 Qizhao Huang
Qizhao Huang Jianjun Hu
Jianjun Hu Jianfang Tang2
Jianfang Tang2 Lifan Xu
Lifan Xu Lilin Ye
Lilin Ye- 1Cancer Center, The General Hospital of Western Theater Command, Chengdu, China
- 2Institute of Immunology, Third Military Medical University, Chongqing, China
During viral infection, virus-specific follicular helper T cells provide important help to cognate B cells for their survival, consecutive proliferation and mutation and eventual differentiation into memory B cells and antibody-secreting plasma cells. Similar to Tfh cells generated in other conditions, the differentiation of virus-specific Tfh cells can also be characterized as a process involved multiple factors and stages, however, which also exhibits distinct features. Here, we mainly focus on the current understanding of Tfh fate commitment, functional maturation, lineage maintenance and memory transition and formation in the context of viral infection.
Introduction
Based on the biological process, viral infections can be divided into two groups: acute viral infection and chronic viral infection. During acute infections, virus is thoroughly eliminated by the orchestration between both innate and adaptive immune cells; whereas, certain types of viruses can effectively evade immune system and persist at a certain level in the host for long time in chronic infections (1). Numbers of specific immune effector mechanisms, coordinating with non-specific defense mechanisms, prevent or eliminate most viral infections. In terms of adaptive immune cells, CD8+ T cell- and CD4+ T cell-mediated immune responses play a critical role in the control of viral infection. During acute viral infection, virus-specific CD8+T cells differentiate into cytotoxic T lymphocytes (CTL) to efficiently eliminate virus-infected target cells and progressively transit into memory CD8+ T cells after viral eradication. Memory CD8+ T cells are maintained for a long time in the absence of antigen and can exert rapid effector functions in response to previously encountered antigens.
After a transit time in the blood, the majority of mature naïve CD4+ T cells produced by the thymus migrate to secondary lymphoid tissues, continually patrolling, and browsing for antigens they can recognize. After entering a lymph node, T cells scan the processed peptide-MHC complexes on the surface of DCs in the paracortex or T-cell zone. DCs that have processed antigen at the sites of infection arrive in the paracortex soon after infection. Upon viral infection, virus-specific CD4+T cells mainly differentiate into Th1 and Tfh (follicular helper T cell) cells, but not other helper subsets, such as Th2, Th17, and Th9 due to the strong type-I inflammation. And the divergence of Tfh and Th1 differentiation fates begin immediately after activation and are faithfully maintained through the life cycle (2). Through interactions between S1P1 receptors and S1P, the Th1 subset leaves the lymph node and travel to sites of infection. And they predominantly function through secreting IL-2, IFN-γ and TNFα and are responsible for many typical cell-mediated effects, including activation of CTL and macrophages. In contrast, virus-specific Tfh cells, characterized by high expression of chemokine receptor CXCR5, are endowed with the ability of migrating into B cell follicles in response to chemokine CXCL13 (3, 4), where they facilitate the maturation of GC B cells by interacting with cognate virus-specific B cells and providing “help” signals such as interleukin 21 (IL-21), IL-4, CD40L and inducible costimulatory molecules (ICOS) (5).
Tfh differentiation is generally characterized as a multistage, multifactorial process (Figure 1) (6). Upon recognition of virus peptide-MHC complex (p-MHC) presented by dendritic cells (DCs), CD4+ T cells adopting Tfh fate upregulate the “master regulator” Bcl-6 (7–9) within 2 or 3 days (10, 11). After engagement with DCs, Tfh cells move to the T-B border by upregulating CXCR5 and down-regulating CCR7 (10, 12). Here, they interact with cognate B cells and get sufficient signals that further support them migrating into B cell follicles and initiating GC reactions (13). During this process, the expression of Bcl-6 is enhanced, propelling the maturation of fully functional Tfh cells (14). Contrarily, Blimp1 (B lymphocyte-induced maturation protein-1), mainly expressed by non-Tfh effector cells, inhibits the expression of Bcl-6 and negatively regulates Tfh cell differentiation (7). Although the majority of Tfh cells originated from precursors in lymphoid tissues, several groups confirmed the existence of circulating CXCR5+ CD4+ T cells in mice or humans with ongoing immune responses, which were termed as peripheral Tfh (pTfh) (15–19). For instance, He et al. (18) demonstrated that pTfh consist of two parts: “effector” pTfh and “resting” pTfh cells, identified as CCR7loPD-1hi and CCR7hiPD-1lo, respectively. They found that CCR7loPD-1hiCXCR5+ CD4+ T cells express large amounts of IL-21, a key cytokine secreted by Tfh cells to support GC responses. And this population is able to further differentiate into mature Tfh cells and initiate GC formation. They believed that CCR7loPD-1hi Tfh precursor cells can circulate to non-draining secondary lymphoid organs and rapidly differentiate into mature Tfh cells to support fast GC formation upon antigen reencounter. However, the underlying mechanisms of the ontology and differentiation of this population remain unsolved.
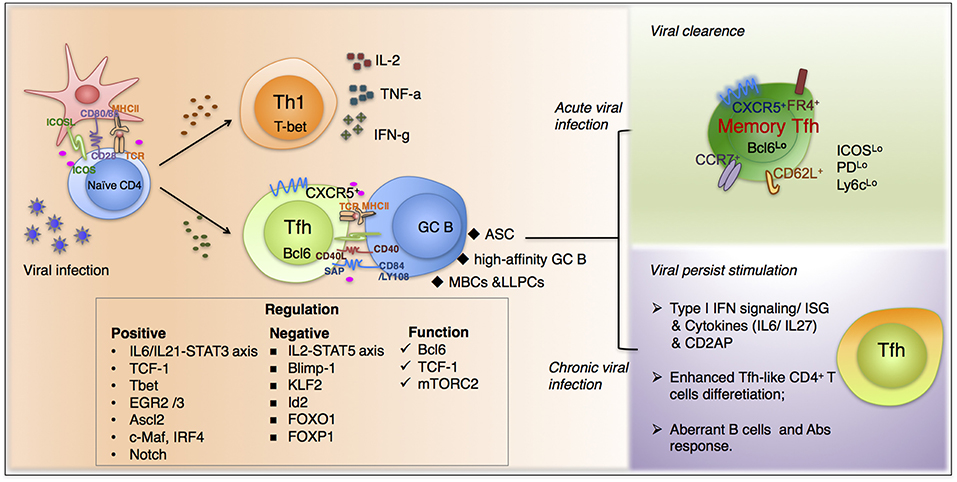
Figure 1. The fate commitment of Tfh cells during viral infection. Upon viral infection, virus-specific CD4+T cells mainly differentiate into Th1 and Tfh cells. Th1 subset predominantly function through secreting IL-2, IFN-γ, and TNFα and is responsible for many typical cell-mediated effects. In contrast, virus-specific Tfh cells, characterized by high expression of chemokine receptor CXCR5, are endowed with the ability of migrating into B cell follicles in response to chemokine CXCL13. The fate commitment and function of Tfh cells require fine-tuned cooperation of cognate p-MHCII molecular interactions, co-stimulation, together with polarizing cytokine signals and other factors. Moreover, upon viral clearance, a proportion of Tfh cells will differentiation in memory cells, which are more active when encountered with the same pathogens. Additionally, during chronic viral infections, Tfh cells accumulate gradually and exhibit a distinguished transcriptional profile compared with that in acute infections. And many factors participate in the unique differentiation pattern during persistent infection, including type I interferon signaling, cytokines from IL-6 family et al.
Tfh cells are essential for antibody-mediated humoral immunity against various pathogens. This review primarily focuses on the current understanding of the fate commitment, functional maturation, and memory formation of Tfh cells during acute viral infection. Moreover, we also focus on the role of Tfh cells during chronic viral infection, especially in HIV infection. Finally, we discuss the potential in boosting viral-specific Tfh cells for improving efficacies of anti-viral vaccines.
The Fate Commitment of Virus-Specific Tfh Cells vs. Th1 Cells
The fine-tuned cooperation of cognate p-MHCII molecular interactions, co-stimulation, together with polarizing cytokine signals initiate the differentiation of functionally divergent CD4+ T helper (Th) cell subsets from their precursors (20). Of note, during acute viral infection, commitment to the Tfh lineage vs. Th1 lineage emerges as early as 24 to 48 h after infection. The dichotomous commitment of Tfh cells vs. Th1 cells is largely linked to reciprocal regulation between key transcription factors Bcl6 and T-bet, and Bcl6 and Blimp-1 (21, 22).
At the priming stage, DCs regulate Tfh cell differentiation by controlling Clec9A expression, which facilitates the formation of a long-term immune synapse between DCs and T cells to promote Tfh differentiation (23, 24). Indeed, published work (25–27) has found that 24 h after T cell activation, T cells carrying high affinity TCRs can form long dynamic immune synapses with DC and are more inclined to differentiate into Tfh but not Th1 cells. In addition to interactions between membrane proteins of APC and Tfh precursors, secreted cytokines interleukin-6 (IL-6) and IL-21 also contribute to Tfh differentiation. Several groups confirmed that IL-6 and IL-21 signaling via the transcription factor STAT3 enhances the upregulated expression of Bcl6, which is the master regulator of Tfh differentiation. Nonetheless, IL-2 suppresses Tfh fates by activating STAT5 and restricting STAT3 binding to the Bcl6 locus and also by promoting the expression of Blimp-1, which divert differentiation away from the Tfh pathway (20, 28–31). Propelled by the antagonism of Bcl6 and Blimp-1, activated CD4+ T cells undergo a bimodal fate decision during acute viral infection: becoming either Tfh (Bcl6+Blimp1−) cells or Th1 (Bcl6−Blimp1+) cells. Notably, the transcription factor TCF-1 (t cell factor 1, coded by gene Tcf-7) has been confirmed to promote the early fate commitment to the Tfh lineage over Th1 lineage during acute viral infection (32–34). Using the LCMV-Armstrong and influenza virus infection model, we (32) found that the expression level of TCF-1 was significantly enhanced in Tfh cells while greatly diminished in Th1 cells. And such divergent expression mode occurred as early as 2 days post infection. TCF-1 potently induced the expression of Bcl-6 but suppressed Blimp1 concomitantly, by directly binding to the Bcl6 promoter region and Prdm1 5' regulatory region, respectively. Accordingly, virus-specific CD4+ T cells deficient in TCF-1 expression almost failed in Tfh differentiation. Notably, TCF-1 seems to specifically regulate Tfh cell differentiation in the context of viral infection, but dispensable for regulating Tfh differentiation during protein immunization (32, 33).
Apart from the master regulator Bcl-6, a network of several other transcription factors also participates in controlling the differentiation of Tfh cells during acute viral infection. For example, it has been confirmed that through two different but complementary mechanisms, the transcription factor KLF2 (Krüppel-like factor 2) functions to restrain Tfh cell generation. Lee et al. (35) found that KLF2 promotes the expression of the trafficking receptor S1PR1, the downregulation of which is essential for efficient Tfh cell differentiation. On the other hand, KLF2 favors the expression of several transcription factors that inhibit Tfh differentiation, such as Blimp1, Tbet, and GATA3. And KLF2 was also reported to suppress the transcription of Cxcr5 by directly binding to its genomic region (36). Importantly, although Tbet is the master transcriptional regulator of Th1 cells, which were thought to inhibit Tfh cell differentiation, Tfh cells do exhibit medium to high levels of Tbet expression in the LCMV infection model (2). Recently, it has been reported that T-bet is virtually essential for the optimal expansion, proliferation, and maintenance of Tfh cells during acute viral infection (37). Besides, Fang et al. (38) demonstrated that at the early stage of CD4+ T cells response, the short-term expression of Tbet is critical for IFN-γ production in Th1-like Tfh cell subset. Additionally, transcription factors of the E-protein and Id families are well-appreciated for their role in T cell development. Shaw et al. (39) found that Tfh cells exhibited lower expression of Id2 than that of Th1 cells during acute viral infection and knockdown of Id2 via shRNA increased the frequency of Tfh cells. Furthermore, Th1 differentiation was significantly blocked by the deficiency of gene Id2 during viral infection. Ogbe et al. (40) found that EGR2 (early growth response gene 2) and EGR3 play a vital role in directing the expression of Bcl6 in Tfh cells. The differentiation of Tfh cells was impaired in Egr2 and Egr3 deficient mice post viral infection because of the defective expression of Bcl-6, resulting in a defective GC reaction and antibody production. Moreover, the overexpression of Bcl-6 in EGR2/3- deficient CD4+ T cells partially rescued the differentiation of Tfh cells and GC formation. Liu et al. (41) found that during influenza virus infection, the deletion of Ascl2 in T cells results in impaired Tfh-cell development and germinal center response. Besides, in protein immunization or other infection models, several other TFs have been confirmed to participate in the regulation of the fate commitment of Tfh cells. For example, c-Maf, IRF4, and Notch signaling pathway has been confirmed to promote Tfh differentiation while FOXO1 and FOXP1 inhibit Tfh fate commitment (21, 42–47). Besides networks mediated by transcriptional factors, other different signaling pathways also control the differentiation and function of Tfh cells. Tfh cell differentiation are closely associated with mTOR-mediated signaling pathways, which exert its effect by sensing and integrating environmental cues. During acute viral infection, the interleukin-2 (IL-2)-mTORC1 signaling axis orchestrates the reciprocal balance between Th1 and Tfh cell fates by promoting Th1 while inhibiting Tfh cell differentiation (20). In contrast, it is reported that mTORC2 was essential for Tfh cell differentiation (48, 49); specifically, mTORC2 mainly functions in the late stage of Tfh differentiation, promoting a Tfh transcriptional program and migratory ability toward B cell follicles (50).
Currently, however, our knowledge about Tfh cells is mainly derived from mouse models, although the gene expression pattern of mouse Tfh cells shares a high percentage of similarities with human Tfh, certain differences do exit between the two species. For instance, in mouse models, the ligand for CXCR5, CXCL13 is mainly expressed by stromal cells but not Tfh cells (6, 51). In humans, however, CXCL13 is primarily generated by Tfh cells, which may promote recruiting GC B cells to the light zone, where most Tfh cells and FDCs reside (52–54). Hence, further research is required for carefully profiling the differences between human and murine Tfh cells, which is critical for translating findings between the two species. Taken together, like Tfh cells developed in other scenarios, the fate commitment of virus-specific Tfh cells also follow a pathway involved a multistep and multifactorial process. Although we have gained a relatively detailed understanding of the ontology and differentiation of Tfh cells during viral infection, there are still important gaps in our knowledge of the Tfh cell differentiation and underlying mechanisms during viral infection both in mouse and humans. In particular, how extrinsic stimuli from APCs and intrinsic factors, such as epigenetic modifications, regulate anti-viral Tfh differentiation await further investigations.
The Functional Maturation and Maintenance of Virus-Specific Tfh Cells
Currently, most licensed anti-viral vaccines protect vaccinated population by inducing long-lived neutralizing antibodies (55). Tfh cells play a critical role in helping B cells to differentiate into neutralizing-antibody-secreting plasma cells. Therefore, it is essential to understand the mechanisms by which Tfh cells co-opt B cell responses during viral infection, so as to effectively facilitate developing a novel vaccine or further improving the efficacies of licensed vaccines. Previous reviews have systematically summarized the functions of Tfh cells (6, 56), we herein mainly focus on the roles of these cells in the case of viral infection. After viral infection, SLAM-associated protein (SAP) expressed by Tfh cells is critical for the formation of germinal centers (57, 58), where Tfh cells facilitate the generation of long-lived memory B cells and plasma cells that produce virus-specific antibodies (57, 59). For example, CD4+ T cells have been confirmed to be essential for the generation of optimal antibody responses during infections with yellow fever virus (60), vaccinia virus (61), coronavirus (62), or vesicular stomatitis virus (VSV) (63). Collectively, Tfh cells play an important role in protective immunity to most, if not all, viruses.
In antiviral humoral immunity, Tfh cells help B cell activation and antibody production in the form of receptor ligand interactions and cytokine signaling. Firstly, Tfh cells highly express CD40 ligand (CD40L), whose interaction with CD40 expressed on B cells is vital to multiple stages and aspects of B cell response. Using LCMV, Pichinde virus, and VSV infection model, Borrow et al. (64) found that CD40L-deficient mice exhibit severely compromised humoral immune responses, supported by low antiviral antibody production, absence of germinal center and memory B cell formation. Consistently, CD40L/CD40 was also reported to be important for generating optimal humoral responses against HSV and influenza virus (65, 66). During LCMV, VSV, and influenza virus infection model, the expression of ICOS (inducible T cell co-stimulator) by Tfh cells has also been reported to be crucial for germinal center formation (6) and optimal induction of humoral responses (67). Other co-stimulatory molecules that promote the T-B conjugates, including SAP and SLAM family are also required for Tfh differentiation as well as Tfh function (6). It is important to appreciate that both defective Tfh cell number and damaged Tfh function can lead to impaired GC response. To more precisely evaluate the effector function of already differentiated Tfh cells during viral infection in vivo, our group combines ERT2cre conditional knockout mice with mature Tfh cells adoptive transfer strategy to determine their “help” ability in promoting the formation of GC and plasma cells (32, 50). The expression of these B-cell helping molecules (CD40L and ICOS) in Tfh cells appears to be coordinated by Bcl-6 (68, 69), TCF-1 (32), and mTORC2 (50). Further studies are needed to determine the importance of additional molecular signals between Tfh cells and B cells in the production of protective antibody responses during viral infection.
The Memory Formation of Virus-Specific Tfh Cells
Most of the virus-specific effector CD4+ T cells will die and only a small portion of them will survive and further differentiate into memory T cells after the elimination of a viral infection. The features of memory lymphocyte generally includes (1) antigen experienced (these cells have undergone antigen-driven expansion); (2) can survive for a long time (undergo homeostatic proliferation) in the absence of antigenic stimulation; (3) self-renewable by homeostatic proliferation; (4) rapidly recall their effector functions in response to re-challenge (70). Memory CD4+ T cells respond much faster than naive T cells, require less synergistic stimulation to respond to low antigen doses, and are more active when challenged by pathogens (71). Recent studies suggest both effector Tfh and Th1 cells can differentiate into memory cells.
Series of studies have clearly demonstrated the existence of memory Tfh cell in both mice and human (Figure 1) (2, 17, 72–75). These studies provide important insights into the characteristics of Tfh cells that can differentiate into long-lived memory-type cells which are endowed with capacity to reboost Tfh-specific effector functions when encountered with the same antigen (76). Meanwhile, considering the long persistence of GC reactions and antigen retention by FDC, it is important to appreciate that GC Tfh cells are not confined to one single GC. Once GC Tfh cells have differentiated and provided help to GC B cells, they can continually enter a different GC or exit GC and emigrate to neighboring follicles (77, 78), where no antigen was presented and Tfh cells acquires a less activated, less polarized phenotype. In this situation, by downregulating Bcl6 expression and upregulating IL-7Ra, Tfh cells gradually transit into a resting memory state (73, 78). In acute viral infection for instance, after the clearance of LCMV in mice, virus-specific CD4+ T cells that survived the contraction phase can be maintained for 60–150 days (2). Among these cells, CXCR5+Ly6clo resting CD4+ T cells shared similarities with effector Tfh cells, both phenotypically and transcriptionally, and can rapidly recall a secondary wave of effector Tfh response even in the absence of B cells, supporting that CXCR5+ memory cells have been imprinted with a Tfh-biased cell program (2). Cell markers that can clearly define memory Tfh cells including high expression of CXCR5, FR4 (79), CCR7, CD62L, and low expression of Bcl-6, ICOS, PD-1, and Ly6c (80). Currently, the differentiation pattern of memory Tfh cell remains controversial. An important question is whether the fate of memory Tfh cells is determined before or after effector phase. Meanwhile, given that Th1 vs. Tfh differentiation are regulated by strength and/or duration of TCR signaling (81, 82), which also influences memory CD4+ T cell differentiation (83), it is possible that Tfh effector population with different TCR strength varies in degrees of lineage commitment to CXCR5+ Tfh memory cells.
Besides, previous study indicated that memory Tfh cells are superior to naive T cells in helping B cells, and they promote faster B cell proliferation, higher antibody production and earlier class-switching reactions than naive CD4+ T cells (76). In many cases, the invasive virus can be quickly recognized and eradicated by pre-existing antibodies. But for viruses that bear high mutation rate (such as influenza virus and HIV), the most important point lies on faster antibody production, as the timely and efficient production of neutralizing antibodies targeting against new variants that escape from previously produced antibodies will be of great significance. Memory Tfh cells maintain a substantial level of CD40L (84) and are retained in the draining lymph nodes for more than 6 months (85). Besides, during several types of viral infections, such as Ebola virus (86), WNV (87), and influenza virus (88), memory Tfh cells can generate higher levels of cytokines as compared with those of naive T cells, which are likely to induce a more-potent B cell responses and to dictate the isotype of antibodies, which play key roles in the antibody responses specific for aforementioned viruses. Nonetheless, we currently know much less as to the molecular mechanisms underlying memory Tfh differentiation than those discovered with effector Tfh cells.
The Differentiation of Virus-Specific Tfh Cells During Chronic Viral Infection
We know far less about how chronic viral infections affect CD4+ T cell responses than we do about CD8+ T cell exhaustion. However, increasing attention has been paid to the impact of persistent viral infection on the function of CD4+ T cells and the importance of CD4+ T cells in chronic viral infection. Compared with acute viral infection, virus-specific CD4+ T cells in LCMV clone 13 persistent infection model exhibit deficiency in production of Th1-type effector cytokines and fail to function optimally following viral re-challenge (89). The loss of CD4+ T cell's ability to respond to persistent antigens may be due to high levels of antigens at the priming stage (90) and appears not to be regulated by the changes in APCs caused by chronic viral pathogens (89).
Similar to CD8+ T cell exhaustion in chronic infection, the virus-specific CD4+ T cell response has been altered profoundly as infection persists. The most significant phenotype of CD4+ T cell response during chronic viral infection is a defect in Th1, while increasing in Tfh response (Figure 1). And both in mouse and human chronic viral infections, the frequency of CXCR5+CD4+ T cells in spleen accumulates gradually, reaching approximately 60~70% of the viral-specific CD4+ T cells by day 30, whereas which were relatively lower at a frequency of 40~50% during Arm infection (91). The increased Tfh differentiation was accompanied by a loss in Th1, including decreased proliferative potential and cytokine production (89). Thereby, to some extent, the immune system promotes antibody responses, which bear less immune-pathological risk compared to cytotoxic and pro-inflammatory T cell responses. Moreover, transcriptional profiling of viral-specific CD4+ T cells in LCMV clone 13 infection identified a loss in Th1 transcriptional signatures, as well as an enrichment of Tfh-associated transcripts (92). Upregulated CXCR5, Bcl-6, ICOS, OX40, and IL-21 expression suggest an enhanced Tfh-like CD4+ T cells phenotype (91). Very importantly, these additional Tfh-like CD4+ T cells are proven to have the ability to help B cells through in vitro culture, suggesting that they are equipped with some key signatures of conceptual Tfh cells and remain suboptimal functions, such as the ability to facilitate coordinate B cell response and production of antibody (91, 93). Although we have not yet got a comprehensive understanding of the biased differentiation of Tfh cells in chronic viral infection, previous research proved that type I IFN signaling may be an important mediator involved in the shift from Th1 to Tfh cells (94–97). Several groups also demonstrated the skewed differentiation toward Tfh cells during chronic LCMV infection is firmly related to cytokines (such as IL-6/ IL-27) signaling through the IL-6 family receptor pathway (98, 99). Recently, Raju et al. (100) found that the deficiency of the signaling adaptor CD2AP (CD2-associated protein) promotes CD4+ T cell differentiation toward Tfh lineage during chronic LCMV infection, leading to better control of viral infection by enhanced GC response. They demonstrated that the strengthened Tfh differentiation is associated with extended duration of TCR signaling and enhanced cytokine production of CD2AP-deficient CD4+ T cells specifically under Th1 conditions. To be noted, the increased CXCR5 level may also contributed by another CD4+ T cell subpopulation, Tfr cells (101, 102), coincide with upregulated Foxp3 expression during chronic infection (103). Although the function of Tfr cells is incompletely understood, especially in chronic infection, the increased Tfr differentiation suggest an active follicular program in chronic infection. And this follicular program may be not only confined to CD4+ T cell lineage, confirmed by newly identified CXCR5+CD8+ T cells described both in mice and human chronic viral infection (104–106).
Although viral persistence redirects a shift in Tfh differentiation, it is not clear to what extent the function of Tfh cells generated during chronic viral infection gets changed. IL-21, canonical Tfh cytokine important for CD8+ T cell function in chronic viral infection (107–110), is increased within Tfh population. However, for humoral immunity, it seems that B cell could not get optimal help from increased Tfh in chronic LCMV infection. Firstly, the generation of neutralizing antibodies are impaired and delayed, whereas, non-neutralizing antibodies to LCMV increased considerably (111). Secondly, persisting viral infections can lead to polyclonal hypergammaglobulinemia and antibody-mediated autoimmunity, results of non-specific B cell activation by Tfh cells (112, 113). The delayed production of neutralizing antibodies and high production of poor quality antibodies indicate that the interaction between Tfh and B cells in chronic LCMV infection is dysregulated, leading to a suboptimal ability of Tfh to help B cells producing high-affinity antibodies.
As has been observed in LCMV, HIV, and SIV infections also have been reported to have increased frequency of CXCR5+CD4+ T cells (114, 115). Besides, compared with uninfected healthy donors, the transcription characteristics of Tfh cells in the SIV infection model were changed. It has been confirmed that the transcriptional signature of Tfh cells derived from SIV-infection models gets remarkably altered compared to those from healthy donors (116). The underlying mechanisms initiating and promoting a Tfh-like program in HIV infection still remain unsolved and the relationship between HIV infection and Tfh differentiation is complicated. During HIV or SIV infections, Tfh cells seem to act in a bilateral manner, both immunological and immunopathogenical: Firstly, Tfh cells are appreciated as an important cellular reservoir of replication-competent HIV virus, contributed by its special phenotype and follicular localization. It is demonstrated that Tfh cells located in B-cell follicles are preferentially targeted by HIV virus to form both long-term latent infection and the enduring generation of virulent particles (110, 117), and the anatomical separation of latently infected Tfh cells might represent a major barrier for HIV-specific CD8+ T cells, which are normally excluded from B-cell follicles, to effectively eradicate HIV infection (118–121). Secondly, Tfh is closely involved in developing antibody-based vaccines for HIV-1 infection, because functional Tfh-B cell interactions are key to production of effective antibodies in vaccination (16, 122). Consistent with mouse chronic infection models, Tfh cells do not provide adequate help to B cells even though these cells are expanded in HIV-infected individuals (increased Tfh frequency dose not result in better B cell response). Instead, similar to mouse chronic infection of LCMV, abnormal B cell activation and hyper-gammaglobulinemia were observed in HIV-1 infection (112, 115, 123), which suggests the dysregulation of Tfh cell-mediated B cell help and disturbed Tfh-B cell interactions. Specifically, data from mass cytometry combine with TCR sequencing confirmed that compared with healthy individuals, Tfh cells in the lymph nodes of HIV+ individuals secreted interleukin-21 but were functionally and clonally restricted and this correlated with impaired isotype switching of B cells in the lymph nodes (124). Given the close relationship with Tfh from lymphoid tissues, circulating or peripheral Tfh cells have also been confirmed to be critical in HIV infection (16, 22, 125). He et al. (18) demonstrated that circulating CXCR5+CD4+ T cells are generated in a SAP independent manner (before they migrate to GC), and CCR7loPD-1hi subset correlated with Tfh cell activity, providing a biomarker to monitor protective humoral immune responses during infection or vaccination. In a related study, combining cytokine production, functional properties as well as gene expression profile, Locci et al. (17) identified pTfh cells related to germinal center Tfh cells as resting CD45RO+PD-1+CXCR5+CXCR3−CD4+ T cells. And they confirmed that the frequency of this population positively correlates with the titers of HIV-specific broadly neutralizing antibodies in a large cohort of HIV-infected patients. Schultz et al. (15) found that during HIV infection, peripheral IL-21+ CD4+ T cells show similarities with lymphoid tissue-resident Tfh cells phenotypically, transcriptionally, and functionally. And they also found that the numbers of HIV-specific IL-21-expressing pTfh cell increased and their number positively correlated with antibody production in the ALVAC priming, AIDSVAX boosting immunization strategy used in the RV144 trial (the only HIV vaccine to demonstrate some signs of efficacy among human patients) when compared with the non-protective DNA prime-Ad5 boosting vaccine trial. Given that the timely development of high-affinity antibodies is central to the prevention and eradication of viral infection (126), further work is needed to understand the detailed mechanism underlying Tfh dysfunction during persistent viral infections.
The key feature of CD8+ T cell exhaustion is upregulated expression of co-inhibitory receptors, such as PD-1, Tim3, 2B4. Although CD4+ T cells sustained the expression of a sets of co-inhibitors (127), however, the specific inhibitory receptors upregulated and the degree of expression between CD4+ and CD8+ T cells differed remarkably (92, 127). For example, the expression of 2B4 is biased toward exhausted CD8+ T cells, while PD-1, CTLA4 are preferentially expressed in CD4+ T cells, particularly in Tfh cells (92). Several groups (128) found that functionally impaired CD4+ T cells derived from HIV patients exhibit significant enhancement in proliferative potential after treatment targeting on CTLA-4 (129, 130), TIM3 (131) or PD-1 signaling blockade (132) in vitro. And the effector function of CD8+ T cells can be rescued through enhancement of CD4+ T cell response during chronic infection with LCMV (133, 134). These findings and others (135, 136) shed new lights on the design of vaccines against chronic viral infections. For Tfh cells, physiologically, PD-1 is assigned to provide inhibitory signals to GC Tfh cells, preventing excess cell proliferation during GC reaction (21). Good-Jacobson et al. (137) demonstrated that upon immunization, the deficiency of PD-1 or PD-1 ligands (PD-L1/PD-L2) results in higher frequency of Tfh cells. Whereas, they also found that the quality of Tfh cells is dramatically impaired by diminishing their capacity to synthesize important cytokines (such as IL-4/IL-21) while not promoting the development of an alternatively polarized T cell type. These results suggest a complex but critical role of PD-1 in Tfh cells. However, despite the above findings, the role of PD-1 in Tfh cells during chronic viral infection remains unclear. Whether the expression of PD-1 on Tfh cells equals exhaustion or whether this is part of their normal regulation and functional differentiation during persistent infection have not yet been fully discovered.
Most of the aforementioned knowledge about CD4+ T cell response during chronic viral infection has been obtained from studies in which animals are infected with a single virus. While valuable for identification of basic principles, this is not reflective of human biology, since human beings undergo repeated viral infections throughout their life span, most notably, multiple herpesviruses. The γ-herpesviruses (Gammaherpesviruses), including EBV (Epstein-Barr virus) and KSHV (Kaposi's sarcoma-associated herpesvirus), are associated with lymphoproliferative diseases and lymphomas and with the majority establishing latency in B lymphocytes (138). In mouse models, intranasal infection of mice with the murine γ-herpesvirus (MHV-68, shares biological and genetic homology with EBV) results in an acute lytic infection in the lung, followed by the establishment of lifelong latency in memory B cells, dendritic cells, and macrophages (104, 139–141). Barton et al. (142) demonstrated that both the proportion and total number of IFNγ+, TNFα+, and IL-2+ CD4+ T cells was increased in mice infected with MHV68 followed with LCMV-Armstrong re-challenge compared to that in mice solely infected with LCMV on day 8 post infection. This result reminded us that MHV68 latency may provide micro-environment in which effector CD4+ T cell responses get enhanced during subsequent infection. Another study confirmed that signals from Tfh cell is critical for B cell latency during MHV68 infection. They found that the absence of these signals lead to a significant reduction in the number of MHV68 latently infected B cells (143). However, whether Tfh cells are selectively up-regulated during MHV68 chronic infection has not yet been fully illustrated. Apart from its fundamental role in supporting B cell latency in MHV68 infection, CD4+ T cells may also control MHV68 replication in a CD8+ T cell dependent or independent manner (138, 144). Recently, several groups (104, 145) identified a specialized group of cytotoxic T cells that expressed high level of the chemokine receptor CXCR5 (Tfc, Follicular cytotoxic T cells), which selectively entered B cell follicles and eradicated infected Tfh cells and B cells during HIV/SIV or EBV infection, respectively. Given that Tfh and Tfc cells have a similar histological location, it will be of interests to determine whether these two subsets have interaction or crosstalk during chronic viral infection.
Collectively speaking, further dissection of unique molecular mechanisms underlying differentiation and functionality of Tfh cells in chronic viral infection will provide opportunities for harnessing this population to prevent and treat chronic viral infection.
Perspective
Currently, the transcriptional regulation of the ontogeny and development of Tfh cell has been extensively investigated. However, the field just starts to dissect the complexities of cellular metabolism within Tfh cell as well as its epigenetic signatures, particularly, in the scenario of viral infections. It is well-acknowledged that the differentiation of Tfh cells is accompanied by unique metabolic alterations required to meet their cellular bioenergetic demands. During acute viral infection, Tfh cells exhibited a relatively quiescent metabolic state when compared to Th1 lineage, characterized by reduced glucose uptake and mitochondrial respiration, as well as lowered maximal respiratory capacity and extracellular acidification. However, despite Tfh cells showing reduced metabolic capacity, they still require glycolysis as well as oxidative phosphorylation to provide sufficient energy and substrates for their specific function (20). Zeng et al. (49) found that mTOR, combining metabolic signals and transcriptional activity, plays as a central control station in Tfh differentiation. Activated by costimulatory molecule ICOS, mTOR acts to drive glycolysis and lipogenesis and subsequently promotes Tfh cell responses during acute viral infection. Given that GC-Tfh cells have a different localization compared to outside Tfh cells. They may have distinct metabolic features influenced by unique cellular and nutritional contact within each microenvironment. Moreover, it is possible that memory Tfh cells differs from effector Tfh cells in metabolism as described in effector vs. memory CD8+ T cells. Whether and how cellular metabolism influence the formation of memory Tfh cells still need further investigation.
Besides metabolic issues, the differentiation of Tfh cells as well as other CD4+ T helper (Th) cells are firmly correlated with specific epigenetic modifications (146, 147). By generating T cell-specific UTX (ubiquitously transcribed tetratricopeptide repeat, X chromosome) deficient mice, Cook et al. (148) found that during chronic but not acute, virus infection, Tfh differentiation were significantly impaired in UTX deficient mice, which in turn leads to suboptimal formation of germinal center and production of virus-specific IgG. Mechanistically, the absence of UTX leads to the upregulation of H3K27 methylation which further results in decreased expression of IL-6R alpha and other Tfh lineage-related genes. Nishizawa et al. (149) demonstrated that Bcl-6 is highly expressed in angioimmunoblastic T-cell lymphoma (AITL) and peripheral T-cell lymphomas (PTCL) containing tumor cells with Tfh features. In their research, hypermethylation of the Bcl6 locus followed by Bcl-6 upregulation, combined with TET2 mutations, was thought to be the key event for lymphoma development which may result in biased Tfh differentiation and eventually contribute to AITL/PTCL development in patients. Apart from these achievements, there are still important gaps in our knowledge of the epigenetic features of Tfh cells. Further studies will be required to draw a comprehensive epigenetic landscape of Tfh cells and identify potential candidate chromatin modifiers that participate in Tfh development. Understanding these issues and dissecting the underlying regulatory mechanisms will advance our knowledge of Tfh cells and shed lights on designing new strategies against those diseases associated with Tfh abnormalities.
Ethics Statement
All of our studies were specifically reviewed and approved by the Institutional Animal Care and Use Committees of the Third Military Medical University.
Author Contributions
QH, JH, and JT wrote and edited the manuscript with LX and LY.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the lab members from LY's lab for discussing this manuscript. This study was supported by grants from the National Key Research Development Plan of China (No. 2016YFA0502202 to LY) and the National Natural Science Foundation of China (No. 31700774 to LX).
References
1. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T cell exhaustion in chronic infection and cancer: opportunities for interventions. Annu Rev Med. (2018) 69:301–18. doi: 10.1146/annurev-med-012017-43208
2. Hale JS, Youngblood B, Latner DR, Mohammed AU, Ye L, Akondy RS, et al. Distinct memory CD4+ T cells with commitment to T follicular helper- and T helper 1-cell lineages are generated after acute viral infection. Immunity (2013) 38:805–17. doi: 10.1016/j.immuni.2013.02.020
3. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. (2000) 192:1545–52. doi: 10.1084/jem.192.11.1545
4. Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. (2000) 192:1553–62. doi: 10.1084/jem.192.11.1553
5. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
6. Crotty S. Follicular helper CD4 T cells (TFH). Ann Rev Immunol. (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
7. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325:1006–10. doi: 10.1126/science.1175870
8. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325:1001–5. doi: 10.1126/science.1176676
9. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31:457–68. doi: 10.1016/j.immuni.2009.07.002
10. Baumjohann D, Okada T, Ansel KM. Cutting edge: distinct waves of BCL6 expression during T follicular helper cell development. J Immunol. (2011) 187:2089–92. doi: 10.4049/jimmunol.1101393
11. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34:932–46. doi: 10.1016/j.immuni.2011.03.023
12. Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol. (2007) 179:5099–108. doi: 10.4049/jimmunol.179.8.5099
13. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496:523–7. doi: 10.1038/nature12058
14. Choi YS, Yang JA, Crotty S. Dynamic regulation of Bcl6 in follicular helper CD4 T (Tfh) cells. Curr Opin Immunol. (2013) 25:366–72. doi: 10.1016/j.coi.2013.04.003
15. Schultz BT, Teigler JE, Pissani F, Oster AF, Kranias G, Alter G, et al. Circulating HIV-specific interleukin-21(+)CD4(+) T cells represent peripheral Tfh cells with antigen-dependent helper functions. Immunity (2016) 44:167–78. doi: 10.1016/j.immuni.2015.12.011
16. Chahroudi A, Silvestri G. HIV and Tfh cells: circulating new ideas to identify and protect. Immunity (2016) 44:16–8. doi: 10.1016/j.immuni.2015.12.016
17. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3-CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity (2013) 39:758–69. doi: 10.1016/j.immuni.2013.08.031
18. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
19. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
20. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi: 10.1016/j.immuni.2015.08.017
21. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Ann Rev Immunol. (2016) 34:335–68. doi: 10.1146/annurev-immunol-041015-055605
22. Qi H. T follicular helper cells in space-time. Nat Rev Immunol. (2016) 16:612–25. doi: 10.1038/nri.2016.94
23. Kato Y, Zaid A, Davey GM, Mueller SN, Nutt SL, Zotos D, et al. Targeting antigen to Clec9A primes follicular Th cell memory responses capable of robust recall. J Immunol. (2015) 195:1006–14. doi: 10.4049/jimmunol.1500767
24. Lahoud MH, Ahmet F, Kitsoulis S, Wan SS, Vremec D, Lee CN, et al. Targeting antigen to mouse dendritic cells via Clec9A induces potent CD4 T cell responses biased toward a follicular helper phenotype. J Immunol. (2011) 187:842–50. doi: 10.4049/jimmunol.1101176
25. Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science (2002) 296:1873–6. doi: 10.126/science.1071065
26. Mempel TR, Henrickson SE, Von Andrian UH. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature (2004) 427:154–9. doi: 10.1038/nature02238
27. Celli S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity (2007) 27:625–34. doi: 10.1016/j.immuni.2007.08.018
28. Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, et al. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS ONE (2011) 6:e17739. doi: 10.1371/journal.pone.0017739
29. Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity (2014) 40:367–77. doi: 10.1016/j.immuni.2014.02.005
30. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. (2012) 209:243–50. doi: 10.1084/jem.20111174
31. Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nat Immunol. (2012) 13:405–11. doi: 10.1038/ni.2242
32. Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol. (2015) 16:991–9. doi: 10.1038/ni.3229
33. Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 is required for the T follicular helper cell response to viral infection. Cell Rep. (2015) 12:2099–110. doi: 10.1016/j.celrep.2015.08.049
34. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. (2015) 16:980–90. doi: 10.1038/ni.3226
35. Lee JY, Skon CN, Lee YJ, Oh S, Taylor JJ, Malhotra D, et al. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity (2015) 42:252–64. doi: 10.1016/j.immuni.2015.01.013
36. Weber JP, Fuhrmann F, Feist RK, Lahmann A, Al Baz MS, Gentz LJ, et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Kruppel-like factor 2. J Exp Med. (2015) 212:217–33. doi: 10.1084/jem.20141432
37. Weinstein JS, Laidlaw BJ, Lu Y, Wang JK, Schulz VP, Li N, et al. STAT4 and T-bet control follicular helper T cell development in viral infections. J Exp Med. (2018) 215:337–55. doi: 10.1084/jem.20170457
38. Fang D, Cui K, Mao K, Hu G, Li R, Zheng M, et al. Transient T-bet expression functionally specifies a distinct T follicular helper subset. J Exp Med. (2018) 215:2705–14. doi: 10.1084/jem.20180927
39. Shaw LA, Belanger S, Omilusik KD, Cho S, Scott-Browne JP, Nance JP, et al. Id2 reinforces TH1 differentiation and inhibits E2A to repress TFH differentiation. Nat Immunol. (2016) 17:834–43. doi: 10.1038/ni.3461
40. Ogbe A, Miao T, Symonds ALJ, Omodho B, Singh R, Bhullar P, et al. Early growth response genes 2 and 3 regulate the expression of Bcl6 and differentiation of T follicular helper cells. J Biol Chem. (2015) 290:20455–65. doi: 10.1074/jbc.M114.634816
41. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature (2014) 507:513–8. doi: 10.1038/nature12910
42. Shi B, Geng J, Wang YH, Wei H, Walters B, Li W, et al. Foxp1 negatively regulates T follicular helper cell differentiation and germinal center responses by controlling cell migration and CTLA-4. J Immunol. (2018) 200:586–94. doi: 10.4049/jimmunol.1701000
43. Stone EL, Pepper M, Katayama CD, Kerdiles YM, Lai CY, Emslie E, et al. ICOS coreceptor signaling inactivates the transcription factor FOXO1 to promote Tfh cell differentiation. Immunity (2015) 42:239–51. doi: 10.1016/j.immuni.2015.01.017
44. Auderset F, Schuster S, Fasnacht N, Coutaz M, Charmoy M, Koch U, et al. Notch signaling regulates follicular helper T cell differentiation. J Immunol. (2013) 191:2344–50. doi: 10.4049/jimmunol.1300643
45. Bollig N, Brustle A, Kellner K, Ackermann W, Abass E, Raifer H, et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc Natl Acad Sci USA. (2012) 109:8664–9. doi: 10.1073/pnas.1205834109
46. Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol. (2009) 10:167–75. doi: 10.1038/ni.1690
47. Krishnamoorthy V, Kannanganat S, Maienschein-Cline M, Cook SL, Chen J, Bahroos N, et al. The IRF4 gene regulatory module functions as a read-write integrator to dynamically coordinate T helper cell fate. Immunity (2017) 47:481–97.e7. doi: 10.1016/j.immuni.2017.09.001
48. Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, et al. Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife (2016) 5:e17936. doi: 10.7554/eLife.17936
49. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45:540–54. doi: 10.1016/j.immuni.2016.08.017
50. Hao Y, Wang Y, Liu X, Yang X, Wang P, Tian Q, et al. The kinase complex mTOR complex 2 promotes the follicular migration and functional maturation of differentiated follicular helper CD4(+) T cells during viral infection. Front Immunol. (2018) 9:1127. doi: 10.3389/fimmu.2018.01127
51. Gunn MD, Ngo VN, Ansel KM, Ekland EH, Cyster JG, Williams LT. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature (1998) 391:799–803. doi: 10.1038/35876
52. Allen CD, Cyster JG. Follicular dendritic cell networks of primary follicles and germinal centers: phenotype and function. Semin Immunol. (2008) 20:14–25. doi: 10.1016/j.smim.2007.12.001
53. Rasheed AU, Rahn HP, Sallusto F, Lipp M, Muller G. Follicular B helper T cell activity is confined to CXCR5(hi)ICOS(hi) CD4 T cells and is independent of CD57 expression. Eur J Immunol. (2006) 36:1892–903. doi: 10.002/eji.200636136
54. Kim CH, Lim HW, Kim JR, Rott L, Hillsamer P, Butcher EC. Unique gene expression program of human germinal center T helper cells. Blood (2004) 104:1952–60. doi: 10.182/blood-2004-03-1206
55. Amanna IJ, Carlson NE, Slifka MK. Duration of humoral immunity to common viral and vaccine antigens. N Eng J Med. (2007) 357:1903–15. doi: 10.1056/NEJMoa066092
56. Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity (2009) 30:324–35. doi: 10.1016/j.immuni.2009.03.003
57. Crotty S, Kersh EN, Cannons J, Schwartzberg PL, Ahmed R. SAP is required for generating long-term humoral immunity. Nature (2003) 421:282–7. doi: 10.1038/nature01318
58. McCausland MM, Yusuf I, Tran H, Ono N, Yanagi Y, Crotty S. SAP regulation of follicular helper CD4 T cell development and humoral immunity is independent of SLAM and Fyn kinase. J Immunol. (2007) 178:817–28. doi: 10.4049/jimmunol.178.2.817
59. Kamperschroer C, Dibble JP, Meents DL, Schwartzberg PL, Swain SL. SAP is required for Th cell function and for immunity to influenza. J Immunol. (2006) 177:5317–27. doi: 10.4049/jimmunol.177.8.5317
60. Liu T, Chambers TJ. Yellow fever virus encephalitis: properties of the brain-associated T-cell response during virus clearance in normal and gamma interferon-deficient mice and requirement for CD4+ lymphocytes. J Virol. (2001) 75:2107–18. doi: 10.1128/jvi.75.5.2107-2118.2001
61. Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, et al. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity (2008) 28:847–58. doi: 10.1016/j.immuni.2008.04.018
62. Chen J, Lau YF, Lamirande EW, Paddock CD, Bartlett JH, Zaki SR, et al. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J Virol. (2010) 84:1289–301. doi: 10.1128/jvi.01281-09
63. Thomsen AR, Nansen A, Andersen C, Johansen J, Marker O, Christensen JP. Cooperation of B cells and T cells is required for survival of mice infected with vesicular stomatitis virus. Int Immunol. (1997) 9:1757–66.
64. Borrow P, Tishon A, Lee S, Xu J, Grewal IS, Oldstone MB, et al. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J Exp Med. (1996) 183:2129–42.
65. Edelmann KH, Wilson CB. Role of CD28/CD80-86 and CD40/CD154 costimulatory interactions in host defense to primary herpes simplex virus infection. J Virol. (2001) 75:612–21. doi: 10.1128/jvi.75.2.612-621.2001
66. Sangster MY, Riberdy JM, Gonzalez M, Topham DJ, Baumgarth N, Doherty PC. An early CD4+ T cell-dependent immunoglobulin A response to influenza infection in the absence of key cognate T-B interactions. J Exp Med. (2003) 198:1011–21. doi: 10.1084/jem.20021745
67. Bertram EM, Tafuri A, Shahinian A, Chan VS, Hunziker L, Recher M, et al. Role of ICOS versus CD28 in antiviral immunity. Eur J Immunol. (2002) 323376–85. doi: 10.1002/1521-4141(200212)32:12<3376::aid-immu3376>3.0.co;2-y
68. Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med. (2015) 212:539–53. doi: 10.1084/jem.20141380
69. Kroenke MA, Eto D, Locci M, Cho M, Davidson T, Haddad EK, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol. (2012) 188:3734–44. doi: 10.4049/jimmunol.1103246
70. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Ann Rev Immunol. (2013) 31:137–61. doi: 10.1146/annurev-immunol-032712-095954
71. McKinstry KK, Strutt TM, Swain SL. The effector to memory transition of CD4 T cells. Immunol Res. (2008) 40:114–27. doi: 10.1007/s12026-007-8004-y
72. Weber JP, Fuhrmann F, Hutloff A. T-follicular helper cells survive as long-term memory cells. Ear J Immunol. (2012) 42:1981–8. doi: 10.1002/eji.201242540
73. Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. (2013) 190:4014–26. doi: 10.4049/jimmunol.1202963
74. Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med. (2012) 209:1841–52. doi: 10.1084/jem.20120219
75. Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. (2013) 5:176ra32. doi: 10.1126/scitranslmed.3005191
76. MacLeod MK, David A, McKee AS, Crawford F, Kappler JW, Marrack P. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J Immunol. (2011) 186:2889–96. doi: 10.4049/jimmunol.1002955
77. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science (2013) 341:673–7. doi: 10.1126/science.1241680
78. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity (2011) 34:961–72. doi: 10.1016/j.immuni.2011.03.025
79. Iyer SS, Latner DR, Zilliox MJ, McCausland M, Akondy RS, Penaloza-Macmaster P, et al. Identification of novel markers for mouse CD4(+) T follicular helper cells. Eur J Immunol. (2013) 43:3219–32. doi: 10.1002/eji.201343469
80. Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol. (2015) 6:16. doi: 10.3389/fimmu.2015.00016
81. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. (2009) 10:375–84. doi: 10.1038/ni.1704
82. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153:785–96. doi: 10.1016/j.cell.2013.04.007
83. Kim C, Wilson T, Fischer KF, Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4(+) memory T cells. Immunity (2013) 39:508–20. doi: 10.1016/j.immuni.2013.08.033
84. Koguchi Y, Thauland TJ, Slifka MK, Parker DC. Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood (2007) 110:2520–7. doi: 10.1182/blood-2007-03-081299
85. Fazilleau N, Eisenbraun MD, Malherbe L, Ebright JN, Pogue-Caley RR, McHeyzer-Williams LJ, et al. Lymphoid reservoirs of antigen-specific memory T helper cells. Nat Immunol. (2007) 8:753–61. doi: 10.1038/ni1472
86. Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, et al. Epitopes involved in antibody-mediated protection from Ebola virus. Science (2000) 287:1664–6. doi: 10.1126/science.287.5458.1664
87. Hofmeister Y, Planitzer CB, Farcet MR, Teschner W, Butterweck HA, Weber A, et al. Human IgG subclasses: in vitro neutralization of and in vivo protection against West Nile virus. J Virol. (2011) 85:1896–9. doi: 10.1128/jvi.02155-10
88. Huber VC, McKeon RM, Brackin MN, Miller LA, Keating R, Brown SA, et al. Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin Vacc Immunol. (2006) 13:981–90. doi: 10.1128/cvi.00156-06
89. Brooks DG, Teyton L, Oldstone MB, McGavern DB. Intrinsic functional dysregulation of CD4 T cells occurs rapidly following persistent viral infection. J Virol. (2005) 79:10514–27. doi: 10.1128/jvi.79.16.10514-10527.2005
90. Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T-cell exhaustion and recovery. Proce Natl Acad Sci USA. (2010) 107:20453–8. doi: 10.1073/pnas.1008437107
91. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. (2011) 208:987–99. doi: 10.1084/jem.20101773
92. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity (2014) 40:289–302. doi: 10.1016/j.immuni.2014.01.005
93. Greczmiel U, Krautler NJ, Pedrioli A, Bartsch I, Agnellini P, Bedenikovic G, et al. Sustained T follicular helper cell response is essential for control of chronic viral infection. Sci Immunol. (2017) 2:aam8686. doi: 10.1126/sciimmunol.aam8686
94. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science (2013) 340:202–7. doi: 10.1126/science.1235208
95. Osokine I, Snell LM, Cunningham CR, Yamada DH, Wilson EB, Elsaesser HJ, et al. Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc Natl Acad Sci USA. (2014) 111:7409–14. doi: 10.1073/pnas.1401662111
96. Snell LM, Brooks DG. New insights into type I interferon and the immunopathogenesis of persistent viral infections. Curr Opin Immunol. (2015) 34:91–8. doi: 10.1016/j.coi.2015.03.002
97. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science (2013) 340:207–11. doi: 10.1126/science.1235214
98. Harker JA, Dolgoter A, Zuniga EI. Cell-intrinsic IL-27 and gp130 cytokine receptor signaling regulates virus-specific CD4(+) T cell responses and viral control during chronic infection. Immunity (2013) 39:548–59. doi: 10.1016/j.immuni.2013.08.010
99. Harker JA, Lewis GM, Mack L, Zuniga EI. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science (2011) 334:825–9. doi: 10.1126/science.1208421
100. Raju S, Kometani K, Kurosaki T, Shaw AS, Egawa T. The adaptor molecule CD2AP in CD4 T cells modulates differentiation of follicular helper T cells during chronic LCMV infection. PLoS Pathog. (2018) 14:e1007053. doi: 10.1371/journal.ppat.1007053
101. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983–8. doi: 10.1038/nm.2426
102. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
103. Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. (2014) 211:1905–18. doi: 10.1084/jem.20132577
104. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat Immunol. (2016) 17:1187–96. doi: 10.1038/ni.3543
105. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
106. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature (2016) 537:412–28. doi: 10.1038/nature19317
107. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science (2009) 324:1569–72. doi: 10.1126/science.1174182
108. Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, et al. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science (2009) 324:1576–80. doi: 10.1126/science.1172815
109. Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science (2009) 324:1572–6. doi: 10.1126/science.1175194
110. Chevalier MF, Julg B, Pyo A, Flanders M, Ranasinghe S, Soghoian DZ, et al. HIV-1-specific interleukin-21+ CD4+ T cell responses contribute to durable viral control through the modulation of HIV-specific CD8+ T cell function. J Virol. (2011) 85:733–41. doi: 10.1128/jvi.02030-10
111. Battegay M, Moskophidis D, Waldner H, Brundler MA, Fung-Leung WP, Mak TW, et al. Impairment and delay of neutralizing antiviral antibody responses by virus-specific cytotoxic T cells. J Immunol. (1993) 151:5408–15.
112. Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, et al. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat Immunol. (2003) 4:343–9. doi: 10.1038/ni911
113. Buchmeier MJ, Oldstone MB. Virus-induced immune complex disease: identification of specific viral antigens and antibodies deposited in complexes during chronic lymphocytic choriomeningitis virus infection. J Immunol. (1978) 120:1297–304.
114. Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest. (2012) 122:3271–80. doi: 10.1172/jci64314
115. Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. (2013) 19:494–9. doi: 10.1038/nm.3109
116. Petrovas C, Yamamoto T, Gerner MY, Boswell KL, Wloka K, Smith EC, et al. CD4 T follicular helper cell dynamics during SIV infection. J Clin Invest. (2012) 122:3281–94. doi: 10.1172/jci63039
117. Xiao M, Chen X, He R, Ye L. Differentiation and function of follicular CD8 T cells during human immunodeficiency virus infection. Front Immunol. (2018) 9:1095. doi: 10.3389/fimmu.2018.01095
118. Kohler SL, Pham MN, Folkvord JM, Arends T, Miller SM, Miles B, et al. Germinal center T follicular helper cells are highly permissive to HIV-1 and alter their phenotype during virus replication. J Immunol. (2016) 196:2711–22. doi: 10.4049/jimmunol.1502174
119. Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, et al. PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat Med. (2016) 22:754–61. doi: 10.1038/nm.4113
120. Perreau M, Savoye AL, De Crignis E, Corpataux JM, Cubas R, Haddad EK, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. (2013) 210:143–56. doi: 10.1084/jem.20121932
121. Hufert FT, van Lunzen J, Janossy G, Bertram S, Schmitz J, Haller O, et al. Germinal centre CD4+ T cells are an important site of HIV replication in vivo. AIDS (1997) 11:849–57.
122. Streeck H, D'Souza MP, Littman DR, Crotty S. Harnessing CD4(+) T cell responses in HIV vaccine development. Nat Med. (2013) 19:143–9. doi: 10.1038/nm.3054
123. Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. (1983) 309:453–8. doi: 10.1056/nejm198308253090803
124. Wendel BS, Del Alcazar D, He C, Del Rio-Estrada PM, Aiamkitsumrit B, Ablanedo-Terrazas Y, et al. The receptor repertoire and functional profile of follicular T cells in HIV-infected lymph nodes. Sci Immunol. (2018) 3:eaan8884. doi: 10.1126/sciimmunol.aan8884
125. Vella LA, Herati RS, Wherry EJ. CD4(+) T cell differentiation in chronic viral infections: the Tfh perspective. Trends Mol Med. (2017) 23:1072–87. doi: 10.1016/j.molmed.2017.10.001
126. Bergthaler A, Flatz L, Verschoor A, Hegazy AN, Holdener M, Fink K, et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. (2009) 7:e1000080. doi: 10.1371/journal.pbio.1000080
127. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. (2012) 188:2957–65. doi: 10.4049/jimmunol.1100038
128. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol. (2017) 18:91–104. doi: 10.1038/nri.2017.112
129. McNeil AC, Shupert WL, Iyasere CA, Hallahan CW, Mican JA, Davey RT Jr, et al. High-level HIV-1 viremia suppresses viral antigen-specific CD4(+) T cell proliferation. Proc Natl Acad Sci USA. (2001) 98:13878–83. doi: 10.1073/pnas.251539598
130. Hryniewicz A, Boasso A, Edghill-Smith Y, Vaccari M, Fuchs D, Venzon D, et al. CTLA-4 blockade decreases TGF-beta, IDO, and viral RNA expression in tissues of SIVmac251-infected macaques. Blood (2006) 108:3834–42. doi: 10.1182/blood-2006-04-010637
131. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. (2008) 205:2763–79. doi: 10.1084/jem.20081398
132. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi: 10.1038/nature05115
133. Aubert RD, Kamphorst AO, Sarkar S, Vezys V, Ha SJ, Barber DL, et al. Antigen-specific CD4 T-cell help rescues exhausted CD8 T cells during chronic viral infection. Proc Natl Acad Sci USA. (2011) 108:21182–7. doi: 10.1073/pnas.1118450109
134. He R, Yang X, Liu C, Chen X, Wang L, Xiao M, et al. Efficient control of chronic LCMV infection by a CD4 T cell epitope-based heterologous prime-boost vaccination in a murine model. Cell Mol Immunol. (2017) 15:815–26. doi: 10.1038/cmi.2017.3
135. Frebel H, Richter K, Oxenius A. How chronic viral infections impact on antigen-specific T-cell responses. Eur J Immunol. (2010) 40:654–63. doi: 10.1002/eji.200940102
136. Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. (2005) 6:873–9. doi: 10.1038/ni1241
137. Good-Jacobson KL, Szumilas CG, Chen L, Sharpe AH, Tomayko MM, Shlomchik MJ. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat Immunol. (2010) 11:535–42. doi: 10.1038/ni.877
138. Barton E, Mandal P, Speck SH. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu Rev Immunol. (2011) 29:351–97. doi: 10.1146/annurev-immunol-072710-81639
139. Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. (2000) 165:1074–81. doi: 10.4049/jimmunol.165.2.1074
140. Stevenson PG, Doherty PC. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J Virol. (1999) 73:1075–9.
141. Decalf J, Godinho-Silva C, Fontinha D, Marques S, Simas JP. Establishment of murine gammaherpesvirus latency in B cells is not a stochastic event. PLoS Pathog. (2014) 10:e1004269. doi: 10.1371/journal.ppat.1004269
142. Barton ES, Rajkarnikar S, Langston PK, Price MJ, Grayson JM. Gammaherpesvirus latency differentially impacts the generation of primary versus secondary memory CD8+ T cells during subsequent infection. J Virol. (2014) 88:12740–51. doi: 10.1128/JVI.02106-14
143. Collins CM, Speck SH. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog. (2014) 10:e1004106. doi: 10.1371/journal.ppat.1004106
144. Sparks-Thissen RL, Braaten DC, Kreher S, Speck SH, Virgin HWt. An optimized CD4 T-cell response can control productive and latent gammaherpesvirus infection. J Virol. (2004) 78:6827–35. doi: 10.1128/JVI.78.13.6827-35.2004
145. Mylvaganam GH, Rios D, Abdelaal HM, Iyer S, Tharp G, Mavigner M, et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc Natl Acad Sci USA. (2017) 114:1976–81. doi: 10.073/pnas.1621418114
146. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. (2009) 9:91–105. doi: 10.1038/nri2487
147. Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity (2009) 30:155–67. doi: 10.1016/j.immuni.2008.12.009
148. Cook KD, Shpargel Karl B, Starmer J, Whitfield-Larry F, Conley B, Allard Denise E, et al. T follicular helper cell-dependent clearance of a persistent virus infection requires T cell expression of the histone demethylase UTX. Immunity (2015) 43:703–14. doi: 10.1016/j.immuni.2015.09.002
Keywords: follicular T helper cells, acute viral infection, chronic viral infection, differentiation, CD4 T cells +
Citation: Huang Q, Hu J, Tang J, Xu L and Ye L (2019) Molecular Basis of the Differentiation and Function of Virus Specific Follicular Helper CD4+ T Cells. Front. Immunol. 10:249. doi: 10.3389/fimmu.2019.00249
Received: 25 July 2018; Accepted: 29 January 2019;
Published: 15 February 2019.
Edited by:
Maria Pia Cicalese, San Raffaele Scientific Institute (IRCCS), ItalyReviewed by:
Francois Villinger, University of Louisiana at Lafayette, United StatesLinrong Lu, Zhejiang University, China
Copyright © 2019 Huang, Hu, Tang, Xu and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lifan Xu, eGxmdG9mdUBzaW5hLmNvbQ==
Lilin Ye, eWVsaWxpbmxjbXZAdG1tdS5lZHUuY24=