 Natalia M. Tijaro-Ovalle
Natalia M. Tijaro-Ovalle Theodoros Karantanos
Theodoros Karantanos Hong-Tao Wang1,2
Hong-Tao Wang1,2 Vassiliki A. Boussiotis
Vassiliki A. Boussiotis- 1Division of Hematology-Oncology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
- 2Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Utilization of the adaptive immune system against malignancies, both by immune-based therapies to activate T cells in vivo to attack cancer and by T-cell therapies to transfer effector cytolytic T lymphocytes (CTL) to the cancer patient, represent major novel therapeutic advancements in oncologic therapy. Allogeneic hematopoietic stem cell (HSC) transplantation (HSCT) is a form of cell-based therapy, which replaces the HSC in the patient's bone marrow but also serves as a T-cell therapy due to the Graft-vs.-leukemia (GVL) effect mediated by donor T cells transferred with the graft. Allogeneic HSCT provides one potentially curative option to patients with relapsed or refractory leukemia but Graft-vs.-Host-Disease (GVHD) is the main cause of non-relapse mortality and limits the therapeutic benefit of allogeneic HSCT. Metabolism is a common cellular feature and has a key role in the differentiation and function of T cells during the immune response. Naïve T cells and memory T cells that mediate GVHD and GVL, respectively, utilize distinct metabolic programs to obtain their immunological and functional specification. Thus, metabolic targets that mediate immunosuppression might differentially affect the functional program of GVHD-mediating or GVL-mediating T cells. Components of the innate immune system that are indispensable for the activation of alloreactive T cells are also subjected to metabolism-dependent regulation. Metabolic alterations have also been implicated in the resistance to chemotherapy and survival of malignant cells such as leukemia and lymphoma, which are targeted by GVL-mediating T cells. Development of novel approaches to inhibit the activation of GVHD-specific naïve T cell but maintain the function of GVL-specific memory T cells will have a major impact on the therapeutic benefit of HSCT. Here, we will highlight the importance of metabolism on the function of GVHD-inducing and GVL-inducing alloreactive T cells as well as on antigen presenting cells (APC), which are required for presentation of host antigens. We will also analyze the metabolic alterations involved in the leukemogenesis which could differentiate leukemia initiating cells from normal HSC, providing potential therapeutic opportunities. Finally, we will discuss the immuno-metabolic effects of key drugs that might be repurposed for metabolic management of GVHD without compromising GVL.
Introduction
Quiescent immune cells use glucose, amino acids, and lipids to meet their bioenergetic demands. ATP, the key energy-transporting molecule, is generated in every cell during the breakdown of such nutrients by glycolysis and OXPHOS. Depending on the functional demands, cell metabolism can be shifted toward anabolic reactions leading to production of molecules involved in biosynthesis necessary for cell growth, or toward catabolic reactions leading to breakdown of macromolecules and the generation of byproducts, which are subsequently used for energy generation or for construction of anabolic pathways. A balance of these anabolic and catabolic processes is mandatory for maintenance of metabolism homeostasis (1).
Glucose is the most abundant extracellular nutrient and, although ATP production during glucose catabolism by glycolysis is significantly lower compared to the ATP generated by OXPHOS reactions, it is faster and more efficient in increasing cellular ATP than mitochondrial metabolism. Glycolysis also supports cell growth because glycolytic intermediates provide a bridge to multiple biosynthetic pathways, including PPP that has an important role in building blocks necessary for nucleotide biosynthesis, rapid generation of metabolic intermediates, and cell growth (2, 3). Additionally, glycolysis fuels production of NADPH, which is mandatory not only for the support of anabolic pathways but also plays a crucial role in decreasing the oxidative stress in rapidly proliferating cells and maintaining the redox state of the cell (4). Pyruvate derived from glucose in glycolysis can be converted into lactate in the cytoplasm or into acetyl-CoA in the mitochondria to subsequently enter the TCA cycle (also known as Krebs cycle). In addition to producing intermediates that feed multiple biosynthetic pathways, the oxidative reactions of the TCA cycle generate NADH and FADH2 which are required for the donation of electrons to the electron-transport chain for OXPHOS.
Rapidly proliferating malignant cells preferentially use glucose to sustain their rapid growth in the hazardous TME (5). The preference of cells to ferment glucose to lactic acid, even in the presence of oxygen that could support OXPHOS, is known as the Warburg effect (6). Although originally observed in cancer cells, it is now known that the Warburg effect is used by most cell types, including immune cells, to generate energy during times of rapid growth, because using glucose for energy generation through glycolysis, spares other nutrients for usage in anabolic reactions.
Metabolic aberration provides a key signature that differentiates malignant hematopoietic cells from normally differentiating hematopoietic progenitors that give rise to committed progenitors and mature myeloid cells (7). As in other cancer types, the Warburg effect dominates the metabolic preference of leukemia cells (7, 8), whereas during normal HSC differentiation glycolysis declines and mitochondrial metabolism and FAO gradually increases (9) (Figure 1). It has been hypothesized that leukemia cells that are resistant to treatment and responsible for relapses, have features of “LSC” that have the ability to reproduce the disease in animal models (10). These LSC, also known as leukemia initiating cells, appear to have unique metabolic features that differentiate them not only from normal HSCs but also from other leukemia cells. These findings underline the significance of metabolism in leukemia initiation and relapse.
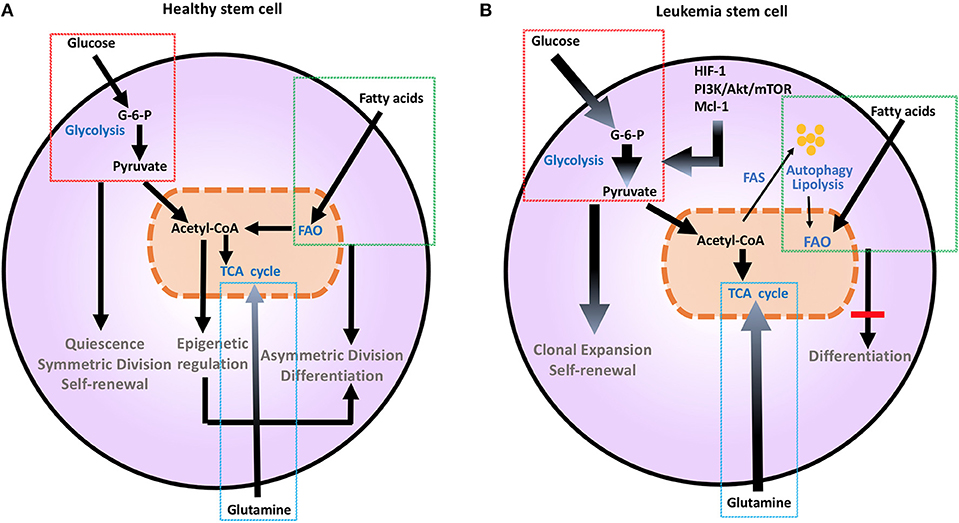
Figure 1. Metabolic changes in healthy (A) and leukemia stem cells (B). Metabolic changes drive stem cell differentiation in healthy subsets and clonal expansion in leukemia stem cells. Glucose uptake and glycolysis supports pluripotency and self-renewal of HSC, and is associated to the persistent ability of HSC to engage glycolysis, converting glucose to G-6-P and pyruvate, while sustaining a low ATP state. During differentiation, HSCs engage mitochondrial metabolic programs, including TCA cycle and FAO. This shift toward more efficient ATP generation is important for maturation and long-term survival. Mutations and aberrant protein expression related to malignant conversion of HSCs, including upregulation of HIF-1, PI3K/Akt/mTOR, and Mcl-1, induce metabolic changes. Leukemia stem cells support their uncontrolled clonal expansion by significantly increasing glucose uptake and metabolism (thick arrows). Although LSCs are highly dependent on glycolysis under steady state conditions, they have a high degree of metabolic plasticity and adaptation potential and can utilize autophagy and catabolic pathways such as OXPHOS and FAO in the setting of energy stress to support their survival and proliferation. Glutamine addiction is also an important feature common to malignant hematopoietic cells (thick arrow). Together these changes support extensive self-renewal without differentiation of LSC. Akt, Protein Kinase B; ATP, Adenosine triphosphate; G-6-P, Glucose-6-phosphate; FAO, Fatty acid oxidation; HIF-1, hypoxia induced factor 1; HSCs, hematopoietic stem cells; Mcl-1, myeloid cell leukemia 1; mTOR, Mechanistic/mammalian target of rapamycin; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinase; TCA, Tricarboxylic acid cycle.
AlloHSCT provides the only curative therapeutic approach for aggressive leukemias and lymphomas that are resistant to chemotherapy and immunotherapy. One of the key benefits of this therapy comes directly from the T cell-mediated offense to cancer, a process known as GVL effect (11). Nonetheless, T lymphocytes can also drive GVHD, the principal cause of non-relapse mortality among alloHSCT recipients. GVHD results from the attack of healthy recipient tissue by donor T cells that recognize host's alloantigens. Detailed, extensive studies have identified that T cells involved in GVHD are substantially different from the ones that mediate GVL (12–14). Specifically, naïve αβ TCR-positive T cells appear to be the main mediators of GVHD upon activation by host antigens (15). Conversely, TMEM subsets have been found to sustain GVL function (12–14), suggesting that the immunologic and metabolic mechanisms implicated in these two effects after allotransplantation are distinct. Moreover, Treg also play a role in GVHD and GVL as they have the ability to suppress GVHD without compromising GVL (16). As a consequence, Tregs have been considered a therapeutic target for the control of GVHD either as a cell-based immunotherapy (17, 18) or as an in vivo therapeutic target by using approaches that induce Treg differentiation and expansion (19, 20).
GVHD is the leading cause of non-relapse mortality after HSCT because its prevention and treatment remain challenging. Global immunosuppression is the mainstay of therapy for GVHD but responses are only partial in most cases. Moreover, complications of chronic immunosuppression are detrimental (21, 22). As an alternative, the administration of T cell depleted donor grafts has been tested, but the high relapse and infection rates seen in patients who receive these graft variants mostly guide against the use of this strategy (23). This renders the discovery of new strategies that can ameliorate GVHD, while preserving the benefits from GVL effect, a real necessity.
Metabolism is an attractive tentative target for therapeutic intervention both in cancer immunotherapy and GVHD. T cell subsets are poised to distinct metabolic pathways that can determine their function and differentiation (24, 25). Upon activation, naïve T cells rely on glycolytic metabolism to rapidly meet the bioenergetic needs required for their proliferation, TCR rearrangement, production of growth factors, and differentiation to TEFF. On the contrary, the function of Treg and TMEM cells depends on enhanced FAO (26, 27). Because distinct T cell subsets mediate GVHD vs. GVL, the dominant metabolic properties of these distinct subsets might serve as new therapeutic targets that can be exploited for prevention or suppression of GVHD without compromising GVL.
Although in the context of GVHD and GVL, emphasis has been placed on T cells, the innate immune cells of the host, particularly macrophages and dendritic cells, have an indispensable role in the activation of alloreactive T cells (28–31). Differentiation, proliferation and function of innate immune cells are also subjected to metabolism-dependent regulation (3). After allogeneic HSCT, these components of the immune system function in the context of the engrafted and rapidly expanding allogeneic HSC, residual leukemia cells potentially remaining at the state of MRD and rapidly dividing cells in host non-hematopoietic tissues that are the targets of GVHD, such as the gut (32, 33).
Based on the above, it is apparent that targeting metabolism for therapy of GVHD will require thorough understanding of the unique metabolic properties and programs of the multiple cellular components involved in GVHD and GVL. In the following sections we will briefly highlight the metabolic features of malignant hematopoietic cells and we will discuss the metabolic features that guide the function of T cells and APCs during processes involved in GVHD and GVL. We will also provide rationale for potential therapeutic interventions by targeting metabolic pathways that guide the differentiation and function of these immune cells in the context of alloHSCT.
Metabolism in Normal and Malignant Hematopoietic Cells
Metabolic changes drive division and differentiation of HSC and MP (9). HSCs are predominantly quiescent, in G0 phase, but divide approximately every 145 days, as a consequence of a cell-cycle-linked maturation process (34, 35). Their dormancy is important to sustain adult HSC pluripotency and to prevent HSC exhaustion (36). In order to maintain this state, HSCs utilize aerobic glycolysis and suppress oxidative phosphorylation, thereby maintaining an environment of low ROS (37). HSCs respond rapidly to stimuli to either maintain themselves via self-renewal by sustaining glycolytic metabolism and symmetric division or give rise to committed progenitors, by shifting their metabolism toward mitochondrial metabolism and activation of TCA cycle or FAO and asymmetric division (9) (Figure 1A). This is supported by the observation that depleting the mitochondrial oxidative phosphatase PTPMT1 blocks the entry into the cell cycle and differentiation of HSC (38). Maturation from a pluripotent state to a committed progenitor state also requires precise epigenetic modifications (39). Defects in DNA methyltransferases Dnmt3a and Dnmt3b that regulate such epigenetic effects are associated with impaired stem cell differentiation, leading to leukemia-inducing events (40).
Similarly to other malignant cell types, anabolic metabolism is the signature of malignant hematopoietic cells including AML, MM, and ALL (7, 41–43). This is mediated by upregulation of glucose transporters and increase of glucose uptake and glycolysis. Such changes are induced by molecular aberrations and inappropriate activation of signaling pathways such as PI3K/Akt/mTOR, enhanced pro-survival mechanisms, and hypoxia (5). Normal and malignant hematopoietic cells also highly depend on the use of glutamine. This is related to the expression of myc, which is proportional to HSC multipotency, cell-maintenance, and self-renewal (44). Upregulation of c-myc in high-grade lymphomas increases glutaminolysis and leads to glutamine dependence and addiction of malignant cells to support their biosynthesis pathways. Anaplerosis via glutamine usage in the TCA cycle may be a c-myc-mediated mechanism critical for survival and growth of malignant hematopoietic cells (45). An additional important anabolic pathway in malignant hematopoietic cells is fatty acid synthesis. Non-Hodgkin B-cell lymphoma cells are particularly sensitive to C-75, a fatty acid synthase inhibitor, supporting the premise that rapidly proliferative lymphoma cells are not only dependent on aerobic glycolysis but on other anabolic pathways for their growth and proliferation (46).
LSCs, which are responsible for survival and persistence of leukemia, are more dependent on aerobic glycolysis (Figure 1B) and display higher expression of the glycolysis enzymes PKM2 and LDH-A compared to normal HSCs. In turn, combined inhibition of PKM2 and LDH-A leads to eradication of LSCs (7). LSCs also rely on catabolic pathways for the production of energy and can utilize fatty acids for FAO in order to escape the detrimental effects of chemotherapy and maintain their survival under conditions of stress (47). Deletion of AMPK, an important sensor of energetic stress that maintains metabolic homeostasis by activating catabolic metabolism and autophagy, synergizes with metabolic stress caused by nutrient restriction in LSCs and profoundly suppresses leukemogenesis (48). In CML, autophagy acts as a possible mechanism of survival and resistance of leukemia to TKI treatment (49). Under these conditions, inhibition of mitochondrial OXPHOS can eradicate TKI-resistant CML LSCs (50). Thus, although LSCs are highly dependent on glycolysis under steady state conditions, they have a high degree of metabolic plasticity and adaptation potential and can utilize catabolic pathways in the setting of energy stress to support their survival and proliferation. The clinical relevance of the increased metabolic plasticity that is pivotal in LSCs is supported by the observation that BCL-2 blockage, which reduces OXPHOS, selectively eliminates this quiescent leukemia subset (51).
Our knowledge regarding the metabolic features of leukemia cells in relapsed or resistant disease in patients who undergo allogeneic HSCT is limited because relevant studies are currently missing. As mentioned above, relapsed or resistant leukemia cells display features of LSC, which are highly depend on glycolysis but also have the metabolic plasticity to adopt other metabolic programs for energy generation, including mitochondrial metabolism, FAO and autophagy. Thus, although therapeutic approaches to target glycolytic metabolism to inhibit activation of GVHD-mediating TEFF cells are expected to suppress or eradicate MRD, it is possible that plasticity and metabolic adaptation will allow LSC to survive by shifting their metabolic preferences. Focused studies are required to address this issue.
Immuno-Metabolic Reprograming and Hematopoietic Stem Cell Transplantation
Role of Metabolism in T Cell Differentiation and Relevance to Alloreactive T Cell Function
Resting T cells rely on mitochondrial respiratory capacity and OXPHOS for their metabolism and bioenergetic demands. Upon activation, they demand higher energetic supply, met mostly by the engagement of glycolytic pathway and mitochondrial OXPHOS (52). Similar to cancer cells, activated T cells predominantly depend on glycolysis for energy production and generation of biosynthetic intermediates while sparing other nutrients for anabolic reactions. Glycolysis has a key role in the differentiation of T effector cells. Conversely, glucose deprivation impairs the ability of CD8+ T cells to express IFN-γ gene, a signature of their differentiation into the effector state (53). Extracellular glucose that T cells uptake during the effector phase, supports fatty acid de novo synthesis and these newly synthesized lipids form the fuel used after the transition and differentiation of TEFF to TMEM cells (27). Environmental cues that promote TMEM cell differentiation, such as IL-15, promote mitochondrial biogenesis and the expression of Cpt1a, which allows entry of long chain fatty acids to the mitochondria and functions as the rate limiting enzyme for FAO. These immune-metabolic properties are associated with longevity and survival in high-stress environments (54). In contrast, pathologically activated lymphocytes, such as those in autoimmune diseases, activate mitochondrial metabolism but utilize glucose for OXPHOS (55).
Metabolic pathways are also linked to the functional differentiation and polarization of T cell subsets. Th1, Th2, Th17 and Tfh preferentially undergo glycolysis by increasing the expression of Glut1 and by activating the PI3K/Akt/mTOR pathway (26). mTOR plays a role as a cell nutrient sensor and is a crucial regulator of T cell metabolism (56) by activating anabolic reactions including glycolysis, but also fatty acid metabolism, by targeting SREBPs (57). Through these coordinated effects, mTORC1 leads to Th1 and Th17 differentiation along with regulation of T cell priming and generation of iTregs, while mTORC2 drives differentiation to Th2 (58). Although Th17 cells are known to depend on glycolysis (59), inhibiting ACC1, a key mediator for de novo fatty acid synthesis, impairs Th17 development in both human and mouse models, favoring the formation of anti-inflammatory Foxp3+ Tregs (60). The significance of these complex effects mediated by mTOR on pathways that regulate glycolysis and fatty acid metabolism are also supported by the implications induced on T cell differentiation and function by AMPK signaling (61) which negatively regulates mTOR-mediated glycolytic metabolism (62). AMPK promotes FAO by multiple mechanisms, including the direct regulation of key lipid metabolizing enzymes, the negative regulation of the mTOR and the intracellular transport of fatty acids (63–65). These coordinated processes, leading to lipid synthesis and utilization, provide two key properties of TMEM cells, namely longevity and immune quiescence (66).
It is therefore apparent that mTOR actively influences the differentiation of all T cell subsets that are involved in GVHD, including Th1, Th2, Th17 and Tfh cells. Th1, Th2 and Th17 have essential roles in the induction of aGVHD, while Tfh cells are pathogenic in cGVHD (67). Intriguingly, Tregs and TMEM cells, which appear to be protective from GVHD, also depend on mTOR for their differentiation and function (68, 69). Due to their overall inhibitory effect on T effector cell function, mTOR antagonists such as sirolimus are routinely used for the prophylaxis or treatment of GVHD in alloHSCT recipients (70). The addition of RGI-2001, a synthetic CD1 ligand that expands Tregs in vivo, to sirolimus results in a greater decrease in GVHD rates, as compared to the ones achieved by either compound alone (71).
Although these results indicate that mTOR antagonists support the activation and differentiation of Treg in vivo, the mechanistic role of mTOR in Treg biology remains controversial. The absence of mTORC1 signaling during T cell differentiation has been associated with lack of Th1/Th2 polarization and enhanced conversion to Treg phenotype (58). Surprisingly, conditional targeting of mTORC1 in Treg cells by deletion of the mTORC1 partner, Raptor, resulted in impaired fatty acid and cholesterol synthesis, leading to defective Treg generation and function (72). Conversely, the absence of mTORC2 signaling by deletion of the mTORC2 partner, Rictor, potentiated the generation of short-lived effector and memory precursor CD8+ T cells (73). The combined administration of the mTOR inhibitor, Rapamycin, and IL-2 not only preserved but also promoted Treg expansion and increased the donor CD4+ CD25+ Foxp3+ Tregs, resulting in decreased aGVHD-related mortality (74). This implies that Treg differentiation and function is positively regulated by mTOR inhibition and mTOR-independent IL-2-mediated signaling. Conversely, cyclosporin, which inhibits IL-2 production by targeting NFAT signaling, compromised Treg proliferation in vivo (68). In light of their specific effects on Treg differentiation and expansion, the mechanisms of these immunosuppressive agents in the prevention and treatment of GVHD should be revisited and their Treg-dependent immunoregulatory effects should be considered when these compounds are used for the prevention or treatment of GVHD.
Metabolism of Alloactivated GVHD-Mediating T Cells
After HSCT, naïve donor T cells are directed to the recipient secondary lymphoid tissues, where they become activated by recipient's alloantigens (32, 33).When this happens, an increase in glycolysis and OXPHOS is induced (75–77). Overall, glycolysis escalates, becoming the principal source of energy for GVHD-causing T cells that under these conditions convert to T effector cells (75). For this mechanism to be efficient, carbohydrate catabolism mediators are also highly upregulated. These metabolic changes alter the functional profile of GVHD-mediating T cells, which are no longer naïve, but undergo differentiation to T effectors, simultaneously with metabolic reprogramming and proliferation in response to alloantigen-mediated stimulation (75) (Figure 2). In comparison to mice that received syngeneic BMT, mice undergoing allogeneic transplant displayed higher ECAR, accumulation of glycolytic intermediates, increased levels of LDH-A, Mct4, Glut1 and Glut3 mRNA, along with higher glucose-6-phosphate levels, all of which imply higher glycolytic activity (75). Consistent with the key role of glycolysis in regulating alloreactive TEFF function, similarly to TEFF of different specificity, overexpression of Glut1 results in superior T cell survival (78).
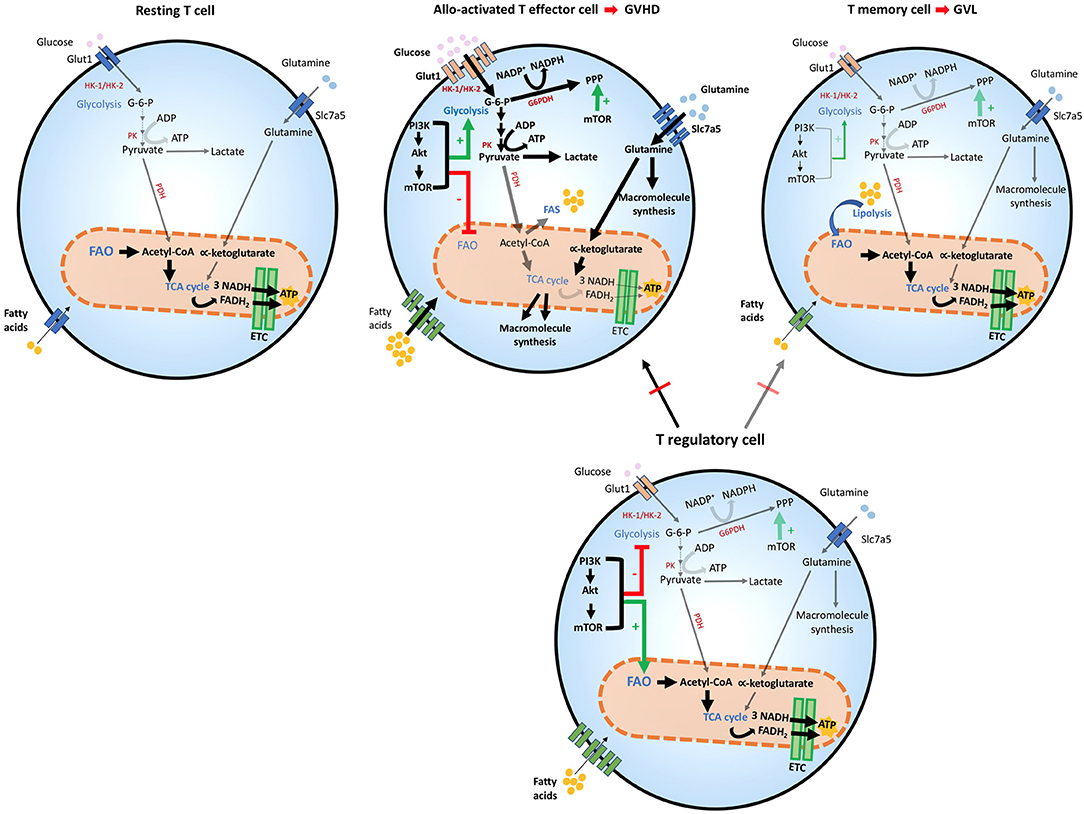
Figure 2. Metabolic reprogramming of T cells after allogeneic stem cell transplantation. T cells adapt to distinct stressors in order to meet their bioenergetic demand. After HSCT, the engagement of distinct metabolic pathways is correlated with T cell role and function. Glycolysis is the preferred pathway for GVHD-mediating alloactivated T naïve cells after exposure to host antigens, which differentiate them to their effector phenotype and increase the expression of Glut1 receptor. Glycolytic reactions also provide a bridge for macromolecule synthesis, redox balance and cell growth, by producing intermediate metabolites that favor PPP. The generation of pyruvate from glucose yields lactate in the cytoplasm or acetyl-CoA, which enters the TCA cycle in the mitochondria. In alloactivated GVHD-mediating T cells, expression of glucose, amino acid, and fatty acid transporters increase leading to enhanced entry of these compounds (thick arrows). The PI3K/Akt/mTOR pathway favors glycolysis and suppresses FAO. mTOR also potentiates PPP in these cells. GVHD-mediating T cells also use amino-acids, especially glutamine, which enters the cell by the increased expression of Slc7a5 transporters. Glutaminolysis yields α-ketoglutarate that enters the TCA cycle in the mitochondria although mitochondrial metabolism does not seem to be the dominant metabolic profile of alloreactive T cells. In T effector memory cells, the main T subset involved in mediating GVL function, FAO is the dominant energy source and mitochondrial metabolism is significantly enhanced, leading to OXPHOS reactions in the ETC, where higher amounts of ATP are produced, thereby providing sustained energy for cell survival. T regulatory cells are protective against GVHD, by inhibiting predominantly alloactivated GVHD-mediating T cells but inhibit GVL-mediating T memory cells to a lesser extent thereby preserving GVL. Treg cells share metabolic features with GVL cells, as they use FAO as their preferred energy source. mTOR regulation appears to have a distinct effect on Treg cells, as it is necessary for optimizing FAO that leads to adequate Treg differentiation and function. ADP, Adenosine diphosphate; ATP, Adenosine triphosphate; Akt, Protein Kinase B; ETC, Electron transport chain; FAD, Flavin adenine dinucleotide (oxidized state); FADH2, Flavin adenine dinucleotide (reduced state); G-6-P, Glucose-6-phosphate; G6PDH, Glucose-6-phosphate dehydrogenase; Glut1, Glucose transporter1; GVHD, graft-vs.-host diseases; GVL, graft-vs.-leukemia; HK-1/2, Hexokinase-1/2; HSCT, hematopoietic stem cell transplant; mTOR, Mechanistic/mammalian target of rapamycin; NAD, Nicotinamide adenine dinucleotide (oxidized state); NADH, Nicotinamide adenine dinucleotide (reduced state); NADP+, Nicotinamide adenine dinucleotide phosphate (oxidized state); NADPH, Nicotinamide adenine dinucleotide phosphate (reduced state); OXPHOS, Oxidative phosphorylation; PI3K, Phosphatidylinositol-4,5-bisphosphate 3-kinase; PPP, Pentose phosphate pathway; PK, Pyruvate kinase; Slc7a5, Solute carrier family 7 member 5; TCA cycle, Tricarboxylic acid cycle.
Glutamine metabolism is also a central component of T cell metabolic reprogramming during activation. T helper cell responses are supported by the upregulation of the glutamine/leucine transporter Slc7a5, and Slc7a5 null cells are unable to complete metabolic reprogramming and fail to undergo differentiation and clonal expansion (79). The role of glutamine in TEFF differentiation is supported by the observations that glutamine availability in the culture media increases IL-2 receptor expression, lymphocyte proliferation and cytokine production (79). Thus, glutamine is another critical source of energy and macromolecule production in activated T cells and might be involved in the development of alloreactive T cell responses and GVHD during alloHSCT (Figure 2).
Although mitochondrial metabolism has a role in the function of alloactivated T cells (76, 77, 80), it was also observed that regulators of fatty acid uptake and FAO are significantly reduced after autologous or allogeneic HSCT, compared to resting T cells. This correlated with metabolic reprogramming of alloreactive T cells to favor glycolytic metabolism and glutaminolysis as the key pathways for energy generation (75). FAO seems to increase in GVHD TEFF cells only after the fifth cell division, around 3 days post BMT (76), suggesting that these metabolic pathways might have distinct roles during the life of alloreactive TEFF cells in vivo. Nevertheless, most experimental evidence indicates that enhanced FAO is critical for TMEM (27) and Treg cell activity (26, 72) and for this reason it would be protective against GVHD (12, 15, 16, 19). Thus, the precise role of FAO in alloreactive T cell function and the details of its regulation in GVHD remain to be determined.
Studies have indicated that administration of metformin, which activates AMPK thereby promoting FAO, might attenuate GVHD by supporting the differentiation of Treg and altering the balance between Th17 and Treg cells (81). This observation potentially provides an opportunity to repurpose metformin for the prevention or treatment of GVHD. However, two important issues should be taken into consideration: First, AMPK does not exclusively affect the function of Treg because TEFF cells from AMPK KO mice display impaired differentiation and metabolic fitness, and impaired glutamine-dependent mitochondrial metabolism that allows TEFF metabolic plasticity and survival under low-glucose conditions (61); Second, in addition to activating AMPK, metformin can inhibit complex I of the electron transport chain (ETC) (82) which may impact the metabolism and function of all T cell subsets independently of AMPK. Thus, glycolysis, FAO and AMPK remain attractive metabolic targets to explore for therapeutic immunomodulation of alloreactive T cells by individual or combinatorial approaches.
Microbiota in T Cell Metabolism and GVHD
The investigation of the role of microbiota in host immunity in health and disease is a highly active topic with major biological relevance. Commensal bacteria are closely related to the host's nutritional status and the function of the immune system. Our understanding about their role in disease pathogenesis is rapidly expanding. It is now well-known that the development, differentiation and polarization of T lymphocytes is affected by gut microbes (83, 84). Commensal microbe-derived SC-FA, butyrate and propionate, can promote the differentiation of Treg cells (85). In mice, Clostridia strains can induce CD4+Foxp3+ Treg differentiation by producing SC-FA (86). Tregs induced by these microbiota can also induce IL-10 and ICOS, affect the intestinal immune function, and prevent colitis and allergic diarrhea (86). Conversely, segmented filamentous bacteria in mice and Bifidobacterium adolescentis in humans, promote Th17 differentiation, enhance Th17 cell survival (87, 88) and exacerbate autoimmune arthritis (88). The latter reinforces the importance of understanding the harmful impact of symbiont-driven T helper cells in the context of inflammatory conditions.
Not unexpectedly, SC-FA can influence the development of GVHD (89). SC-FA regulate both TEFF and Treg cells accumulation by increasing histone H3 acetylation in the locus of Foxp3 and activating the mTOR pathway (90, 91). Consequently, Butyrate restoration in intestinal epithelial cells, implemented to overcome the reduction caused by the inflammatory cascade seen in alloHSCT, promotes histone acetylation and correlates with lower GVHD clinical scores (92). These findings indicate that microbial-derived metabolic products have a potential use in GVHD, probably due their impact on T cell subset differentiation and survival.
Metabolism of Leukemia-Activated GVL Effector Cells
In mice, GVL effect is driven by CD4+ effector TMEM that require cognate interaction with MHC-II and leukemia antigens (12). Although mouse models do not fully recapitulate human TMEM life-long repertoire and CD4/CD8 ratio, it should be noted that the infusion of CD4+ TMEM to recipients of T cell-depleted human allografts effectively enhanced GVL and immune reconstitution without increasing GVHD (93). This observation provided an important insight on the potential use of sorted T cell populations to promote GVL, instead of administering unfractionated donor lymphocyte infusions, which are associated with GVHD (94). The therapeutic efficacy of this approach was explored by implementing selective depletion of T naïve cells from allografts given to high-risk leukemia patients. This modification of the allografts resulted in comparable rates of aGVHD but significantly improved responsiveness of aGVHD to steroid treatment. In addition, these patients had decreased cGVHD rates and improved immune reconstitution characterized by rapid T cell recovery and transfer of protective anti-viral immunity (93). Thus, selective utilization of donor TMEM cells might be the most preferred approach to preserve immunity while decreasing GVHD-mediated morbidity.
The reason for the differential action of T naïve and TMEM cells after allogeneic HSCT, has been hypothesized to rely on their differential responses. Unlike T naïve cells, T effector memory alloreactive cells cannot expand or sustain high magnitude responses and while they are less likely to induce cGVHD, they are sufficient to mediate GVL function (12, 15). Moreover, cytokine production by memory T cells is also suboptimal, compared to naïve T cells that rapidly increase aGVHD-associated cytokines TNF-α, IL-1, IL-6, and IFN-γ or the cGVHD-associated cytokine IL-17 (95). Because T naive cells that convert to effectors and T memory cells engage different metabolic pathways to meet their energetic demands, the distinct nature of GVL-specific and GVHD-specific alloreactive T cell populations might provide an excellent opportunity to introduce selective metabolism-targeting therapies, to optimize GVL and prevent the development of GVHD. Moreover, the differentiation of Treg cells that have the ability to suppress GVHD but not GVL (16, 17) are also supported by metabolic pathways similar to those engaged by TMEM cells such as oxidative metabolism and FAO (26, 54). This metabolic program of TMEM cells is supported by utilization of LC-FA for FAO (54). Although the mitochondrial transporter of LC-FAs, Cpt1a, is involved in this mechanism (54), subsequent studies discovered that pharmacologic inhibition or genetic ablation of Cpt1a did not affect the generation of TMEM (96), suggesting that T cells may metabolize short-chain fatty acid, in the absence of Cpt1a activity. Further, in Cpt1a KO T cells, the use the Cpt1a inhibitor, etomoxir, used in concentrations significantly higher than those required to inhibit Cpt1a, suppressed the generation of Tregs in vitro, suggesting an a Cpt1a-independent action (96). Thus, differentiation of TMEM and Treg depends on FAO that is regulated by CPT1a-dependent and independent mechanisms. FAO might be a tentative therapeutic target to induce TMEM and Treg differentiation in order to prevent GVHD and preserve GVL.
Metabolism of Antigen Presenting Cells and Relevance to Allogeneic-HSCT
The role of APCs, both from host and donor, in the setting of GVHD and GVL is a growing research focus during the past few years, given the recent understanding of their key role in both processes in alloHSCT (33, 97, 98). Recipient APCs are also important mediators of graft rejection, due to their potential to activate graft-infiltrating T cells (99). Today, it is well-known that the activation of the innate arm of the immune system is essential for the unfolding of GVHD, as APCs mediate T-cell priming and imprinting to GVHD target organs after transplantation (30, 98). Professional APCs, comprised DCs, B cells and macrophages, are capable of processing and presenting antigens to T cells through MHC proteins, promoting the formation of the immunological synapse that allows development of adaptive immune responses (98). During HSCT, host bone marrow APCs are mostly ablated by the conditioning regimen (100). Under these circumstances, skin macrophages and, to a lesser extent, dendritic cells engage in most of the antigen-presenting activities, mainly due to their resistance to myeloablative regimens (101, 102). Host and donor APCs have different roles in the development of GVHD. Host APCs seem to be involved in the induction of aGVHD, while donor macrophages contribute to cGVHD by cross-priming alloactivated CD8+ T cells (98, 103, 104). Replacing host APCs with donor APCs reverses T cell activation, as it decreases the interaction between GVHD-related host APCs and donor CD8+ T cells (28). Additionally, depletion of host liver and spleen-resident APCs results in decreased recruitment of allogeneic CD8+ T cells, thereby suppressing hepatic aGVHD but not skin involvement (29). Paradoxically, host APCs also take part in GVL function, whereas donor APCs only have a limited role in this process (105).
APCs activate different metabolic pathways, depending on the engagement of specific accessory surface receptors, cytokine stimulation and other microenvironmental cues. DCs increase their glycolytic activity upon their TLR activation as a means to produce enough pyruvate that can activate TCA cycle reactions and OXPHOS (3, 106). IFN-γ-mediated signals can direct macrophages into the classic M1 proinflammatory phenotype, in which glucose uptake via GLUT1 mediated influx predominates. LPS expressed on the outer membrane of gram-negative bacteria, after interacting with TLR4 on M1 macrophages, induces glycolysis, leading to lactate accumulation and production of TCA cycle metabolites, particularly succinate, which induces the IL1-β production and inflammation (107). Conversely, IL-10, IL-4, and IL-13, induce the alternative anti-inflammatory M2 phenotype, which relies mostly on mitochondrial respiration and instead of inducing tissue inflammation, promotes resolution of inflammation, tissue remodeling, and repair (108–110).
These extensive studies indicate that the metabolic profile of M1 macrophages has similarities to that exhibited by activated effector-like T cells (such as those inducing GVHD), while the metabolic phenotype of M2 macrophages parallels that of memory-like T cells (such as those inducing GVL). Thus, concomitant metabolic reprogramming of APCs and T cells will have an important role in the net outcome of GVHD and GVL and these outcomes might vary dependent on the metabolic polarization of one or both these immune populations. For instance, the preferential engagement of APCs and T cells in glycolytic metabolism will allow immune cells to sustain inflammatory GVHD-mediating functions by promoting the generation of GVHD-inducing M1 macrophages and effector-like T cells. Conversely, the metabolic shift of these cell populations toward oxidative phosphorylation might selectively promote the differentiation of GVL-inducing memory-like T cells while supporting M2 differentiation and resolution of inflammation thereby preventing or suppressing GVHD. Indeed, inhibition of FAO by etomoxir suppressed M2 polarization of macrophages (111) and TMEM cell differentiation in a Cpt1a dependent and independent manner (96). It should be noted that immune cell polarization in vivo is not an all or nothing event but rather a continuum that leads to an immune signature depending on the dominating metabolic balance, thereby providing an opportunity for therapeutic intervention through implementation of subtle metabolic changes that will influence both APCs and T cells. Future studies are needed to investigate whether targeting glycolytic metabolism will have a similar simultaneous effect to suppress both M1 macrophage polarization and generation of alloreactive T effector cells that mediate GVHD.
Therapeutic Relevance of Metabolism in Allogeneic Hematopoietic Stem Cell Transplantation
The central goal of post-transplant therapeutic immunomodulation is the prevention or treatment of GVHD, without diminishing GVL activity. As outlined in the previous sections, the distinct metabolic pathways in these processes point to potential new therapeutic targets. Although FAO and glutamine metabolism might have a role in the activation of GVHD-inducing alloreactive T cells, elegant work has provided evidence that glycolysis is the dominant metabolic pathway of alloactivated donor T cells and inhibition of glycolysis by targeting HK-2 or the rate-limiting PFKFB3 prevents alloreactivity in vivo and attenuates GVHD in HSCT recipient mice (75). Blocking PFKFB3 in cancer cells also downregulates glucose influx, thereby interfering with tumor growth and disease progression (112). Thus, targeting PFKB3 might control GVHD while suppressing metabolic activity and growth of residual leukemia cells. Other means of reducing glycolytic activity also translate into amelioration of GVHD severity. The role of glucose metabolism in GVHD is also supported by the observation that Glut1 transporter-deficient murine CD4+ effector T cells are unable to expand and induce GVHD in vivo, while, in this context, Treg population increases, showing independence from Glut1 (113). Notably, inhibiting glycolysis by using 2-deoxyglucose, not only diminished the expansion of TEFF cells but enhanced the differentiation of CD8+ TMEM cells (114). Similarly, IL-15-driven overexpression of Cpt1a induced TMEM cell production and supported their survival (54). Such modifications in the abundance of T cell subsets by targeting glycolysis might selectively prevent GVHD while preserving GVL.
Targeting PI3K/AKT/mTOR pathway has been explored in the context of alloHSCT, because this pathway is central to the activation, expansion and differentiation of TEFF, TMEM, and Treg cells. Inhibition of PI3K/AKT/mTOR with BEZ235, a dual PI3K/mTOR inhibitor, resulted in decreased T cell activation and diminished GVHD grade (115). Importantly, using rapamycin, an mTORC1 inhibitor, enforced FAO and increased TMEM cell differentiation (116). Because rapamycin also promotes the differentiation of Treg (69), such approach might selectively suppress GVHD and promote GVL by inducing TMEM and Treg. Thus, rapamycin might be repurposed and used not simply as an immunosuppressant but also as an immunomodulator to alter metabolism-driven differentiation of T cell subsets in recipients of alloHSCT.
Utilization of mitochondrial F1F0-ATPase inhibitor, Bz-423, promotes apoptosis of alloactivated cells, thereby reducing GVHD rates and improving survival without impairing immune reconstitution (77). The Cpt1a inhibitor, etomoxir, was also reported to decrease GVHD severity in mice after day 30 post-transplant, without impairing immune reconstitution (76). As mentioned above, etomoxir, used in concentrations significantly higher than those required to inhibit Cpt1a, suppressed the generation of Tregs in vitro, suggesting a Cpt1a-independent action (96). Such Cpt1a-independent effect of etomoxir was also observed in bone marrow derived macrophages from Cpt1/2 KO mice, in which etomoxir retained the ability to disrupt IL-4-mediated M2 macrophage polarization possibly by causing depletion of intracellular coA (111). Thus, the combined effects of such metabolism-targeting compounds might have implications in the components of the innate and adoptive immune system resulting in clinical effects on GVHD, GVL and immune reconstitution that are driven by the altered function of more than one immune population or by previously unidentified selective effects on a certain immune cell population. This is also supported by the observation that AEB071, an inhibitor of protein kinase C-θ that preferentially halts Treg differentiation and activation, preserves graft survival and GVL but prevents IFN-γ production and GVHD by enhancing the function of Treg (117).
Together, the results of targeting studies in various mouse models (54, 75, 77, 113, 115–117) are of direct clinical relevance and indicate that therapeutic targeting of selective components of signaling and metabolic pathways might have distinct outcomes on T cell differentiation and distinct clinical implications in the prevention and treatment of GVHD and GVL. Because possibly distinct metabolic mechanisms dominate during different phases of alloreactive T cell lifespan, it will be critical to determine the metabolic signatures of alloreactive GVHD- and GVL-specific T cells during various times after alloHSCT. Such knowledge will allow the design of proper therapeutic combinatorial therapies to selectively induce the desired metabolism-driven immune cell differentiation.
A major challenge when targeting metabolism for therapy of GVHD, will be to preserve the metabolic properties of pathogen-specific TEFF cells, which are mandatory for their function under conditions of stress and response to pathogens. Future work is required to identify and dissect the potential metabolic differences of pathogen-specific vs. host antigen-specific T cells that induce GVHD. Identifying pathways that dominate in each of these populations during their lifespan will allow the development of experimental approaches and clinical trials to implement and evaluate metabolic interventions in these distinct TEFF cell populations in parallel to studies dissecting the effects of such approaches on GVHD vs. GVL.
Concluding Remarks
Metabolism is a rapidly growing subject in immunology and malignant hematology. LSC that survive under intensive chemotherapy are responsible for MRD and relapse. These LSC use both anabolic and catabolic pathways, depending on the environmental cues. Our knowledge regarding the metabolic features of leukemia cells in relapsed or resistant disease in patients who undergo allogeneic HSCT is limited because relevant studies are currently missing. Based on current data, relapsed or resistant leukemia cells display features of LSC, which are highly dependent on glycolysis, but also have the metabolic plasticity to adopt other metabolic programs for energy generation, including mitochondrial metabolism, FAO and autophagy. Thus, although therapeutic approaches to target glycolytic metabolism employed to suppress GVHD-mediating TEFF cells are expected to suppress or eradicate leukemia cells, it is possible that the high degree of plasticity and metabolic adaptation of LSC may provide them the means to survive by shifting their metabolic preferences. Identifying LSC dominant pathways upfront and modulating them by metabolism-targeting interventions, together with chemotherapy, will be highly beneficial because will eradicate LSC, thereby minimizing the risk for relapse. It is particularly intriguing and hopeful to attempt achieving this objective, together with selective metabolism-driven differentiation of immune cell subsets, with the goal to minimize GVHD and enhance GVL after allogeneic HSCT.
Author Contributions
NT-O: wrote the main body of the manuscript and prepared figures; TK: wrote several sections of the manuscript and provided relevant citations; H-TW: wrote sections of the manuscript and provided relevant citations; VB: supervised NT-O, TK, and H-TW and was responsible for the overall preparation of the manuscript. All authors read and approved the content of the manuscript.
Funding
This work was supported by NIH/NCI grants: RO1CA183605-01, R01 CA229784-01, and RO1CA212605.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACC1, acetyl-CoA carboxylase 1; acetyl-CoA, acetyl coenzyme A; AEB071, Sotrastaurin; aGVHD, Acute graft-vs.-host-disease; ALL, Acute lymphoblastic leukemia; AML, Acute myelogenous leukemia; alloHSCT, Allogeneic hematopoietic stem-cell transplantation; AKT, Protein Kinase B; AMPK, AMP-activated protein kinase; APC, Antigen-presenting cell; ATP, Adenosine triphosphate; BEZ235, Dactolisib; BCL-2, B-cell lymphoma 2; Bz-423, Benzodiazepine (Bz)-423; C75, trans-4-Carboxy-5-octyl-3-methylene-butyrolactone; cGVHD, Chronic graft-vs.-host-disease; CML, Chronic myelogenous leukemia; CoA, Coenzyme A; CPT1a, carnitine palmitoyl transferase; DC, Dendritic cell; ECAR, extracellular acidification rate; F1F0-ATPase, F1 portion and a transmembrane FO portion of ATP synthase; FADH2, Flavin adenine dinucleotide; FAO, Fatty acid oxidation; FAS, Fatty acid synthesis; FOXP3, Forkhead-Box P3; Glut1, Glucose transporter 1; Glut 3, Glucose transporter 3; GVL, Graft-vs.-leukemia; GVHD, Graft-vs.-host-disease; HIF-1α, Hypoxia-inducible factor 1α; HK2, Hexokinase 2; HSC, hematopoietic stem cells; HSCT, hematopoietic stem cell transplantation; ICOS, Inducible T-cell costimulator; IFNγ, Interferon γ; IL-1/2/4/6/7/10/13/15, Interleukin 1/2/4/6/7/10/13/15; iTreg, inducible regulatory T cell; KO, knock out; LC-FA, Long-chain fatty acids; LPS, Lipopolisacharide; LSC, leukemia stem cells; LDH-A, Lactate dehydrogenase-A; M1, Classically activated macrophage; M2, Alternatively activated macrophage; MHC, Major Histocompatibility Complex; MHC-II, Major Histocompatibility Complex type 2; MM, Multiple myeloma; MRD, Minimal residual disease; MP, Myeloid progenitors; Mct4, Monocarboxylate transporter 4; mTOR, Mechanistic/mammalian target of rapamycin; mTORC1, Mechanistic/mammalian target of rapamycin complex 1; mTORC2, Mechanistic/mammalian target of rapamycin complex 2; Myc, Myc proto-oncogene; NADH, Nicotinamide adenine dinucleotide; NADPH, Nicotinamide adenine dinucleotide phosphate; NFAT, Nuclear factor of activated T cells; NOS, Nitric oxide synthase; OXPHOS, Oxidative phosphorylation; PFKFB3, 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase 3; PI3K, Phosphatidylinositol-4, 5-bisphosphate 3-kinase; PKM2, Pyruvate kinase M2; PPP, Pentose phosphate pathway; ROS, Reactive oxygen species; SREBPs, Sterol regulatory element-binding proteins; SC-FA, Short-chain fatty acids; SLC7A5, Solute Carrier Family 7 member 5; TCA cycle, Tricarboxylic acid cycle; TCR, T cell receptor; TEFF, T effector cells; Th, T helper cells; Th1, T helper 1; Th2, T helper 2; Th17, T helper 17; Tfh, follicular T helper; TLR, Toll-Like Receptor; TKI, Tyrosine kinase inhibitors; TME, Tumor microenvironment; TMEM, T memory; TNF α, Tumor necrosis factor α; Treg, Regulatory T cells.
References
1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
2. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
3. O'Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. (2016) 213:15–23. doi: 10.1084/jem.20151570
4. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
5. Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. (2016) 5:29. doi: 10.1186/s40169-016-0110-9
7. Wang Y-H, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. (2014) 158:1309–23. doi: 10.1016/j.cell.2014.07.048
8. Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, et al. Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation. Leuk Lymphoma. (2010) 51:2112–9. doi: 10.3109/10428194.2010.512966
9. Ito K, Ito K. Hematopoietic stem cell fate through metabolic control. Exp Hematol. (2018) 64:1–11. doi: 10.1016/j.exphem.2018.05.005
10. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. (1994) 367:645–8. doi: 10.1038/367645a0
11. Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. (1990) 75:555–62.
12. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood. (2008) 111:2476–84. doi: 10.1182/blood-2007-08-109678
13. Chen BJ, Cui X, Sempowski GD, Liu C, Chao NJ. Transfer of allogeneic CD62L- memory T cells without graft-versus-host disease. Blood. (2004) 103:1534–41. doi: 10.1182/blood-2003-08-2987
14. Dutt S, Baker J, Kohrt HE, Kambham N, Sanyal M, Negrin RS, et al. CD8+CD44hi but not CD4+CD44hi memory T cells mediate potent graft antilymphoma activity without GVHD. Blood. (2011) 117:3230–9. doi: 10.1182/blood-2010-10-312751
15. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest. (2003) 112:101–8. doi: 10.1172/JCI17601
16. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. (2003) 9:1144–50. doi: 10.1038/nm915
17. Taylor PA, Lees CJ, Blazar BR. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft-versus-host disease lethality. Blood. (2002) 99:3493–9. doi: 10.1182/blood.V99.10.3493
18. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood. (2011) 117:1061–70. doi: 10.1182/blood-2010-07-293795
19. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. New Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
20. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med. (2013) 5:179ra43. doi: 10.1126/scitranslmed.3005265
21. Inamoto Y, Flowers MED, Lee SJ, Carpenter PA, Warren EH, Deeg HJ, et al. Influence of immunosuppressive treatment on risk of recurrent malignancy after allogeneic hematopoietic cell transplantation. Blood. (2011) 118:456–63. doi: 10.1182/blood-2011-01-330217
22. Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat Rev Immunol. (2012) 12:443–58. doi: 10.1038/nri3212
23. Saad A, Lamb LS. Ex vivo T-cell depletion in allogeneic hematopoietic stem cell transplant: past, present and future. Bone Marrow Transplant. (2017) 52:1241–8. doi: 10.1038/bmt.2017.22
24. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Semin Immunol. (2016) 28:514–24. doi: 10.1016/j.smim.2016.10.009
25. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
26. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
27. O'Sullivan D, van der Windt GJW, Ching-Cheng Huang S, Curtis JD, Chang C-H, Buck MD, et al. Memory CD8+ T cells use cell intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
28. Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. (1999) 285:412–5.
29. Zhang Y, Shlomchik WD, Joe G, Louboutin J-P, Zhu J, Rivera A, et al. APCs in the liver and spleen recruit activated allogeneic CD8+ T cells to elicit hepatic graft-versus-host disease. J Immunol. (2002) 169:7111–8. doi: 10.4049/jimmunol.169.12.7111
30. Zhang Y, Louboutin J-P, Zhu J, Rivera AJ, Emerson SG. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell–mediated acute graft-versus-host disease. J Clin Invest. (2002) 109:1335–44. doi: 10.1172/JCI14989
31. Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, et al. Non-self recognition by monocytes initiates allograft rejection. J Clin Invest. (2014) 124:3579–89. doi: 10.1172/JCI74370
32. Beilhack A, Schulz S, Baker J, Beilhack GF, Wieland CB, Herman EI, et al. In vivo analyses of early events in acute graft-versus-host disease reveal sequential infiltration of T-cell subsets. Blood. (2005) 106:1113–22. doi: 10.1182/blood-2005-02-0509
33. MacDonald KP, Shlomchik WD, Reddy P. Biology of graft-versus-host responses: recent Insights. Biol Blood Marrow Transplant. (2013) 19:S10–4. doi: 10.1016/j.bbmt.2012.11.005
34. Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. (2008) 135:1118–29. doi: 10.1016/j.cell.2008.10.048
35. Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. (2008) 132:631–44. doi: 10.1016/j.cell.2008.01.025
36. Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. (2008) 9:115–28. doi: 10.1038/nrg2269
37. Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. (2015) 21:392–402. doi: 10.1016/j.cmet.2015.02.002
38. Yu W-M, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. (2013) 12:62–74. doi: 10.1016/j.stem.2012.11.022
39. Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic stem cells count and remember self-renewal divisions. Cell. (2016) 167:1296–309.e10. doi: 10.1016/j.cell.2016.10.022
40. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. (2011) 44:23–31. doi: 10.1038/ng.1009
41. McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M. Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood. (2012) 119:4686–97. doi: 10.1182/blood-2011-09-377846
42. Frolova O, Samudio I, Benito JM, Jacamo R, Kornblau SM, Markovic A, et al. Regulation of HIF-1α signaling and chemoresistance in acute lymphocytic leukemia under hypoxic conditions of the bone marrow microenvironment. Cancer Biol Ther. (2012) 13:858–70. doi: 10.4161/cbt.20838
43. Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. (2014) 124:1645–54. doi: 10.1182/blood-2014-02-554204
44. Laurenti E, Varnum-Finney B, Wilson A, Ferrero I, Blanco-Bose WE, Ehninger A, et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell. (2008) 3:611–24. doi: 10.1016/j.stem.2008.09.005
45. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
46. Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP, et al. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc Natl Acad Sci USA. (2012) 109:11818–23. doi: 10.1073/pnas.1205995109
47. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. (2016) 19:23–37. doi: 10.1016/j.stem.2016.06.001
48. Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell. (2015) 17:585–96. doi: 10.1016/j.stem.2015.08.019
49. Ianniciello A, Dumas P-Y, Drullion C, Guitart A, Villacreces A, Peytour Y, et al. Chronic myeloid leukemia progenitor cells require autophagy when leaving hypoxia-induced quiescence. Oncotarget. (2017) 8:96984–92. doi: 10.18632/oncotarget.18904
50. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. (2017) 23:1234–40. doi: 10.1038/nm.4399
51. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
52. Chang C-H, Curtis JD, Maggi LB, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
53. Cham CM, Gajewski TF. Glucose availability regulates IFN- production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. (2005) 174:4670–7. doi: 10.4049/jimmunol.174.8.4670
54. van der Windt GJW, Everts B, Chang C-H, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
55. Wahl DR, Petersen B, Warner R, Richardson BC, Glick GD, Opipari AW. Characterization of the metabolic phenotype of chronically activated lymphocytes. Lupus. (2010) 19:1492–501. doi: 10.1177/0961203310373109
56. Powell JD, Delgoffe GM. The mammalian target of rapamycin (mTOR) provides a critical link between T cell differentiation, function and metabolism. Immunity. (2010) 33:301–11. doi: 10.1016/j.immuni.2010.09.002
57. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. (2010) 39:171–83. doi: 10.1016/j.molcel.2010.06.022
58. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. (2009) 30:832–44. doi: 10.1016/j.immuni.2009.04.014
59. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
60. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
61. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
62. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
63. Jäger S, Handschin C, St.-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci USA. (2007) 104:12017–22. doi: 10.1073/pnas.0705070104
64. Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA. AMPKα1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol. (2013) 43:889–96. doi: 10.1002/eji.201243008
65. MacIver NJ, Blagih J, Saucillo DC, Tonelli L, Griss T, Rathmell JC, et al. The liver kinase B1 is a central regulator of T cell development, activation, and metabolism. J Immunol. (2011) 187:4187–98. doi: 10.4049/jimmunol.1100367
66. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
67. Forcade E, Kim HT, Cutler C, Wang K, Alho AC, Nikiforow S, et al. Circulating T follicular helper cells with increased function during chronic graft-versus-host disease. Blood. (2016) 127:2489–97. doi: 10.1182/blood-2015-12-688895
68. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. (2008) 111:453–62. doi: 10.1182/blood-2007-06-094482
69. Battaglia M, Stabilini A, Roncarolo M-G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. (2005) 105:4743–8. doi: 10.1182/blood-2004-10-3932
70. Lutz M, Mielke S. New perspectives on the use of mTOR inhibitors in allogeneic haematopoietic stem cell transplantation and graft-versus-host disease. Br J Clin Pharmacol. (2016) 82:1171–9. doi: 10.1111/bcp.13022
71. Chen Y-B, Efebera YA, Johnston L, Ball ED, Avigan D, Lekakis LJ, et al. Increased Foxp3+Helios+ Regulatory T cells and decreased acute graft-versus-host disease after allogeneic bone marrow transplantation in patients receiving sirolimus and RGI-2001, an activator of invariant natural killer T cells. Biol Blood Marrow Transplant. (2017) 23:625–34. doi: 10.1016/j.bbmt.2017.01.069
72. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. (2013) 499:485–90. doi: 10.1038/nature12297
73. Zhang L, Tschumi BO, Lopez-Mejia IC, Oberle SG, Meyer M, Samson G, et al. Mammalian Target of rapamycin complex 2 controls CD8 T cell memory differentiation in a foxo1-dependent manner. Cell Rep. (2016) 14:1206–17. doi: 10.1016/j.celrep.2015.12.095
74. Shin H-J, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 reduce lethal acute graft-versus-host disease associated with increased expansion of donor type CD4+CD25+Foxp3+ regulatory T cells. Blood. (2011) 118:2342–50. doi: 10.1182/blood-2010-10-313684
75. Nguyen HD, Chatterjee S, Haarberg KMK, Wu Y, Bastian D, Heinrichs J, et al. Metabolic reprogramming of alloantigen-activated T cells after hematopoietic cell transplantation. J Clin Invest. (2016) 126:1337–52. doi: 10.1172/JCI82587
76. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. (2013) 122:3230–7. doi: 10.1182/blood-2013-04-495515
77. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Transl Med. (2011) 3:67ra8. doi: 10.1126/scitranslmed.3001975
78. Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
79. Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
80. Glick GD, Rossignol R, Lyssiotis CA, Wahl D, Lesch C, Sanchez B, et al. Anaplerotic metabolism of alloreactive T cells provides a metabolic approach to treat graft-versus-host disease. J Pharmacol Exp Ther. (2014) 351:298–307. doi: 10.1124/jpet.114.218099
81. Park M-J, Lee S-Y, Moon S-J, Son H-J, Lee S-H, Kim E-K, et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl Res. (2016) 173:115–30. doi: 10.1016/j.trsl.2016.03.006
82. El-Mir MY, Nogueira V, Fontaine E, Avéret N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. (2000) 275:223–8. doi: 10.1074/jbc.275.1.223
83. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
84. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. (2012) 336:1268–73. doi: 10.1126/science.1223490
85. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
86. Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. (2013) 500:232–6. doi: 10.1038/nature12331
87. Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. (2015) 163:367–80. doi: 10.1016/j.cell.2015.08.058
88. Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, et al. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci USA. (2016) 113:E8141–50. doi: 10.1073/pnas.1617460113
89. Teshima T, Reddy P, Zeiser R. Acute graft-versus-host disease: novel biological insights. Biol Blood Marrow Transplant. (2016) 22:11–6. doi: 10.1016/j.bbmt.2015.10.001
90. Corrêa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MAR. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunol. (2016) 5:e73. doi: 10.1038/cti.2016.17
91. Short-Chain Fatty Acids Induce Both Effector and Regulatory T Cells by Suppression of Histone Deacetylases and Regulation of the mTOR-S6K Pathway. Available online at: https://www.ncbi.nlm.nih.gov/pubmed/24917457 (Accessed October 24, 2018).
92. Mathewson ND, Jenq R, Mathew AV, Koenigsknecht M, Hanash A, Toubai T, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol. (2016) 17:505–13. doi: 10.1038/ni.3400
93. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. (2015) 125:2677–89. doi: 10.1172/JCI81229
94. Or R, Hadar E, Bitan M, Resnick IB, Aker M, Ackerstein A, et al. Safety and efficacy of donor lymphocyte infusions following mismatched stem cell transplantation. Biol Blood Marrow Transplant. (2006) 12:1295–301. doi: 10.1016/j.bbmt.2006.07.014
95. Henden AS, Hill GR. Cytokines in graft-versus-host disease. J Immunol. (2015) 194:4604–12. doi: 10.4049/jimmunol.1500117
96. Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, et al. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. (2018) 28:504–15.e7. doi: 10.1016/j.cmet.2018.06.002
97. Kwan W-H, Hashimoto D, Paz-Artal E, Ostrow K, Greter M, Raedler H, et al. Antigen-presenting cell-derived complement modulates graft-versus-host disease. J Clin Invest. (2012) 122:2234–38. doi: 10.1172/JCI61019
98. Chakraverty R, Sykes M. The role of antigen-presenting cells in triggering graft-versus-host disease and graft-versus-leukemia. Blood. (2007) 110:9–17. doi: 10.1182/blood-2006-12-022038
99. Zhuang Q, Liu Q, Divito SJ, Zeng Q, Yatim KM, Hughes AD, et al. Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat Commun. (2016) 7:12623. doi: 10.1038/ncomms12623
100. Ogonek J, Kralj Juric M, Ghimire S, Varanasi PR, Holler E, Greinix H, et al. Immune Reconstitution after allogeneic hematopoietic stem cell transplantation. Front Immunol. (2016) 7:507. doi: 10.3389/fimmu.2016.00507
101. Auffermann-Gretzinger S, Eger L, Bornhäuser M, Schäkel K, Oelschlaegel U, Schaich M, et al. Fast appearance of donor dendritic cells in human skin: dynamics of skin and blood dendritic cells after allogeneic hematopoietic cell transplantation. Transplantation. (2006) 81:866–73. doi: 10.1097/01.tp.0000203318.16224.57
102. Durakovic N, Bezak KB, Skarica M, Radojcic V, Fuchs EJ, Murphy GF, et al. Host-derived langerhans cells persist after MHC-matched allografting independent of donor T cells and critically influence the alloresponses mediated by donor lymphocyte infusions. J Immunol. (2006) 177:4414–25. doi: 10.4049/jimmunol.177.7.4414
103. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med. (2004) 10:987–92. doi: 10.1038/nm1089
104. Alexander KA, Flynn R, Lineburg KE, Kuns RD, Teal BE, Olver SD, et al. CSF-1–dependant donor-derived macrophages mediate chronic graft-versus-host disease. J Clin Invest. (2014) 124:4266–80. doi: 10.1172/JCI75935
105. Reddy P, Maeda Y, Liu C, Krijanovski OI, Korngold R, Ferrara JLM. A crucial role for antigen-presenting cells and alloantigen expression in graft-versus-leukemia responses. Nat Med. (2005) 11:1244–9. doi: 10.1038/nm1309
106. Everts B, Pearce EJ. Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front Immunol. (2014) 5:203. doi: 10.3389/fimmu.2014.00203
107. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
108. Galván-Peña S, O'Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
109. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. (2011) 6:275–97. doi: 10.1146/annurev-pathol-011110-130138
110. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. (1992) 176:287–92.
111. Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. (2018) 28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001
112. Lu L, Chen Y, Zhu Y. The molecular basis of targeting PFKFB3 as a therapeutic strategy against cancer. Oncotarget. (2017) 8:62793–802. doi: 10.18632/oncotarget.19513
113. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
114. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. (2013) 123:4479–88. doi: 10.1172/JCI69589
115. Herrero-Sánchez MC, Rodríguez-Serrano C, Almeida J, San Segundo L, Inogés S, Santos-Briz Á, et al. Targeting of PI3K/AKT/mTOR pathway to inhibit T cell activation and prevent graft-versus-host disease development. J Hematol Oncol. (2016) 9:113. doi: 10.1186/s13045-016-0343-5
116. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. (2009) 460:108–12. doi: 10.1038/nature08155
Keywords: T cells, antigen presenting cells (APCs), GVHD, metabolism, GVL
Citation: Tijaro-Ovalle NM, Karantanos T, Wang H-T and Boussiotis VA (2019) Metabolic Targets for Improvement of Allogeneic Hematopoietic Stem Cell Transplantation and Graft-vs.-Host Disease. Front. Immunol. 10:295. doi: 10.3389/fimmu.2019.00295
Received: 14 November 2018; Accepted: 05 February 2019;
Published: 05 March 2019.
Edited by:
Brian Christopher Betts, University of Minnesota Twin Cities, United StatesReviewed by:
Robert James Salmond, University of Leeds, United KingdomMark A. Schroeder, Washington University in St. Louis, United States
Craig Alan Byersdorfer, University of Pittsburgh, United States
Copyright © 2019 Tijaro-Ovalle, Karantanos, Wang and Boussiotis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vassiliki A. Boussiotis, dmJvdXNzaW9AYmlkbWMuaGFydmFyZC5lZHU=