-specific functional assays (56). While these assays are likely to be the most informative in the assessment of therapeutic efficacy, it is unlikely that current assay results will be directly comparable between independent trials. Therefore, the inclusion of adequate control cohorts and reference groups in the assays, considering the specific cell therapy approaches and disease characteristics, remains an objective to achieve feasible comparisons (107).
Final conclusion
Tolerance-inducing cellular therapies have great potential. Several cell types are now in early-stage clinical trials, including various types of Treg and tolAPC (including tolDC and Mreg) (Tables 1, 2). At the present time it is unclear which of these cell types will prove most suitable as a cell-based therapy; each likely has particular advantages that may be suitable for one particular disease or another. In Table 3, the main specific limitations to be considered for the treatment of autoimmunity or transplantation with tolerance-inducing cell therapies are summarized. It becomes clear that although we might treat these diseases with the same manufactured cell product, the optimal treatment regime could be quite different.
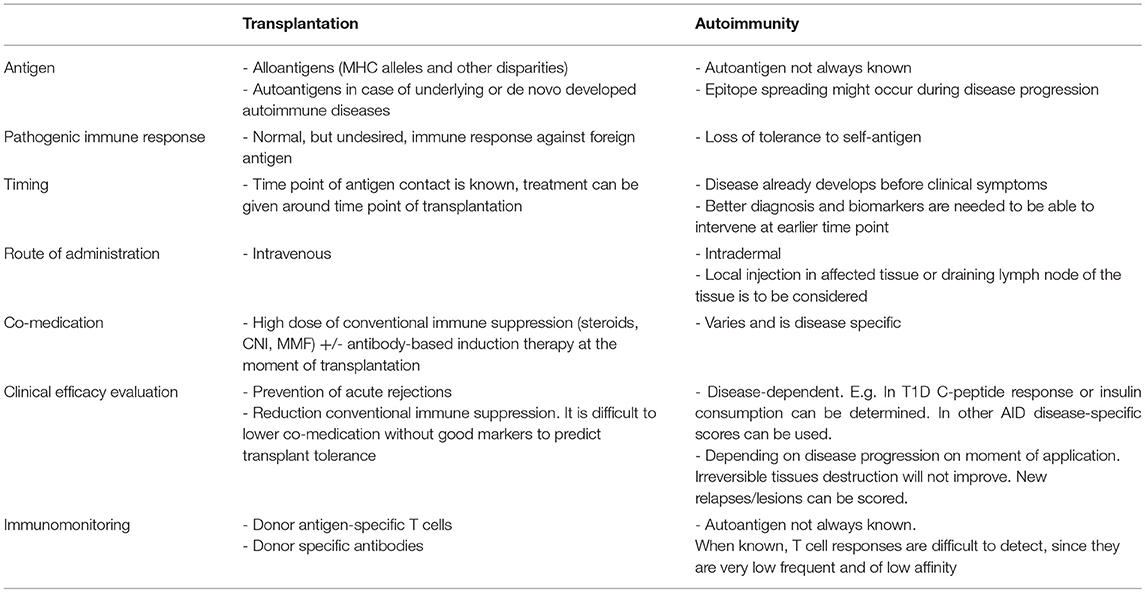
Table 3. Main specific differences in tolerance-inducing cell treatment between transplantation and autoimmune disease setting.
Immunomonitoring is an indispensable aspect of current and future tolerogenic cell-based therapies that will provide fundamental information to understand and optimize cell therapies. Monitoring will also aid in identifying biomarkers with the capacity for early identification of therapy responders and non-responders and patient-tailoring of therapy. As part of the overall strategy to increase implementation of ATMPs, it will be critical to harmonize GMP manufacturing protocols, product characterization and immunomonitoring. Minimal information models such as MITAP and MiTREG (132, 151) will serve as important tools in this respect.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This article is based upon work from COST Action A FACTT (BM1305: www.afactt.eu), supported by COST (European Cooperation in Science and Technology) (www.cost.eu). COST is a funding agency for research and innovation networks. COST Actions help connect research initiatives across Europe and enable scientists to grow their ideas by sharing them with their peers. This boosts their research, career and innovation. COST is supported by the EU Framework Programme Horizon 2020. Part of the work discussed is funded by The ONE Study, EU FP7 Funding Program, BIO-DrIM, EU FP7 Funding Program and ReSToRe, EU H2020 Funding Program. PT is supported by the National Center for Research and Development, PL (grant no: STRATEGMED1/233368/1/NCBR/2014). EM-C acknowledges the support by projects PI14/01175, PI16/01737 and PI17/01521, integrated in the Plan Nacional de I+D+I and co-supported by the ISCIII-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER). NC and EM-C acknowledge the support by project 140191 from the Institute for the Promotion of Innovation by Science and Technology (IWT-TMB) in Flanders (Belgium).
Conflict of Interest Statement
PT Medical University of Gdańsk–owns a patent related to presented content.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been supported by positive discussion by all AFACTT members during A FACTT network meetings. We specifically like to thank Juan Navarro Barriuso and Tanja Nikolic for their input on the manuscript.
References
1. Ten Brinke A, Hilkens CM, Cools N, Geissler EK, Hutchinson JA, Lombardi G, et al. Clinical use of tolerogenic dendritic cells-harmonization approach in European collaborative effort. Mediators Inflamm. (2015) 2015:471719. doi: 10.1155/2015/471719
2. Trzonkowski P, Bacchetta R, Battaglia M, Berglund D, Bohnenkamp HR, Ten Brinke A, et al. Hurdles in therapy with regulatory T cells. Sci Transl Med. (2015) 7:304ps318. doi: 10.1126/scitranslmed.aaa7721
3. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science (1993) 259:974–7. doi: 10.1126/science.8094901
4. Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. (2002) 80:477–83. doi: 10.1046/j.1440-1711.2002.01115.x
5. Thomson AW, Robbins PD. Tolerogenic dendritic cells for autoimmune disease and transplantation. Ann Rheum Dis. (2008) 67(Suppl. 3):iii90–6. doi: 10.1136/ard.2008.099176
6. Hilkens CM, Isaacs JD, Thomson AW. Development of dendritic cell-based immunotherapy for autoimmunity. Int Rev Immunol. (2010) 29:156–83. doi: 10.3109/08830180903281193
7. Raich-Regue D, Grau-Lopez L, Naranjo-Gomez M, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E, et al. Stable antigen-specific T-cell hyporesponsiveness induced by tolerogenic dendritic cells from multiple sclerosis patients. Eur J Immunol. (2012) 42:771–82. doi: 10.1002/eji.201141835
8. Nikolic T, Roep BO. Regulatory multitasking of tolerogenic dendritic cells - lessons taken from vitamin d3-treated tolerogenic dendritic cells. Front Immunol. (2013) 4:113. doi: 10.3389/fimmu.2013.00113
9. Van Brussel I, Lee WP, Rombouts M, Nuyts AH, Heylen M, De Winter BY, et al. Tolerogenic dendritic cell vaccines to treat autoimmune diseases: can the unattainable dream turn into reality? Autoimmun Rev. (2014) 13:138–50. doi: 10.1016/j.autrev.2013.09.008
10. Raker VK, Domogalla MP, Steinbrink K. Tolerogenic dendritic cells for regulatory T cell induction in man. Front Immunol. (2015) 6:569. doi: 10.3389/fimmu.2015.00569
11. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
12. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity? Trends Immunol. (2002) 23:445–9. doi: 10.1016/S1471-4906(02)02281-0
13. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
14. Riquelme P, Geissler EK, Hutchinson JA. Alternative approaches to myeloid suppressor cell therapy in transplantation: comparing regulatory macrophages to tolerogenic DCs and MDSCs. Transplant Res. (2012) 1:17. doi: 10.1186/2047-1440-1-17
15. Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care (2011) 34:2026–32. doi: 10.2337/dc11-0472
16. Benham H, Nel HJ, Law SC, Mehdi AM, Street S, Ramnoruth N, et al. Citrullinated peptide dendritic cell immunotherapy in HLA risk genotype-positive rheumatoid arthritis patients. Sci. Transl. Med. (2015) 7:290ra87. doi: 10.1126/scitranslmed.aaa9301
17. Bell GM, Anderson AE, Diboll J, Reece R, Eltherington O, Harry RA, et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann Rheum Dis. (2017) 76:227–34. doi: 10.1136/annrheumdis-2015-208456
18. Joo YB, Park J-E, Choi C-B, Choi J, Heo M, Kim H-Y, et al. Phase I study of immunotherapy using autoantigen-loaded dendritic cells in patients with anti-citrullinated peptide antigen positive rheumatoid arthritis. In: Proceedings of the ACR/ARHP Annual Meeting, Abstract 946. Boston, MA (2014).
19. Jauregui-Amezaga A, Cabezon R, Ramirez-Morros A, Espana C, Rimola J, Bru C, et al. Intraperitoneal administration of autologous tolerogenic dendritic cells for refractory Crohn's disease: a phase I study. J Crohns Colitis (2015) 9:1071–8. doi: 10.1093/ecco-jcc/jjv144
20. Geissler EK. The ONE Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplant Res. (2012) 1:11. doi: 10.1186/2047-1440-1-11
21. Hutchinson JA, Riquelme P, Brem-Exner BG, Schulze M, Matthai M, Renders L, et al. Transplant acceptance-inducing cells as an immune-conditioning therapy in renal transplantation. Transpl Int. (2008) 21:728–41. doi: 10.1111/j.1432-2277.2008.00680.x
22. Hutchinson JA, Brem-Exner BG, Riquelme P, Roelen D, Schulze M, Ivens K, et al. A cell-based approach to the minimization of immunosuppression in renal transplantation. Transpl Int. (2008) 21:742–54. doi: 10.1111/j.1432-2277.2008.00692.x
23. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. (2001) 193:233–8. doi: 10.1084/jem.193.2.233
24. Dhodapkar MV, Steinman RM. Antigen-bearing immature dendritic cells induce peptide-specific CD8(+) regulatory T cells in vivo in humans. Blood (2002) 100:174–7. doi: 10.1182/blood.V100.1.174
25. Kryczanowsky F, Raker V, Graulich E, Domogalla MP, Steinbrink K. IL-10-modulated human dendritic cells for clinical use: identification of a stable and migratory subset with improved tolerogenic activity. J Immunol. (2016) 197:3607–17. doi: 10.4049/jimmunol.1501769
26. Tureci O, Bian H, Nestle FO, Raddrizzani L, Rosinski JA, Tassis A, et al. Cascades of transcriptional induction during dendritic cell maturation revealed by genome-wide expression analysis. FASEB J. (2003) 17:836–47. doi: 10.1096/fj.02-0724com
27. Naranjo-Gomez M, Raich-Regue D, Onate C, Grau-Lopez L, Ramo-Tello C, Pujol-Borrell R, et al. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med. (2011) 9:89. doi: 10.1186/1479-5876-9-89
28. Raich-Regue D, Naranjo-Gomez M, Grau-Lopez L, Ramo C, Pujol-Borrell R, Martinez-Caceres E, et al. Differential effects of monophosphoryl lipid A and cytokine cocktail as maturation stimuli of immunogenic and tolerogenic dendritic cells for immunotherapy. Vaccine (2012) 30:378–87. doi: 10.1016/j.vaccine.2011.10.081
29. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4+ and CD8+ anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood (2002) 99:2468–76. doi: 10.1182/blood.V99.7.2468
30. Apostolou I, Von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. (2004) 199:1401–8. doi: 10.1084/jem.20040249
31. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, Von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. (2005) 6:1219–27. doi: 10.1038/ni1265
32. Boks MA, Kager-Groenland JR, Haasjes MS, Zwaginga JJ, Van Ham SM, Ten Brinke A. IL-10-generated tolerogenic dendritic cells are optimal for functional regulatory T cell induction - a comparative study of human clinical-applicable DC. Clin Immunol. (2012) 142:332–42. doi: 10.1016/j.clim.2011.11.011
33. Li H, Shi B. Tolerogenic dendritic cells and their applications in transplantation. Cell Mol Immunol. (2015) 12:24–30. doi: 10.1038/cmi.2014.52
34. Keir ME, Francisco LM, Sharpe AH. PD-1 and its ligands in T-cell immunity. Curr Opin Immunol. (2007) 19:309–14. doi: 10.1016/j.coi.2007.04.012
35. Wu J, Horuzsko A. Expression and function of immunoglobulin-like transcripts on tolerogenic dendritic cells. Hum Immunol. (2009) 70:353–6. doi: 10.1016/j.humimm.2009.01.024
36. Hoves S, Krause SW, Herfarth H, Halbritter D, Zhang HG, Mountz JD, et al. Elimination of activated but not resting primary human CD4+ and CD8+ T cells by Fas ligand (FasL/CD95L)-expressing Killer-dendritic cells. Immunobiology (2004) 208:463–75. doi: 10.1078/0171-2985-00293
37. Schutz C, Hoves S, Halbritter D, Zhang HG, Mountz JD, Fleck M. Alloantigen specific deletion of primary human T cells by Fas ligand (CD95L)-transduced monocyte-derived killer-dendritic cells. Immunology (2011) 133:115–22. doi: 10.1111/j.1365-2567.2011.03417.x
38. Izawa T, Kondo T, Kurosawa M, Oura R, Matsumoto K, Tanaka E, et al. Fas-independent T-cell apoptosis by dendritic cells controls autoimmune arthritis in MRL/lpr mice. PLoS ONE (2012) 7:e48798. doi: 10.1371/journal.pone.0048798
39. Wakkach A, Fournier N, Brun V, Breittmayer JP, Cottrez F, Groux H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity (2003) 18:605–17. doi: 10.1016/S1074-7613(03)00113-4
40. Anderson AE, Sayers BL, Haniffa MA, Swan DJ, Diboll J, Wang XN, et al. Differential regulation of naive and memory CD4+ T cells by alternatively activated dendritic cells. J Leukoc Biol. (2008) 84:124–33. doi: 10.1189/jlb.1107744
41. Speck S, Lim J, Shelake S, Matka M, Stoddard J, Farr A, et al. TGF-beta signaling initiated in dendritic cells instructs suppressive effects on Th17 differentiation at the site of neuroinflammation. PLoS ONE (2014) 9:e102390. doi: 10.1371/journal.pone.0102390
42. Anderson AE, Swan DJ, Wong OY, Buck M, Eltherington O, Harry RA, et al. Tolerogenic dendritic cells generated with dexamethasone and vitamin D3 regulate rheumatoid arthritis CD4(+) T cells partly via transforming growth factor-beta1. Clin Exp Immunol. (2017) 187:113–23. doi: 10.1111/cei.12870
43. Beringer DX, Kleijwegt FS, Wiede F, Van Der Slik AR, Loh KL, Petersen J, et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat Immunol. (2015) 16:1153–61. doi: 10.1038/ni.3271
44. Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood (2010) 116:935–44. doi: 10.1182/blood-2009-07-234872
45. Carretero-Iglesia L, Bouchet-Delbos L, Louvet C, Drujont L, Segovia M, Merieau E, et al. Comparative study of the immunoregulatory capacity of in vitro generated tolerogenic dendritic cells, suppressor macrophages, and myeloid-derived suppressor cells. Transplantation (2016) 100:2079–89. doi: 10.1097/TP.0000000000001315
46. Riquelme P, Haarer J, Kammler A, Walter L, Tomiuk S, Ahrens N, et al. TIGIT(+) iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun. (2018) 9:2858. doi: 10.1038/s41467-018-05167-8
47. Navarro-Barriuso J, Mansilla MJ, Naranjo-Gomez M, Sanchez-Pla A, Quirant-Sanchez B, Teniente-Serra A, et al. Comparative transcriptomic profile of tolerogenic dendritic cells differentiated with vitamin D3, dexamethasone and rapamycin. Sci Rep. (2018) 8:14985. doi: 10.1038/s41598-018-33248-7
48. Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. (2010) 69:2042–50. doi: 10.1136/ard.2009.126383
49. Nikolic T, Woittiez NJC, Van Der Slik A, Laban S, Joosten A, Gysemans C, et al. Differential transcriptome of tolerogenic versus inflammatory dendritic cells points to modulated T1D genetic risk and enriched immune regulation. Genes Immun. (2017) 18:176–83. doi: 10.1038/gene.2017.18
50. Xue Z, Zhang X, Chen M, Lu X, Deng R, Ma Y. Dendritic cells transduced with single immunoglobulin IL-1-related receptor exhibit immature properties and prolong islet allograft survival. Front Immunol. (2017) 8:1671. doi: 10.3389/fimmu.2017.01671
51. Agrawal S, Ganguly S, Hajian P, Cao JN, Agrawal A. PDGF upregulates CLEC-2 to induce T regulatory cells. Oncotarget (2015) 6:28621–32. doi: 10.18632/oncotarget.5765
52. Schinnerling K, Soto L, Garcia-Gonzalez P, Catalan D, Aguillon JC. Skewing dendritic cell differentiation towards a tolerogenic state for recovery of tolerance in rheumatoid arthritis. Autoimmun Rev. (2015) 14:517−27. doi: 10.1016/j.autrev.2015.01.014
53. Schinnerling K, Garcia-Gonzalez P, Aguillon JC. Gene expression profiling of human monocyte-derived dendritic cells - searching for molecular regulators of tolerogenicity. Front Immunol. (2015) 6:528. doi: 10.3389/fimmu.2015.00528
54. Navarro-Barriuso J, Mansilla MJ, Martinez-Caceres EM. Searching for the transcriptomic signature of immune tolerance induction-biomarkers of safety and functionality for tolerogenic dendritic cells and regulatory macrophages. Front Immunol. (2018) 9:2062. doi: 10.3389/fimmu.2018.02062
55. Lord P, Spiering R, Aguillon JC, Anderson AE, Appel S, Benitez-Ribas D, et al. Minimum information about tolerogenic antigen-presenting cells (MITAP): a first step towards reproducibility and standardisation of cellular therapies. PeerJ (2016) 4:e2300. doi: 10.7717/peerj.2300
56. Ten Brinke A, Marek-Trzonkowska N, Mansilla MJ, Turksma AW, Piekarska K, Iwaszkiewicz-Grzes D, et al. Monitoring T-cell responses in translational studies: optimization of dye-based proliferation assay for evaluation of antigen-specific responses. Front Immunol. (2017) 8:1870. doi: 10.3389/fimmu.2017.01870
57. Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, et al. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther. (2015) 21:222–30. doi: 10.1111/cns.12342
58. Mansilla MJ, Contreras-Cardone R, Navarro-Barriuso J, Cools N, Berneman Z, Ramo-Tello C, et al. Cryopreserved vitamin D3-tolerogenic dendritic cells pulsed with autoantigens as a potential therapy for multiple sclerosis patients. J Neuroinflammation (2016) 13:113. doi: 10.1186/s12974-016-0584-9
59. Verginis P, Li HS, Carayanniotis G. Tolerogenic semimature dendritic cells suppress experimental autoimmune thyroiditis by activation of thyroglobulin-specific CD4+CD25+ T cells. J Immunol. (2005) 174:7433–9. doi: 10.4049/jimmunol.174.11.7433
60. Stoop JN, Harry RA, Von DA, Isaacs JD, Robinson JH, Hilkens CM. Therapeutic effect of tolerogenic dendritic cells in established collagen-induced arthritis is associated with a reduction in Th17 responses. Arthritis Rheum. (2010) 62:3656–65. doi: 10.1002/art.27756
61. Yang J, Yang Y, Ren Y, Xie R, Zou H, Fan H. A mouse model of adoptive immunotherapeutic targeting of autoimmune arthritis using allo-tolerogenic dendritic cells. PLoS ONE (2013) 8:e77729. doi: 10.1371/journal.pone.0077729
62. Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. (2002) 2:85–95. doi: 10.1038/nri724
63. Grau-Lopez L, Raich D, Ramo-Tello C, Naranjo-Gomez M, Davalos A, Pujol-Borrell R, et al. Specific T-cell proliferation to myelin peptides in relapsing-remitting multiple sclerosis. Eur J Neurol. (2011) 18:1101–4. doi: 10.1111/j.1468-1331.2010.03307.x
64. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci. Transl. Med. (2013) 5:188ra175. doi: 10.1126/scitranslmed.3006168
65. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC, et al. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology (2018) 90:e955–62. doi: 10.1212/WNL.0000000000005118
66. Ingulli E. Mechanism of cellular rejection in transplantation. Pediatr Nephrol. (2010) 25:61–74. doi: 10.1007/s00467-008-1020-x
67. Marin E, Cuturi MC, Moreau A. Tolerogenic dendritic cells in solid organ transplantation: where do we stand? Front Immunol. (2018) 9:274. doi: 10.3389/fimmu.2018.00274
68. Anderson AE, Swan DJ, Sayers BL, Harry RA, Patterson AM, Von Delwig A, et al. LPS activation is required for migratory activity and antigen presentation by tolerogenic dendritic cells. J Leukoc Biol. (2009) 85:243–50. doi: 10.1189/jlb.0608374
69. Nuyts AH, Ponsaerts P, Van Tendeloo VF, Lee WP, Stein B, Nagels G, et al. Except for C-C chemokine receptor 7 expression, monocyte-derived dendritic cells from patients with multiple sclerosis are functionally comparable to those of healthy controls. Cytotherapy (2014) 16:1024–30. doi: 10.1016/j.jcyt.2014.02.016
70. Grover A, Kim GJ, Lizee G, Tschoi M, Wang G, Wunderlich JR, et al. Intralymphatic dendritic cell vaccination induces tumor antigen-specific, skin-homing T lymphocytes. Clin Cancer Res. (2006) 12:5801–8. doi: 10.1158/1078-0432.CCR-05-2421
71. Radomski M, Zeh HJ, Edington HD, Pingpank JF, Butterfield LH, Whiteside TL, et al. Prolonged intralymphatic delivery of dendritic cells through implantable lymphatic ports in patients with advanced cancer. J Immunother Cancer (2016) 4:24. doi: 10.1186/s40425-016-0128-y
72. De Laere M, Derdelinckx J, Hassi M, Kerosalo M, Oravamaki H, Van Den Bergh J, et al. Shuttling tolerogenic dendritic cells across the blood-brain barrier in vitro via the introduction of de novo C-C chemokine receptor 5 expression using messenger RNA electroporation. Front Immunol. (2017) 8:1964. doi: 10.3389/fimmu.2017.01964
73. Garrod KR, Chang CK, Liu FC, Brennan TV, Foster RD, Kang SM. Targeted lymphoid homing of dendritic cells is required for prolongation of allograft survival. J Immunol. (2006) 177:863–8. doi: 10.4049/jimmunol.177.2.863
74. Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, et al. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. (2001) 194:769–80. doi: 10.1084/jem.194.6.769
75. Dhodapkar MV, Sznol M, Zhao B, Wang D, Carvajal RD, Keohan ML, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl. Med. (2014) 6:232ra251. doi: 10.1126/scitranslmed.3008068
76. Hotaling NA, Tang L, Irvine DJ, Babensee JE. Biomaterial strategies for immunomodulation. Annu Rev Biomed Eng. (2015) 17:317–49. doi: 10.1146/annurev-bioeng-071813-104814
77. Tostanoski LH, Gosselin EA, Jewell CM. Engineering tolerance using biomaterials to target and control antigen presenting cells. Discov Med. (2016) 21:403–10.
78. Ochando J, Braza MS. Nanoparticle-based modulation and monitoring of antigen-presenting cells in organ transplantation. Front Immunol. (2017) 8:1888. doi: 10.3389/fimmu.2017.01888
79. Capini C, Jaturanpinyo M, Chang HI, Mutalik S, Mcnally A, Street S, et al. Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol. (2009) 182:3556–65. doi: 10.4049/jimmunol.0802972
80. Pujol-Autonell I, Serracant-Prat A, Cano-Sarabia M, Ampudia RM, Rodriguez-Fernandez S, Sanchez A, et al. Use of autoantigen-loaded phosphatidylserine-liposomes to arrest autoimmunity in type 1 diabetes. PLoS ONE (2015) 10:e0127057. doi: 10.1371/journal.pone.0127057
81. Pujol-Autonell I, Mansilla MJ, Rodriguez-Fernandez S, Cano-Sarabia M, Navarro-Barriuso J, Ampudia RM, et al. Liposome-based immunotherapy against autoimmune diseases: therapeutic effect on multiple sclerosis. Nanomedicine (2017) 12:1231–42. doi: 10.2217/nnm-2016-0410
82. Lan YY, Wang Z, Raimondi G, Wu W, Colvin BL, De Creus A, et al. “Alternatively Activated” dendritic cells preferentially secrete IL-10, expand Foxp3+CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-Ig. J Immunol. (2006) 177:5868–77. doi: 10.4049/jimmunol.177.9.5868
83. Ezzelarab MB, Zahorchak AF, Lu L, Morelli AE, Chalasani G, Demetris AJ, et al. Regulatory dendritic cell infusion prolongs kidney allograft survival in nonhuman primates. Am J Transplant (2013) 13:1989–2005. doi: 10.1111/ajt.12310
84. Baas MC, Kuhn C, Valette F, Mangez C, Duarte MS, Hill M, et al. Combining autologous dendritic cell therapy with CD3 antibodies promotes regulatory T cells and permanent islet allograft acceptance. J Immunol. (2014) 193:4696–703. doi: 10.4049/jimmunol.1401423
85. Stanko K, Iwert C, Appelt C, Vogt K, Schumann J, Strunk FJ, et al. CD96 expression determines the inflammatory potential of IL-9-producing Th9 cells. Proc Natl Acad Sci USA. (2018) 115:E2940–9. doi: 10.1073/pnas.1708329115
87. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. (2008) 8:523–32. doi: 10.1038/nri2343
88. Trzonkowski P, Bieniaszewska M, Juścinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol. (2009) 133:22–6. doi: 10.1016/j.clim.2009.06.001
89. Brunstein CG, Miller J, Cao Q, Mckenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
90. Brunstein CG, Blazar BR, Miller JS, Cao Q, Hippen KL, Mckenna DH, et al. Adoptive transfer of umbilical cord blood-derived regulatory T cells and early viral reactivation. Biol Blood Marrow Transplant. (2013) 19:1271–3. doi: 10.1016/j.bbmt.2013.06.004
91. Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood (2011) 117:3921–8. doi: 10.1182/blood-2010-10-311894
92. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood (2014) 124:638–44. doi: 10.1182/blood-2014-03-564401
93. Edinger M, Hoffmann P. Regulatory T cells in stem cell transplantation: strategies and first clinical experiences. Curr Opin Immunol. (2011) 23:679–84. doi: 10.1016/j.coi.2011.06.006
94. Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy (2015) 17:473–86. doi: 10.1016/j.jcyt.2014.11.005
95. Bacchetta R, Lucarelli B, Sartirana C, Gregori S, Lupo Stanghellini MT, Miqueu P, et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front Immunol. (2014) 5:16. doi: 10.3389/fimmu.2014.00016
96. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, et al. Administration of CD4+CD25highCD127- regulatory T cells preserves beta-cell function in type 1 diabetes in children. Diabetes Care (2012) 35:1817–20. doi: 10.2337/dc12-0038
97. Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol. (2014) 153:23–30. doi: 10.1016/j.clim.2014.03.016
98. Marek-Trzonkowska N, Mysliwiec M, Iwaszkiewicz-Grzes D, Gliwinski M, Derkowska I, Zalinska M, et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J Transl Med. (2016) 14:332. doi: 10.1186/s12967-016-1090-7
99. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. (2015) 7:315ra189. doi: 10.1126/scitranslmed.aad4134
100. Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn's disease. Gastroenterology (2012) 143:1207–17.e02. doi: 10.1053/j.gastro.2012.07.116
101. Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS: a phase I, first-in-human study. Neurol Neuroimmunol Neuroinflamm. (2018) 5:e465. doi: 10.1212/NXI.0000000000000465
102. Gliwinski M, Iwaszkiewicz-Grzes D, Trzonkowski P. Cell-based therapies with T regulatory cells. BioDrugs (2017) 31:335–47. doi: 10.1007/s40259-017-0228-3
103. Chandran S, Tang Q, Sarwal M, Laszik ZG, Putnam AL, Lee K, et al. Polyclonal regulatory T cell therapy for control of inflammation in kidney transplants. Am J Transplant. (2017) 17:2945–54. doi: 10.1111/ajt.14415
104. Mathew JM, Jessica HV, Lefever A, Konieczna I, Stratton C, He J, et al. A phase I clinical trial with ex vivo expanded recipient regulatory T cells in living donor kidney transplants. Sci Rep. (2018) 8:7428. doi: 10.1038/s41598-018-25574-7
105. Streitz M, Miloud T, Kapinsky M, Reed MR, Magari R, Geissler EK, et al. Standardization of whole blood immune phenotype monitoring for clinical trials: panels and methods from the ONE study. Transplant Res. (2013) 2:17. doi: 10.1186/2047-1440-2-17
106. Geissler EK, Tullius SG, Chong AS. Establishment of a global virtual laboratory for transplantation: a symposium report. Transplantation (2015) 99:381–4. doi: 10.1097/TP.0000000000000560
107. Kverneland AH, Streitz M, Geissler E, Hutchinson J, Vogt K, Boes D, et al. Age and gender leucocytes variances and references values generated using the standardized ONE-Study protocol. Cytometry A (2016) 89:543–64. doi: 10.1002/cyto.a.22855
108. Safinia N, Vaikunthanathan T, Fraser H, Thirkell S, Lowe K, Blackmore L, et al. Successful expansion of functional and stable regulatory T cells for immunotherapy in liver transplantation. Oncotarget (2016) 7:7563–77. doi: 10.18632/oncotarget.6927
109. Akimova T, Kamath BM, Goebel JW, Meyers KE, Rand EB, Hawkins A, et al. Differing effects of rapamycin or calcineurin inhibitor on T-regulatory cells in pediatric liver and kidney transplant recipients. Am J Transplant. (2012) 12:3449–61. doi: 10.1111/j.1600-6143.2012.04269.x
110. Scotta C, Fanelli G, Hoong SJ, Romano M, Lamperti EN, Sukthankar M, et al. Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica (2016) 101:91–100. doi: 10.3324/haematol.2015.128934
111. Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest. (2008) 118:3619–28. doi: 10.1172/JCI33185
112. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant. (2013) 13:3010–20. doi: 10.1111/ajt.12433
113. Macdonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
114. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. (2017) 17:931–43. doi: 10.1111/ajt.14185
115. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am J Transplant. (2017) 17:917–30. doi: 10.1111/ajt.14175
116. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell (2016) 167:1067–78.e16. doi: 10.1016/j.cell.2016.09.050
117. Lei H, Kuchenbecker L, Streitz M, Sawitzki B, Vogt K, Landwehr-Kenzel S, et al. Human CD45RA(-) FoxP3(hi) memory-type regulatory T cells show distinct TCR repertoires with conventional T cells and play an important role in controlling early immune activation. Am J Transplant. (2015) 15:2625–35. doi: 10.1111/ajt.13315
118. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
119. Whitehouse G, Gray E, Mastoridis S, Merritt E, Kodela E, Yang JHM, et al. IL-2 therapy restores regulatory T-cell dysfunction induced by calcineurin inhibitors. Proc Natl Acad Sci USA. (2017) 114:7083–8. doi: 10.1073/pnas.1620835114
120. Von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis. (2016) 75:1407–15. doi: 10.1136/annrheumdis-2015-207776
121. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol. (2015) 15:283–94. doi: 10.1038/nri3823
122. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
123. Hendrikx Thijs K, Velthuis Jurjen HL, Klepper M, Van Gurp E, Geel A, Schoordijk W, et al. Monotherapy rapamycin allows an increase of CD4+ CD25bright+ FoxP3+ T cells in renal recipients. Transplant Int. (2009) 22:884–91. doi: 10.1111/j.1432-2277.2009.00890.x
124. Scottà C, Esposito M, Fazekasova H, Fanelli G, Edozie FC, Ali N, et al. Differential effects of rapamycin and retinoic acid on expansion, stability and suppressive qualities of human CD4+CD25+FOXP3+ Tregs subpopulations. Heamatologica (2013) 98:1291–9. doi: 10.3324/haematol.2012.074088
125. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments tregs yet transiently impairs β-cell function. Diabetes (2012) 61:2340–8. doi: 10.2337/db12-0049
126. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A rapamycin-based GMP-compatible process for the isolation and expansion of regulatory T cells for clinical trials. Mol Ther Methods Clin Dev. (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
127. Baron U, Floess S, Wieczorek G, Baumann K, Grützkau A, Dong J, et al. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur J Immunol. (2007) 37:2378–89. doi: 10.1002/eji.200737594
128. Marek N, Bieniaszewska M, Krzystyniak A, Juscinska J, Mysliwska J, Witkowski P, et al. The time is crucial for ex vivo expansion of T regulatory cells for therapy. Cell Transplant. (2011) 20:1747–58. doi: 10.3727/096368911X566217
129. Gołab K, Krzystyniak A, Marek-Trzonkowska N, Misawa R, Wang LJ, Wang X, et al. Impact of culture medium on CD4+ CD25highCD127lo/neg Treg expansion for the purpose of clinical application. Int Immunopharmacol. (2013) 16:358–63. doi: 10.1016/j.intimp.2013.02.016
130. Marek-Trzonkowska N, Piekarska K, Filipowicz N, Piotrowski A, Gucwa M, Vogt K, et al. Mild hypothermia provides Treg stability. Sci Rep. (2017) 7:11915. doi: 10.1038/s41598-017-10151-1
131. Trzonkowski P, Dukat-Mazurek A, Bieniaszewska M, Marek-Trzonkowska N, Dobyszuk A, Juścinska J, et al. Treatment of graft-versus-host disease with naturally occurring T regulatory cells. BioDrugs (2013) 27:605–14. doi: 10.1007/s40259-013-0050-5
132. Fuchs A, Gliwinski M, Grageda N, Spiering R, Abbas AK, Appel S, et al. Minimum information about T regulatory cells: a step toward reproducibility and standardization. Front Immunol. (2018) 8:1844. doi: 10.3389/fimmu.2017.01844
133. Verbij FC, Turksma AW, De Heij F, Kaijen P, Lardy N, Fijnheer R, et al. CD4+ T cells from patients with acquired thrombotic thrombocytopenic purpura recognize CUB2 domain-derived peptides. Blood (2016) 127:1606–9. doi: 10.1182/blood-2015-10-668053
134. Crespo E, Lucia M, Cruzado JM, Luque S, Melilli E, Manonelles A, et al. Pre-transplant donor-specific T-cell alloreactivity is strongly associated with early acute cellular rejection in kidney transplant recipients not receiving T-cell depleting induction therapy. PLoS ONE (2015) 10:e011761. doi: 10.1371/journal.pone.0117618
135. Sindhi R, Ashokkumar C, Higgs BW, Levy S, Soltys K, Bond G, et al. Profile of the Pleximmune blood test for transplant rejection risk prediction. Expert Rev Mol Diagn. (2016) 16:387–93. doi: 10.1586/14737159.2016.1139455
136. Andreola G, Chittenden M, Shaffer J, Cosimi AB, Kawai T, Cotter P, et al. Mechanisms of donor-specific tolerance in recipients of haploidentical combined bone marrow/kidney transplantation. Am J Transplant. (2011) 11:1236–47. doi: 10.1111/j.1600-6143.2011.03566.x
137. Taubert R, Danger R, Londono MC, Christakoudi S, Martinez-Picola M, Rimola A, et al. Hepatic infiltrates in operational tolerant patients after liver transplantation show enrichment of regulatory T cells before proinflammatory genes are downregulated. Am J Transplant. (2016) 16:1285–93. doi: 10.1111/ajt.13617
138. Newell KA, Adams AB, Turka LA. Biomarkers of operational tolerance following kidney transplantation - the immune tolerance network studies of spontaneously tolerant kidney transplant recipients. Hum Immunol. (2018) 79:380–7. doi: 10.1016/j.humimm.2018.02.007
139. Newell KA, Asare A, Kirk AD, Gisler TD, Bourcier K, Suthanthiran M, et al. Identification of a B cell signature associated with renal transplant tolerance in humans. J Clin Invest. (2010) 120:1836–47. doi: 10.1172/JCI39933
140. Bohne F, Martinez-Llordella M, Lozano JJ, Miquel R, Benitez C, Londono MC, et al. Intra-graft expression of genes involved in iron homeostasis predicts the development of operational tolerance in human liver transplantation. J Clin Invest. (2012) 122:368–82. doi: 10.1172/JCI59411
141. Rebollo-Mesa I, Nova-Lamperti E, Mobillo P, Runglall M, Christakoudi S, Norris S, et al. Biomarkers of tolerance in kidney transplantation: are we predicting tolerance or response to immunosuppressive treatment? Am J Transplant. (2016) 16:3443–57. doi: 10.1111/ajt.13932
142. Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, et al. Cutting edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. (2011) 187:2072–8. doi: 10.4049/jimmunol.1100762
143. Managh AJ, Hutchinson RW, Riquelme P, Broichhausen C, Wege AK, Ritter U, et al. Laser ablation-inductively coupled plasma mass spectrometry: an emerging technology for detecting rare cells in tissue sections. J Immunol. (2014) 193:2600–8. doi: 10.4049/jimmunol.1400869
144. Hutchinson RW, Mclachlin KM, Riquelme P, Haarer J, Broichhausen C, Ritter U, et al. Laser ablation inductively coupled plasma mass spectrometry: an emerging technology for multiparametric analysis of tissue antigens. Transplant Direct (2015) 1:e32. doi: 10.1097/TXD.0000000000000541
145. Sharif-Paghaleh E, Sunassee K, Tavare R, Ratnasothy K, Koers A, Ali N, et al. In vivo SPECT reporter gene imaging of regulatory T cells. PLoS ONE (2011) 6:e25857. doi: 10.1371/journal.pone.0025857
146. Lee HW, Gangadaran P, Kalimuthu S, Ahn BC. Advances in molecular imaging strategies for in vivo tracking of immune cells. Biomed Res Int. (2016) 2016:1946585. doi: 10.1155/2016/1946585
147. Theil A, Wilhelm C, Kuhn M, Petzold A, Tuve S, Oelschlagel U, et al. T cell receptor repertoires after adoptive transfer of expanded allogeneic regulatory T cells. Clin Exp Immunol. (2017) 187:316–24. doi: 10.1111/cei.12887
148. Malek M, Taghiyar MJ, Chong L, Finak G, Gottardo R, Brinkman RR. flowDensity: reproducing manual gating of flow cytometry data by automated density-based cell population identification. Bioinformatics (2015) 31:606–7. doi: 10.1093/bioinformatics/btu677
149. Schlickeiser S, Streitz M, Sawitzki B. Standardized multi-color flow cytometry and computational biomarker discovery. Methods Mol Biol. (2016) 1371:225–38. doi: 10.1007/978-1-4939-3139-2_15
150. Rahim A, Meskas J, Drissler S, Yue A, Lorenc A, Laing A, et al. High throughput automated analysis of big flow cytometry data. Methods (2018) 134–5:164–76. doi: 10.1016/j.ymeth.2017.12.015
Keywords: tolerance, dendritic cells, regulatory T cells, autoimmunity, transplantation, cell therapy, clinic, immunomonitoring
Citation: ten Brinke A, Martinez-Llordella M, Cools N, Hilkens CMU, van Ham SM, Sawitzki B, Geissler EK, Lombardi G, Trzonkowski P and Martinez-Caceres E (2019) Ways Forward for Tolerance-Inducing Cellular Therapies- an AFACTT Perspective. Front. Immunol. 10:181. doi: 10.3389/fimmu.2019.00181
Received: 24 October 2018; Accepted: 21 January 2019;
Published: 22 February 2019.
Edited by:
Duncan Howie, University of Oxford, United KingdomReviewed by:
Sylvaine You, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCiriana Orabona, University of Perugia, Italy
Copyright © 2019 ten Brinke, Martinez-Llordella, Cools, Hilkens, van Ham, Sawitzki, Geissler, Lombardi, Trzonkowski and Martinez-Caceres. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja ten Brinke, YS50ZW5icmlua2VAc2FucXVpbi5ubA==