 Chan-Wang J. Lio1*
Chan-Wang J. Lio1* Anjana Rao1,2,3
Anjana Rao1,2,3- 1Division of Signaling and Gene Expression, La Jolla Institute, La Jolla, CA, United States
- 2Department of Pharmacology and Moores Cancer Center, University of California, San Diego, La Jolla, CA, United States
- 3Sanford Consortium for Regenerative Medicine, San Diego, CA, United States
DNA methylation is an abundant and stable epigenetic modification that allows inheritance of information from parental to daughter cells. At active genomic regions, DNA methylation can be reversed by TET (Ten-eleven translocation) enzymes, which are responsible for fine-tuning methylation patterns. TET enzymes oxidize the methyl group of 5-methylcytosine (5mC) to yield 5-hydroxymethylcytosine (5hmC) and other oxidized methylcytosines, facilitating both passive and active demethylation. Increasing evidence has demonstrated the essential functions of TET enzymes in regulating gene expression, promoting cell differentiation, and suppressing tumor formation. In this review, we will focus on recent discoveries of the functions of TET enzymes in the development and function of lymphoid and myeloid cells. How TET activity can be modulated by metabolites, including vitamin C and 2-hydroxyglutarate, and its potential application in shaping the course of immune response will be discussed.
Introduction
Cells rely on the proper propagation and preservation of epigenetic information in order to regulate gene expression appropriately. 5-methylcytosine (5mC), described as the 5th base of DNA, is a chemically stable modification that is one of the most reliable ways of transmitting epigenetic information. In most cells, 5mC is present primarily at symmetrically-methylated CG dinucleotides in DNA, although methylation of cytosines in other contexts (CH=CA, CT, CC) has been reported in stem cells and in neurons (1). During DNA replication, methylated CGs are replaced by unmodified cytosines in the newly synthesized DNA strand, and the resulting hemimethylated CGs are recognized by a complex of UHRF1 and the maintenance methyl-transferase DNMT1 (2–4). The remethylation of hemi-methylated CpGs in newly replicated DNA is complete within 20 min, accounting for the stable inheritance of DNA methylation (5). In contrast to DNMT1, which depends on 5mC deposition at CpG motifs for maintenance DNA methylation, the de novo methyltransferases DNMT3A and DNMT3B can methylate unmodified cytosines in both CG and CH sequence contexts. While the writers for DNA methylation (DNMTs) have been known for decades, how DNA methylation is removed remained unclear until the discovery of TET (Ten-Eleven Translocation) enzymes and their ability to oxidize 5mC to 5-hydroxymethyl-cytosine (5hmC) [(6); reviewed in (3, 4)].
5hmC, the so-called 6th base, is a stable epigenetic modification that accounts for 1–10% of 5mC depending on the cell type: ~10% in embryonic stem cells (6) and as high as 40% in Purkinje neurons (7). While 5hmC or related modifications have been known to exist in simpler organisms including T-even phages for more than half a century (8), it was not until 2009 that 5hmC was rediscovered in mammalian cells (6, 7). The mammalian enzymes responsible for generating this modification are the three TET dioxygenases (TET1, TET2, and TET3) that utilize the co-factors α-ketoglutarate (αKG), reduced iron (Fe2+), and molecular oxygen to oxidize the methyl group at the 5 position of 5mC (6). TET proteins can be found in every metazoan organism that contains DNMTs, even simple organisms such as comb jellies (9–11).
Besides being a potential epigenetic mark, 5hmC is the key intermediate for TET-mediated active (replication-independent) and passive (replication-dependent) DNA demethylation (Figure 1). TET enzymes iteratively oxidize 5mC and 5hmC into other oxidized cytosines (oxi-mCs) including 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (12); in active DNA demethylation, 5fC and 5caC are recognized and excised by thymine DNA glycosylase (TDG), repaired by the base-excision repair system, and replaced by unmodified C, thus resulting in DNA demethylation (13). In replication-dependent passive DNA demethylation, the DNMT1/UHRF1 complex does not recognize hemi-modified CGs with 5hmC, 5fC, or 5caC and thus the cytosine on the newly synthesized DNA strand is not methylated (5, 14, 15). Thus, the interplay between DNMT and TET proteins sculpts the DNA methylation landscape and enables the flow of epigenetic information across cell generations.
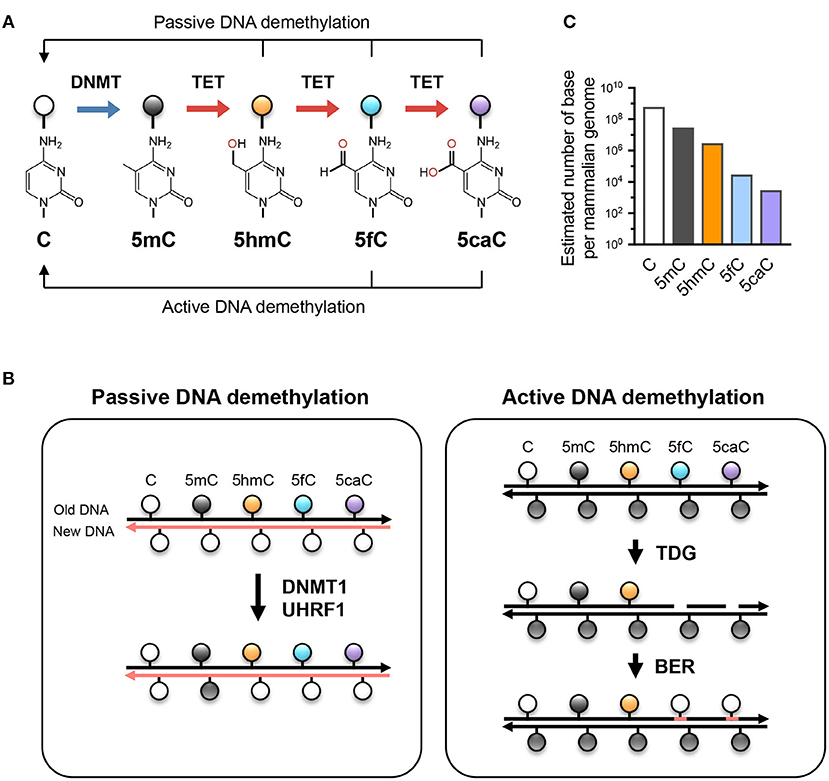
Figure 1. TET-mediated DNA modifications and demethylation. (A) Unmodified cytosine (C) is methylated by DNA methyltransferases (DNMTs) at the 5 position to become 5-methylcytosine (5mC). TET proteins oxidize 5mC into 5-hydroxymethylcytosine (5hmC), a stable epigenetic mark, and subsequently to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). TET can demethylate DNA via replication-dependent (passive) or replication-independent (active) mechanisms. (B) Left, passive DNA demethylation. DNMT1/UHRF1 complex recognizes 5mC at the hemi-methylated CpG motif during DNA replication and methylates the unmodified cytosine on the newly synthesized DNA strand (left; pink strand). However, the oxidized methylcytosines 5hmC, 5fC, and 5caC (together, oxi-mC) are not recognized by DNMT1/UHRF1, resulting in unmodified cytosine on the new DNA strand. Further DNA replication in the presence of continuing TET activity will result in progressive dilution of 5mC in the daughter cells. Right panel, active DNA demethylation. While 5hmC is stable and persists in the genome, 5fC and 5caC can be recognized and excised by thymine DNA glycosylase (TDG), and the resulting abasic sites are repaired as unmodified C by base excision repair (BER). Other mechanisms (e.g., decarboxylation of 5caC) have been suggested but have not yet been proven to exist. (C) The approximate abundance of unmodified and modified cytosines in the haploid human/mouse genome. About 5% of cytosine is methylated (5mC); in most cells, the vast majority of 5mC is present at CG dinucleotides although it is low at CpG islands. 5hmC amounts to about 1-10% of 5mC (estimated at 10% here as in embryonic stem cells), while the levels of 5fC and 5caC are each about an order of magnitude lower than the previous oxidative modification. It is not known whether the low levels of 5fC and 5caC are due to features of TET enzymes that cause them to arrest at 5hmC, or to their continuing removal by TDG or other mechanisms.
DNA modification by TET proteins is essential for gene regulation (Figure 2). TET3 is expressed in the oocyte and the zygote; all three TET proteins are expressed in blastocysts; TET1 and TET2 are expressed in embryonic stem (ES) cells; and TET2 and TET3 are expressed ubiquitously in differentiated cells (3, 4). The three TET enzymes appear to have overlapping but distinct targets in the genome. For instance, in mouse ES cells, TET2 rather than TET1 is responsible for the vast majority of 5hmC generation, and TET1 preferentially facilitates promoter demethylation while TET2 and TET3 act on enhancers (16, 17). The longstanding association of high-level gene transcription with low levels of promoter methylation may be explained by TET-mediated conversion of 5mC to 5hmC at promoters, and subsequent DNA demethylation.
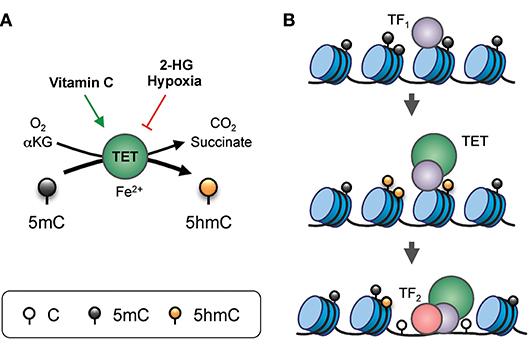
Figure 2. Gene regulation by TET proteins. (A) Enzymatic activity of TET. TET proteins, with the co-factors Fe2+ and α-ketoglutarate (αKG), use oxygen to oxidize 5mC into 5hmC, generating CO2 and succinate as by-products. The enzymatic activity of TET can be modulated by additional factors. For instance, vitamin C (ascorbate) can enhance TET activity, potentially via reduction of the iron ion. On the other hand, the “oncometabolite” 2-hydroxyglutarate (2-HG), generated in acute myeloid leukemia and glioblastoma by recurrent dominant-active mutants of isocitrate dehydrogenase 1 or 2 (IDH1/2), inhibits TET activity. Furthermore, lack of oxygen in hypoxia also inhibits TET function. (B) Model of TET-mediated enhancer regulation. Prior to the commissioning of an enhancer, pioneer transcription factor (indicated as TF1) binds to nucleosomal DNA and recruits TET which oxidizes the surrounding 5mC into 5hmC (and/or other oxi-mCs), facilitating DNA demethylation. TET proteins and, TF1 promote enhancer accessibility by recruiting nucleosome remodeling complexes, thus allowing binding of secondary transcription factors (indicated here as TF2) that are otherwise inhibited by DNA methylation or the presence of nucleosomes.
The genome-wide distribution of 5hmC reflects the strong association of TET enzymes with gene transcription. 5hmC is enriched at the most active enhancers and the gene bodies of the most highly transcribed genes (18). Moreover, multiple transcription factors important in cell differentiation and lineage specification, including NANOG, SALL4A, WT1, EBF1, PU.1, and E2A, have been shown to recruit TET proteins to specific genomic loci (primarily enhancers) for 5hmC modification, in most cases marking them for subsequent demethylation (19–24). As a result, TET function is particularly essential for gene transcription during cell activation and lineage specification, and deficiencies of TET protein expression or activity result in skewed or arrested cell differentiation in multiple lineages, including those in neural and hematopoietic systems (25–30).
TET loss-of-function is strongly connected to oncogenesis (31, 32). Especially in the hematopoietic system, arrested or skewed cell differentiation is often associated with cell transformation (22, 26). In humans, TET2 is one of the most frequently mutated genes in hematopoietic cancers of both myeloid and lymphoid origin (26). Using mouse models, we and other groups have shown that deletion of Tet2 alone, or deletion of both Tet2 and Tet3 (the two TET enzymes with the greatest overlap in expression and function), leads to myeloid or lymphoid expansion and the development of aggressive cancers with 100% penetrance (22, 25, 33). For instance, a striking example is the inducible deletion of both Tet2 and Tet3 in adult mice, which leads to acute myeloid leukemia with the mice succumbing as early as 3 weeks post-deletion (25). Since the role of TET proteins in malignancies has been reviewed extensively (26, 34–36), we will focus here on their roles in immune cell development and function. In the sections below, we outline our current understanding of the roles of TET proteins in regulating the adaptive and innate immune systems. The major findings are summarized in Figures 3, 4.
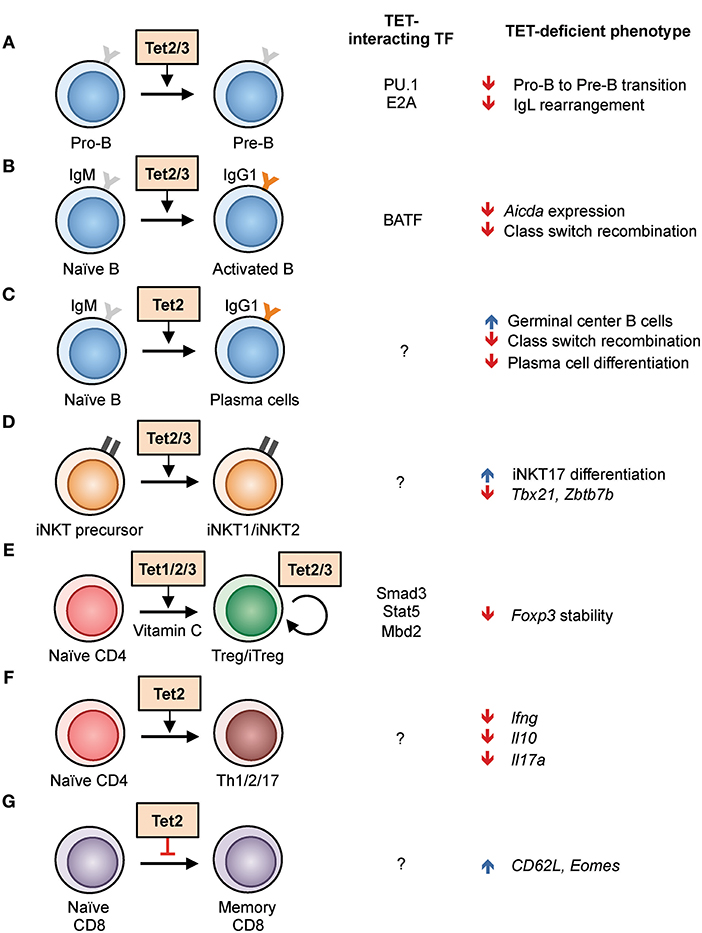
Figure 3. Regulation of lymphoid development and function by TET proteins in the mouse. (A–G) List of known TET functions in lymphoid cells. The interacting transcription factors and the phenotypes found in TET-deficient mice are shown in the right columns. (A) The use of Mb1-cre Tet2/3-deficient mice showed that Tet2 and Tet3 regulate the pro-B to pre-B cell transition, in part by enhancing the rearrangement of immunoglobulin light chains (22, 37). (B) Acute deletion of Tet2/3 using CreERT2 in B cells resulted in decreased Aicda expression and thus class switch recombination (28). (C) Deletion of Tet2 using Vav-Cre and Cd19-Cre resulted in hyperplasia of germinal center B cells. Vav-Cre-driven Tet2 deletion resulted in decreased plasma cell differentiation (38). (D) Cd4-cre Tet2/3-deficient mice exhibited skewed differentiation toward iNKT17 cells, partly due to decreased expression of Tbx21 and Zbtb7b expression, and a massive T-cell-receptor-dependent expansion of affected T cells (33). (E) Tet proteins facilitate the in vitro differentiation of naïve CD4 T cells to iTreg cells by demethylating Foxp3 enhancer CNS2, a process enhanced by the presence of vitamin C. All three TET proteins have a role in stabilizing the expression of Foxp3 in Treg cells in vivo (39, 40). (F) CD4 T cells from Cd2-cre Tet2-deficient mice showed impaired Th1, Th2, and Th17 differentiation and cytokine production (41). (G) Increased differentiation of CD8 memory cells from Cd4-cre Tet2-deficient mice in response to lymphocytic choriomeningitis virus infection (42).
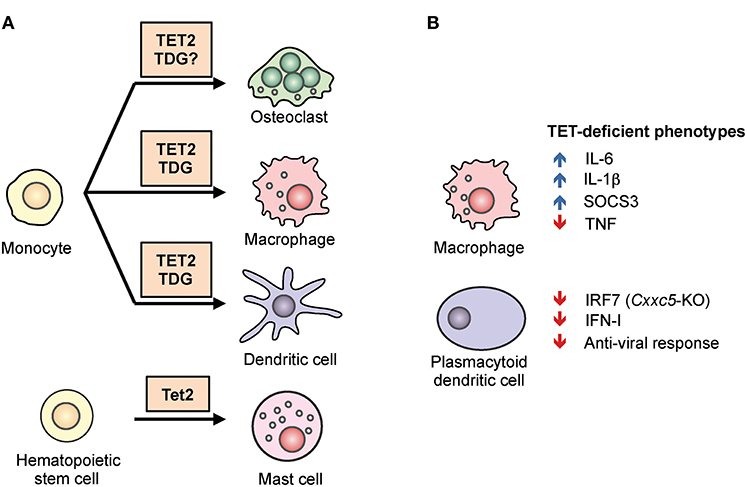
Figure 4. The role of TET2 in myeloid differentiation and function. (A) TET2 regulates myeloid cell differentiation. TET2, together with the thymine DNA glycosylase (TDG), facilitates active DNA demethylation and promotes lineage-specific gene expression during the differentiation of osteoclasts, macrophages, and dendritic cells from human monocytes. In mice, TET2 is required for the differentiation of mast cells in vitro and in vivo (43, 44). (B) TET2 regulates the function of myeloid cells. In mouse and human macrophages, TET2 repressed expression of the pro-inflammatory cytokines IL-1β and IL-6 (45–48). TET2 was shown to associate with Iκbζ, bind to the Il6 promoter, recruit HDAC2, and repress Il6 expression (46). Tet2-deficient macrophages also expressed a high level of Socs3 mRNA, which in normal mice was suggested to be demethylated by TET2 and subsequently degraded by ADAR1 (44). In plasmacytoid dendritic cells, CXXC5 recruited TET2 to an intragenic CpG island in Irf7, facilitating the demethylation and maintaining basal expression. As a result, loss of Cxxc5, and to a lesser extent Tet2, resulted in decreased levels of IRF7, decreased type I interferon (IFN-I) production, and decreased anti-viral responses (49).
TET Proteins in T and B Cell Differentiation and Function
During development and immune responses, T and B cells continuously receive signals from antigen and cytokine receptors. These external signals converge and are interpreted by combinations of ubiquitously expressed and cell type-specific transcription factors, which function together with chromatin regulators to remodel the epigenome. The epigenetic changes associated with immune cell activation and differentiation include DNA and histone modifications, which allow information to be stored and/or inherited by daughter cells. As noted above, analyses of genome-wide 5hmC distribution reveal a close relationship between 5hmC and gene transcription. In thymic and peripheral T cell subsets, the level of 5hmC at gene bodies shows a striking positive correlation with the level of gene expression, as well as occupancy by RNA polymerase II and the level of H3K36me3 histone modification, an epigenetic mark reflective of RNA transcription into the gene body (17, 18, 50, 51). Similarly, in lymphoid cells, 5hmC showed a strong positive correlation with enhancer activity, denoted by the level of H3K27 acetylation (H3K27ac), suggesting that TET is important for regulating enhancer function (16, 18, 52). Indeed, recent studies of T and B cells from our lab (see below) and others demonstrated that one of the functions of TET proteins is to facilitate chromatin accessibility at enhancers (22, 28, 33, 37). TET-mediated conversion of 5mC to 5hmC potentially disrupts the binding of 5mC-binding proteins including MeCP2 and MBD (Methyl-CpG-binding domain) proteins, facilitating nucleosome remodeling and the binding of transcription factors (53, 54). These changes in the epigenetic status of enhancers are likely transmitted to daughter cells, thus facilitating the establishment of lineage identity.
TET Proteins in B Cell Development and Function
From bone marrow progenitors to peripheral memory and plasma cells, the B cell genome undergoes progressive demethylation following differentiation (55). Furthermore, TET2 is one of the most frequently mutated genes (6–12%) in diffuse large B cell lymphoma, a malignancy originating from germinal center B cells (56–58). These observations suggest that TET proteins play an important role in B cell biology. Indeed, when Tet2 and Tet3 were deleted in early B cells during bone marrow development using Mb1-Cre, B cell differentiation was arrested at the transition from the pro-B to the pre-B stage (22, 37) (Figure 3A). One function of these TET proteins during early B cell development is to regulate the arrangement of the Ig kappa (Igκ) light chain genes, that pairs with the rearranged Ig heavy chain to form the complete B cell receptor. TET proteins regulate Igκ rearrangement by oxidizing 5mC at Igκ enhancers and facilitating their DNA demethylation and chromatin accessibility.
Mechanistically, TET proteins appear to be recruited to enhancers by “pioneer” transcription factors, defined by their ability to associate with their binding motifs on nucleosome-bound DNA. Our data indicate that in pro-B cells, the pioneer transcription factor PU.1 binds to the Igκ enhancers prior to light chain rearrangement as a placeholder and recruits Tet proteins for DNA demethylation, facilitating the binding of additional B cell transcription factors including E2A (Figure 3A). Tet proteins also regulate the expression of IRF4 and IRF8, both of which are important for Igκ rearrangement (22). Similar to the expansion phenotype observed in T cells (described in more detail below), Mb1-Cre Tet2/3-deficient mice developed massive expansion of immature B cells resembling acute lymphoblastic lymphoma (22). Therefore, Tet proteins are essential for B cell development by controlling the expression of multiple key genes.
In mature B cells, TET proteins are important for the antibody response. Recently we showed that B cell activation induced Tet protein expression and changes in the genome-wide distribution of 5hmC (the hydroxymethylome): lipopolysaccharide (LPS) and IL-4 stimulation induced a progressive TET-dependent hydroxymethylation at ~8,000 regions in the span of 3 days (28). Functionally, the two major members in naïve B cells TET2 and TET3 are crucial for antibody class switch recombination (CSR) (Figure 3B). Acute deletion of Tet2 and Tet3 by CreERT2 resulted in an ~50% decrease the expression of AID (Activation Induced Deaminase; encoded by Aicda), the critical enzyme for CSR; reconstitution of catalytically active AID in Tet2/3-deficient B cells restored CSR. Interestingly, the CSR phenotype is reminiscent of that resulting from Aicda haploinsufficiency (59, 60), suggesting that TET proteins are required for optimal expression of Aicda. Mechanistically, we showed that the transcription factor BATF recruits Tet proteins to the Aicda superenhancer, facilitating hydroxymethylation and chromatin accessibility of two Tet-responsive elements, TetE1 and TetE2, within the superenhancer and augmenting the expression of Aicda (28) (Figure 3B).
Recently, Vav-Cre and Cd19-Cre, which are expressed in the entire hematopoietic system and during B cell development, respectively, were used to show that disruption of Tet2 resulted in germinal center hyperplasia (38) (Figure 3C). However, germinal center B cells appeared to be normal in Tet2 deletion driven by Cγ1-Cre, which is expressed in germinal center B cells. Consistent with our findings, Tet2 was shown to be required for CSR and affinity maturation of antibody (Figure 3C). More importantly, TET2 positively regulated the expression of the transcription factor Prdm1 (encoding BLIMP1), and plasma cell differentiation was impaired in Tet2-deficient mice. Interestingly, the gene signature of TET2-deficient DLBCL resembles that of cells with mutations in the histone acetyltransferase CREBBP, suggesting that TET2 and CREBBP may cooperate to regulate enhancer H3K27 acetylation. Taken together, these observations demonstrate that TET proteins regulate multiple processes in B cells by preferentially strengthening the activity of enhancers, including individual enhancer elements located within super-enhancers (Igκ and Aicda) (61).
TET Proteins in T Cell Development
Tet2 and Tet3 are expressed at higher levels than Tet1 in thymocytes and peripheral T cells, and are responsible for the majority of 5hmC modification in these cells. Deletion of Tet2 alone in the germline, in the hematopoietic system using Cd2-cre, or in T cells (Cd4-cre) did not lead to any obvious defect in T cell development (41, 42, 62), suggesting that Tet3 was able to compensate for the loss of Tet2. Indeed, data from our lab showed that the deletion of both Tet2 and Tet3 in T cells using Cd4-cre caused a massive lymphoproliferative phenotype with enlarged spleen and lymph nodes, and the mice succumbed by 8 weeks of age (33). At 3–4 weeks of age, young Tet2/3 Cd4-cre DKO mice showed decreased thymic cellularity, a lower percentage of CD4+CD8+ double positive cells, and an increased percentage of CD4+ and CD8+ single positive cells, phenotypes reminiscent of thymic atrophy induced by stress or inflammation. Further examination showed that the expanded cells in the periphery were invariant natural killer T (iNKT or NKT) cells that expressed the transcription factor Rorγt and produced IL-17 (Figure 3D). These cells thus resemble the NKT17 subset, one of the three subsets of NKT cells besides NKT1 (T-bet-expressing) and NKT2 (Gata3-expressing). In contrast, NKT cells from wildtype mice are primarily of the NKT1 and NKT2 subsets (63).
Genome-wide analyses provided mechanistic explanations for the lineage skewing observed in Tet2/3 Cd4-cre DKO mice. Briefly, the profiles of transcriptome, whole-genome methylome, and chromatin accessibility showed that Tet2/3 deficiency resulted in decreased expression of Tbx21 (encoding T-bet) and Zbtb7b (encoding Th-POK), likely because of hypermethylation at the corresponding regulatory elements (Figure 3D). Both T-bet and Th-POK repress Rorc (encoding Rorγt) expression, thus the decreased levels of T-bet and Th-POK transcription factors in Tet2/3-deficient cells permitted increased Rorγt expression and skewed the cells to the NKT17 lineage (33). Interestingly, the Tet2/3-deficient iNKT cells were able to expand upon transfer to fully immunocompetent, wild-type (WT) but not Cd1d-deficient recipient mice (33), suggesting (i) that the expansion was secondary to recognition of “self” antigens presented by CD1d and (ii) that expansion was not suppressible by WT regulatory T (Treg) cells (see below). Together, these observations indicate that TET enzymes are important to maintain the proper expression of lineage-specifying transcription factors, and to limit the differentiation and proliferation of overly self-reactive cells including iNKT cells.
Maintenance of Foxp3+ Treg Cells Requires TET Proteins
TET enzymes are important for the homeostasis of T regulatory (Treg) cells, which are distinguished from other T cell lineages by their expression of the transcription factor FOXP3. In Treg cells, TET2 and TET3 are required for stable Foxp3 expression through their ability to demethylate two intronic enhancers, termed conserved non-coding sequence (CNS) 1 and CNS2 (39, 64) (Figure 3E). Bisulfite sequencing showed that the Foxp3 CNS1 and CNS2 enhancers were hypermethylated in Treg cells from Tet2/3 Cd4-cre DKO mice (39). Moreover, overexpression of the TET1 catalytic domain in CD4 cells induced to differentiate into Foxp3-expressing induced Treg cells (iTreg) in vitro and resulted in partial demethylation of CNS2 (65), suggesting that TET enzymes may be in constant balance with the methylation machinery. Hypermethylation at Foxp3 CNS2 was also observed in Tet1/2-deficient mice, suggesting all three Tet proteins may function redundantly in regulating Foxp3 (40).
Several proteins have been identified to partner with TET proteins in regulating Foxp3 CNS2. For instance, loss of the DNA methyl-binding protein MBD2 also resulted in hypermethylation of CNS2 (also termed TSDR), potentially because of decreased TET2 binding (66). How MBD2 cooperates with TET to demethylate CNS2 remained to be determined. Besides MBD2, the transcription factors SMAD3 and STAT5, induced by TGFβ and IL-2 respectively, recruit TET proteins to Foxp3 CNS2 and facilitate DNA demethylation (40). In addition, the level of TCR and cytokine stimulation has been linked to the degree of DNA demethylation at Foxp3 CNS2 (67). Since there is only one functional allele of Foxp3 per Treg cell, this observation implies that stronger a TCR stimulation might increase the probability of TET-mediated DNA demethylation at Foxp3 CNS2 and concomitantly, the stability of Foxp3 expression.
TET Proteins Link Metabolism to Foxp3 Expression
The enzymatic activity of TET can be influenced by various factors, including the level of co-factors αKG, oxygen, and vitamin C (Figure 2). In a chemical screen using mouse embryonic stem cells, vitamin C was found to enhance the expression of gene expression in germ cells and ES cells by facilitating TET-mediated DNA demethylation at their promoters (68). Vitamin C treatment also facilitated TET-mediated demethylation of Foxp3 CNS2 and stabilized Foxp3 expression in differentiating induced Treg (iTreg) cells (Figure 3E). Inhibition of the vitamin C transporter by sulfinpyrazone confirmed the role of vitamin C and TET proteins in CNS2 demethylation and the generation of peripheral Treg cells in vivo (69). In addition, vitamin C facilitated the conversion of mouse and human naïve CD4 T cells into iTreg cells induced by TGFβ and retinoic acid with improved stability and suppressive function (39). Besides vitamin C, another metabolite hydrogen sulfide (H2S) was shown to be required for Treg cell differentiation, at least in part by increasing Tet1 and Tet2 expression (40).
TET activity can be inhibited by the “oncometabolite” 2-hydroxyglutarate (2-HG), a competitive inhibitor of αKG-dependent dioxygenases including TET (70, 71) (Figure 2). 2-HG is a normal metabolite that exists as two stereoisomers, R-2-HG and S-2-HG; the latter is considerably more potent at inhibiting TET activity (72). In the past few years, it has become clear that 2-HG can be generated via multiple pathways; for instance, recurrent mutations of isocitrate dehydrogenase 1 and 2 (IDH1/2) give rise to dominant-active enzymes with the novel property of converting isocitrate to the R enantiomer of 2-HG (R-2-HG) (70, 71). A recent study identified a compound, (aminooxy)acetic acid (AOA), that is able to reprogram differentiating Th17 cells into Foxp3-expressing iTreg cells (73). Metabolic profiling identified the target of AOA in Th17 cells as GOT1 (glutamate oxaloacetate transaminase 1), an enzyme that catalyzes the conversion of glutamate to αKG. Th17 cells express a high level of GOT1 compared to iTreg cells, consistent with their elevated level of αKG. However, instead of facilitating the function of TET enzymes and other dioxygenases, the αKG is converted by wild-type IDH1/2 into R-2-HG, which inhibits TET activity, promotes increased methylation at Foxp3 CNS2, and represses Foxp3 expression. By targeting GOT1, the small molecule AOA effectively decreased the intracellular level of R-2-HG and allowed TET proteins to demethylate CNS2, favoring differentiation to iTreg cells at the expense of the Th17 lineage (73). Therefore, these observations suggest that, besides conveying signals from cell surface receptors, TET proteins also integrate environmental cues into the epigenome.
TET Proteins Regulate Peripheral T Cell Differentiation and Function
After stimulation and depending on the extracellular signal received, naïve CD4 T cells can differentiate into multiple lineages, including Th1, Th2, Th17, follicular T helper cells (Tfh), and Treg. Analysis of 5hmC distribution in peripheral T cells showed a positive correlation between gene expression level and 5hmC modification at gene bodies, including those of the lineage-specific transcription factor Tbx21 and Gata3 for Th1 and Th2 cells, respectively. This observation suggests that TET proteins may regulate the differentiation of peripheral T cells (18, 41). Similar lineage-specific 5hmC modifications during Th1 and Th2 polarization were also reported in human CD4 T cells (74). Indeed, Tet2-deficient murine CD4 T cells produced less IFNγ and IL-17 when polarized in vitro to Th1 and Th17, respectively (41) (Figure 3F). Compared to WT cells, adoptively transferred Tet2-deficient CD4 T cells were more pathogenic in an experimental autoimmune encephalomyelitis (EAE) model, and immunization with myelin oligodendrocyte glycoprotein (MOG) peptide induced significantly less IFNγ and IL-10 but a similar level of IL-17 (41). These observations reinforce the idea that Tet proteins are important for proper lineage differentiation and gene expression.
Analysis of Tet2-deficient (Tet2fl/fl Cd4-cre) CD8 T cells responding to infection with lymphocytic choriomeningitis virus (LCMV) showed increased LCMV gp33-specific memory precursor cells (KLRG1− CD127+) and decreased short-lived effector cells (KLRG1+ CD127−) on day 8 post-infection (42) (Figure 3G). These memory-like cells expressed CD27, CD62L, and CXCR3, a phenotype similar to central memory cells, and persisted for at least 45 days post-infection with a higher level of Eomes compared to WT. Transfer of Tet2-deficient memory cells conferred better protection against gp33-expressing Listeria monocytogenes compared to WT memory cells, strongly suggesting that TET2 represses memory cell differentiation (42). In addition to TCR-induced TET protein expression (42), TET activity can also be modulated by physiologically produced 2-HG. CD8 T cells generate substantial levels of the potent 2-HG enantiomer “oncometabolite” S-2-HG as early as day two after TCR stimulation, coinciding with the decrease in 5hmC (75). Similar to genetic ablation of Tet2, S-2-HG treatment of CD8 T cells induced higher expression of Eomes and CD62L, markers for central memory cells. Surprisingly, OT-I CD8 T cells cultured in the presence of S-2-HG in vitro displayed enhanced survival and tumor clearance upon adoptive transfer in vivo (75), suggesting the effect of S-2-HG is long lasting by stably remodeling the epigenome.
In humans, TET loss-of-function was shown to have a major potentiating role in a case of cancer immunotherapy against B cell malignancy using T cells bearing the anti-CD19 chimeric antigen receptor (CAR). The patient bore a hypomorphic mutation in one allele of TET2, and the CAR lentivirus serendipitously became integrated into the other TET2 allele. The resulting profound loss of function of TET2 resulted in an almost monoclonal expansion of this particular CAR-T cell, and the patient went into complete remission (76). Thus the loss of TET2 activity resulting from insertional mutation of one TET2 allele due to lentiviral integration, combined with the preexisting hypomorphic mutation in the other TET2 allele, led to superior anti-tumor function and again conferred a central memory phenotype on the expanded CAR-T cells. Together, these observations show that TET proteins are important in regulating peripheral T cell differentiation.
TET Proteins in Myeloid Differentiation and Function
TET2 Regulates Myeloid Differentiation
TET2 mutation has been closely linked to myeloid malignancies including myelodysplastic syndrome and acute myeloid leukemia in human (26). In mice, germline disruption of the Tet2 gene decreased the global level of 5hmC in hematopoietic stem cells (HSCs), enhanced HSC survival and proliferation, inhibited T, B, and erythroid differentiation, and biased differentiation toward the myeloid lineage (62). Similarly, knockdown of TET2 in human cord blood CD34+ progenitor cells decreased total 5hmC in the cells and skewed their differentiation toward the granulomonocytic lineage, specifically monocytes, at the expense of both lymphoid and erythroid lineages (77). These and other studies suggested that, compared to other lineages, the myeloid lineage requires less reconfiguration of the DNA methylome during differentiation and therefore is relatively unaffected in the absence of TET2.
Beyond HSC, TET2 also regulates the differentiation of mast cells (Figure 4A). In a model of in vitro mast cell differentiation in which bone marrow progenitors were cultured with IL-3, loss of Tet2 inhibited mast cell differentiation, decreased cytokine production, and induced aberrant hyperproliferation (43). Two major transcription factors involved in myeloid development, C/EBPα and C/EBPε, were up-regulated in Tet2-deficient mast cells, and both contributed to the observed defect in differentiation. Similar to another observation in macrophages (see below), both catalytically active and inactive TET2 could partially rescue these phenotypic defects, suggesting that part of the function of TET2 is to maintain the structure of a repressive protein complex (43). In vivo, Tet2 is important for the expansion of mast cells induced by parasites (44).
Human monocytes can differentiate into macrophages (MACs), dendritic cells (DCs), and osteoclasts (OCs) in vitro depending on cytokine signals, and the epigenetic regulation of this process has been studied extensively (78). During post-mitotic differentiation of DCs from monocytes, stimulation with cytokines GM-CSF and IL-4 induced DNA demethylation. Since these cells do not proliferate prior to differentiation, the mechanism of demethylation is assumed to involve an active replication-independent process. Similar observations were made during MAC and OC differentiation (20, 79–81). TET2-mediated oxidation of 5mC into 5hmC preceded and was required for DNA demethylation, which was accompanied by the presence of active histone modifications (H3K4me1, H3K4me3, H3/H4 acetylation) (Figure 4A). In general, the degree of DNA demethylation at distal elements or promoters showed a loose positive correlation with gene expression with numerous exceptions, suggesting that additional mechanisms contributed to gene regulation, such as H3K27 methylation by the polycomb complex (82). In monocyte to DC differentiation, IL-4-activated STAT6 induced TET2-dependent demethylation, and this was important for acquiring the proper cell identity and priming the expression of inducible genes (e.g., IL1B, CCL20) (81). During monocyte to OC or to MAC differentiation, the transcription factor PU.1 was found to associate with both hypo- and hypermethylated regions and to directly bind to TET2 as well as to the DNA methyltransferase DNMT3B (20). TET2 functioned together with thymine-DNA glycosylase (TDG), and to a lesser extent with activation-induced deaminase (AID), to hydroxymethylate and demethylate DNA, facilitating the establishment of cell-type-specific gene expression programs (83). The same study also showed that TET2 was responsible for recruiting the histone H3K4 methyltransferase SETD1A, and for increasing H3K4me3 modification at cell-type specific genes examined (83).
Together, these in vitro human studies showed that post-mitotic myeloid cells utilize TET2 and TDG for replication-independent, active DNA demethylation to establish cell-specific gene expression patterns or to prime gene for subsequent induction. Besides regulating lymphoid development, Tet proteins are required for the differentiation of multiple lineages of myeloid cells.
TET Proteins Regulate Immune Responses by Myeloid Cells
One function of TET proteins in normal myeloid cells appears to be the repression of inflammatory gene expression (Figure 4B). For instance, Tet2-deficient macrophages and dendritic cells expressed a higher level of IL-6 in response to stimulation (45, 46). Mechanistically, TET2 was shown to associate with Iκbζ and bind to the Il6 promoter, recruiting the histone deacetylase HDAC2 and repressing Il6 expression. As discussed for mast cells above, the repression appeared to be independent of TET2 catalytic activity, suggesting that TET2 provided a structural scaffold for the formation of a repressive complex (46). Compared to WT controls, Tet2-deficient mice were more susceptible to endotoxin-induced septic shock and dextran sulfate sodium (DSS)-induced colitis, coincident with an increased IL-6 level (46).
TET proteins also repressed another inflammatory cytokine, IL-1β (47, 48). Moreover, loss of Tet2 accelerated atherosclerosis development in a mouse model of low-density lipoprotein receptor (Ldrl) deficiency (47). Tet2-deficient macrophages increased IL-1β secretion via the NLRP3 inflammasome, the inhibition of which protects mice from atherosclerosis (47). Interestingly, IL-1R/MyD88 signaling was shown to induce Tet2 mRNA and protein expression in bone marrow-derived macrophages (84), suggesting a potential negative feedback loop controlling IL-1β expression by TET proteins. Lastly, TET2 facilitated immunosuppression by tumor-infiltrating myeloid cells in a melanoma model and loss of TET2 in myeloid cells inhibited melanoma growth in vivo (84), consistent with the role of TET proteins in suppressing inflammation in myeloid cells. TET proteins contribute to osteoclast differentiation and suppress inflammation, and osteoclast activation has been linked to rheumatoid arthritis (RA) (85), warranting further detailed investigation of the role of TET proteins in autoimmune and auto-inflammatory diseases.
TET proteins appear to have different functions in myeloid cells depending on the circumstances. For instance, TET proteins have been reported to promote myeloid immune responses and production of inflammatory cytokines rather than suppressing inflammation. Plasmacytoid dendritic cells (pDCs) are fast responders to infection and are able to produce a large quantity of type I interferon. This ability has previously been attributed to their high basal level of the transcription factor IRF7, the expression of which is regulated by an intronic CpG island (CGI) (49). TET2 is recruited to this locus by the zinc-finger protein CXXC5, and is required to maintain the demethylated status of the CGI (Figure 4B). As a result, mice deficient in Cxxc5, or to a lesser extent Tet2, were more vulnerable to infection by herpes simplex virus and vesicular stomatitis virus due to an impaired interferon response (49). Similarly, in a model of abdominal sepsis, Tet2 deficiency was shown to reduce infection-induced myelopoiesis with a decreased level of TNFα and chemokines (44). The authors suggested that instead of oxidizing DNA, TET2 repressed Socs3 expression by oxidizing methylcytosine in the 3′ untranslated region of Socs3 RNA, thereby facilitating ADAR1-mediated destabilization of the mRNA in a manner independent of the normal RNA-editing function of ADAR1 (44) (Figure 4B). Although TET proteins are capable of oxidizing methylcytosine on RNA (86, 87), whether TETs can demethylate RNA (i.e., replace 5mC with unmodified C) is still an open question as neither passive nor active mechanisms for DNA demethylation would apply in RNA (Figure 1).
Finally, it is worth noting that the phenotypes in Tet2-deficient mice may be complicated due to environmental influences. Whole-body Tet2 deficiency was shown to result in a compromised intestinal barrier, allowing bacteria to translocate from the intestinal lumen to internal organs and induce IL-6 production and inflammation; in turn, the pro-inflammatory signal facilitated pre-leukemic myeloproliferation (88). Therefore, depending on the microbiota at a given facility, Tet2-deficient mice may display differing basal levels of inflammation, a feature that may account for the variable reported phenotypes of different strains of Tet2-deficient mice (26). Since most TET2 mutations in human are acquired somatically rather than through the germline, the extent to which inflammation plays a role in human myeloid neoplasms remains to be determined. Taken together, these studies provide clear evidence that TET proteins regulate innate immune responses in myeloid cells.
Our Current Understanding of TET-Mediated Gene Regulation
TET Regulation of Transcription Factor Expression in Immune System
Transcription factors have emerged as one of the major targets of TET-mediated regulation. For instance, TET2 is important for inducing Blimp1 expression in peripheral B cells by demethylating intronic CpGs (38). On the other hand, TET proteins may be required for repressing BCL6 expression. In the human BCL6 locus, DNA methylation of intragenic CpG islands at the first intron prevents CTCF binding and promotes BCL6 expression. DNA demethylation at these CpG islands allowed CTCF binding, resulting in repressed BCL6 expression (89). However, whether TET proteins regulate BCL6 expression remains to be demonstrated.
Many loci encoding transcription factors are heavily hydroxymethylated, including Tbx21, Zbtb7b, and Gata3 in iNKT and T cells (18, 33, 41). Loss of Tet2 alone, however, has no significant effect on Tbx21 expression in CD4 and CD8 T cells (41, 42). It is likely that other TET proteins such as TET3 can compensate, since Tbx21 expression is decreased in iNKT cells that are deficient in both Tet2 and Tet3 (33). In contrast to Tbx21 which is decreased in TET-deficient iNKT cells, loss of TET activity, either by gene targeting or inhibition by 2-HG, facilitates Eomes expression in iNKT and CD8 T cells (33, 42, 75). Whether TET proteins directly regulate Tbx21 and Eomes expression by binding to regulatory elements in the Tbx21 and Eomes loci remains to be determined.
TET-Mediated Regulation of Enhancers
Consistent with the functions of TET proteins in gene regulation, enhancers are usually enriched in 5hmC. TET proteins can be recruited to specific regulatory elements through interaction with multiple transcription factors including NANOG, SALL4A, WT-1, PU.1, E2A, and EBF1 (19–24). The pleiotropic interaction between TET proteins and transcription factors is reminiscent of histone acetyltransferase p300, which interacts with hundreds of transcription factors (90). Once recruited to enhancers, TET proteins can oxidize 5mC into 5hmC, marking enhancers for DNA demethylation.
TET-dependent DNA modifications potentially affect gene expression via at least two non-mutually exclusive mechanisms. First, 5hmC, other oxi-mCs, and the ensuing DNA demethylation increase chromatin accessibility (22, 28, 33). In this scenario, unmodified C and oxi-mC potentially relieve the nucleosome rigidity caused by DNA methylation (91, 92); additionally, TET proteins may recruit nucleosome remodeling complexes to displace nucleosomes from enhancers. Second, TET-generated oxi-mC modifications may exert immediate effects on gene expression by modulating transcription factor binding, and TET proteins may also exert more long-term effects. Specifically, 5mC and oxi-mCs are known to modify the binding of several transcription factors with CG or TG dinucleotides in their recognition sequences (54). The methyl group of thymine is located at the 5th position, corresponding to the methyl group of 5mC. Thus, transcription factors with TG dinucleotides in their preferred binding sequences often also bind the same sequences with methylated CGs (93), and their DNA binding is likely to be modified by the presence of oxi-mCs. Other transcription factors, including WT1, can bind sequences containing 5caC in a CG context with higher affinity than the corresponding sequence with unmodified CG (94). The exact mechanisms of enhancer regulation by TET enzymes and oxi-mCs remain to be delineated.
TET-Mediated DNA Oxidation and Demethylation
TET proteins can oxidize 5mC into oxi-mCs and mediate DNA demethylation. Depending on the conditions, TET2 can iteratively oxidize 5mC to 5hmC and then to all other oxidized cytosines in a single encounter (95). However, in the genome, most 5mC oxidation appears to pause at 5hmC and to a lesser extent 5fC (Figure 1C), a notion supported by mass spectrometric analyses showing that both 5hmC and 5fC are rather stable in cells (96, 97). It remains to be determined why 5hmC is the most abundant of the oxi-mCs. Two mechanisms (not mutually exclusive) may be involved: (i) TET-mediated oxidation preferentially arrests at 5hmC or 5fC; (ii) 5fC and 5caC, but not 5hmC, are continuously removed by TDG/ BER or by other mechanisms (Figure 1B). Regardless of the mechanism, the modified cytosines can facilitate active or passive demethylation and affect gene regulation. In addition, 5hmC may act as a bookmark to label CpG sites in cis-elements such as promoters, enhancers and insulators marked by CTCF binding (5, 98, 99) for subsequent demethylation upon cell division, thus affecting gene expression patterns in the daughter cells (a latent effect).
Potential Co-transcriptional 5hmC Modification
5hmC distribution at gene bodies is positively correlated with gene expression levels, suggesting that TET activity is coupled to transcription by RNA polymerase II (RNA pol II) (18). One of the possible links between TET and RNA pol II is via their mutual association with the histone H3K4 methyltransferase Set1/COMPASS complex (100). Another possible link between 5hmC and RNA transcription is via the gene body histone mark H3K36me3: the levels of 5hmC and H3K36me3 in gene bodies are positively correlated with one another and with gene expression. During transcription, the methyltransferase SETD2 associates with the phosphorylated C-terminal domain of RNA pol II and co-transcriptionally methylates H3K36 to yield H3K36me3 (94). H3K36me3 is subsequently recognized by the de novo DNA methyltransferases DNMT3B, and to a lesser extent DNMT3A, via the PWWP domain (101–103), mediating gene body DNA methylation. Since all three TET proteins have been shown to co-immunoprecipitate with the maintenance methyltransferase DNMT1, and all three DNMT proteins co-immunoprecipitate with TET2 (104), the extensive interaction between TET and DNMT may provide a possible mechanism for transcription-coupled 5hmC modification. The biological significance of gene body 5hmC modification remains to be determined.
Potential Model for TET-Mediated Asymmetric Cell-Fate Decision
Hypothetically, it may also be possible to facilitate asymmetric gene regulation by engineering an asymmetric distribution of DNA methylation between two daughter cells via strand-biased 5hmC modifications. In one potential scenario, 5mC bases at CpG motifs on one strand at a given locus are preferentially oxidized by TET into 5hmC, while the complementary strand remains as 5mC (e.g., the template strand during transcription). As a result, after cell division, the CpG motifs at the locus in one of the daughter cell will remain methylated because the DNMT1/UHRF1 complex restores symmetrical methylation; the CpG motifs in the other daughter cell will contain 5hmC and unmodified C. This is an attractive putative mechanism by which TET enzymes could regulate cell fate decisions.
Harnessing the Power of the Dark Side for the Light Side
TET loss-of-function, either through genetic mutations or catalytic inhibition, has shown a strong causal relationship with multiple malignancies (31, 32). TET deficiency appears to enhance cell survival and increase “stemness,” as in the case of TET-deficient HSCs which could be passaged for a much longer period of time in vitro and out-competed WT HSCs after transplantation in vivo. Interestingly, at least some of the phenotypes are reversible by re-introducing TET or enhancing the remaining TET activity by vitamin C (105), raising the possibility of temporarily inhibiting TET activity to enhance immune responses. In fact, two recent studies of human and mouse CD8 T cells provided supporting evidence for this approach. In both cases, TET2-deficiency facilitated the differentiation and expansion of CD8 T cells with central memory phenotype that could provide long-lasting protection against tumor and virus (discussed above). Using non-specific inhibitors such as the oncometabolite 2-HG or other TET-specific inhibitors that remain to be developed, it should be possible to inhibit TET activity and boost antigen-specific responses and immune cell expansion during vaccination or infusion of cancer-specific T cells. It would be of great interest to borrow the trick of losing TET function from cancer cells to arm immune cells with the superpower to fight against the cancer cells themselves and other pathogens.
Concluding Remarks
TET proteins and 5hmC were identified/rediscovered almost 10 years ago. Numerous studies have shown their importance in gene regulation, tumor suppression, and cell differentiation. Yet, much remains to be learned about TET and 5hmC. For instance, how do TET enzymes suppress cancer progression? How does TET-mediated DNA modification affect cell identity? What is the relative contribution of enzymatic activity-dependent and –independent (structural) mechanisms to the functions of TET? Besides being intermediates for DNA demethylation, what is the function of 5hmC and other oxidized methylcytosines as potential epigenetic marks? Who are the “readers” of these epigenetic marks? Also, given their seemingly opposite functions, why do mutations of Tet and Dnmt3a/b result in similar phenotypes in hematopoiesis? Besides all these fundamental questions, modulating the activity of epigenetic regulating enzymes including TET proteins may provide a promising way to alter and to achieve the desired magnitude and direction of immune responses.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH grants R35 CA210043, AI109842, AI128589 and Translation Research Project grants (6187-12 and 6464-15) from the Leukemia and Lymphoma Society (to AR). C-WL was supported by the Independent Investigator Fund (Kyowa Hakko Kirin/La Jolla Institute) and an Irvington Postdoctoral Fellowship from the Cancer Research Institute.
Conflict of Interest Statement
AR is a member of the Scientific Advisory Board of Cambridge Epigenetix.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Xiaojing Yue for discussion and critical reading of the manuscript.
References
1. Lister, R., Pelizzola, M., Dowen, R.H., Hawkins, R.D., Hon, G., Tonti-Filippini, J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322. doi: 10.1038/nature08514
2. Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. (2018) 19:81–92. doi: 10.1038/nrg.2017.80
3. Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. (2013) 14:341–56. doi: 10.1038/nrm3589
4. WuX Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. (2017) 18:517–34. doi: 10.1038/nrg.2017.33
5. Xu C, Corces VG. Nascent DNA methylome mapping reveals inheritance of hemimethylation at CTCF/cohesin sites. Science (2018) 359:1166–70. doi: 10.1126/science.aan5480
6. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324:930–5. doi: 10.1126/science.1170116
7. Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science (2009) 324:929–30. doi: 10.1126/science.1169786
8. Wyatt GR, Cohen SS. The bases of the nucleic acids of some bacterial and animal viruses: the occurrence of 5-hydroxymethylcytosine. Biochem J. (1953) 55:774–82. doi: 10.1042/bj0550774
9. Iyer LM, Abhiman S, de Souza RF, Aravind L. Origin and evolution of peptide-modifying dioxygenases and identification of the wybutosine hydroxylase/hydroperoxidase. Nucleic Acids Res. (2010) 38:5261–79. doi: 10.1093/nar/gkq265
10. Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle (2009) 8:1698–710. doi: 10.4161/cc.8.11.8580
11. Moroz LL, Kocot KM, Citarella MR, Dosung S, Norekian TP, Povolotskaya IS, et al. The ctenophore genome and the evolutionary origins of neural systems. Nature (2014) 510:109–14. doi: 10.1038/nature13400
12. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science (2011) 333:1300–3. doi: 10.1126/science.1210597
13. Dalton SR, Bellacosa A. DNA demethylation by TDG. Epigenomics (2012) 4:459–67. doi: 10.2217/epi.12.36
14. Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature (2013) 502:472–9. doi: 10.1038/nature12750
15. Otani J, Kimura H, Sharif J, Endo TA, Mishima Y, Kawakami T, et al. Cell cycle-dependent turnover of 5-hydroxymethyl cytosine in mouse embryonic stem cells. PLoS ONE (2013) 8:e82961. doi: 10.1371/journal.pone.0082961
16. Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell (2014) 56:286–97. doi: 10.1016/j.molcel.2014.08.026
17. Huang Y, Chavez L, Chang X, Wang X, Pastor WA, Kang J, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Nat Acad Sci USA. (2014) 111:1361–6. doi: 10.1073/pnas.1322921111
18. Tsagaratou A, Aijo T, Lio CW, Yue X, Huang Y, Jacobsen SE, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Nat Acad Sci USA. (2014) 111:E3306–3315. doi: 10.1073/pnas.1412327111
19. Costa Y, Ding J, Theunissen TW, Faiola F, Hore TA, Shliaha PV, et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature (2013) 495:370–4. doi: 10.1038/nature11925
20. de la Rica L, Rodriguez-Ubreva J, Garcia M, Islam AB, Urquiza JM, Hernando H, et al. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. (2013) 14:R99. doi: 10.1186/gb-2013-14-9-r99
21. Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, et al. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun. (2013) 4:2166. doi: 10.1038/ncomms3166
22. Lio CJ, Zhang J, Gonzalez-Avalos E, Hogan PG, Chang X, Rao A. Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. Elife (2016) 5:e18290. doi: 10.7554/eLife.18290
23. Wang Y, Xiao M, Chen X, Chen L, Xu Y, Lv L, et al. WT1 recruits TET2 to regulate its target gene expression and suppress leukemia cell proliferation. Mol Cell (2015) 57:662–73. doi: 10.1016/j.molcel.2014.12.023
24. Xiong J, Zhang Z, Chen J, Huang H, Xu Y, Ding X, et al. Cooperative action between SALL4A and TET proteins in stepwise oxidation of 5-methylcytosine. Mol Cell (2016) 64:913–25. doi: 10.1016/j.molcel.2016.10.013
25. An J, Gonzalez-Avalos E, Chawla A, Jeong M, Lopez-Moyado IF, Li W, et al. Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun. (2015) 6:10071. doi: 10.1038/ncomms10071
26. Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers. Immunol Rev. (2015) 263:6–21. doi: 10.1111/imr.12239
27. Li X, Yue X, Pastor WA, Lin L, Georges R, Chavez L, et al. Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc Nat Acad Sci USA. (2016) 113:E8267–76. doi: 10.1073/pnas.1617802113
28. Lio C-WJ, Shukla V, Samaniego-Castruita D, González-Avalos E, Chakraborty A, Yue X, et al. TET enzymes augment AID expression via 5hmC modifications at the Aicda superenhancer (2018). bioRxiv. doi: 10.1101/438531
29. Madzo J, Liu H, Rodriguez A, Vasanthakumar A, Sundaravel S, Caces DBD, et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. (2014) 6:231–44. doi: 10.1016/j.celrep.2013.11.044
30. Santiago M, Antunes C, Guedes M, Sousa N, Marques CJ. TET enzymes and DNA hydroxymethylation in neural development and function—How critical are they? Genomics (2014) 104:334–40. doi: 10.1016/j.ygeno.2014.08.018
31. Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. (2014) 30:464–74. doi: 10.1016/j.tig.2014.07.005
32. Ko M, An J, Rao A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr Opin Cell Biol. (2015) 37:91–101. doi: 10.1016/j.ceb.2015.10.009
33. Tsagaratou A, Gonzalez-Avalos E, Rautio S, Scott-Browne JP, Togher S, Pastor WA, et al. TET proteins regulate the lineage specification and TCR-mediated expansion of iNKT cells. Nat Immunol. (2016) 18:45–53. doi: 10.1038/ni.3630
34. Bowman RL, Levine RL. TET2 in normal and malignant hematopoiesis. Cold Spring Harb Perspect Med. (2017) 7:a026518. doi: 10.1101/cshperspect.a026518.
35. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. (2016) 30:733–50. doi: 10.1101/gad.276568.115
36. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer (2012) 12:599–612. doi: 10.1038/nrc3343
37. Orlanski S, Labi V, Reizel Y, Spiro A, Lichtenstein M, Levin-Klein R, et al. Tissue-specific DNA demethylation is required for proper B-cell differentiation and function. Proc Nat Acad Sci USA. (2016) 113:5018–23. doi: 10.1073/pnas.1604365113
38. Dominguez PM, Ghamlouch H, Rosikiewicz W, Kumar P, Beguelin W, Fontan L, et al. TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Dis. (2018) 8:1632–53. doi: 10.1158/2159-8290.CD-18-0657
39. Yue X, Trifari S, Aijo T, Tsagaratou A, Pastor WA, Zepeda-Martinez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med. (2016) 213:377–97. doi: 10.1084/jem.20151438
40. Yang R, Qu C, Zhou Y, Konkel JE, Shi S, Liu Y, et al. Hydrogen sulfide promotes Tet1- and Tet2-mediated Foxp3 demethylation to drive regulatory T cell differentiation and maintain immune homeostasis. Immunity (2015) 43:251–63. doi: 10.1016/j.immuni.2015.07.017
41. Ichiyama K, Chen T, Wang X, Yan X, Kim BS, Tanaka S, et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity (2015) 42:613–26. doi: 10.1016/j.immuni.2015.03.005
42. Carty SA, Gohil M, Banks LB, Cotton RM, Johnson ME, Stelekati E, et al. The loss of TET2 promotes CD8+ T cell memory differentiation. J Immunol. (2017) 200:82–91. doi: 10.4049/jimmunol.1700559
43. Montagner S, Leoni C, Emming S, Della Chiara G, Balestrieri C, Barozzi I, et al. TET2 regulates mast cell differentiation and proliferation through catalytic and non-catalytic activities. Cell Rep. (2016) 15:1566–79. doi: 10.1016/j.celrep.2016.04.044
44. Shen Q, Zhang Q, Shi Y, Shi Q, Jiang Y, Gu Y, et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature (2018) 554:123–7. doi: 10.1038/nature25434
45. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. (2017) 55:56–70.e13. doi: 10.1016/j.exphem.2017.08.001
46. Zhang Q, Zhao K, Shen Q, Han Y, Gu Y, Li X, et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature (2015) 525:389–93. doi: 10.1038/nature15252
47. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (2017) 355:842–7. doi: 10.1126/science.aag1381
48. Neves-Costa A, Moita LF. TET1 is a negative transcriptional regulator of IL-1beta in the THP-1 cell line. Mol Immunol. (2013) 54:264–70. doi: 10.1016/j.molimm.2012.12.014
49. Ma S, Wan X, Deng Z, Shi L, Hao C, Zhou Z, et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J Exp Med. (2017) 214:1471–91. doi: 10.1084/jem.20161149
50. Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. (2013) 3:291–300. doi: 10.1016/j.celrep.2013.01.011
51. Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. (2011) 25:679–84. doi: 10.1101/gad.2036011
52. Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. (2011) 12:R54. doi: 10.1186/gb-2011-12-6-r54
53. Du Q, Luu P-L, Stirzaker C, Clark SJ. Methyl-CpG-binding domain proteins: readers of the epigenome. Epigenomics (2015) 7:1051–73. doi: 10.2217/epi.15.39
54. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. (2016) 17:551–65. doi: 10.1038/nrg.2016.83
55. Kulis M, Merkel A, Heath S, Queiros AC, Schuyler RP, Castellano G, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. (2015) 47:746–56. doi: 10.1038/ng.3291
56. Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica (2013) 98:1912–20. doi: 10.3324/haematol.2013.088740
57. Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell (2017) 171:481–94.e15. doi: 10.1016/j.cell.2017.09.027
58. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
59. Sernandez IV, de Yebenes VG, Dorsett Y, Ramiro AR. Haploinsufficiency of activation-induced deaminase for antibody diversification and chromosome translocations both in vitro and in vivo. PLoS ONE (2008) 3:e3927. doi: 10.1371/journal.pone.0003927
60. Takizawa M, Tolarova H, Li Z, Dubois W, Lim S, Callen E, et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. (2008) 205:1949–57. doi: 10.1084/jem.20081007
61. Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon K-R, Resch W, et al. B cell super-enhancers and regulatory clusters recruit aid tumorigenic activity. Cell (2014) 159:1524–37. doi: 10.1016/j.cell.2014.11.013
62. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Nat Acad Sci USA. (2011) 108:14566–71. doi: 10.1073/pnas.1112317108
63. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol. (2013) 14:1146–54. doi: 10.1038/ni.2731
64. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature (2010) 463:808–12. doi: 10.1038/nature08750
65. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda KI, Akanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol. (2017) 29:365–75. doi: 10.1093/intimm/dxx049
66. Wang L, Liu Y, Han R, Beier UH, Thomas RM, Wells AD, et al. Mbd2 promotes foxp3 demethylation and T-regulatory-cell function. Mol Cell Biol. (2013) 33:4106–15. doi: 10.1128/MCB.00144-13
67. Wakamatsu E, Omori H, Kawano A, Ogawa S, Abe R. Strong TCR stimulation promotes the stabilization of Foxp3 expression in regulatory T cells induced in vitro through increasing the demethylation of Foxp3 CNS2. Biochem Biophy Res Commun. (2018) 503:2597–602. doi: 10.1016/j.bbrc.2018.07.021
68. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martinez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature (2013) 500:222–6. doi: 10.1038/nature12362
69. Sasidharan Nair V, Song MH, Oh KI. Vitamin C facilitates demethylation of the Foxp3 enhancer in a tet-dependent manner. J Immunol. (2016) 196:2119–31. doi: 10.4049/jimmunol.1502352
70. Dang L, Su S-SM. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: from basic discovery to therapeutics development. Ann Rev Biochem. (2017) 86:305–31. doi: 10.1146/annurev-biochem-061516-044732
71. Ye D, Guan KL, Xiong Y. Metabolism, activity, and targeting of D- and L-2-hydroxyglutarates. Trends Cancer (2018) 4:151–65. doi: 10.1016/j.trecan.2017.12.005
72. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell (2011) 19:17–30. doi: 10.1016/j.ccr.2010.12.014
73. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature (2017) 548:228–33. doi: 10.1038/nature23475
74. Nestor CE, Lentini A, Hagg Nilsson C, Gawel DR, Gustafsson M, Mattson L, et al. 5-Hydroxymethylcytosine remodeling precedes lineage specification during differentiation of human CD4(+) T Cells. Cell Rep. (2016) 16:559–70. doi: 10.1016/j.celrep.2016.05.091
75. Tyrakis PA, Palazon A, Macias D, Lee KL, Phan AT, Velica P, et al. S-2-hydroxyglutarate regulates CD8(+) T-lymphocyte fate. Nature (2016) 540:236–41. doi: 10.1038/nature20165
76. Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature (2018) 558:307–12. doi: 10.1038/s41586-018-0178-z
77. Pronier E, Almire C, Mokrani H, Vasanthakumar A, Simon A, da Costa Reis Monte Mor B, et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood (2011) 118:2551–5. doi: 10.1182/blood-2010-12-324707
78. Alvarez-Errico D, Vento-Tormo R, Sieweke M, Ballestar E. Epigenetic control of myeloid cell differentiation, identity and function. Nat Rev Immunol. (2015) 15:7–17. doi: 10.1038/nri3777
79. Klug M, Heinz S, Gebhard C, Schwarzfischer L, Krause SW, Andreesen R, et al. Active DNA demethylation in human postmitotic cells correlates with activating histone modifications, but not transcription levels. Genome Biol. (2010) 11:R63. doi: 10.1186/gb-2010-11-6-r63
80. Klug M, Schmidhofer S, Gebhard C, Andreesen R, Rehli M. 5-Hydroxymethylcytosine is an essential intermediate of active DNA demethylation processes in primary human monocytes. Genome Biol. (2013) 14:R46. doi: 10.1186/gb-2013-14-5-r46
81. Vento-Tormo R, Company C, Rodriguez-Ubreva J, de la Rica L, Urquiza JM, Javierre BM, et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. (2016) 17:4. doi: 10.1186/s13059-015-0863-2
82. Wiles ET, Selker EU. H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev. (2017) 43:31–7. doi: 10.1016/j.gde.2016.11.001
83. Garcia-Gomez A, Li T, Kerick M, Catala-Moll F, Comet NR, Rodriguez-Ubreva J, et al. TET2- and TDG-mediated changes are required for the acquisition of distinct histone modifications in divergent terminal differentiation of myeloid cells. Nucleic Acids Res. (2017) 45:10002–17. doi: 10.1093/nar/gkx666
84. Pan W, Zhu S, Qu K, Meeth K, Cheng J, He K, et al. The DNA methylcytosine dioxygenase Tet2 sustains immunosuppressive function of tumor-infiltrating myeloid cells to promote melanoma progression. Immunity (2017) 47:284–97.e5. doi: 10.1016/j.immuni.2017.07.020
85. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
86. Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science (2016) 351:282–5. doi: 10.1126/science.aac5253
87. Guallar D, Bi X, Pardavila JA, Huang X, Saenz C, Shi X, et al. RNA-dependent chromatin targeting of TET2 for endogenous retrovirus control in pluripotent stem cells. Nat Genet. (2018) 50:443–51. doi: 10.1038/s41588-018-0060-9
88. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z
89. Lai AY, Fatemi M, Dhasarathy A, Malone C, Sobol SE, Geigerman C, et al. DNA methylation prevents CTCF-mediated silencing of the oncogene BCL6 in B cell lymphomas. J Exp Med. (2010) 207:1939–50. doi: 10.1084/jem.20100204
90. Dyson HJ, Wright PE. Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding Protein (CBP) and p300. J Biol Chem. (2016) 291:6714–22. doi: 10.1074/jbc.R115.692020
91. Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y, et al. Relationship between nucleosome positioning and DNA methylation. Nature (2010) 466:388–92. doi: 10.1038/nature09147
92. Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee T-H. DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc. (2010) 132:1782–3. doi: 10.1021/ja910264z
93. Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science (2017) 356:eaaj2239. doi: 10.1126/science.aaj2239
94. Hashimoto H, Olanrewaju YO, Zheng Y, Wilson GG, Zhang X, Cheng X. Wilms tumor protein recognizes 5-carboxylcytosine within a specific DNA sequence. Genes Dev. (2014) 28:2304–13. doi: 10.1101/gad.250746.114
95. Crawford DJ, Liu MY, Nabel CS, Cao X-J, Garcia BA, Kohli RM. Tet2 catalyzes stepwise 5-methylcytosine oxidation by an iterative and de novo mechanism. J Am Chem Soc. (2016) 138:730–3. doi: 10.1021/jacs.5b10554
96. Bachman M, Uribe-Lewis S, Yang X, Burgess HE, Iurlaro M, Reik W, et al. 5-formylcytosine can be a stable DNA modification in mammals. Nat Chem Biol. (2015) 11:555–7. doi: 10.1038/nchembio.1848
97. Bachman M, Uribe-Lewis S, Yang X, Williams M, Murrell A, Balasubramanian S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nature chemistry (2014) 6:1049–55. doi: 10.1038/nchem.2064
98. Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature (2016) 529:110–4. doi: 10.1038/nature16490
99. Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG, Cheng X. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol Cell (2017) 66:711–20.e3. doi: 10.1016/j.molcel.2017.05.004
100. Deplus R, Delatte B, Schwinn MK, Defrance M, Méndez J, Murphy N, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. (2013) 32:645–55.
101. Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature (2015) 520:243–7. doi: 10.1038/nature14176
102. Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. (2010) 285:26114–20. doi: 10.1074/jbc.M109.089433
103. Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K, et al. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife (2015) 4:e06205. doi: 10.7554/eLife.06205
104. Zhang YW, Wang Z, Xie W, Cai Y, Xia L, Easwaran H, et al. Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol Cell (2017) 65:323–35. doi: 10.1016/j.molcel.2016.12.013
Keywords: 5hmC, 5 hydroxymethylcytosine, ten eleven translocation (TET), DNA modification, epigenetics (methylation/demethylation), gene regulation and expression
Citation: Lio C-WJ and Rao A (2019) TET Enzymes and 5hmC in Adaptive and Innate Immune Systems. Front. Immunol. 10:210. doi: 10.3389/fimmu.2019.00210
Received: 24 October 2018; Accepted: 24 January 2019;
Published: 12 February 2019.
Edited by:
Keiko Ozato, National Institutes of Health (NIH), United StatesReviewed by:
Jianzhu Chen, Massachusetts Institute of Technology, United StatesWarren Leonard, National Institutes of Health (NIH), United States
Copyright © 2019 Lio and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chan-Wang J. Lio, bGlvQGxqaS5vcmc=