 Silvia Menegatti
Silvia Menegatti Elisabetta Bianchi
Elisabetta Bianchi Lars Rogge
Lars Rogge- 1Immunoregulation Unit, Department of Immunology, Institut Pasteur, Paris, France
- 2Unité Mixte de Recherche, Institut Pasteur/AP-HP Hôpital Cochin, Paris, France
- 3Université Paris Diderot, Sorbonne Paris Cité, Paris, France
Immune-mediated inflammatory diseases (IMIDs), such as spondyloarthritis (SpA), psoriasis, Crohn's disease (CD), and rheumatoid arthritis (RA) remain challenging illnesses. They often strike at a young age and cause lifelong morbidity, representing a considerable burden for the affected individuals and society. Pioneering studies have revealed the presence of a TNF-dependent proinflammatory cytokine cascade in several IMIDs, and the introduction of anti-TNF therapy 20 years ago has proven effective to reduce inflammation and clinical symptoms in RA, SpA, and other IMID, providing unprecedented clinical benefits and a valid alternative in case of failure or intolerable adverse effects of conventional disease-modifying antirheumatic drugs (DMARDs, for RA) or non-steroidal anti-inflammatory drugs (NSAIDs, for SpA). However, our understanding of how TNF inhibitors (TNFi) affect the immune system in patients is limited. This question is relevant because anti-TNF therapy has been associated with infectious complications. Furthermore, clinical efficacy of TNFi is limited by a high rate of non-responsiveness (30–40%) in RA, SpA, and other IMID, exposing a substantial fraction of patients to side-effects without clinical benefit. Despite the extensive use of TNFi, it is still not possible to determine which patients will respond to TNFi before treatment initiation. The recent introduction of antibodies blocking IL-17 has expanded the therapeutic options for SpA, as well as psoriasis and psoriatic arthritis. It is therefore essential to develop tools to guide treatment decisions for patients affected by SpA and other IMID, both to optimize clinical care and contain health care costs. After a brief overview of the biology of TNF, its receptors and currently used TNFi in the clinics, we summarize the progress that has been made to increase our understanding of the action of TNFi on the immune system in patients. We then summarize efforts dedicated to identify biomarkers that can predict treatment responses to TNFi and we conclude with a section dedicated to the recently introduced inhibitors of IL-17A and IL-23 in SpA and related diseases. The focus of this review is on SpA, however, we also refer to RA on topics for which only limited information is available on SpA in the literature.
Introduction
Immune-Mediated Inflammatory Diseases—An Overview
Immune-mediated inflammatory diseases (IMID) is a term used to define a group of clinically heterogeneous, unrelated conditions that share common inflammatory pathways and derive from aberrant immune responses of the human adaptive or innate immune system. Overall, the estimated incidence of IMIDs in Western populations approximates 5–7% (1) and encompasses over 100 different clinical disorders such as rheumatoid arthritis (RA), inflammatory bowel disease (IBD), spondyloarthritis (SpA) or ankylosing spondylitis (AS), systemic lupus erythematosus (SLE), and psoriasis. The immune dysregulation in IMIDs causes significant morbidity and is a considerable burden for the patients in terms of pain, limited mobility and diminished quality of life, as well as for the society, because of the associated high health-care costs, and the loss of productivity. Our understanding of the pathogenic mechanisms involved in these diseases remains very limited but recent advances revealed that they are likely to derive from a complex interplay between extrinsic environmental triggers and genetic risk factors (2). Several environmental factors have been recognized to play an important role in the risk of developing immune-mediated inflammatory diseases, including smoking, diet, excess alcohol, antibiotic intake, infections, and socioeconomic status (3, 4). However, there is limited evidence of their causality with respect to IMIDs.
Recent genome-wide association studies (GWAS) performed with thousands of patients and controls from different populations have provided detailed information about the genetic variants associated with immune-mediated inflammatory diseases (5). These studies have brought to the forefront many genes linked to signaling pathways that were not known to be involved in the pathogenesis, pointing to new directions in the study of disease mechanisms. At present, more than 600 loci affecting susceptibility to chronic inflammation and/or autoimmune disorders have been mapped by GWAS, revealing many loci that are common to several immune-mediated disorders, suggesting that these conditions may share pathways (6–8). A recent meta-analysis of multiple sclerosis (MS) combining independent GWAS results and genotyping of 80,000 cases and controls, revealed 110 non-MHC risk loci, the majority of which were mapped in the proximity of genes involved in different immune processes. Moreover, the genetic risk loci identified for MS have prominent intersections with loci for other chronic inflammatory diseases, such as IBD, ulcerative colitis, Crohn's disease, celiac disease, rheumatoid arthritis and psoriasis (7, 9, 10). Another recently published meta-analysis of IBD identified a total of 163 genetic risk loci, of which one third was found to overlap with loci previously identified in other inflammatory and autoimmune diseases (7, 11). Therefore, GWAS provided fundamental evidence for a key role of the immune system in the pathogenesis of these diseases, as many of the identified loci map to genes involved in different immune processes. However, for most single nucleotide polymorphisms (SNPs), the mechanisms by which they affect pathogenesis and the targeted cell populations are still unknown.
Frequently, multiple immune-mediated inflammatory disorders co-exist within the same patient. This was observed in a large study involving 3,287 AS patients, of which the 39% also developed uveitis, 16% psoriasis, and 8% inflammatory bowel disease (12). In addition, different IMIDs may co-exist within the same family (13). Considerable progress in the classification of these different disorders in the same group of diseases derives from the introduction in the clinic of tumor necrosis factor-α (TNFα) inhibitors, demonstrating clinical benefit in a number of different diseases, such as rheumatoid arthritis, Crohn's disease, psoriasis, and AS and this concept has been used to establish a cytokine-based disease taxonomy (7, 14). Taken together, this information provides evidence that immune-mediated inflammatory diseases are complex disorders that may share pathogenic mechanisms and triggers, such as environmental factors and genetic susceptibility, so that different diseases may be present in the same patient. Our limited understanding of the pathogenic mechanisms involved in these diseases currently hinders early diagnosis and the development of more specific and effective therapies.
Biology of TNF-α
Cloned and characterized by Pennica et al. (15), Tumor Necrosis Factor alpha (TNF-α) is a potent pro-inflammatory cytokine secreted by different immune cells, such as activated NK and T-cells, macrophages, monocytes, and neutrophils. TNF-α is also produced by non-immune cells, including fibroblasts and endothelial cells (16). Monocytes and macrophages are the primary source of TNF-α in response to inflammatory stimuli (17). At the transcriptional level, TNF mRNA is induced by the cooperation of AP-1 transcription factors with nuclear factor associated with activated T cells (NFAT) and nuclear factor-kB (NF-kB), which can bind directly to the promoter of the TNF gene (18).
The first studies on TNF-α characterized its biological function as a potent tumoricidal, in particular as an inducer of tumor hemorrhagic necrosis in vivo and a promoter of programmed cell death (apoptosis) in vitro (19). Subsequent studies have shown that TNF-α is implicated in a wide spectrum of biological effects. In the immune system, these include: (i) promoting monocyte/macrophage differentiation (20, 21); (ii) enhancing activated B cell proliferation (22, 23); (iii) inducing inflammation, often acting together with IL-1β (24), to protect against viral and bacterial infections (i.e., Mycobacteria tuberculosis) (25). Other functions of TNF include mediation of cachexia, apoptosis, regulation of cell proliferation and maturation of myeloid cells [reviewed by (26)].
The pleiotropic effects of TNF can be understood by the complexity of the signaling pathways activated. Like most of the TNF super family members, TNF-α is synthesized as a 26 kDa type II transmembrane protein (tmTNF), which is subsequently cleaved in the extracellular domain by the metalloprotease TNF-converting enzyme (TACE, also called ADAM-17), resulting in the release of the mature soluble TNF monomer (sTNF), a protein of 17 kDa (27, 28). The same enzyme can cleave TNF receptors (TNFRs) from the cell surface, a mechanism that downregulates receptor expression and releases circulating TNFRs that may act as inhibitors. Both soluble and mTNF can be found as monomers, or assembled in biologically active trimers of 51 kDa.
TNF-α exerts its activity by binding to two different receptors that differ in cellular localization and signaling mechanisms (Figure 1). Like their ligands, both TNFR1 and TNFR2 receptors are trimerized in biological active complexes through a conserved domain in their extracellular region that mediates ligand-independent receptor assembly. While TNFR1 is engaged by both soluble and membrane-bound TNF, TNFR2 is thought to be mainly activated by mTNF (29, 30). However, both receptors are co-expressed on immune cell types, and it has been suggested that they could also signal cooperatively (31, 32).
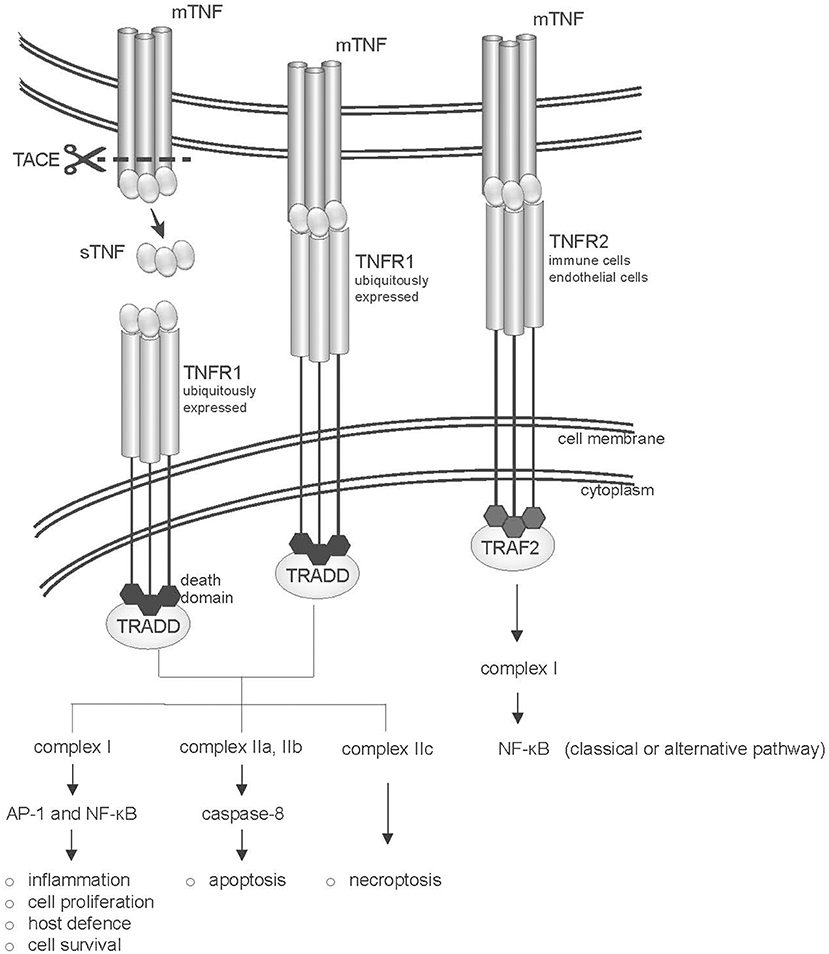
Figure 1. Structure of the TNF-TNFR system. The two TNF receptors (TNFR1 and TNFR2) are shown. TNFR1 and TNFR2 bind both soluble (sTNF) and transmembrane-TNF (mTNF) trimers, however TNFR2 is mainly activated by mTNF. TNFR1 is ubiquitously expressed and in its intracellular portion bears a “death domain” motif (dd), which recruits the adaptor protein TNFR1-associated death domain protein (TRADD). Binding of TNF to TNFR1 leads to the activation of several pathways, including inflammation, tissue degeneration, cell survival and proliferation or alternatively apoptosis or necroptosis. TNFR2 recruits TNFR-associated factor 2 (TRAF2) via its TRAF domain, activating the classical or alternative NF-kB pathways.
The 55-kDa TNFR1 (also known as p55 or CD120a, encoded by TNFRSF1A) is ubiquitously expressed (except for erythrocytes), and is characterized by the presence in its intracellular portion of a “death domain” motif. Binding of TNF-α to TNFR1 initiates a complex network of downstream events that may result in both the induction of apoptosis and of acute inflammation. Upon activation, TNFR1 recruits TNFR1-associated death domain protein (TRADD) to the plasma membrane, followed by the assembly of a scaffolding signaling complex (complex I) that includes TNF receptor associated factor 2 (TRAF2), and that results in the activation of AP-1 and NF-kB transcription factors (33, 34). The activation of these transcriptional pathways leads to the expression of genes involved in the defense against pathogens, inflammation, cell proliferation and survival (see https://www.bu.edu/nf-kb/gene-resources/target-genes/) (35). Among the genes induced by NF-kB are many chemokines and cytokines (including IL-6, IL-1β, and IFNγ), as well as several anti-apoptotic factors, such as cIAP-1, cIAP-2, cFLIP, TRAF1, and TRAF2 (36), that suppress caspase 8 activation. On the other hand, TNF binding also induces internalization of TNFR1 in the endocytic compartment, which is crucial for the recruitment to the receptor of TRADD-FADD containing complexes (complexes IIa, b, and c). Complexes IIa and IIb promote cleavage of caspase 8 and apoptosis (35, 37, 38). Complexe IIc activates the process of necroptosis, that is, programmed cell death associated with rupture of the plasma membrane and release of molecules that elicit inflammation (39).
The relevance of TNFR1 for immune-mediate diseases is supported by the association of genetic variations at the TNFRSF1A locus with AS, primary biliary cirrhosis and MS (https://www.immunobase.org/). A genetic variant identified selectively in multiple sclerosis has been demonstrated to affect splicing and induce expression of a soluble form of the receptor, which can block TNF. Of note, this variant has not been identified in diseases that are ameliorated by inhibition of TNF, such RA, psoriasis, and Crohn's disease (40).
The 75-kDa TNFR2 (also known as p75/p80 or CD120b, encoded by TNFRSF1B) is mainly expressed on lymphocytes, endothelial cells and astrocytes or oligodendrocytes (33), and undergoes transcriptional and posttranscriptional regulation in response to external stimuli (41). TNFR2 preferentially binds mTNF (29), and lacks a death domain. The intracellular portion of TNFR2 presents a transmembrane immunoglobulin and mucin (TIM) domain, which interacts with TRAF adaptor proteins (33, 34). Signaling occurs through the assembly of a complex I containing as a major adaptor protein TRAF2. This leads to the activation of the JNK kinase and of the AP1 transcriptional complex, and of NF-κB signaling through both the classical and alternative pathways (42, 43). Signaling through TNFR2 is subject to an autoregulatory loop, where TRAF2 undergoes ubiquitin-dependent degradation, following TNFR2 activation (44).
A biallelic polymorphism in exon 6 of TNFR2 has been described to result in a non-conservative amino acid substitution (methionine to arginine at codon 196) in the membrane proximal region of TNFR2 (45). This variant has been associated with chronic inflammatory disorders, such as systemic lupus erythematosus, familial rheumatoid arthritis, and ulcerative colitis (46, 47). The mutated receptor shows reduced recruitment of TRAF2 upon TNF-α stimulation and reduced NF-kB activation (48).
The important pathogenetic role of TNF-α in chronic inflammatory diseases is supported by the therapeutic efficacy of anti-TNF agents.
The Advent of TNF Inhibitors
As mentioned above, TNF-α was initially considered as a potential onco-therapeutic agent. However, despite the name, administration of recombinant TNF-α to patients with malignant diseases resulted in disease progression and severe side effects, rather than disease improvement (49). Phase I clinical trials showed dose-dependent acute toxicities including fevers, chills, nausea, and confusion. Once its role as an early and primary cytokine implicated in the inflammatory immune response had been established, several studies on animal models demonstrated the central importance of TNF-α in the pathogenesis of a wide range of immune-mediated inflammatory diseases, suggesting its blockade as a therapeutic approach. Beutler and colleagues showed that neutralizing antibodies against TNF protected the animals against TNF-mediated endotoxemia (50). The idea of using compounds blocking TNF-α was further supported by the increased levels of TNF-α in the serum or tissues of patients with inflammatory diseases or infections, and in individuals affected by sepsis (51, 52). The concept that blocking a single pro-inflammatory cytokine such as TNF-α could restore homeostasis of a complex network and ameliorate signs and symptoms of chronic inflammatory diseases was a real breakthrough in medical practice. Originally developed for the treatment of rheumatoid arthritis (RA), the therapeutic employment of anti-TNF agents was extended to the treatment of ankylosing spondylitis in the early 2000 (53–55).
Initially, treatment options for IMID were limited to non-steroidal anti-inflammatory drugs (NSAIDs) and physical therapy. At present, NSAIDs such as ibuprofen, naproxen, diclofenac, celecoxib, highly effective in reducing stiffness and back pain in axial SpA, are recommended by the ASAS/EULAR guidelines as first-line treatment (56). When patients do not respond to, or do not tolerate NSAIDs, anti-TNF agents are approved as a second step of medical intervention (57). TNF-neutralization has been very successful for the treatment of SpA in the past decade, however, as for other chronic inflammatory diseases, 30–40% of SpA patients do not respond or respond inadequately to the therapy. In clinical practice, non-responsive patients are treated with various molecules until an effective therapeutic agent is identified. However, this procedure is expensive and may take a long time, during which the patient is not appropriately treated and is exposed to side effects without clinical benefit.
The most common side effect observed during anti-TNF therapy is the increased risk for serious chronic infections, in particular affecting the respiratory tract. Reactivation of latent tuberculosis (TB) remains indeed the major concern (58). Other infections are also recurrent, such as histoplasmosis, Pneumocystis pneumonia, influenza, and adenovirus infections, latent viral infections (varicella-zoster, herpes-zoster), skin and soft tissue infections and urinary tract infections. Cases of exacerbated legionella were also reported. More rare side effects include severe hepatic reactions, nervous system disorders, hypersensitivity reactions and leucopenia. Some meta-analyses have been performed to assess the increased risk of malignancies following anti-TNF therapy and there is no proven increase of any malignancy so far. An increased risk of non-melanoma skin cancer during therapy with TNF-blockers has been reported (59, 60). Increased frequencies of anti-double-stranded DNA antibodies have been observed in some patients (61).
TNF Inhibitors Currently in Clinical Use
Natural and engineered anti-TNF antibodies share a similar structure (Figure 2), constituted of 2 heavy (H) chains and 2 light (L) chains connected by disulphide bonds at the hinge region. The two chains contains a variable region (V) in the N-terminal domains (VH and VL), that recognizes the target and a C-terminal constant region (Fc). The engineered monoclonal antibody monomer comprises a variable region that can be either murine or human, and a constant region (Fc), usually human to preserve favorable pharmacokinetic properties.
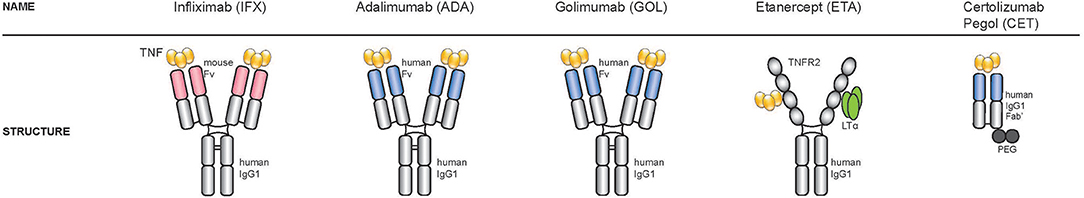
Figure 2. Structure of the five TNF-inhibitors approved for the treatment of spondyloarthritis. Starting from the left: three TNF-inhibitors are full-length bivalent IgG monoclonal antibodies (infliximab, adalimumab, golimumab), one is a soluble receptor (etanercept) and one a PEGylated Fab fragment of a monoclonal antibody (certolizumab).
At present, there are five TNF-inhibitors approved for the treatment of spondyloarthritis in the UE, the USA and many other countries (57). Three are full-length bivalent IgG monoclonal antibodies (adalimumab, golimumab, infliximab), one is a soluble receptor (etanercept) and one a PEGylated Fab fragment of a monoclonal antibody (certolizumab).
The first TNF blocker introduced in the clinic for the treatment of RA was the monoclonal antibody infliximab (trade name Remicade) (62). Infliximab is a chimeric human IgG1 antibody linked to murine immunoglobulin variable regions with specificity for human TNF (63, 64). Produced by hybridoma cells, infliximab can bind both soluble and membrane-bound TNF-α, therefore preventing the interaction of TNF with its receptors. Infliximab is administered intravenously and as a consequence requires “day hospital” treatment.
Adalimumab (commercial name Humira) and golimumab (Simponi) (65), are fully human monoclonal antibodies. Adalimumab was developed using phage display, golimumab is obtained from the immunization with TNF of transgenic mice expressing human IgGs (66) and both antibodies are potentially less immunogenic than infliximab.
A different type of anti-TNF compound is etanercept (commercial name Enbrel), an engineered dimer composed of 2 extra-cellular portions of the human p75 TNF-α receptor (TNFR2) fused to a C-terminal human IgG1 Fc domain (67). Etanercept was the first recombinant receptor:Ig fusion protein to be approved for therapeutic use (26). Like adalimumab and golimumab, etanercept is administered subcutaneously. It is produced in Chinese hamster ovary (CHO) mammalian cells, has increased affinity for soluble TNF-α and decreased serum half-life, compared to the monoclonal antibodies (68–71). Whether etanercept also binds to transmembrane TNF-α (mTNF) is still a matter of debate. Some groups showed that the binding affinities/avidities of monoclonal antibodies and etanercept to mTNF were similar (68, 71, 72), whereas other studies reported that infliximab and adalimumab bind to mTNF-α with 3-fold greater avidity than etanercept or certolizumab. However, a more recent study from Kaymakcalan and colleagues, demonstrated that infliximab, adalimumab, and etanercept bind sTNF and mTNF with similar characteristics (69) in transfected cell lines and primary peripheral blood mononuclear cells (PBMCs) isolated from healthy donors, highlighting the ability of etanercept to also interact with the transmembrane form of TNF-α. In addition to TNF-α, etanercept binds to lymphotoxin (LT-α or TNFSF1), a related member of the TNF family that also binds to TNF receptors (TNFR1 and TNFR2) (16). Whether the concomitant blockade of lymphotoxin by etanercept improves clinical responses in patients has still to be fully elucidated. In a large cohort of RA patients enrolled in the CORRONA study, the authors observed that drug response or remission outcomes were similar for patients treated with etanercept and patients treated with anti-TNF-α antibodies (adalimumab, infliximab), except for the fact that response, remission and persistence rates were lower for patients who switched anti-TNF (73). In addition, RA patients treated with pateclizumab, an anti-lymphotoxin-α antibody, did not show statistically significant therapeutic responses as compared to placebo. Response rates were much lower than in RA patients treated with adalimumab in the same clinical study, suggesting that TNF-α blockade has a prevalent role in the improvement of signs and symptoms of RA over LT-α blockade (74). One clinical case involving only one RA patient, reports that the primary non-responder patient initially treated with infliximab, obtained a better response after switching to etanercept. The authors discussed that the responsiveness to etanercept was due to the presence of high levels of LT-α in the synovial tissue as assessed in a biopsy specimen and that resistance to TNF blockade might happen when TNF-α is not the dominant inflammatory cytokine (75). Interestingly, while infliximab and adalimumab have demonstrated efficacy for Crohn's disease (76), etanercept has not shown therapeutic benefits in this disease (77). Differences in agent design and affinity to TNF-α could also explain the differences in infection rates between patients treated with etanercept or with monoclonal antibodies (78–80). There is no evidence that the concomitant blockade of TNF-α and LT-α by etanercept increases mycobacterial infection rates. In a French study including patients with different IMID and treated with TNFi, the risk of tuberculosis was rather higher for those receiving monoclonal antibodies than the soluble-receptor etanercept (80). The differences in design and affinity might perhaps also explain why some patients do not respond to one type of TNF blocker but can achieve significant clinical response by switching to a different type (81).
Certolizumab pegol is the most recent anti-TNF compound introduced in the clinic. Certolizumab is a humanized anti-TNF monoclonal antibody that contains murine and human amino-acid sequences within the VH and VL domains. The hinge region of certolizumab is modified by polyethylene glycol, which reduces immunogenicity, improves solubility and the in vivo half-life (68).
When anti-TNF antibodies bind to tmTNF, they may also induce Fc-mediated effects, such as antibody-dependent cellular cytotoxicity (ADCC) or complement-dependent (CDC), although these effects have never been demonstrated in patients (68, 72, 82). All anti-TNF agents, except certolizumab (68), can induce ADCC, whereas etanercept, in contrast to monoclonal antibodies, lacks CDC activities (72). In a process called “reverse signaling” TNF inhibitors may also trigger in tmTNF-positive cells diverse intracellular signals, that inhibit cell proliferation, induce apoptosis, affect inflammatory cytokines and chemokines production, or, conversely, promote cell activation [reviewed by (83)].
Given the complexity of the cellular pathways involved, the molecular mechanism of action of anti-TNF agents is still not fully elucidated. In general, the biological effects of anti-TNF agents can be summarized by these main functions: (i) binding of soluble TNF-α and LT-α, reducing the signaling cascades downstream of these cytokines; (ii) binding to FcR-expressing cells, inducing antibody-mediated cellular cytotoxicity; (iii) binding to membrane-bound TNF (tmTNF), triggering reverse signaling.
Effects of anti-TNF therapy on the immune system
Given the pleiotropic functions of TNFα, its blockade has long ranging effects on a variety of immune cells. Tables 1, 2 summarize a few of the many studies conducted in rheumatic diseases on the effect of anti-TNF treatment, with a focus on changes in cell populations in Table 1 and secreted cytokines in Table 2. A large number of these studies have analyzed T cell function, since it has been largely demonstrated that these cells are affected by exposure to TNF (158, 159). In experimental models and in humans, long-term exposure to TNFα induced downregulation of components of the T cell receptor (TCR) complex, with consequent reduced responses to TCR-mediated stimulation (160, 161). TNF-blockade was found to reverse the functional impairment, and promote T cell proliferation (160), explaining the increase in memory (91, 127) or effector T cell populations observed in patients treated with TNF inhibitors (see Tables 1, 2) (144).
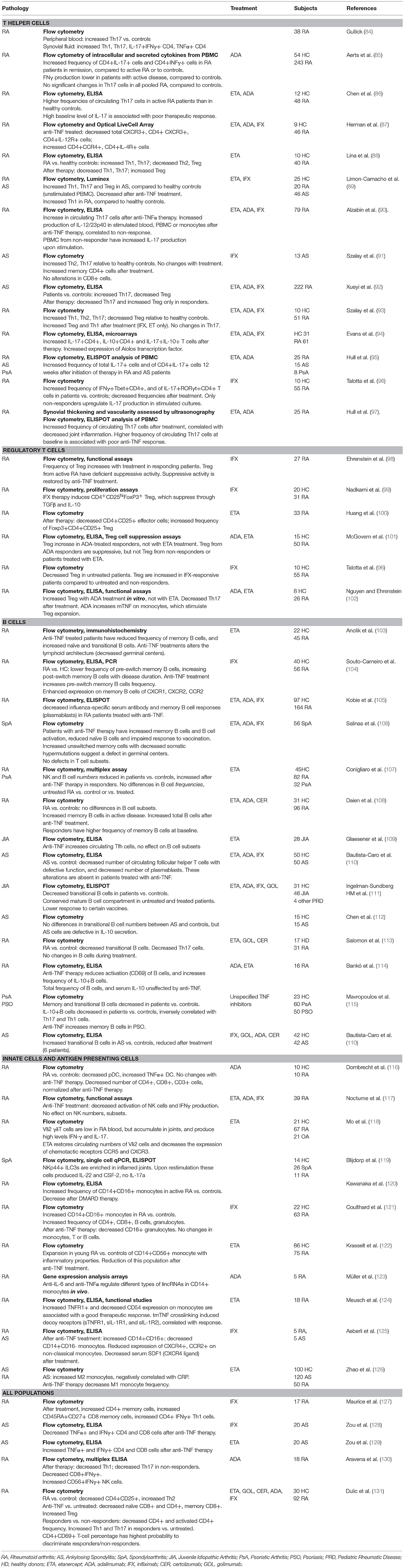
Table 1. Immune cell subsets and anti-TNF treatment.
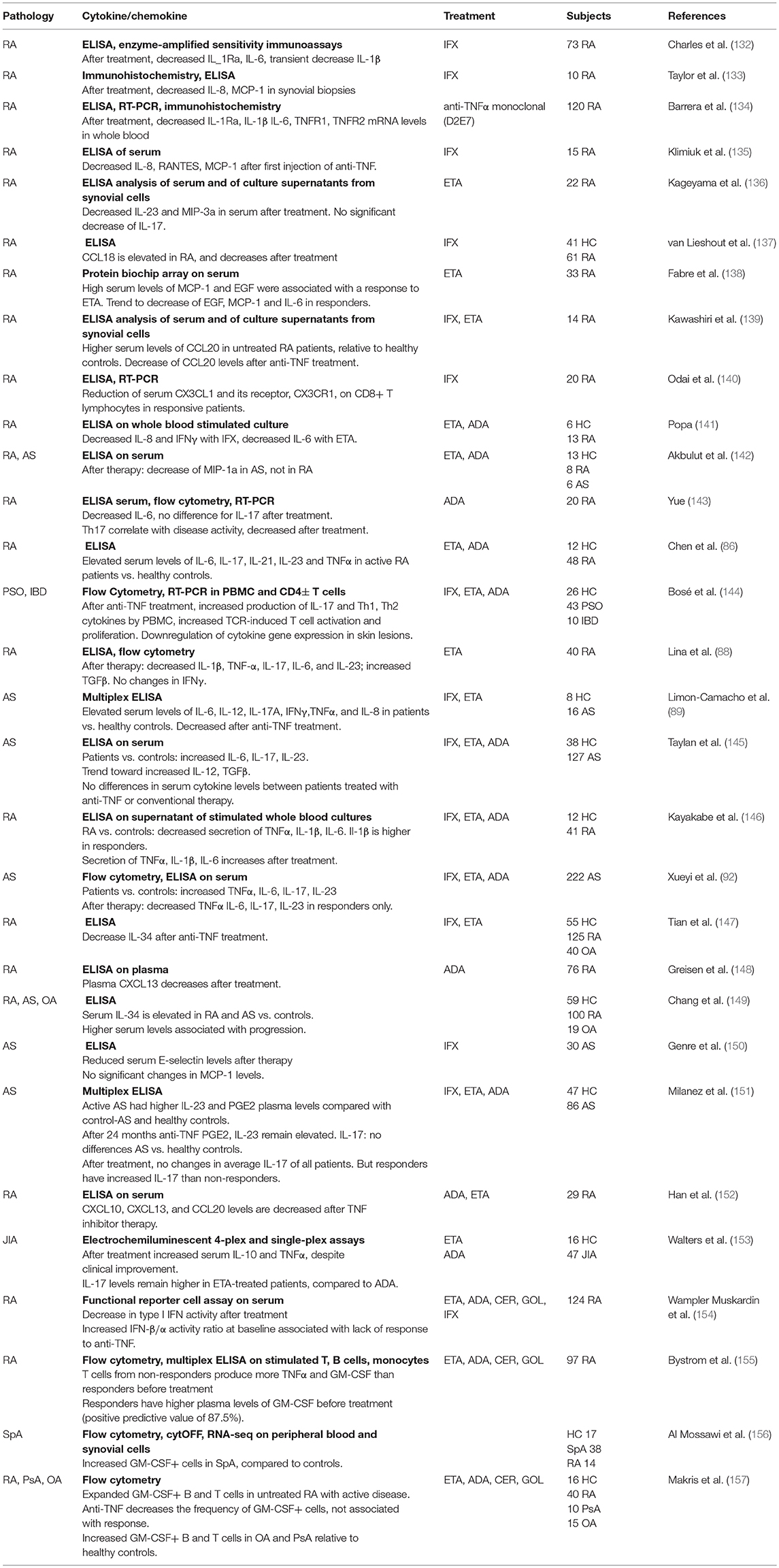
Table 2. Cytokines/chemokines and anti-TNF treatment.
However, the effects of TNF inhibition on immune cell populations are not always consistent in different studies, and may vary depending on the disease setting, the treatment (often studies group patients treated with different inhibitors) and whether the patients respond to therapy.
Anti-TNF and CD4+ T Cell Subsets
The ability of the immune system to mount efficient responses against an array of pathogens depends on the differentiation of naïve CD4+ T cells into functionally distinct T helper (Th) subsets, characterized by the secretion of specific “cytokine signatures.” Th1 cells secrete IFNγ and are important for host defense against intracellular pathogens, while Th2 cells produce IL-4, IL-5, IL-13, and are involved in the protection against parasitic infections. The more recently identified Th17 cells secrete IL-17, IL-21, IL-22, and IL-26 in humans, and contribute to immune responses against extracellular bacteria and fungi. Both Th1 and Th17 responses have been associated with autoimmune disease (162), and the interaction between Th1 and Th17-secreted cytokines may drive disease phenotypes. As a demonstration of the complexity of the cytokine network activated in inflammation, IFNγ was shown to inhibit Th17 differentiation (163, 164) and to affect susceptibility to IL-17-induced experimental arthritis in mouse models (165, 166). In humans, IFNγ production has been found to increase after TNF inhibition (85, 127, 167, 168), however both increased (85, 93, 127) and decreased (88, 89, 96) Th1 frequencies have been reported after anti-TNF therapy. The underlying causes for such discrepancies are not clear, and better patient stratification may allow to provide a better understanding of the mechanism of function of anti-TNF agents. As an example, Th1 cells were significantly increased after adalimumab treatment only in patients in remission, compared to patients with active RA (85, 93, 127). This effect was not visible when the global treated population was compared to the untreated one. Individual drugs may also have specific mechanisms of action: two studies conducted by the same laboratory have found both increased (128) and decreased (128) Th1 cells in the blood of AS patients treated with etanercept or infliximab, respectively. The authors suggested that this differential effect may be linked to the different efficacy of these compounds in the treatment of Crohn's disease, and in the distinct rates of tuberculosis infections following these treatments.
Similar contrasting data have been observed for Th17 cells, with the only consensus of increased levels of Th17 (and Th1) frequencies in patients vs. healthy controls (see Tables 1, 2). High baseline frequency of Th17 cells may be associated with poor response to anti-TNF in RA (86, 90, 97), suggesting that disease in these patients could be driven by a different cytokine network.
The increase of Th17 cells after TNF-blockade has also been correlated with lack of response to treatment in AS (92) or RA (86), although increased Th17 and Th1 have also been found in responders to adalimumab in RA (85). An expansion of Th17 cells is also present in animal models of chronic inflammatory diseases (158). In a model of collagen-induced arthritis, TNF-blockade caused increased expression of the cytokine p40 subunit shared by IL-12 and IL-23, and these cytokines, in turn, induced the expansion of Th1 and Th17 cells, respectively (169). Albazin et al. demonstrated a similar increase in IL-12/IL-23p40 in stimulated blood and lymphocytes from RA patients with poor responses to anti-TNF treatment, suggesting that a similar mechanism could be active in human disease (90).
In several studies, the decrease in Th subsets was mirrored by an increase in regulatory T cells (Treg), indicating that the balance between effector T cells and Treg is important for the re-establishment of immune homeostasis. Altered Treg functions have been described in IMID, and Treg frequency is often decreased in patients, compared to healthy donors. These defects can be reversed by anti-TNF therapy, depending on the agent used (11, 102, 158) (Table 1). Adalimumab was found to restore Treg function in RA patients by increasing mTNF expression on the surface of monocytes. The resulting interaction with TNFR2 on Treg promoted their expansion (102).
Anti-TNF and Cytokines/Chemokines
Anti-TNF treatment downregulates the production of a large range of inflammatory cytokines/chemokine, including IL-6, IL-1β, IL-8, RANTES, and MCP-1 (Table 2).
More recently, granulocyte macrophage-colony stimulating factor (GM-CSF), a myelopoietic cytokine that induces myeloid cells activation and differentiation, has emerged as a potential target in the treatment of rheumatic diseases (170, 171).
GM-CSF gene expression tends to occur locally in inflamed tissues and can be induced by inflammatory cytokines such as IL-6, IL-1β, TNF-α in many different cell types, both immune (T cells, monocytes) and not (fibroblasts, chondrocytes and endothelial cells) (172). GM-CSF receptors are found mostly on the surface of myeloid cells but are also expressed by non-hematopoietic cells, including fibroblasts (172). GM-CSF may affect adaptive immune responses indirectly, by supporting differentiation and function of antigen-presenting cell, and regulating Th cell development (112, 173).
Overexpression of GM-CSF and the GM-CSF receptor has been found in synovial joints of RA patients (173, 174) and in SpA (156). In RA joints, GM-CSF is mainly produced by IFN-γ+CD4+ T cells (175). In SpA blood and synovial fluid, CD4+ T cells were the main source of GM-CSF, which they produced alone or in combination with IL-17. GM-CSF was also produced by CD8+, NK, and innate lymphoid cells (156). The frequency of circulating GM-CSF+ cells was decreased in RA patients after anti-TNF treatment (157). GM-CSF is currently being investigated as a target for treatment of RA patients with inadequate responses to DMARDs (176).
It should be noted that the immune balance observed in peripheral blood may not reflect what occurs in the inflamed tissues, where pathogenic immune cells accumulate. One of the mechanisms of action through which TNF inhibitors decrease joint inflammation is by inhibiting immune cell trafficking (177, 178), by regulating the expression of adhesion and chemotactic molecules, and their receptors (127, 179). Chemokines (such as IL-8 and MCP-1) have an important role in recruiting immune cells to the synovia. Th17 cells express the chemokine receptor CCR6, which is important for their recruitment to the inflamed tissues (180). RA patients responding to adalimumab had significantly lower CCR6 expression than patients with active disease (85). These data are consistent with the decreased serum expression of the CCR6 ligand, CCL20, in RA patients undergoing anti-TNF therapy (139), and suggest that TNF-blockade may induce rerouting of Th17 cells from inflamed tissues to peripheral blood. Another example of TNF action on trafficking is the reduction of CX3CL1, produced by the endothelium, and its receptor CX3CR1 on CD8+ T cells in RA patients under infliximab, which may result in reduced T cell recruitment to the synovia (140). Additional chemo attractants that are decreased by anti-TNF therapy are CCL18, a product of dendritic cells (137), CXCL10, a ligand for CXCR3 on Th1 lymphocytes and monocytes, and CXCL13, a chemokine that attracts B lymphocytes and follicular T helper cells. In RA patients CXCL10 may also be important for chemotaxis of B cells toward inflamed tissues, as suggested by their increased expression of the receptor CXCR3 compared to healthy controls, and their ability to migrate toward CXCL10 in a chemotaxis assay in vitro (181). Consistently, CXCL10, as well as CXCL13 were found elevated in RA patients, compared to healthy or inflammatory controls, and in particular CXCL10 levels correlated with disease severity (182, 183). Notably, CXCL10 and CXCL13 levels were decreased specifically in patients responding to anti-TNF treatment, supporting the notion that disruption of lymphocyte recruitment is one of the keys of success for anti-TNF therapy (137).
Anti-TNF and Other T Cell Subsets
The analysis of the inflamed joints has also revealed the importance of additional lymphocytic populations in the pathogenesis of rheumatic inflammation. γδ T cells carrying the Vδ2 receptor chain have been shown to accumulate in inflamed synovia from RA patients, and to produce high levels of IL-17 and IFNγ. Interestingly, synovial enrichment of Vδ2 T cells went in parallel with depleted numbers of these cells in the periphery, which were restored by anti-TNF treatment. TNF-α blockade strongly downregulates expression of CCR5 and CXCR3 on Vδ2 T cell, interfering with trafficking of these cells to peripheral tissues (184). Although few studies have analyzed innate immune cell populations, these data suggest that innate cells may also be important in the pathogenesis of chronic rheumatic inflammation.
B Cells
B cells play an important role in IMIDs, by producing antibodies and a range of cytokines with pro- (TNF, IL-6, IL-17) or anti-inflammatory (IL-10) functions (185).
B cells exit the bone marrow as “transitional” cells that give rise to mature/naïve B cells, memory (CD27+) B cells and antibody-secreting plasmacells. In secondary lymphoid tissue follicles B cells can be activated to class-switch the B Cell Receptor from IgM/IgD to IgG, IgA, or IgE (186).
Several studies have reported an alteration in the number of B cells and the distribution of B cell subsets in rheumatic diseases (Table 1). Alterations have been reported in the frequency of various subsets of memory B cells, or plasmablasts (104, 108, 187, 188). A decrease in the immature transitional subsets has been reported in RA, PsA, and JIA patients (111, 115), but not in AS patients (110). Mauri and colleagues demonstrated that the transitional subset identified by the CD19+CD24highCD38high markers contains the largest fraction of IL-10-producing B cells upon CD40 ligation in human peripheral blood (189). These cells have been called “regulatory B cells” (Breg), and can have an anti-inflammatory role, by inhibiting Th1 and Th17 responses, and inducing the expansion of Type 1 regulatory T cells (Tr1) (190).
As for T cells, many discrepancies have been observed in the various studies addressing the phenotype of B cells in IMID (Table 1). These could be explained by many factors, including the different markers used to characterized B cell populations, the cohort size, and patients' characteristics, including type of pathology, disease duration, and treatment. Daien et al. demonstrated that patients under steroid treatment have higher frequencies of memory B cells, in particular post-switch memory B cells, and fewer naïve B cells, compared to patients not treated with steroids. Age and sex were also important confounders: gender significantly influenced the composition of CD27−IgD+ naïve, CD27+IgD− post-switch memory, and CD27−IgD− B cells, while age was negatively correlated with CD27+IgD+ pre-switch memory B cells (108). These authors found that few of the differences in B-cell composition between RA patients and controls were confirmed after adjusting for age, gender, and steroid dose.
The reports in the literature on the effects of anti-TNF therapy on B cells are also controversial. In this case, the patients' different response to therapy, and the possible redistribution of cell subsets between inflamed tissue and circulation may also affect the balance of cell subsets.
In some, but not all, cases, anti-TNF therapy caused an increase in total B cells (107), or in memory B cells (106, 115). In the blood, however, this increase can coexist with profound defects in B cell function, as demonstrated by the defective antibody response to vaccination (105, 106, 111, 191, 192). Weak responses are mainly reported for T-cell dependent vaccines, suggesting an indirect effect of TNF blockade on antibody production. Consistently, profound alterations of germinal center structure have been observed in etanercept-treated RA patients (103), which may be explained by the ability of the TNFR2-Ig decoy to neutralize both TNFα and Lymphotoxin α. This could result in reduced isotype switching and hypersomatic mutations, leading to impaired B cell maturation (106).
Anti-TNF and Ectopic Lymphoid Structures (ELS)
Lymphoid infiltrates that form in peripheral organs in conditions of chronic inflammation may assume organized features of ELS, characterized by the segregation of T and B cell areas, the presence of high endothelial venules and of a network of follicular dendritic cells [reviewed in (193, 194)]. ELS formation is driven by a number of cytokines, such as member of the TNF superfamily (TNF-α and LT-α) (195–197) and chemokines, including CXCL13, CCL19, and CCL21, which may be found enriched in the inflamed synovia of RA and SpA patients (198–200). These structures are reminiscent of germinal centers and have been proposed to promote tissue-specific B cell responses, by facilitating class-switch and affinity maturation of locally produced antibodies in the inflamed synovia of RA patients (201). However, whether ELS are active initiator of disease or a consequence of the ongoing chronic inflammation is still debated. In particular, it has been observed that synovial lymphoid neogenesis often lacks some of the features of germinal centers, possibly due to the lack of expression of essential chemokines, such as CCL21 (199).
ELS are particularly prominent in severe forms of RA, Sjogren disease and systemic lupus erythematosus, but have also been described in patients with “sero-negative” inflammation, such as osteoarthritis, crystal-induced arthritis (199), and diseases of the SpA spectrum. Follicular lymphoid-like structures were observed in 2 out of 7 surgical hip specimens from patients with advanced ankylosing spondylitis (202), and in 13 of 27 synovial specimens from PsA patients with no correlation with disease severity or the extent of joint involvement. However, a regression of ELS was observed in PsA or RA patients successfully treated with etanercept or anti-TNFα antibodies, suggestive of a role for TNF-induced inflammation in the generation of these structures (198). In RA patients, in particular, the presence of synovial ELS was also shown to be an independent negative predictor of response to anti-TNF agents (198).
Prediction of Immune Responses to Anti-TNF Therapy
Baseline Patient Clinical Characteristics as Predictors of Therapeutic Response to Anti-TNF Therapy
One of the major challenges in SpA care remains the development of better clinical or biological markers able to aid in disease diagnosis, describe disease activity, and predict structural damage, and therapeutic responses to biological treatments.
In this review we will limit our discussion to predictors of clinical outcome to anti-TNF therapy (see Table 3). Two scoring systems are used to assess disease activity in SpA; the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) (220) and the Ankylosing Spondylitis Disease Activity Score (ASDAS) (221). The BASDAI is based exclusively on patient reporting. It does not include laboratory measurements of inflammation, such as CRP level, but is widely used in clinical practice. In contrast, the ASDAS is a combination of patient reporting and measurements of inflammation [C-reactive protein (CRP) or erythrocyte sedimentation rate (ESR)]. Of note, ranking of patients according to the disease activity determined with the BASDAI or the ASDAS is not identical and it has been suggested more recently that the BASDAI should be replaced with the ASDAS (222).
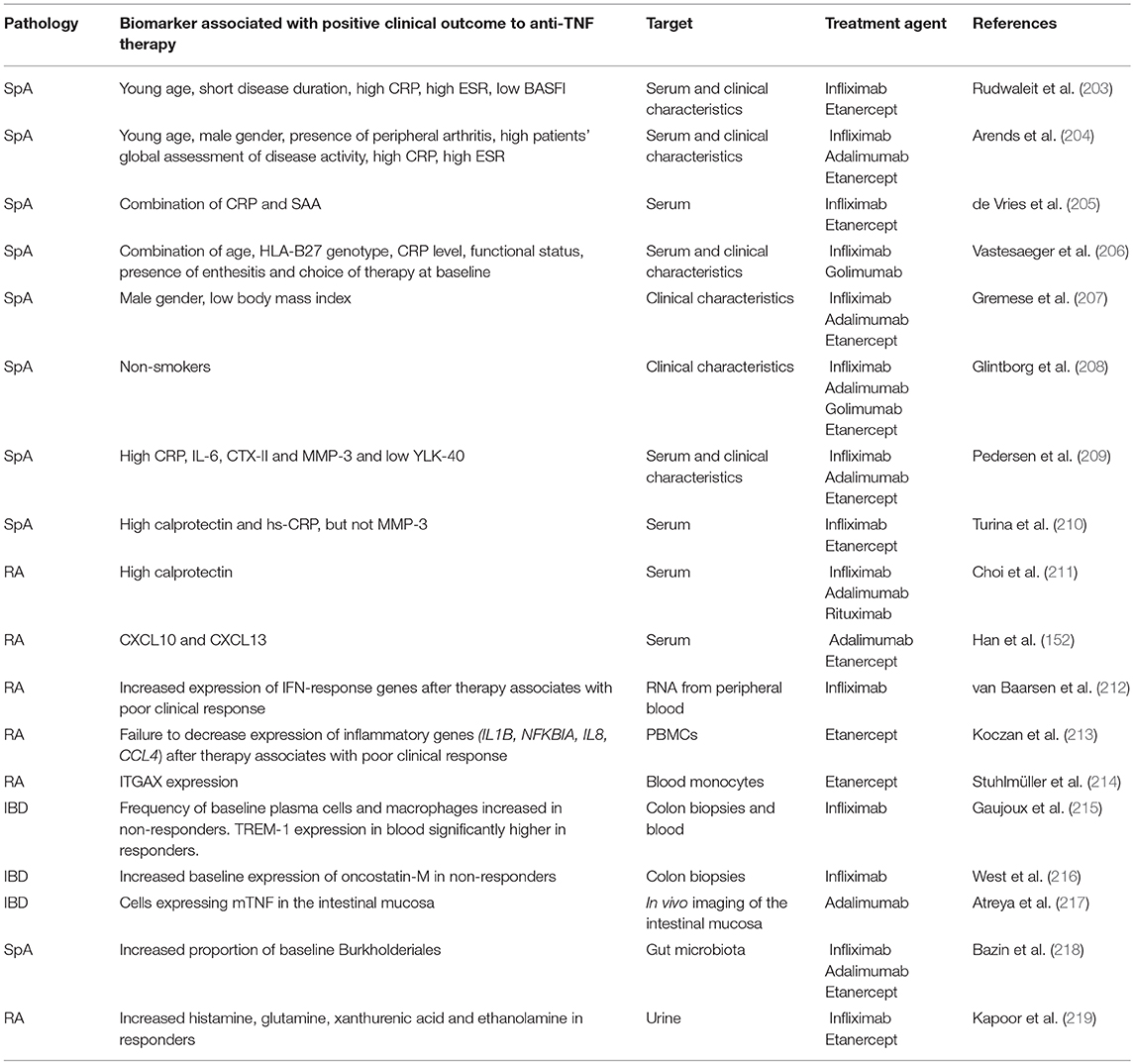
Table 3. Biomarkers predicting therapeutic responses to TNF-blockers.
CRP or ESR are commonly used in clinical practice to help diagnose SpA, evaluate disease activity and predict therapeutic outcome to TNF inhibitors. However, both have low sensitivity and specificity (223). CRP levels are also increased by aging, anemia, infections and presence of immunoglobulins (224, 225). Furthermore, elevated CRP or ESR are only present in about 40–50% of patients with SpA (203) and a normal CRP or ESR does not exclude a diagnosis of SpA. In fact, normal levels of these markers are seen in some SpA patients with active disease.
Rudwaleit and colleagues reported the first systematic analysis of the parameters correlated with clinical responses to anti-TNF therapy in active ankylosing spondylitis patients in 2004. Analysis of clinical parameters in 99 AS patients treated with infliximab or etanercept revealed that the likelihood of achieving good responses to TNF blockers was significantly associated with young age and shorter disease duration, with increased markers of acute phase and higher disease activity (203).
A study performed on 220 SpA patients indicated that the highest response rate during anti-TNF therapy was achieved in those with elevated CRP or ESR levels at baseline, younger age, male gender, presence of peripheral arthritis and higher patient's assessment of disease activity (204). In a large prospective cohort of AS patients, De Vries and colleagues confirmed the predictive value of inflammatory markers, such as CRP and serum amyloid A (SAA), to monitor the efficacy of anti-TNF treatment, and to select the patients more likely to respond to anti-TNF therapy. The study was performed on 155 SpA patients (117 treated with etanercept, 38 with infliximab) and clearly demonstrated that all the markers of inflammation tested, and the BASDAI score were significantly decreased after anti-TNF therapy. However, only elevated baseline levels of CRP and SAA were shown to correlate to positive therapeutic outcome and to be a valuable instrument for prediction. A combination of elevated baseline levels of CRP and SAA resulted in the best prediction of clinical response (81%), which was not improved by the inclusion of baseline ESR levels (205). The same study also reported that high sensitivity CRP (hs-CRP) did not provide advantage over standard CRP measurement.
More recently, a report from Vastesaeger defined an algorithm in which the combination of HLA-B27 genotype, age, functional status, CRP level, the presence of enthesitis, and choice of therapy at baseline enabled good prediction of anti-TNF outcome in two large cohorts of SpA patients. Interestingly, the authors demonstrated that although each factor can be an independent predictor, the combination of the six selected variables outperformed the single predictive value (226). This study also suggested that single predictors may not be strong enough for decision-making in the individual patient.
Additional baseline characteristics that have been correlated to therapeutic outcome in SpA patients are body weight and smoking. A report confirmed the lower probability to respond to TNF inhibitors in female SpA patients, and found an association with high body mass index (BMI) and poor response to therapy, in particular for infliximab treated patients (207). In a large observational cohort of SpA patients established in Denmark, in which more than half of the patients were current or previous smokers, the authors reported that current and previous smokers had significantly poorer anti-TNF treatment responses as compared to non-smokers, and suggested that the negative impact of smoking may be linked to increased systemic inflammation, accelerated radiographic progression, decreased functional activity and reduced lung capacity (208). This study confirmed a previous report in RA patients, in which a lower rate of response to anti-TNF therapy among current smokers was observed, particularly in patients receiving infliximab (227).
The clinical characteristics described so far may help clinicians to guide treatment decisions in daily practice, however none of them has strong specificity.
Predictive Markers in Blood and Tissues
Different strategies have been proposed in the past 15 years with the aim to identify markers predicting clinical responsiveness to anti-TNF therapy. However, results reported in the literature have not always been validated in subsequent studies. Many different strategies have been pursued to identify biomarkers, such as measuring soluble molecules in serum or applying a variety of molecular and genomic techniques. More recently, alternative approaches including molecular imaging with fluorescent antibodies, analysis composition of the gut microbiota and the hunt for biomarkers in tissues have been proposed. Since there are only limited numbers of studies performed in SpA patients, we will extend our discussion to other chronic inflammatory diseases, the results of which might also be relevant for SpA.
A report from Pedersen and colleagues investigated circulating biomarkers of inflammation including CRP, IL-6 and YKL-40, vascular endothelial growth factor (VEGF, a marker of angiogenesis), C-terminal crosslinking telopeptide of type II collagen (CTX-II), matrix metalloproteinase 3 (MMP-3), total aggrecan, cartilage oligomeric matrix protein and bone turnover in axSpA patients undergoing anti-TNF treatment. Patients that reached a major improvement in ASDAS at week 22 had higher baseline CRP, IL-6, CTX-II, and MMP-3 and lower YLK-40 levels as compared to partial or non-responders. BASDAI responders had higher baseline CRP and VEGF, and lower YLK-40 as compared to BASDAI non-responders (209).
Higher MMP3 and other matrix metalloproteinases such as MMP8 and MMP9 levels were repeatedly shown to reflect disease activity and response to treatment in SpA patients but the results were not always concordant when comparing different studies. Arends and colleagues showed that although serum MMP-3 significantly decreased after etanercept treatment in SpA patients, baseline levels were not superior to the predictive accuracy of ESR or CRP (228). In line with this result, Turina et al. more recently reported that calprotectin and hs-CRP, but not MMP-3, were good biomarkers for treatment response in SpA patients, confirming these results in a replication cohort (210, 229). From this report and other studies it appeared that the most reliable marker correlating with clinical parameters in SpA identified so far is calprotectin, an heterodimeric protein complex consisting of S100A8 and S100A9 subunits (previously called myeloid-related protein 8 and 14, respectively), which bind calcium and zinc in the cytosol of monocytes and granulocytes (230, 231). Myeloid-related protein 8 and 14 are damage-associated molecular pattern molecules (DAMPs), and have been reported to be upregulated in several autoimmune disorders (232, 233). Produced by myeloid cells and neutrophils, calprotectin is secreted in the transmigration of these cells through the endothelium to the inflamed tissues and promotes inflammation by activating leucocytes and endothelial cells (234). As shown in animal models, it is also a key factor in the pathogenicity of autoreactive IL-17–producing CD8+ T cells (235).
Elevated serum levels of calprotectin were reported in axSpA and reactive arthritis patients compared to controls, and treatment with anti-TNF agents significantly decreased calprotectin and hs-CRP levels already 2 weeks after infliximab treatment in SpA patients, but not in the placebo group (229). In addition to calprotectin, several biomarkers (hs-CRP, IL-6, pentraxin-3, α-2-macroglobulin, MMP-3, and VEGF) were assessed at baseline for their ability to predict response to anti-TNF therapy. Importantly, serum levels of calprotectin were the most accurate and reliable marker of treatment response in axSpA and peripheral SpA, and outperformed hs-CRP in some analyses.
In RA patients, baseline serum levels of myeloid-related proteins 8 and 14 were significantly higher in responders compared to non-responders. The study was done in three prospective cohorts, one treated with adalimumab, one with infliximab and one with a different therapy, the anti-CD20 antibody rituximab. MRP 8/14 levels were consistently higher in responders to targeted treatment, independently of the mechanism of action of the biologics (211). Interestingly, the predictive accuracy of baseline MRP 8/14 levels in the three groups was higher as compared to other baseline patient characteristics such as CRP, ESR, and DAS28. The ROC curve AUC for baseline MRP 8/14 levels was 0.688 in the adalimumab-treated patients, 0.791 in the infliximab group and 0.984 in the rituximab group. Another important point of the study was that while in good and moderate responders there was a significant decrease of calprotectin levels 4- and 16-weeks after initiation of anti-TNF therapy, the levels of the protein remained unchanged in the non-responder group. This suggests that serial measurement of MRP 8/14 serum complexes may be useful for monitoring the early changes after biological treatment, and may predict the clinical response over time. The advantage of measuring MRP 4/18 serum levels, apart from the low cost and accessibility of the complex in the serum, is that the protein is relatively stable at room temperature, and, contrary to other cytokines, can be measured in serum without cold storage of samples.
Other markers correlated to anti-TNF therapy outcome were reported in a small cohort of RA patients, in which serum levels of CXCL10, CXCL13, and CCL20 were measured by ELISA. Baseline levels of CXCL10 and CXCL13, but not CCL20 were significantly higher in responders as compared to non-responders and both markers were reduced after therapy in the responder group, however the results were not validated in independent cohorts of patients (152).
Most treatment prediction studies have focused on markers measured in serum. Multiplex analysis of cytokines and chemokines in patients' serum samples often result in measurements close or below the lower limit of quantification of the secreted proteins, giving rise to conflicting results. These molecules are often secreted in low abundance in cells at the steady state and are therefore difficult to measure in robust and reproducible assays.
Few studies have been performed at the transcriptomic level, to identify genes associated with clinical outcome to anti-TNF therapy. Van Barseen and colleagues reported that poor clinical response to anti-TNF therapy was associated with an increased IFN gene expression signature one to 2 months post-therapy in peripheral blood of RA patients. Higher level of expression of IFN response gene set such as OAS1, LGALS3BP, MX2, OAS2, and SERPING1 were found in the poor responder group of a validation set of patients, and this was most evident at the 2-months post therapy time point. Interestingly, the combination of the five genes into one IFN response gene set improved the prediction accuracy over the single genes (212). In another study done in RA patients, a failure to downregulate key inflammatory genes (IL1B, NFKBIA, IL8, CCL4) in PBMCs after TNF-α blockade was associated with poor long-term clinical outcome (213). Investigation of blood monocytes in RA patients revealed molecular differences between responders and non-responders, both before and after anti-TNF therapy. Of note, expression of the integrin alpha X encoding gene (ITGAX, encoding CD11c) was significantly higher in responders at baseline and was able to predict the therapeutic outcome with 100% sensitivity and 91.7% specificity. Clinical responders revealed an almost complete reset to normal levels of inflammatory monocyte markers whereas in non-responders the levels of those genes remained elevated (214).
It was recently shown that the majority of the genes in a signature for response to anti-TNF in IBD patients show higher expression in immune cell subsets, compared to other cells present in the biopsy tissues, suggesting that resident or infiltrating leucocyte populations represent a good target to investigate responses to anti-TNF therapy. However, a clear cell population has not yet been implicated in the response (215).
A combined statistical deconvolution and meta-analysis methodology of IBD patients naïve to anti-TNF therapy showed that the genes most involved in response to therapy are expressed in the myeloid, B and T-cell subsets. Gaujoux et al. demonstrated that baseline proportions of intestinal plasmacells and inflammatory macrophages were significantly increased in non-responders, and this difference was maintained after treatment initiation. Interestingly, a good response to anti-TNF was associated with a strong decrease in both cell populations. The predictive power of baseline abundance of macrophages and plasmacells associated with failure to anti-TNF therapy obtained by the computational deconvolution was validated in plasma samples from an independent cohort of IBD patients. At the transcriptomic level, the chemokine ligand 7 (CCL7) and the chemokine receptor 2 (CCR2) were upregulated in biopsies obtained from non-responders, whereas no significant difference for these two genes was observed in blood. Increased levels of genes in the CCL7-CCR2 pathway suggest that in non-responders there is an increased recruitment of inflammatory TNF-secreting macrophages to the inflamed tissuers, which contributes to increased plasmacell abundance ultimately impacting clinical outcome. Interestingly, one of the upstream regulators of the CCL7-CCR2 axis, TREM-1, the expression of which was measured in the blood of 22 CD patients prior to anti-TNF therapy, was significantly downregulated in non-responders, and showed a high prediction accuracy.
Still in the context of IBD, a recent publication from the Powrie lab has demonstrated that oncostatin M (OSM) levels are higher in inflamed intestinal tissue of IBD patients with deep mucosal ulcerations (severe disease). Of particular interest was the observation that OSM gene expression levels were substantially higher in colonic biopsies of IBD patients refractory to anti-TNF therapy. These findings identified OSM as a potential biomarker of disease activity and therapeutic response to anti-TNF therapy. In addition, OSM could be an interesting novel therapeutic target for patients not adequately responding to TNF-blockers (216). However, it should be noted that this work has been performed with biopsies, material which is not available for most patients affected by axial SpA.
A study by Neurath and colleagues has recently used in vivo molecular imaging of the intestinal mucosa of Crohns' disease patients to identify mTNF-expressing cells. After topical application of labeled adalimumab through a standard spray catheter onto the most inflamed region of the bowel during colonoscopy, in vivo imaging of the intestinal mucosa showed a specific fluorescence signal of mTNF+ cells. Interestingly, cells expressing mTNF were mainly lamina propria CD14+ macrophages and some CD4+ T lymphocytes. The authors correlated the number of immune cells expressing mTNF with clinical outcome to adalimumab, and demonstrated that responders had a significantly higher mean number of mTNF-expressing cells than patients refractory to the therapy. This study was based on molecular imaging with fluorescent antibodies, which constitutes a new approach for the identification of patients responding to therapy, both in chronic inflammatory and autoimmune disorders, and in cancer (217).
More recently, several studies have addressed the role of metabolism and gut microbiome in patients with chronic inflammatory diseases and some reports have identified associations between baseline metabolites or microbiota composition and clinical responses to anti-TNF treatment. A recent study performed on a small cohort of axSpA patients reported an increased proportion of baseline Burkholderiales in clinical responders to anti-TNF therapy, and a high proportion of Dialister sp. at month 3 post-therapy (218).
Moreover, the screening of baseline urine metabolic profiles in a cohort of RA and PsA patients undergoing anti-TNF therapy allowed the identification of several metabolites associated with positive therapeutic outcome (219). The authors used different bioinformatics strategies to select the metabolites that better discriminate between responders and non-responders, and found that three different methods of analysis identified histamine, glutamine, xanthurenic acid, and ethanolamine as increased in urine samples from good responders to anti-TNF therapy, while ethanolamine, p-hydroxyphenylpyruvic acid, and phosphocreatine were lower in patients with a good response.
These studies suggest that the molecular analysis of serum or tissues may be a promising approach to identify biomarkers of response. However, a robustly validated and easily applicable biomarker is not available yet, in particular for SpA.
Genetic Predictors of Treatment Response to Anti-TNF Therapy
The success of GWAS in identifying new disease susceptibility loci and indicating cellular pathways involved in pathogenesis has sparked interest to use the same approach to identify genetic determinants associated with treatment responses. Most of these studies have been performed in RA and only limited data are currently available for SpA.
In order to achieve optimal clinical outcome, treatment of RA needs to be initiated as soon as possible in newly diagnosed patients (236). According to the EULAR recommendations (236), most RA patients are treated with a conventional synthetic disease-modifying antirheumatic drug (csDMARD), such as methotrexate as first-line therapy, although this treatment is effective only in ~50% of patients. Patients who do not adequately respond or tolerate this treatment are subsequently switched to biologic drugs, such as TNF inhibitors (TNFi). Approximately 30% of RA patients do not respond to TNFi and are then prescribed another biologic therapy. This “trial and error” approach may take a long time, during which the patient is not appropriately treated but exposed to side effects without clinical benefit. More importantly, this approach is not compatible with the notion that treatment of RA needs to be initiated early in the disease. Therefore, identifying the most effective treatment for each individual patient at diagnosis is of critical importance to improve patient care. As biologic therapies are expensive, there is also a strong economic incentive to prescribe the most appropriate treatment early on.
Implementing “precision medicine” in rheumatic diseases will require robust tools that can predict responsiveness of patients to a specific drug. Genetic variants associated with therapeutic responses would be an ideal biomarker because these assays can be performed at diagnosis, are very robust, fast and inexpensive. A substantial number of studies have therefore been performed to identify genetic variants associated with treatment responses in RA (237). Some of these collaborative efforts employed candidate gene approaches to test if RA susceptibility alleles were also associated with therapeutic responses to TNF-blockers. Cui et al. tested the association of 31 RA risk alleles with therapeutic responses to TNFi in 1,253 RA patients from an international collaborative consortium of 9 different RA cohorts. They found that only the RA susceptibility allele (rs10919563) at the PTPRC locus (encoding CD45) was also associated with response to TNFi, but none of the other RA-associated risk alleles had an effect on treatment responses (238). These findings were replicated in a cohort of 1,115 patients from the UK (239). A third study, however, involving a meta-analysis of 1,516 patients did not find significant associations of PTPRC with response to anti-TNF therapy (240). Only few GWAS have revealed loci associated with treatment responses to anti-TNF therapy at accepted levels of genome-wide significance (p < 5 × 10−8). Cui et al. have performed a GWAS meta-analysis on 2,706 RA patients. 733 of these patients were treated with the soluble TNFR-Fc fusion etanercept, while 1,071 or 894 patients were treated with the monoclonal anti-TNF antibodies adalimumab or infliximab, respectively. They identified in the etanercept group, but not in the two other treatment groups a SNP in the 3′-untranslated region of the CD84 gene, which may disrupt a transcription factor binding site. The allele associated with better treatment response was associated with higher gene expression levels of CD84 and explained 2.6% variance in response to treatment with etanercept (241). The reason why this specific SNP at CD84 was found to be associated only with therapeutic responses to etanercept but not to infliximab or adalimumab, is currently not known.
An innovative approach to identify a validated genetic predictor of anti-TNF response in RA has been performed in the context of the RA responder DREAM challenge (http://www.synapse.org/RA_Challenge). Genotyping data from 2,706 RA patients treated with anti-TNF were given to challenge participants to develop predictive models of treatment responses to TNFi. This challenge ran for 8 months in 2014 and 73 research teams submitted computer code covering a wide range of state-of-the-art modeling methodologies. In the validation phase, these models were evaluated with 591 anti-TNF-treated RA patients from an independent cohort. Despite the remarkable assembly of expertise in this crowdsourcing approach, no significant genetic predictors of treatment responses could be identified. The authors concluded that genetic information does not significantly enhance prediction of therapeutic responses over standard clinical assessments and suggested to embark on other research strategies to identify biomarkers (242).
From IL-23/IL-17 biology to novel treatments of SpA and related diseases
GWAS data, together with mouse models of autoimmune disease, demonstrated that CD4+ inflammatory Th17 cells, which produce IL-17, play a pivotal role in the initiation of inflammatory diseases (243–245). IL-23 is important for the expansion and the functional activity of the Th17 cell subset (246). More recent studies have pointed to an additional role of IL-17-producing innate immune cells, which express the IL-23 receptor (IL-23R) in inflammatory disease. In particular, innate lymphoid cells (ILCs) were shown to drive IL-23-dependent intestinal inflammation in mice (247), and were enriched in the intestine of patients affected by inflammatory bowel disease (IBD) (248). In addition, a subpopulation of γδ T cells that produce IL-17 contributes to experimental autoimmune encephalomyelitis (EAE) in mice (249). IL-23R-expressing γδ T cells are also enriched in the peripheral blood of SpA patients (250). A direct link between IL-23 and tissue inflammation has been established in a mouse model of SpA. Sherlock et al. demonstrated that IL-23 mediates enthesal inflammation, the hallmark of SpA, by acting on a small population of CD3+CD4−CD8−IL-23R+RORγt+ enthesal resident T cells (251). The implication of the IL-23/IL-17 axis is also supported by the finding that at least 6 of the non-MHC loci genetically linked with axSpA are associated with genes in this pathway (RUNX3, IL23R, IL6R, IL1R2, IL12B, TYK2) (252). Taken together, these data suggest that the inflammatory response in SpA may be the result of a complex interplay of different immune cell types and that the IL-23/IL-17 pathway is likely to play a key role in chronic inflammation. Understanding the cellular and molecular mechanisms that regulate this network of innate and adaptive immune responses is therefore of critical importance for the design of rational therapies.
To address this question, our lab and others have investigated the impact of genetic polymorphisms in genes of the IL-23 signaling pathway on the effector functions of CD4+ T cells from SpA patients (253–255). We have measured the expression levels of Th17 and Th1 cytokine genes and transcription factors in CD4+ T cells isolated from SpA patients, and we correlated them with the patients' genotype at loci genetically associated with SpA. We showed that SpA patients carrying risk-associated alleles of genes in the IL-23/IL-17 pathway expressed high levels of genes involved in the differentiation and function of Th17 and Th1 cells, whereas the presence of protective alleles was associated with low-level expression of these genes. In contrast, variation at loci genetically linked to SpA, but not associated with the IL-23 pathway (such as ERAP1 and ANTXR2), did not correlate with expression of Th17 and Th1 genes, suggesting that these SNPs may contribute to SpA pathogenesis through distinct cellular mechanisms. These data showed that genetic variation at multiple loci within the IL-23/Th17 pathway, such as IL23R, IL12B, and CCR6, affects CD4+ effector functions in SpA patients. Of note, the effect of genetic variation on CD4+ T cell function could be detected in activated, but not in resting T cells, consistent with the context-dependent action of expression quantitative trait loci (eQTL) observed in several studies (256–258). We also showed that the combinatorial action of multiple SNPs at distinct loci, rather than a single genetic variant, determined the immune cell functions of SpA patients and we have established a hierarchy among the SNPs with respect to their effect on regulating the expression of effector molecules using multivariate analysis. These results demonstrate a link between disease-associated genetic variants and defined functions of immune cell populations involved in the pathogenesis of chronic inflammatory diseases.
A large number of clinical trials have been performed in the past years to evaluate if targeting IL-23 and IL-17 is beneficial for the treatment of SpA and related diseases (259).
As mentioned above, the only treatment options for SpA patients not adequately or not tolerating treatment with NSAIDs have been TNF-blockers. The rationale to test IL-17A blockade were the strong genetic association of loci linked to the IL-23/IL-17 axis and the expansion of circulating CD4+IL-17+ cells in AS (260, 261), including KIR3DL2-expressing T cells responding to cell-surface HLA-B27 homodimers (262) and IL-17-producing γδ T cells expressing the IL-23R (250). Baeten and colleagues performed a phase 2 study to determine the efficacy and safety of secukinumab, a monoclonal antibody blocking IL-17A, in patients with active AS. This proof-of-concept study showed that inhibiting IL-17A rapidly reduced clinical and biological signs of active AS when compared to placebo and was well tolerated (263). Two subsequent phase 3 trials confirmed that inhibition of IL-17A significantly reduced signs and symptoms of AS (264), and this treatment is now recommended for the treatment of axSpA not adequately responding to TNF-inhibitors (265). Phase 3 trials have also documented efficacy of anti-IL-17A therapy for the treatment of psoriasis and psoriatic arthritis (266–268).
In contrast, inhibition of IL-17A with secukinumab was not effective and higher rates of adverse events were noted compared with placebo in a proof-of-concept study of Crohn's disease (269). A subsequent phase 2 study evaluated safety and efficacy of brodalumab, a human monoclonal antibody targeting the IL-17RA that blocks the biologic activity of multiple IL-17 cytokines including IL-17A, IL-17F, and the IL-17A/F heterodimer, in patients with moderate-to-severe Crohn's disease. This study was terminated early because a disproportionate number of cases of worsening of Crohn's disease in the treatment groups compared with placebo was observed (270). The precise mechanisms for the unexpected failure of IL-17A inhibitors in Crohn's disease are currently not known. However, Lee and colleagues recently reported that the dominant function of IL-17A in a mouse model of colitis is to preserve the integrity of the intestinal barrier by inducing expression of the tight junction protein occluding during epithelial injury (271).
To start to define the mechanism of action of IL-17A inhibitors, van Mens and colleagues have analyzed the effects of IL-17A inhibition on the immunopathology of target lesions and systemic immune responses in peripheral SpA. They reported that clinical improvement in joint counts was associated with a histologic decrease in synovial sublining macrophages and neutrophils, as well as with decreased synovial expression of IL17A but not of TNF transcripts. Systemically, anti-IL-17A treatment decreased inflammatory markers such as CRP and ESR and MMP-3 production in whole-blood stimulation assays using SEB and zymosan as stimuli. However, with exception of IL-17A itself, the capacity of peripheral blood cells to produce a broad panel of cytokines and chemokines upon stimulation with microbial antigens was not affected. This mechanism-of-action study was conducted in 20 peripheral SpA patients and indicated that clinical improvement upon anti-IL-17A treatment was paralleled by immunomodulation of inflamed target tissues without compromising systemic immune responses (272).
The crucial role of IL-17A in the pathogenesis of SpA has been demonstrated in clinical trials of the anti-IL-17A antibody secukinumab, however the cellular source of this proinflammatory cytokine in this disease remained controversial. Group 3 innate lymphoid cells (ILC3s) have been identified in several tissues as potent producers of proinflamatory cytokines, including IL-17A and IL-22. In collaboration with the team of D. Baeten we have recently characterized the presence and composition of ILCs and investigated if these cells are an important source of IL-17A in the synovial tissue of patients with peripheral SpA. We analyzed matched synovial tissue (ST), synovial fluid, and peripheral blood from SpA patients with actively inflamed knee joints. We found that ILCs, and in particular NKp44+ ILC3s, were expanded in inflamed arthritic joints. Single-cell gene expression analysis demonstrated that ST ILCs were clearly distinguishable from ST T cells and from their peripheral blood counterparts. We detected expression of the Th17 signature transcripts RORC, AHR and IL23R in a large fraction of ST ILC3s. These cells were capable to induce IL-22 and CSF2 but not IL-17A expression in response to in vitro re-stimulation. This study demonstrated that ILC3s were absolutely and relatively enriched in the synovial joint of patients with SpA, however these cells are not a significant source of IL-17A in this pathology (119). Thus, further studies are needed to define the cellular sources of IL-17A in this disease.
With respect to IL-23, a phase 2 clinical study tested the safety and efficacy of a fully human monoclonal antibody (ustekinumab) targeting the p40 subunit shared by IL-12 and IL-23 in psoriasis (273). This study revealed a 75% improvement in the psoriasis area-and-severity index (PASI) at week 12 in up to 80% of patients and a 90% improvement in 50% of anti-IL-12p40-treated patients (273). These remarkable results were confirmed in two subsequent phase 3 studies (274, 275). Of note, blocking IL-12/IL-23 by ustekinumab was more effective for the treatment of psoriasis than treatment with the TNF-blocker etanercept (276) and has shifted the treatment paradigm for this disease affecting 2–3% of the population. Treatment with ustekinumab blocks the bioactivity of both IL-12 and IL-23. Subsequent studies with a monoclonal antibody targeting the p19 subunit of IL-23 (risankizumab; neutralizing selectively IL-23 bioactivity) demonstrated an even higher efficacy in the treatment of psoriasis than blocking both IL-12 and IL-23 (277). Although neither IL-17A inhibitors nor IL-23 blockers result in a cure of psoriasis, the impact of these new drugs on the quality of life of patients cannot be overstated. It is also important to note that in this case the clinical observations in patients confirmed previous results obtained in experimental mouse models. Mice with a deletion of the IL-23p19 subunit, but not mice with a deletion of the IL-12p35 subunit were protected from disease in several experimental models of autoimmunity, such as experimental autoimmune encephalomyelitis (EAE) (244, 245), collagen-induced arthritis (CIA) (278), and inflammatory bowel disease (IBD) (279, 280).
Blocking the activity of IL-12/23 and of IL-23 has also been tested in moderately to severely active Crohn's disease and patients receiving either of these monoclonal antibodies had significantly higher response rates than did those receiving placebo (281, 282). In contrast, a phase 2 study testing the IL-23 inhibitor risankizumab did not demonstrate any evidence of clinically meaningful improvements compared to placebo in patients with active ankylosing spondylitis, although a significant reduction of the inflammatory marker CRP was observed (283). These findings were unexpected for several reasons. First, genetic variants in IL23R have been associated with AS (284) and this finding had been replicated in several GWAS (285, 286). Second, treatment of AS with IL-17A inhibitors has proven to be effective (263, 264) and IL-17A was shown to be downstream of IL-23 (243). Finally, overexpression of IL-23 in a mouse model induced a SpA-like phenotype (251). The reasons underlying the failure of IL-23 blockade in SpA are currently unclear. It is possible that IL-23-independent of sources of IL-17A, such as mucosal-associated invariant T (MAIT) cells play important roles in SpA pathology (287), or that IL-23 is important at the initiation of the disease, but not in established disease, as has recently been demonstrated in HLA-B27 transgenic rats, an experimental model of SpA (288). Clearly, more work needs to be done to resolve this intriguing issue.
Conclusions
The clinical relevance of a “personalized” approach in medicine is well-accepted. However, despite the well-known individual variability in the susceptibility to infections and inflammatory diseases, disease progression and response to therapy, medical practice and public health policies typically take a “one size fits all” approach to disease management. This is due to a lack of understanding of what determines the individual predisposition to disease and the mechanisms associated with the response to a specific therapy. Biological strategies that block specific immune mediators such as TNF, IL-6, IL-17A, IL-23, or JAKs are effective only in a subpopulation of patients and can be associated with serious side effects. To improve clinical outcome, tools that allow prediction of treatment responses are needed. In addition, a better understanding of the pathogenic mechanisms will permit a more efficient use of existing therapies, as well as the development of novel targeted therapies. In a context in which new treatments for chronic inflammatory diseases have recently been introduced in the clinics, we highlight the importance of establishing a path toward personalized medicine by defining immunological correlates associated with therapeutic responses to anti-TNF therapy in SpA and related diseases.
The limitations of developing reliable biomarkers that can be used in daily practice derive from several factors. Many of the studies reviewed here have been performed in different centers and in relatively small cohorts of patients, limiting the unbiased identification of robust and validated biomarkers. In this sense, increased collaboration between centers and “merging” of cohorts of patients affected by IMIDs to increase sample size and power may help to define more reliably define biomarkers. “Consortium science” has been key to the success of large GWA studies aimed at identifying risk alleles for various diseases and should be reinforced as a valuable strategy in translational research.
Due to limited patient numbers, several studies have grouped patients not homogeneous for disease status, treatment agent, response to therapy, and other patient characteristics. Most of these variables have significant effects on the measurable parameters of patient immune functions, and are important confounders for the analysis of the impact of TNF-blockade on the immune system. This may be the cause underlining the many controversial findings reported in the literature about the modifications in immune function imparted by TNFi. Accurate patient stratification may allow a better understanding of the molecular and functional consequences of anti-TNF therapy, and improve the chances to identify strong biomarkers of response.
With respect to axial SpA, an additional challenge is provided by the difficult access to inflamed axial tissues. Sequential sampling during treatment in large cohorts of patients is almost impossible to perform, except for SpA patients with a predominant peripheral inflammation. In this particular case, peripheral blood still remains the only easily accessible material to study pathophysiologic processes and mechanisms contributing to the response to therapies.
Finally, recent clinical trials testing the safety and efficacy of monoclonal antibodies targeting IL-17A and IL-23 in SpA, psoriasis, psoriatic arthritis and Crohn's disease have been very encouraging and have increased the therapeutic options for these diseases. At the same time, their introduction into the clinics has increased the urgency to implement personalized treatment strategies. A trial and error approach, as it is currently applied in most cases, is simply not compatible with optimal patient care and efficient use of resources. However, defining objective criteria to guide treatment decisions for each individual patient remains a major challenge. Furthermore, clinical studies testing blockade of IL-17A and IL-23 have not been without surprises. The unexpected failures of anti-IL-17A in Crohn's disease and of anti-IL-23 in AS are stark reminders of our limited understanding of the pathogenic mechanisms of these diseases and are a call to action to all of us in the biomedical research community.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
SM was a scholar of the Pasteur-Paris University (PPU) International Ph.D program and supported by a grant from the Fondation pour la Recherche Médicale. Work in the authors' laboratory was supported by grants from Institut Pasteur, FOREUM Foundation for Research in Rheumatology, the Fondation Arthritis, MSD Avenir (Project iCARE-SpA), and a Bourse Passerelle from Pfizer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Corinne Miceli-Richard and Maxime Dougados for helpful discussions.
References
1. El-Gabalawy H, Guenther LC, Bernstein CN. Epidemiology of immune-mediated inflammatory diseases: incidence, prevalence, natural history, and comorbidities. J Rheumatol Suppl. (2010) 85:2–10. doi: 10.3899/jrheum.091461
2. Banchereau R, Cepika AM, Banchereau J, Pascual V. Understanding human autoimmunity and autoinflammation through transcriptomics. Annu Rev Immunol. (2017) 35:337–70. doi: 10.1146/annurev-immunol-051116-052225
3. Ananthakrishnan AN. Environmental risk factors for inflammatory bowel diseases: a review. Digest Dis Sci. (2015) 60:290–8. doi: 10.1007/s10620-014-3350-9
4. Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I. Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol. (2015) 14:263–73. doi: 10.1016/S1474-4422(14)70267-4
5. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. (2013) 14:661–73. doi: 10.1038/nrg3502
6. Cotsapas C, Voight BF, Rossin E, Lage K, Neale BM, Wallace C, et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. (2011) 7:e1002254. doi: 10.1371/journal.pgen.1002254
7. Langlais D, Fodil N, Gros P. Genetics of infectious and inflammatory diseases: overlapping discoveries from association and exome-sequencing studies. Annu Rev Immunol. (2017) 35:1–30. doi: 10.1146/annurev-immunol-051116-052442
8. Wu X, Chen H, Xu H. The genomic landscape of human immune-mediated diseases. J Hum Genet. (2015) 60:675–81. doi: 10.1038/jhg.2015.99
9. International Multiple Sclerosis Genetics Consortium, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. (2013) 45:1353–60. doi: 10.1038/ng.2770
10. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
11. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
12. Brophy S, Pavy S, Lewis P, Taylor G, Bradbury L, Robertson D, et al. Inflammatory eye, skin, and bowel disease in spondyloarthritis: genetic, phenotypic, and environmental factors. J Rheumatol. (2001) 28:2667–73.
13. Somers EC, Thomas SL, Smeeth L, Hall AJ. Autoimmune diseases co-occurring within individuals and within families: a systematic review. Epidemiology. (2006) 17:202–17. doi: 10.1097/01.ede.0000193605.93416.df
14. Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF. How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med. (2013) 19:822–4. doi: 10.1038/nm.3260
15. Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, et al. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. (1984) 312:724–9. doi: 10.1038/312724a0
16. Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. (2001) 104:487–501. doi: 10.1016/S0092-8674(01)00237-9
17. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. (2003) 3:745–56. doi: 10.1038/nri1184
18. Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. (1990) 10:1498–506.
19. Haranaka K, Carswell EA, Williamson BD, Prendergast JS, Satomi N, Old LJ. Purification, characterization, and antitumor activity of nonrecombinant mouse tumor necrosis factor. Proc Natl Acad Sci USA. (1986) 83:3949–53. doi: 10.1073/pnas.83.11.3949
20. Takeda K, Iwamoto S, Sugimoto H, Takuma T, Kawatani N, Noda M, et al. Identity of differentiation inducing factor and tumour necrosis factor. Nature. (1986) 323:338–40. doi: 10.1038/323338a0
21. Trinchieri G, Kobayashi M, Rosen M, Loudon R, Murphy M, Perussia B. Tumor necrosis factor and lymphotoxin induce differentiation of human myeloid cell lines in synergy with immune interferon. J Exp Med. (1986) 164:1206–25. doi: 10.1084/jem.164.4.1206
22. Jelinek DF, Lipsky PE. Enhancement of human B cell proliferation and differentiation by tumor necrosis factor-alpha and interleukin 1. J Immunol. (1987) 139:2970–6.
23. Kehrl JH, Miller A, Fauci AS. Effect of tumor necrosis factor alpha on mitogen-activated human B cells. J Exp Med. (1987) 166:786–91. doi: 10.1084/jem.166.3.786
24. Dinarello CA, Cannon JG, Wolff SM, Bernheim HA, Beutler B, Cerami A, et al. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J Exp Med. (1986) 163:1433–50. doi: 10.1084/jem.163.6.1433
25. Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity. (1995) 2:561–72. doi: 10.1016/1074-7613(95)90001-2
26. Sedger LM, McDermott MF. TNF and TNF-receptors: from mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. (2014) 25:453–72. doi: 10.1016/j.cytogfr.2014.07.016
27. Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. (1988) 53:45–53. doi: 10.1016/0092-8674(88)90486-2
28. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. (1997) 385:733–6. doi: 10.1038/385733a0
29. Grell M, Douni E, Wajant H, Lohden M, Clauss M, Maxeiner B, et al. The transmembrane form of tumor-necrosis-factor is the prime activating ligand of the 80 Kda tumor-necrosis-factor receptor. Cell. (1995) 83:793–802. doi: 10.1016/0092-8674(95)90192-2
30. Krippner-Heidenreich A, Tubing F, Bryde S, Willi S, Zimmermann G, Scheurich P. Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J Biol Chem. (2002) 277:44155–63. doi: 10.1074/jbc.M207399200
31. Mukhopadhyay A, Suttles J, Stout RD, Aggarwal BB. Genetic deletion of the tumor necrosis factor receptor p60 or p80 abrogates ligand-mediated activation of nuclear factor-kappa B and of mitogen-activated protein kinases in macrophages. J Biol Chem. (2001) 276:31906–12. doi: 10.1074/jbc.M105252200
32. Weiss T, Grell M, Siemienski K, Muhlenbeck F, Durkop H, Pfizenmaier K, et al. TNFR80-dependent enhancement of TNFR60-induced cell death is mediated by TNFR-associated factor 2 and is specific for TNFR60. J Immunol. (1998) 161:3136–42.
33. Cabal-Hierro L, Lazo PS. Signal transduction by tumor necrosis factor receptors. Cell Signal. (2012) 24:1297–305. doi: 10.1016/j.cellsig.2012.02.006
34. Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. (2016) 12:49–62. doi: 10.1038/nrrheum.2015.169
35. Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. (2015) 15:362–74. doi: 10.1038/nri3834
36. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. (1998) 281:1680–3. doi: 10.1126/science.281.5383.1680
37. Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. (2004) 21:415–28. doi: 10.1016/j.immuni.2004.08.017
38. Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. (2015) 33:79–106. doi: 10.1146/annurev-immunol-032414-112248
39. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. (2015) 517:311–20. doi: 10.1038/nature14191
40. Gregory AP, Dendrou CA, Attfield KE, Haghikia A, Xifara DK, Butter F, et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature. (2012) 488:508–11. doi: 10.1038/nature11307
41. Thoma B, Grell M, Pfizenmaier K, Scheurich P. Identification of a 60-kD tumor necrosis factor (TNF) receptor as the major signal transducing component in TNF responses. J Exp Med. (1990) 172:1019–23. doi: 10.1084/jem.172.4.1019
42. Haridas V, Darnay BG, Natarajan K, Heller R, Aggarwal BB. Overexpression of the p80 TNF receptor leads to TNF-dependent apoptosis, nuclear factor-kappa B activation, and c-Jun kinase activation. J Immunol. (1998) 160:3152–62.
43. Reinhard C, Shamoon B, Shyamala V, Williams LT. Tumor necrosis factor alpha-induced activation of c-jun N-terminal kinase is mediated by TRAF2. EMBO J. (1997) 16:1080–92. doi: 10.1093/emboj/16.5.1080
44. Wu CJ, Conze DB, Li X, Ying SX, Hanover JA, Ashwell JD. TNF-alpha induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J. (2005) 24:1886–98. doi: 10.1038/sj.emboj.7600649
45. Pantelidis P, Lympany PA, Foley PJ, Fanning GC, Welsh KI, du Bois RM. Polymorphic analysis of the high-affinity tumor necrosis factor receptor 2. Tissue Antigens. (1999) 54:585–91. doi: 10.1034/j.1399-0039.1999.540608.x
46. Barton A, John S, Ollier WE, Silman A, Worthington J. Association between rheumatoid arthritis and polymorphism of tumor necrosis factor receptor II, but not tumor necrosis factor receptor I, in Caucasians. Arthritis Rheumat. (2001) 44:61–5. doi: 10.1002/1529-0131(200101)44:1<61::AID-ANR9>3.0.CO;2-Q
47. Dieude P, Petit E, Cailleau-Moindrault S, Osorio J, Pierlot C, Martinez M, et al. Association between tumor necrosis factor receptor II and familial, but not sporadic, rheumatoid arthritis: evidence for genetic heterogeneity. Arthritis Rheumat. (2002) 46:2039–44. doi: 10.1002/art.10101
48. Till A, Rosenstiel P, Krippner-Heidenreich A, Mascheretti-Croucher S, Croucher PJ, Schafer H, et al. The Met-196 -> Arg variation of human tumor necrosis factor receptor 2 (TNFR2) affects TNF-alpha-induced apoptosis by impaired NF-kappaB signaling and target gene expression. J Biol Chem. (2005) 280:5994–6004. doi: 10.1074/jbc.M411541200
49. Lenk H, Tanneberger S, Müller U, Ebert J, Shiga T. Phase II clinical trial of high-dose recombinant human tumor necrosis factor. Cancer Chemother Pharmacol. (1989) 24:391–2. doi: 10.1007/BF00257449
50. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science. (1985) 229:869–71. doi: 10.1126/science.3895437
51. Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, et al. Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature. (1987) 330:662–4. doi: 10.1038/330662a0
52. Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nat Rev Immunol. (2002) 2:364–71. doi: 10.1038/nri802
53. Brandt J, Haibel H, Cornely D, Golder W, Gonzalez J, Reddig J, et al. Successful treatment of active ankylosing spondylitis with the anti-tumor necrosis factor alpha monoclonal antibody infliximab. Arthritis Rheum. (2000) 43:1346–52. doi: 10.1002/1529-0131(200006)43:6<1346::AID-ANR18>3.0.CO;2-E
54. Braun J, Sieper J, Breban M, Collantes-Estevez E, J Davis RI, Marzo-Ortega H, et al. Anti-tumour necrosis factor α therapy for ankylosing spondylitis: international experience. Ann Rheum Dis. (2002) 61:51–60. doi: 10.1136/ard.61.suppl_3.iii51
55. Gorman JD, Sack KE, Davis JCJ. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. (2002) 346:1349–56. doi: 10.1056/NEJMoa012664
56. Ward MM, Deodhar A, Akl EA, Lui A, Ermann J, Gensler LS, et al. American College of Rheumatology/Spondylitis Association of America/spondyloarthritis research and treatment network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. (2016) 68:282–98. doi: 10.1002/art.39298
57. Sieper J, Poddubnyy D. New evidence on the management of spondyloarthritis. Nat Rev Rheumatol. (2016) 12:282–95. doi: 10.1038/nrrheum.2016.42
58. Baeten D, Kruithof E, Van den Bosch F, Van den Bossche N, Herssens A, Mielants H, et al. Systematic safety follow up in a cohort of 107 patients with spondyloarthropathy treated with infliximab: a new perspective on the role of host defence in the pathogenesis of the disease? Ann Rheum Dis. (2003) 62:829–34. doi: 10.1136/ard.62.9.829
59. Assassi S. Rheumatoid arthritis, TNF inhibitors, and non-melanoma skin cancer. Bmj. (2016) 352:i472. doi: 10.1136/bmj.i472
60. van Lumig PP, Menting SP, van den Reek JM, Spuls PI, van Riel PL, van de Kerkhof PC, et al. An increased risk of non-melanoma skin cancer during TNF-inhibitor treatment in psoriasis patients compared to rheumatoid arthritis patients probably relates to disease-related factors. J Eur Acad Dermatol Venereol. (2015) 29:752–60. doi: 10.1111/jdv.12675
61. De Rycke L, Baeten D, Foell D, Kruithof E, Veys EM, Roth J, et al. Differential expression and response to anti-TNFα treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J Pathol. (2005) 206:17–27. doi: 10.1002/path.1758
62. Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. (1994) 344:1105–10. doi: 10.1016/S0140-6736(94)90628-9
63. Knight DM, Trinh H, Le J, Siegel S, Shealy D, McDonough M, et al. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol Immunol. (1993) 30:1443–53. doi: 10.1016/0161-5890(93)90106-L
64. Siegel S, Shealy DJ, Nakada MT, Le J, Woulfe DS, Probert L, et al. The mouse/human chimeric monoclonal antibody cA2 neutralizes TNF in vitro and protects transgenic mice from cachexia and TNF lethality in vivo. Cytokine. (1995) 7:15–25. doi: 10.1006/cyto.1995.1003
65. Hutas G. Golimumab as the first monthly subcutaneous fully human anti-TNF-alpha antibody in the treatment of inflammatory arthropathies. Immunotherapy. (2010) 2:453–60. doi: 10.2217/imt.10.34
66. Shealy DJ, Cai A, Staquet K, Baker A, Lacy ER, Johns L, et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. mAbs. (2010) 2:428–39. doi: 10.4161/mabs.12304
67. Weinblatt ME, Kremer JM, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Fox RI, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor:Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. (1999) 340:253–9. doi: 10.1056/NEJM199901283400401
68. Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor α agents. Inflamm Bowel Dis. (2007) 13:1323–32. doi: 10.1002/ibd.20225
69. Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. (2009) 131:308–16. doi: 10.1016/j.clim.2009.01.002
70. Mitoma H, Horiuchi T, Tsukamoto H. Binding activities of infliximab and etanercept to transmembrane tumor necrosis factor-alpha. Gastroenterology. (2004) 126:934–5; author reply 935–6. doi: 10.1053/j.gastro.2004.01.036
71. Van den Brande JM, Braat H, van den Brink GR, Versteeg HH, Bauer CA, Hoedemaeker I, et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology. (2003) 124:1774–85. doi: 10.1016/S0016-5085(03)00382-2
72. Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, et al. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor α-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. (2008) 58:1248–57. doi: 10.1002/art.23447
73. Greenberg JD, Reed G, Decktor D, Harrold L, Furst D, Gibofsky A, et al. A comparative effectiveness study of adalimumab, etanercept and infliximab in biologically naive and switched rheumatoid arthritis patients: results from the US CORRONA registry. Ann Rheum Dis. (2012) 71:1134–42. doi: 10.1136/annrheumdis-2011-150573
74. Kennedy WP, Simon JA, Offutt C, Horn P, Herman A, Townsend MJ, et al. Efficacy and safety of pateclizumab (anti-lymphotoxin-alpha) compared to adalimumab in rheumatoid arthritis: a head-to-head phase 2 randomized controlled study (The ALTARA Study). Arthritis Res Ther. (2014) 16:467. doi: 10.1186/s13075-014-0467-3
75. Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P. True infliximab resistance in rheumatoid arthritis: a role for lymphotoxin alpha? Ann Rheum Dis. (2004) 63:1344–6. doi: 10.1136/ard.2003.014878
76. Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. (1997) 337:1029–35. doi: 10.1056/NEJM199710093371502
77. Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, et al. Etanercept for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. (2001) 121:1088–94. doi: 10.1053/gast.2001.28674
78. Di Costanzo L, Ayala F, Megna M, Gaudiello F, Patri A, Balato N. The risk of herpes zoster in the anti-TNF-alpha era: a case report and review of the literature. J Dermatol Case Rep. (2013) 7:1–4. doi: 10.3315/jdcr.2013.1126
79. Dixon WG, Hyrich KL, Watson KD, Lunt M, Galloway J, Ustianowski A, et al. Drug-specific risk of tuberculosis in patients with rheumatoid arthritis treated with anti-TNF therapy: results from the British Society for Rheumatology Biologics Register (BSRBR). Ann Rheum Dis. (2010) 69:522–8. doi: 10.1136/ard.2009.118935
80. Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Breban M, et al. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: the three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum. (2009) 60:1884–94. doi: 10.1002/art.24632
81. Coates LC, Cawkwell LS, Ng NWF, Bennett AN, Bryer DJ, Fraser AD, et al. Real life experience confirms sustained response to long-term biologics and switching in ankylosing spondylitis. Rheumatology. (2008) 47:897–900. doi: 10.1093/rheumatology/ken094
82. Scallon BJ, Moore MA, Trinh H, Knight DM, Ghrayeb J. Chimeric anti-TNF-alpha monoclonal antibody cA2 binds recombinant transmembrane TNF-alpha and activates immune effector functions. Cytokine. (1995) 7:251–9.
83. Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. (2008) 117:244–79. doi: 10.1016/j.pharmthera.2007.10.001
84. Gullick NJ, Evans HG, Church LD, Jayaraj DM, Filer A, Kirkham BW, et al. Linking power Doppler ultrasound to the presence of th17 cells in the rheumatoid arthritis joint. PLoS ONE. (2010) 5:e12516. doi: 10.1371/journal.pone.0012516
85. Aerts NE, De knop KJ, Leysen J, Ebo DG, Bridts CH, Weyler JJ, et al. Increased IL-17 production by peripheral T helper cells after tumour necrosis factor blockade in rheumatoid arthritis is accompanied by inhibition of migration-associated chemokine receptor expression. Rheumatology. (2010) 49:2264–72. doi: 10.1093/rheumatology/keq224
86. Chen D-Y, Chen Y-M, Chen H-H, Hsieh C-W, Lin C-C, Lan J-L. Increasing levels of circulating Th17 cells and interleukin-17 in rheumatoid arthritis patients with an inadequate response to anti-TNF-α therapy. Arthritis Res Ther. (2011) 13:R126. doi: 10.1186/ar3431
87. Herman S, Zurgil N, Machlav S, Shinberg A, Langevitz P, Ehrenfeld M, et al. Distinct effects of anti-tumor necrosis factor combined therapy on TH1/TH2 balance in rheumatoid arthritis patients. Clin Vacc Immunol. (2011) 18:1077–82. doi: 10.1128/CVI.00061-11
88. Lina C, Conghua W, Nan L, Ping Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J Clin Immunol. (2011) 31:596–605. doi: 10.1007/s10875-011-9542-6
89. Limón-Camacho L, Vargas-Rojas MI, Vázquez-Mellado J, Casasola-Vargas J, Moctezuma JF, Burgos-Vargas R, et al. In vivo peripheral blood proinflammatory T cells in patients with ankylosing spondylitis. J Rheumatol. (2012) 39:830–5. doi: 10.3899/jrheum.110862
90. Alzabin S, Abraham SM, Taher TE, Palfreeman A, Hull D, McNamee K, et al. Incomplete response of inflammatory arthritis to TNFα blockade is associated with the Th17 pathway. Ann Rheum Dis. (2012) 71:1741–8. doi: 10.1136/annrheumdis-2011-201024
91. Szalay B, Mészáros G, Cseh Á, Ács L, Deák M, Kovács L, et al. Adaptive immunity in ankylosing spondylitis: phenotype and functional alterations of T-cells before and during infliximab therapy. Clin Dev Immunol. (2012) 2012:808724. doi: 10.1155/2012/808724
92. Xueyi L, Lina C, Zhenbiao W, Qing H, Qiang L, Zhu P. Levels of circulating Th17 cells and regulatory T cells in ankylosing spondylitis patients with an inadequate response to anti-TNF-α therapy. J Clin Immunol. (2013) 33:151–61. doi: 10.1007/s10875-012-9774-0
93. Szalay B, Vásárhelyi B, Cseh A, Tulassay T, Deák M, Kovács L, et al. The impact of conventional DMARD and biological therapies on CD4+ cell subsets in rheumatoid arthritis: a follow-up study. Clin. Rheumatol. (2014) 33:175–85. doi: 10.1007/s10067-013-2352-x
94. Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, et al. TNF-alpha blockade induces IL-10 expression in human CD4+ T cells. Nat Commun. (2014) 5:3199. doi: 10.1038/ncomms4199
95. Hull DN, Williams RO, Pathan E, Alzabin S, Abraham S, Taylor PC. Anti-tumour necrosis factor treatment increases circulating T helper type 17 cells similarly in different types of inflammatory arthritis. Clin Exp Immunol. (2015) 181:401–6. doi: 10.1111/cei.12626
96. Talotta R, Berzi A, Atzeni F, Batticciotto A, Clerici M, Sarzi-Puttini P, et al. Paradoxical expansion of Th1 and Th17 lymphocytes in rheumatoid arthritis following infliximab treatment: a possible explanation for a lack of clinical response. J Clin Immunol. (2015) 35:550–7. doi: 10.1007/s10875-015-0182-0
97. Hull DN, Cooksley H, Chokshi S, Williams RO, Abraham S, Taylor PC. Increase in circulating Th17 cells during anti-TNF therapy is associated with ultrasonographic improvement of synovitis in rheumatoid arthritis. Arthritis Res Ther. (2016) 18:303. doi: 10.1186/s13075-016-1197-5
98. Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFalpha therapy. J Exp Med. (2004) 200:277–85. doi: 10.1084/jem.20040165
99. Nadkarni S, Mauri C, Ehrenstein MR. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med. (2007) 204:33–9. doi: 10.1084/jem.20061531
100. Huang Z, Yang B, Shi Y, Cai B, Li Y, Feng W, et al. Anti-TNF-alpha therapy improves Treg and suppresses Teff in patients with rheumatoid arthritis. Cell Immunol. (2012) 279:25–9. doi: 10.1016/j.cellimm.2012.09.001
101. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheumat. (2012) 64:3129–38. doi: 10.1002/art.34565
102. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med. (2016) 213:1241–53. doi: 10.1084/jem.20151255
103. Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner ECB, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. (2008) 180:688–92. doi: 10.4049/jimmunol.180.2.688
104. Souto-Carneiro MM, Mahadevan V, Takada K, Fritsch-Stork R, Nanki T, Brown M, et al. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res Ther. (2009) 11:R84. doi: 10.1186/ar2718
105. Kobie JJ, Zheng B, Bryk P, Barnes M, Ritchlin CT, Tabechian DA, et al. Decreased influenza-specific B cell responses in rheumatoid arthritis patients treated with anti-tumor necrosis factor. Arthritis Res Ther. (2011) 13:R209. doi: 10.1186/ar3542
106. Salinas GF, De Rycke L, Barendregt B, Paramarta JE, Hreggvidstdottir H, Cantaert T, et al. Anti-TNF treatment blocks the induction of T cell-dependent humoral responses. Ann Rheum Dis. (2013) 72:1037–43. doi: 10.1136/annrheumdis-2011-201270
107. Conigliaro P, Triggianese P, Perricone C, Chimenti MS, Di Muzio G, Ballanti E, et al. Restoration of peripheral blood natural killer and B cell levels in patients affected by rheumatoid and psoriatic arthritis during etanercept treatment. Clin Exp Immunol. (2014) 177:234–43. doi: 10.1111/cei.12335
108. Daien CI, Gailhac S, Mura T, Audo R, Combe B, Hahne M, et al. Regulatory B10 cells are decreased in patients with rheumatoid arthritis and are inversely correlated with disease activity. Arthritis Rheumatol. (2014) 66:2037–46. doi: 10.1002/art.38666
109. Glaesener S, Quach TD, Onken N, Weller-Heinemann F, Dressler F, Huppertz HI, et al. Distinct effects of methotrexate and etanercept on the B cell compartment in patients with juvenile idiopathic arthritis. Arthritis Rheumatol. (2014) 66:2590–600. doi: 10.1002/art.38736
110. Bautista-Caro M-B, de Miguel E, Peiteado D, Plasencia-Rodríguez C, Villalba A, Monjo-Henry I, et al. Increased frequency of circulating CD19+CD24hiCD38hi B cells with regulatory capacity in patients with Ankylosing spondylitis (AS) naïve for biological agents. PLoS ONE. (2017) 12:e0180726. doi: 10.1371/journal.pone.0180726
111. Ingelman-Sundberg HM, Laestadius Å, Chrapkowska C, Mördrup K, Magnusson B, Sundberg E, et al. Diverse effects on vaccine-specific serum IgG titres and memory B cells upon methotrexate and anti-TNF-α therapy in children with rheumatic diseases: a cross-sectional study. Vaccine. (2016) 34:1304–11. doi: 10.1016/j.vaccine.2016.01.027
112. Chen M, Zhang L, Ren Y, Zhang K, Yang Y, Fang Y, et al. Defective function of CD24(+)CD38(+) regulatory B cells in ankylosing spondylitis. DNA Cell Biol. (2016) 35:88–95. doi: 10.1089/dna.2015.3046
113. Salomon S, Guignant C, Morel P, Flahaut G, Brault C, Gourguechon C, et al. Th17 and CD24hiCD27+ regulatory B lymphocytes are biomarkers of response to biologics in rheumatoid arthritis. Arthritis Res Ther. (2017) 19:33. doi: 10.1186/s13075-017-1244-x
114. Banko Z, Pozsgay J, Gati T, Rojkovich B, Ujfalussy I, Sarmay G. Regulatory B cells in rheumatoid arthritis: alterations in patients receiving anti-TNF therapy. Clin Immunol. (2017) 184:63–9. doi: 10.1016/j.clim.2017.05.012
115. Mavropoulos A, Varna A, Zafiriou E, Liaskos C, Alexiou I, Roussaki-Schulze A, et al. IL-10 producing Bregs are impaired in psoriatic arthritis and psoriasis and inversely correlate with IL-17- and IFNgamma-producing T cells. Clin Immunol. (2017) 184:33–41. doi: 10.1016/j.clim.2017.04.010
116. Dombrecht EJ, Aerts NE, Schuerwegh AJ, Hagendorens MM, Ebo DG, Van Offel JF, et al. Influence of anti-tumor necrosis factor therapy (Adalimumab) on regulatory T cells and dendritic cells in rheumatoid arthritis. Clin Exp Rheumatol. (2006) 24:31–7.
117. Nocturne G, Boudaoud S, Ly B, Pascaud J, Paoletti A, Mariette X. Impact of anti-TNF therapy on NK cells function and on immunosurveillance against B-cell lymphomas. J Autoimmunity. (2017) 80:56–64. doi: 10.1016/j.jaut.2017.02.001
118. Mo WX, Yin SS, Chen H, Zhou C, Zhou JX, Zhao LD, et al. Chemotaxis of Vdelta2 T cells to the joints contributes to the pathogenesis of rheumatoid arthritis. Ann Rheum Dis. (2017) 76:2075–84. doi: 10.1136/annrheumdis-2016-211069
119. Blijdorp ICJ, Menegatti S, Van Mens LJJ, van de Sand MGH, Chen S, Hreggvidsdottir HS, et al. IL-22- and GM-CSF-expressing but not IL-17A-expressing group 3 innate lymphoid cells are expanded in the inflamed spondyloarthritis joint. Arthritis Rheumatol. (2019) 71:392–402. doi: 10.1002/art.40736
120. Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheumat. (2002) 46:2578–86. doi: 10.1002/art.10545
121. Coulthard LR, Geiler J, Mathews RJ, Church LD, Dickie LJ, Cooper DL, et al. Differential effects of infliximab on absolute circulating blood leucocyte counts of innate immune cells in early and late rheumatoid arthritis patients. Clin Exp Immunol. (2012) 170:36–46. doi: 10.1111/j.1365-2249.2012.04626.x
122. Krasselt M, Baerwald C, Wagner U, Rossol M. CD56+ monocytes have a dysregulated cytokine response to lipopolysaccharide and accumulate in rheumatoid arthritis and immunosenescence. Arthritis Res Ther. (2013) 15:R139. doi: 10.1186/ar4321
123. Muller N, Doring F, Klapper M, Neumann K, Schulte DM, Turk K, et al. Interleukin-6 and tumour necrosis factor-alpha differentially regulate lincRNA transcripts in cells of the innate immune system in vivo in human subjects with rheumatoid arthritis. Cytokine. (2014) 68:65–8. doi: 10.1016/j.cyto.2014.03.004
124. Meusch U, Krasselt M, Rossol M, Baerwald C, Klingner M, Wagner U. In vitro response pattern of monocytes after tmTNF reverse signaling predicts response to anti-TNF therapy in rheumatoid arthritis. J Transl Med. (2015) 13:256. doi: 10.1186/s12967-015-0620-z
125. Aeberli D, Kamgang R, Balani D, Hofstetter W, Villiger PM, Seitz M. Regulation of peripheral classical and non-classical monocytes on infliximab treatment in patients with rheumatoid arthritis and ankylosing spondylitis. RMD Open. (2016) 2:e000079. doi: 10.1136/rmdopen-2015-000079
126. Zhao J, Yuan W, Tao C, Sun P, Yang Z, Xu W. M2 polarization of monocytes in ankylosing spondylitis and relationship with inflammation and structural damage. Acta Pathol Microbiol Immunol Scand. (2017) 125:1070–5. doi: 10.1111/apm.12757
127. Maurice MM, van der Graaff WL, Leow A, Breedveld FC, van Lier RA, Verweij CL. Treatment with monoclonal anti-tumor necrosis factor alpha antibody results in an accumulation of Th1 CD4+ T cells in the peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum. (1999) 42:2166–73.
128. Zou J, Rudwaleit M, Brandt J, Thiel A, Braun J, Sieper J. Down-regulation of the nonspecific and antigen-specific T cell cytokine response in ankylosing spondylitis during treatment with infliximab. Arthritis Rheum. (2003) 48:780–90. doi: 10.1002/art.10847
129. Zou J, Rudwaleit M, Brandt J, Thiel A, Braun J, Sieper J. Up regulation of the production of tumour necrosis factor alpha and interferon gamma by T cells in ankylosing spondylitis during treatment with etanercept. Ann Rheumat Dis. (2003) 62:561–4.
130. Aravena O, Pesce B, Soto L, Orrego N, Sabugo F, Wurmann P, et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-gamma-producing NK cell and regulatory T cell subsets. Immunobiology. (2011) 216:1256–63. doi: 10.1016/j.imbio.2011.07.006
131. Dulic S, Vasarhelyi Z, Sava F, Berta L, Szalay B, Toldi G, et al. T-cell subsets in rheumatoid arthritis patients on long-term anti-TNF or IL-6 receptor blocker therapy. Mediators Inflamm. (2017) 2017:6894374. doi: 10.1155/2017/6894374
132. Charles P, Elliott MJ, Davis D, Potter A, Kalden JR, Antoni C, et al. Regulation of cytokines, cytokine inhibitors, and acutephase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J Immunol. (1999) 163:1521–8.
133. Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheumat. (2000) 43:38–47. doi: 10.1002/1529-0131(200001)43:1<38::AID-ANR6>3.0.CO;2-L
134. Barrera P, Joosten LA, den Broeder AA, van de Putte LB, van Riel PL, van den Berg WB. Effects of treatment with a fully human anti-tumour necrosis factor alpha monoclonal antibody on the local and systemic homeostasis of interleukin 1 and TNFalpha in patients with rheumatoid arthritis. Ann Rheumat Dis. (2001) 60:660–9. doi: 10.1136/ard.60.7.660
135. Klimiuk PA, Sierakowski S, Domyslawska I, Chwiecko J. Regulation of serum chemokines following infliximab therapy in patients with rheumatoid arthritis. Clin Exp Rheumatol. (2006) 24:529–33.
136. Kageyama Y, Ichikawa T, Nagafusa T, Torikai E, Shimazu M, Nagano A. Etanercept reduces the serum levels of interleukin-23 and macrophage inflammatory protein-3 alpha in patients with rheumatoid arthritis. Rheumatol Int. (2007) 28:137–43. doi: 10.1007/s00296-007-0388-4
137. van Lieshout AW, Fransen J, Flendrie M, Eijsbouts AM, van den Hoogen FH, van Riel PL, et al. Circulating levels of the chemokine CCL18 but not CXCL16 are elevated and correlate with disease activity in rheumatoid arthritis. Ann Rheum Dis. (2007) 66:1334–8. doi: 10.1136/ard.2006.066084
138. Fabre S, Dupuy AM, Dossat N, Guisset C, Cohen JD, Cristol JP, et al. Protein biochip array technology for cytokine profiling predicts etanercept responsiveness in rheumatoid arthritis. Clin Exp Immunol. (2008) 153:188–95. doi: 10.1111/j.1365-2249.2008.03691.x
139. Kawashiri S, Kawakami A, Iwamoto N, Fujikawa K, Aramaki T, Tamai M, et al. Proinflammatory cytokines synergistically enhance the production of chemokine ligand 20 (CCL20) from rheumatoid fibroblast-like synovial cells in vitro and serum CCL20 is reduced in vivo by biologic disease-modifying antirheumatic drugs. J Rheumatol. (2009) 36:132. doi: 10.3899/jrheum.090132
140. Odai T, Matsunawa M, Takahashi R, Wakabayashi K, Isozaki T, Yajima N, et al. Correlation of CX3CL1 and CX3CR1 levels with response to infliximab therapy in patients with rheumatoid arthritis. J Rheumatol. (2009) 36:1158–65. doi: 10.3899/jrheum.081074
141. Popa C, Barrera P, Joosten LA, van Riel PL, Kullberg BJ, van der Meer JW, et al. Cytokine production from stimulated whole blood cultures in rheumatoid arthritis patients treated with various TNF blocking agents. Eur Cytokine Netw. (2009) 20:88–93. doi: 10.1684/ecn.2009.0150
142. Akbulut H, Koca SS, Ozgen M, Isik A. Anti-tumor necrosis factor therapies reduce serum macrophage inflammatory protein-1alpha in ankylosing spondylitis. J Rheumatol. (2010) 37:1073–4. doi: 10.3899/jrheum.091469
143. Yue C, You X, Zhao L, Wang H, Tang F, Zhang F, et al. The effects of adalimumab and methotrexate treatment on peripheral Th17 cells and IL-17/IL-6 secretion in rheumatoid arthritis patients. Rheumatol Int. (2010) 30:1553–7. doi: 10.1007/s00296-009-1179-x
144. Bosè F, Raeli L, Garutti C, Frigerio E, Cozzi A, Crimi M, et al. Dual role of anti-TNF therapy: enhancement of TCR-mediated T cell activation in peripheral blood and inhibition of inflammation in target tissues. Clin Immunol. (2011) 139:164–76. doi: 10.1016/j.clim.2011.01.015
145. Taylan, Sari I, Akinci B, Bilge S, Kozaci D, Akar S, et al. Biomarkers and cytokines of bone turnover: extensive evaluation in a cohort of patients with ankylosing spondylitis. BMC Musculoskelet Disorders. (2012) 13:191. doi: 10.1186/1471-2474-13-191
146. Kayakabe K, Kuroiwa T, Sakurai N, Ikeuchi H, Kadiombo T, Sakairi T, et al. Interleukin-1beta measurement in stimulated whole blood cultures is useful to predict response to anti-TNF therapies in rheumatoid arthritis. Rheumatology. (2012) 51:1639–43. doi: 10.1093/rheumatology/kes094
147. Tian Y, Shen H, Xia L, Lu J. Elevated serum and synovial fluid levels of interleukin-34 in rheumatoid arthritis: possible association with disease progression via interleukin-17 production. J Interfer Cytokine Res. (2013) 33:398–401. doi: 10.1089/jir.2012.0122
148. Greisen SR, Schelde KK, Rasmussen TK, Kragstrup TW, Stengaard-Pedersen K, Hetland ML, et al. CXCL13 predicts disease activity in early rheumatoid arthritis and could be an indicator of the therapeutic ‘window of opportunity’. Arthritis Res Ther. (2014) 16:434. doi: 10.1186/s13075-014-0434-z
149. Chang SH, Choi BY, Choi J, Yoo JJ, Ha Y-J, Cho HJ, et al. Baseline serum interleukin-34 levels independently predict radiographic progression in patients with rheumatoid arthritis. Rheumatol Int. (2015) 35:71–9. doi: 10.1007/s00296-014-3056-5
150. Genre F, Lopez-Mejias R, Miranda-Filloy JA, Ubilla B, Mijares V, Carnero-Lopez B, et al. Anti-TNF-alpha therapy reduces endothelial cell activation in non-diabetic ankylosing spondylitis patients. Rheumatol Int. (2015) 35:2069–78. doi: 10.1007/s00296-015-3314-1
151. Milanez FM, Saad CG, Viana VT, Moraes JC, Perico GV, Sampaio-Barros PD, et al. IL-23/Th17 axis is not influenced by TNF-blocking agents in ankylosing spondylitis patients. Arthritis Res Ther. (2016) 18:52. doi: 10.1186/s13075-016-0949-6
152. Han BK, Kuzin I, Gaughan JP, Olsen NJ, Bottaro A. Baseline CXCL10 and CXCL13 levels are predictive biomarkers for tumor necrosis factor inhibitor therapy in patients with moderate to severe rheumatoid arthritis: a pilot, prospective study. Arthritis Res Ther. (2016) 10:1–7. doi: 10.1186/s13075-016-0995-0
153. Walters HM, Pan N, Lehman TJ, Adams A, Kalliolias GD, Zhu YS, et al. The impact of disease activity and tumour necrosis factor-alpha inhibitor therapy on cytokine levels in juvenile idiopathic arthritis. Clin Exp Immunol. (2016) 184:308–17. doi: 10.1111/cei.12782
154. Wampler Muskardin T, Vashisht P, Dorschner JM, Jensen MA, Chrabot BS, Kern M, et al. Increased pretreatment serum IFN-β/a ratio predicts nonresponse to tumour necrosis factor a inhibition in rheumatoid arthritis. Ann Rheumat Dis. (2016) 75:1757–62. doi: 10.1136/annrheumdis-2015-208001
155. Bystrom J, Clanchy FI, Taher TE, Al-Bogami MM, Muhammad HA, Alzabin S, et al. Response to treatment with TNFa inhibitors in rheumatoid arthritis is associated with high levels of GM-CSF and GM-CSF+ T lymphocytes. Clin Rev Allergy Immunol. (2017) 53:265–76. doi: 10.1007/s12016-017-8610-y
156. Al-Mossawi MH, Chen L, Fang H, Ridley A, De Wit J, Yager N, et al. Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat Commun. (2017) 8:1–11. doi: 10.1038/s41467-017-01771-2
157. Makris A, Adamidi S, Koutsianas C, Tsalapaki C, Hadziyannis E, Vassilopoulos D. Increased frequency of peripheral B and T cells expressing granulocyte monocyte colony-stimulating factor in rheumatoid arthritis patients. Front Immunol. (2017) 8:1967. doi: 10.3389/fimmu.2017.01967
158. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. TNFα in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine. (2018) 101:4–13. doi: 10.1016/j.cyto.2016.09.001
159. Davignon J-L, Rauwel B, Degboé Y, Constantin A, Boyer J-F, Kruglov A, et al. Modulation of T-cell responses by anti-tumor necrosis factor treatments in rheumatoid arthritis: a review. Arthritis Res Ther. (2018) 20:229. doi: 10.1186/s13075-018-1725-6
160. Cope AP, Liblau RS, Yang X-D, Congia M, Laudanna C, Schreiber RD, et al. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J Exp Med. (1997) 185:1573–84.
161. Zhang Z, Gorman CL, Vermi A-C, Monaco C, Foey A, Owen S, et al. TCRzetadim lymphocytes define populations of circulating effector cells that migrate to inflamed tissues. Blood. (2007) 109:4328–4335. doi: 10.1182/blood-2006-12-064170
162. Steinman L. A brief history of TH17, the first major revision in the TH1/TH2 hypothesis of T cell–mediated tissue damage. Nat Med. (2007) 13:139–45. doi: 10.1038/nm1551
163. Feng G, Gao W, Strom TB, Oukka M, Francis RS, Wood KJ, et al. Exogenous IFN-gamma ex vivo shapes the alloreactive T-cell repertoire by inhibition of Th17 responses and generation of functional Foxp3+ regulatory T cells. Eur J Immunol. (2008) 38:2512–27. doi: 10.1002/eji.200838411
164. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
165. Chu C-Q, Swart D, Alcorn D, Tocker J, Elkon KB. Interferon-gamma regulates susceptibility to collagen-induced arthritis through suppression of interleukin-17. Arthritis Rheum. (2007) 56:1145–51. doi: 10.1002/art.22453
166. Kelchtermans H, Schurgers E, Geboes L, Mitera T, Van Damme J, Van Snick J, et al. Effector mechanisms of interleukin-17 in collagen-induced arthritis in the absence of interferon-gamma and counteraction by interferon-gamma. Arthritis Res Ther. (2009) 11:R122. doi: 10.1186/ar2787
167. Nissinen R, Leirisalo-Repo M, Peltomaa R, Palosuo T, Vaarala O. Cytokine and chemokine receptor profile of peripheral blood mononuclear cells during treatment with infliximab in patients with active rheumatoid arthritis. Ann Rheum. Dis. (2004) 63:681–7. doi: 10.1136/ard.2003.008599
168. Schuerwegh AJ, Van Offel JF, Stevens WJ, Bridts CH, De Clerck LS. Influence of therapy with chimeric monoclonal tumour necrosis factor-alpha antibodies on intracellular cytokine profiles of T lymphocytes and monocytes in rheumatoid arthritis patients. Rheumatology. (2003) 42:541–8. doi: 10.1093/rheumatology/keg171
169. Notley CA, Inglis JJ, Alzabin S, McCann FE, McNamee KE, Williams RO. Blockade of tumor necrosis factor in collagen-induced arthritis reveals a novel immunoregulatory pathway for Th1 and Th17 cells. J Exp Med. (2008) 205:2491–7. doi: 10.1084/jem.20072707
170. Hamilton JA. Plasminogen activator/plasmin system in arthritis and inflammation: friend or foe? Arthritis Rheum. (2008) 58:645–8. doi: 10.1002/art.23269
171. Worth C, Bowness P, Hussein Al-Mossawi M. Novel therapeutic targets in axial spondyloarthritis. Curr Treatm Opt Rheumatol. (2018) 4:174–82. doi: 10.1007/s40674-018-0095-1
172. Griffin JD, Cannistra SA, Sullivan R, Demetri GD, Ernst TJ, Kanakura Y. The biology of GM-CSF: regulation of production and interaction with its receptor. Int J Cell Cloning. (1990) 8(Suppl. 1):35–44; discussion 44–5.
173. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:899–907. doi: 10.1136/annrheumdis-2014-206578
174. Greven DEA, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1924–30. doi: 10.1136/annrheumdis-2014-205234
175. Yamada H, Haraguchi A, Sakuraba K, Okazaki K, Fukushi J-I, Mizu-Uchi H, et al. Th1 is the predominant helper T cell subset that produces GM-CSF in the joint of rheumatoid arthritis. RMD Open. (2017) 3:e000487. doi: 10.1136/rmdopen-2017-000487
176. Burmester GR, McInnes IB, Kremer JM, Miranda P, Vencovský J, Godwood A, et al. Mavrilimumab, a fully human granulocyte-macrophage colony-stimulating factor receptor α monoclonal antibody: long-term safety and efficacy in patients with rheumatoid arthritis. Arthritis Rheumatol. (2018) 70:679–89. doi: 10.1002/art.40420
177. Tak PP, Taylor PC, Breedveld FC, Smeets TJ, Daha MR, Kluin PM, et al. Decrease in cellularity and expression of adhesion molecules by anti-tumor necrosis factor alpha monoclonal antibody treatment in patients with rheumatoid arthritis. Arthritis Rheum. (1996) 39:1077–81. doi: 10.1002/art.1780390702
178. Banks RE. Measurement of cytokines in clinical samples using immunoassays: problems and pitfalls. Crit Rev Clin Lab Sci. (2000) 37:131–82. doi: 10.1080/10408360091174187
179. Aeberli D, Seitz M, Jüni P, Villiger PM. Increase of peripheral CXCR3 positive T lymphocytes upon treatment of RA patients with TNF-alpha inhibitors. Rheumatology. (2005) 44:172–5. doi: 10.1093/rheumatology/keh437
180. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. (2007) 204:2803–12. doi: 10.1084/jem.20071397
181. Strieter RM, Burdick MD, Gomperts BN, Belperio JA, Keane MP. CXC chemokines in angiogenesis. Cytokine Growth Factor Rev. (2005) 16:593–609. doi: 10.1016/j.cytogfr.2005.04.007
182. Erdem H, Pay S, Musabak U, Simsek I, Dinc A, Pekel A, et al. Synovial angiostatic non-ELR CXC chemokines in inflammatory arthritides: does CXCL4 designate chronicity of synovitis? Rheumatol Int. (2007) 27:969–73. doi: 10.1007/s00296-007-0317-6
183. Sucur A, Jajic Z, Artukovic M, Matijasevic MI, Anic B, Flegar D, et al. Chemokine signals are crucial for enhanced homing and differentiation of circulating osteoclast progenitor cells. Arthritis Res Ther. (2017) 19:142. doi: 10.1186/s13075-017-1337-6
184. Aggarwal A, Sarangi AN, Gaur P, Shukla A, Aggarwal R. Gut microbiome in children with enthesitis-related arthritis in a developing country and the effect of probiotic administration. Clin Exp Immunol. (2017) 187:480–9. doi: 10.1111/cei.12900
185. Pala O, Diaz A, Blomberg BB, Frasca D. B Lymphocytes in rheumatoid arthritis and the effects of anti-TNF-α agents on B lymphocytes: a review of the literature. Clin Ther. (2018) 40:1034–45. doi: 10.1016/j.clinthera.2018.04.016
186. Kaminski DA, Wei C, Qian Y, Rosenberg AF, Sanz I. Advances in human B cell phenotypic profiling. Front Immunol. (2012) 3:302. doi: 10.3389/fimmu.2012.00302
187. Bautista-Caro M-B, Arroyo-Villa I, Castillo-Gallego C, de Miguel E, Peiteado D, Plasencia-Rodríguez C, et al. Decreased frequencies of circulating follicular helper T cell counterparts and plasmablasts in ankylosing spondylitis patients Naïve for TNF blockers. PLoS ONE. (2014) 9:e107086. doi: 10.1371/journal.pone.0107086
188. Fekete A, Soos L, Szekanecz Z, Szabo Z, Szodoray P, Barath S, et al. Disturbances in B- and T-cell homeostasis in rheumatoid arthritis: suggested relationships with antigen-driven immune responses. J Autoimmun. (2007) 29:154–63. doi: 10.1016/j.jaut.2007.07.002
189. Mauri C, Menon M. The expanding family of regulatory B cells. Int Immunol. (2015) 27:479–86. doi: 10.1093/intimm/dxv038
190. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. CD19+CD24hiCD38hi B cells maintain regulatory T cells while limiting TH1 and TH17 differentiation. Sci Transl Med. (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
191. Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, et al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. (2009) 45:124–31. doi: 10.1016/j.cyto.2008.11.008
192. Salemi S, Picchianti-Diamanti A, Germano V, Donatelli I, Di Martino A, Facchini M, et al. Influenza vaccine administration in rheumatoid arthritis patients under treatment with TNFalpha blockers: safety and immunogenicity. Clin Immunol. (2010) 134:113–20. doi: 10.1016/j.clim.2009.09.014
193. Bombardieri M, Lewis M, Pitzalis C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nature reviews. Rheumatology. (2017) 13:141–54. doi: 10.1038/nrrheum.2016.217
194. Corsiero E, Nerviani A, Bombardieri M, Pitzalis C. Ectopic lymphoid structures: powerhouse of autoimmunity. Front Immunol. (2016) 7:430. doi: 10.3389/fimmu.2016.00430
195. Drayton DL, Ying X, Lee J, Lesslauer W, Ruddle NH. Ectopic LT alpha beta directs lymphoid organ neogenesis with concomitant expression of peripheral node addressin and a HEV-restricted sulfotransferase. J Exp Med. (2003) 197:1153–63. doi: 10.1084/jem.20021761
196. Furtado GC, Pacer ME, Bongers G, Benezech C, He Z, Chen L, et al. TNFalpha-dependent development of lymphoid tissue in the absence of RORgammat(+) lymphoid tissue inducer cells. Mucos Immunol. (2014) 7:602–14. doi: 10.1038/mi.2013.79
197. Kratz A, Campos-Neto A, Hanson MS, Ruddle NH. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J Exp Med. (1996) 183:1461–72. doi: 10.1084/jem.183.4.1461
198. Canete JD, Celis R, Moll C, Izquierdo E, Marsal S, Sanmarti R, et al. Clinical significance of synovial lymphoid neogenesis and its reversal after anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann Rheumat Dis. (2009) 68:751–6. doi: 10.1136/ard.2008.089284
199. Cantaert T, Kolln J, Timmer T, van der Pouw Kraan TC, Vandooren B, Thurlings RM, et al. B lymphocyte autoimmunity in rheumatoid synovitis is independent of ectopic lymphoid neogenesis. J Immunol. (2008) 181:785–94. doi: 10.4049/jimmunol.181.1.785
200. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen-specific immune responses within tissues. Immunology. (2016) 147:141–51. doi: 10.1111/imm.12554
201. Humby F, Bombardieri M, Manzo A, Kelly S, Blades MC, Kirkham B, et al. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. (2009) 6:e1. doi: 10.1371/journal.pmed.0060001
202. Feng X, Xu X, Wang Y, Zheng Z, Lin G. Ectopic germinal centers and IgG4-producing plasmacytes observed in synovia of HLA-B27+ ankylosing spondylitis patients with advanced hip involvement. Int J Rheumatol. (2015) 2015:316421. doi: 10.1155/2015/316421
203. Rudwaleit M, Listing J, Brandt J, Braun J, Sieper J. Prediction of a major clinical response (BASDAI 50) to tumour necrosis factor alpha blockers in ankylosing spondylitis. Ann Rheum Dis. (2004) 63:665–70. doi: 10.1136/ard.2003.016386
204. Arends S, Brouwer E, van der Veer E, Groen H, Leijsma MK, Houtman PM, et al. Baseline predictors of response and discontinuation of tumor necrosis factor-alpha blocking therapy in ankylosing spondylitis: a prospective longitudinal observational cohort study. Arthritis Res Ther. (2011) 13:R94. doi: 10.1186/ar3369
205. de Vries MK, van Eijk IC, van der Horst-Bruinsma IE, Peters MJ, Nurmohamed MT, Dijkmans BA, et al. Erythrocyte sedimentation rate, C-reactive protein level, and serum amyloid a protein for patient selection and monitoring of anti-tumor necrosis factor treatment in ankylosing spondylitis. Arthritis Rheumat. (2009) 61:1484–90. doi: 10.1002/art.24838
206. Vastesaeger N, Cruyssen BV, Mulero J, Gratacos Masmitja J, Zarco P, Almodovar R, et al. ASDAS high disease activity versus BASDAI elevation in patients with ankylosing spondylitis as selection criterion for anti-TNF therapy. Reumatol Clin. (2014) 10:204–9. doi: 10.1016/j.reuma.2013.12.006
207. Gremese E, Bernardi S, Bonazza S, Nowik M, Peluso G, Massara A, et al. Body weight, gender and response to TNF-α blockers in axial spondyloarthritis. Rheumatology. (2014) 53:875–81. doi: 10.1093/rheumatology/ket433
208. Glintborg B, Højgaard P, Lund Hetland M, Steen Krogh N, Kollerup G, Jensen J, et al. Impact of tobacco smoking on response to tumour necrosis factor-alpha inhibitor treatment in patients with ankylosing spondylitis: results from the Danish nationwide DANBIO registry. Rheumatology. (2016) 55:659–68. doi: 10.1093/rheumatology/kev392
209. Pedersen SJ, Sorensen IJ, Garnero P, Johansen JS, Madsen OR, Tvede N, et al. ASDAS, BASDAI and different treatment responses and their relation to biomarkers of inflammation, cartilage and bone turnover in patients with axial spondyloarthritis treated with TNFalpha inhibitors. Ann Rheumat Dis. (2011) 70:1375–81. doi: 10.1136/ard.2010.138883
210. Turina MC, Yeremenko N, Paramarta JE, De Rycke L, Baeten D. Calprotectin (S100A8/9) as serum biomarker for clinical response in proof-of-concept trials in axial and peripheral spondyloarthritis. Arthritis Res Ther. (2014) 16:413. doi: 10.1186/s13075-014-0413-4
211. Choi IY, Gerlag DM, Herenius MJ, Thurlings RM, Wijbrandts CA, Foell D, et al. MRP8/14 serum levels as a strong predictor of response to biological treatments in patients with rheumatoid arthritis. Ann Rheum Dis. (2015) 74:499–505. doi: 10.1136/annrheumdis-2013-203923
212. van Baarsen LG, Wijbrandts CA, Rustenburg F, Cantaert T, van der Pouw Kraan TC, Baeten DL, et al. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther. (2010) 12:R11. doi: 10.1186/ar2912
213. Koczan D, Drynda S, Hecker M, Drynda A, Guthke R, Kekow J, et al. Molecular discrimination of responders and nonresponders to anti-TNF alpha therapy in rheumatoid arthritis by etanercept. Arthritis Res Ther. (2008) 10:R50. doi: 10.1186/ar2419
214. Stuhlmüller B, Häupl T, Hernandez MM, Grützkau A, Kuban R-J, Tandon N, et al. CD11c as a transcriptional biomarker to predict response to anti-TNF monotherapy with adalimumab in patients with rheumatoid arthritis. Clin Pharmacol Ther. (2010) 87:311–21. doi: 10.1038/clpt.2009.244
215. Gaujoux R, Starosvetsky E, Maimon N, Vallania F, Bar-Yoseph H, Pressman S, et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IB. Gut. (2018) 2018:1–11.
216. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat Med. (2017) 23:579–89. doi: 10.1038/nm.4307
217. Atreya R, Neumann H, Neufert C, Waldner MJ, Billmeier U, Zopf Y, et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn's disease. Nat Med. (2014) 20:313–8. doi: 10.1038/nm.3462
218. Bazin T, Hooks KB, Barnetche T, Truchetet ME, Enaud R, Richez C, et al. Microbiota composition may predict anti-Tnf alpha response in spondyloarthritis patients: an exploratory study. Sci Rep. (2018) 8:5446. doi: 10.1038/s41598-018-23571-4
219. Kapoor SR, Filer A, Fitzpatrick MA, Fisher BA, Taylor PC, Buckley CD, et al. Metabolic profiling predicts response to anti-tumor necrosis factor α therapy in patients with rheumatoid arthritis. Arthritis Rheum. (2013) 65:1448–56. doi: 10.1002/art.37921
220. Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A new approach to defining disease status in ankylosing spondylitis: the Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. (1994) 21:2286–91.
221. Machado P, Landewé R, Lie E, Kvien TK, Braun J, Baker D, et al. Ankylosing Spondylitis Disease Activity Score (ASDAS): defining cut-off values for disease activity states and improvement scores. Ann Rheum Dis. (2011) 70:47–53. doi: 10.1136/ard.2010.138594
222. Machado P, Landewe R. Spondyloarthritis: is it time to replace BASDAI with ASDAS? Nat Rev Rheumatol. (2013) 9:388–90. doi: 10.1038/nrrheum.2013.93
223. Spoorenberg A, van der Heijde D, de Klerk E, Dougados M, de Vlam K, Mielants H, et al. Relative value of erythrocyte sedimentation rate and C-reactive protein in assessment of disease activity in ankylosing spondylitis. J Rheumatol. (1999) 26:980–4.
224. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. (1999) 340:448–54. doi: 10.1056/NEJM199902113400607
225. Ruof J, Stucki G. Validity aspects of erythrocyte sedimentation rate and C-reactive protein in ankylosing spondylitis: a literature review. J Rheumatol. (1999) 26:966–70.
226. Vastesaeger N, van der Heijde D, Inman RD, Wang Y, Deodhar A, Hsu B, et al. Predicting the outcome of ankylosing spondylitis therapy. Ann Rheumat Dis. (2011) 70:973–81. doi: 10.1136/ard.2010.147744
227. Hyrich KL, Watson KD, Silman AJ, Symmons DPM. Predictors of response to anti-TNF-α therapy among patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Rheumatology. (2006) 45:1558–65. doi: 10.1093/rheumatology/kel149
228. Arends S, van der Veer E, Groen H, Houtman PM, Jansen TL, Leijsma MK, et al. Serum MMP-3 level as a biomarker for monitoring and predicting response to etanercept treatment in ankylosing spondylitis. J Rheumatol. (2011) 38:1644–50. doi: 10.3899/jrheum.101128
229. Turina MC, Sieper J, Yeremenko N, Conrad K, Haibel H, Rudwaleit M, et al. Calprotectin serum level is an independent marker for radiographic spinal progression in axial spondyloarthritis. Ann Rheumat Dis. (2014) 73:1746–8. doi: 10.1136/annrheumdis-2014-205506
230. Hobbs JAR, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, et al. Identification of fungi with potential micotoxigenic in fish meals used for preparation. Mol Cell Biol. (2003) 23:2564–76.
231. Leukert N, Vogl T, Strupat K, Reichelt R, Sorg C, Roth J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J Mol Biol. (2006) 359:961–72. doi: 10.1016/j.jmb.2006.04.009
232. Foell D, Roth J. Proinflammatory S100 proteins in arthritis and autoimmune disease. Arthritis Rheumat. (2004) 50:3762–71. doi: 10.1002/art.20631
233. Van Lent PL, Grevers L, Blom AB, Sloetjes A, Mort JS, Vogl T, et al. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann rheumat Dis. (2008) 67:1750–8. doi: 10.1136/ard.2007.077800
234. Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, Van Zoelen MAD, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. (2007) 13:1042–9. doi: 10.1038/nm1638
235. Loser K, Vogl T, Voskort M, Lueken A, Kupas V, Nacken W, et al. The toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+T cells. Nat Med. (2010) 16:713–7. doi: 10.1038/nm.2150
236. Smolen JS, Landewe R, Bijlsma J, Burmester G, Chatzidionysiou K, Dougados M, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheumat Dis. (2017) 76:960–77. doi: 10.1136/annrheumdis-2016-210715
237. Ranganathan P. Rheumatoid arthritis: biomarkers of response to TNF inhibition in RA. Nat Rev Rheumatol. (2015) 11:446–8. doi: 10.1038/nrrheum.2015.83
238. Cui J, Saevarsdottir S, Thomson B, Padyukov L, van der Helm-van Mil AH, Nititham J, et al. Rheumatoid arthritis risk allele PTPRC is also associated with response to anti-tumor necrosis factor alpha therapy. Arthritis Rheumat. (2010) 62:1849–61. doi: 10.1002/art.27457
239. Plant D, Prajapati R, Hyrich KL, Morgan AW, Wilson AG, Isaacs JD, et al. Replication of association of the PTPRC gene with response to anti-tumor necrosis factor therapy in a large UK cohort. Arthritis Rheumat. (2012) 64:665–70. doi: 10.1002/art.33381
240. Pappas DA, Oh C, Plenge RM, Kremer JM, Greenberg JD. Association of rheumatoid arthritis risk alleles with response to anti-TNF biologics: results from the CORRONA registry and meta-analysis. Inflammation. (2013) 36:279–84. doi: 10.1007/s10753-012-9544-4
241. Cui J, Stahl EA, Saevarsdottir S, Miceli C, Diogo D, Trynka G, et al. Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet. (2013) 9:e1003394. doi: 10.1371/journal.pgen.1003394
242. Sieberts SK, Zhu F, Garcia-Garcia J, Stahl E, Pratap A, Pandey G, et al. Crowdsourced assessment of common genetic contribution to predicting anti-TNF treatment response in rheumatoid arthritis. Nat Commun. (2016) 7:12460. doi: 10.1038/ncomms12460
243. Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. (2003) 278:1910–4. doi: 10.1074/jbc.M207577200
244. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. (2003) 421:744–8. doi: 10.1038/nature01355
245. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
246. McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. (2009) 10:314–24. doi: 10.1038/ni.1698
247. Buonocore S, Ahern PP, Uhlig HH, Ivanov II, Littman DR, Maloy KJ, et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature. (2010) 464:1371–5. doi: 10.1038/nature08949
248. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
249. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
250. Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, et al. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive gamma/delta T cells in patients with active ankylosing spondylitis. Arthritis Rheum. (2012) 64:1420–9. doi: 10.1002/art.33507
251. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat(+) CD3(+)CD4(-)CD8(-) entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
252. Brown MA, Kenna T, Wordsworth BP. Genetics of ankylosing spondylitis-insights into pathogenesis. Nat Rev Rheumatol. (2016) 12:81–91. doi: 10.1038/nrrheum.2015.133
253. Coffre M, Roumier M, Rybczynska M, Sechet E, Law HK, Gossec L, et al. Combinatorial control of Th17 and Th1 cell functions by genetic variations in genes associated with the interleukin-23 signaling pathway in spondyloarthritis. Arthritis Rheum. (2013) 65:1510–21. doi: 10.1002/art.37936
254. Di Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, Villanova F, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE. (2011) 6:e17160. doi: 10.1371/journal.pone.0017160
255. Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci USA. (2011) 108:9560–5. doi: 10.1073/pnas.1017854108
256. Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. (2014) 343:1246949. doi: 10.1126/science.1246949
257. Knight JC. Approaches for establishing the function of regulatory genetic variants involved in disease. Genome Med. (2014) 6:92. doi: 10.1186/s13073-014-0092-4
258. Piasecka B, Duffy D, Urrutia A, Quach H, Patin E, Posseme C, et al. Distinctive roles of age, sex, and genetics in shaping transcriptional variation of human immune responses to microbial challenges. Proc Natl Acad Sci USA. (2018) 115:E488–97. doi: 10.1073/pnas.1714765115
259. Fragoulis GE, Siebert S, McInnes IB. Therapeutic targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu Rev Med. (2016) 67:337–53. doi: 10.1146/annurev-med-051914-021944
260. Jandus C, Bioley G, Rivals JP, Dudler J, Speiser D, Romero P. Increased numbers of circulating polyfunctional Th17 memory cells in patients with seronegative spondylarthritides. Arthritis Rheum. (2008) 58:2307–17. doi: 10.1002/art.23655
261. Shen H, Goodall JC, Hill Gaston JS. Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. (2009) 60:1647–56. doi: 10.1002/art.24568
262. Bowness P, Ridley A, Shaw J, Chan AT, Wong-Baeza I, Fleming M, et al. Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J Immunol. (2011) 186:2672–80. doi: 10.4049/jimmunol.1002653
263. Baeten D, Baraliakos X, Braun J, Sieper J, Emery P, van der Heijde D, et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: a randomised, double-blind, placebo-controlled trial. Lancet. (2013) 382:1705–13. doi: 10.1016/S0140-6736(13)61134-4
264. Baeten D, Sieper J, Braun J, Baraliakos X, Dougados M, Emery P, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. (2015) 373:2534–48. doi: 10.1056/NEJMoa1505066
265. van der Heijde D, Ramiro S, Landewe R, Baraliakos X, Van den Bosch F, Sepriano A, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. (2017) 76:978–91. doi: 10.1136/annrheumdis-2016-210770
266. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
267. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2015) 386:1137–46. doi: 10.1016/S0140-6736(15)61134-5
268. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. (2015) 373:1329–39. doi: 10.1056/NEJMoa1412679
269. Hueber W, Sands BE, Lewitzky S, Vandemeulebroecke M, Reinisch W, Higgins PD, et al. Secukinumab in Crohn's Disease Study, Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn's disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. (2012) 61:1693–700. doi: 10.1136/gutjnl-2011-301668
270. Targan SR, Feagan B, Vermeire S, Panaccione R, Melmed GY, Landers C, et al. A randomized, Double-blind, placebo-controlled phase 2 study of brodalumab in patients with moderate-to-severe crohn's disease. Am J Gastroenterol. (2016) 111:1599–607. doi: 10.1038/ajg.2016.298
271. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity. (2015) 43:727–38. doi: 10.1016/j.immuni.2015.09.003
272. van Mens LJJ, van de Sande MGH, Menegatti S, Chen S, Blijdorp ICJ, de Jong HM, et al. Brief report: interleukin-17 blockade with secukinumab in peripheral spondyloarthritis impacts synovial Immunopathology without compromising systemic immune responses. Arthritis Rheumatol. (2018) 70:1994–2002. doi: 10.1002/art.40581
273. Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. (2007) 356:580–92. doi: 10.1056/NEJMoa062382
274. Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet. (2008) 371:1665–74. doi: 10.1016/S0140-6736(08)60725-4
275. Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet. (2008) 371:1675–84. doi: 10.1016/S0140-6736(08)60726-6
276. Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. (2010) 362:118–28. doi: 10.1056/NEJMoa0810652
277. Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. (2017) 376:1551–60. doi: 10.1056/NEJMoa1607017
278. Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. (2003) 198:1951–7. doi: 10.1084/jem.20030896
279. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med. (2006) 203:2473–83. doi: 10.1084/jem.20061099
280. Yen D, Cheung J, Scheerens H, Poulet F, McClanahan T, McKenzie B, et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J Clin Invest. (2006) 116:1310–6. doi: 10.1172/JCI21404
281. Feagan BG, Sandborn WJ, D'Haens G, Panes J, Kaser A, Ferrante M, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn's disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. (2017) 389:1699–709. doi: 10.1016/S0140-6736(17)30570-6
282. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as Induction and Maintenance Therapy for Crohn's Disease. N Engl J Med. (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
283. Baeten D, Ostergaard M, Wei JC, Sieper J, Jarvinen P, Tam LS, et al. Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann Rheum Dis. (2018) 77:1295–302. doi: 10.1136/annrheumdis-2018-213328
284. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. (2007) 39:1329–37. doi: 10.1038/ng.2007.17
285. Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. (2016) 48:510–8. doi: 10.1038/ng.3528
286. International Genetics of Ankylosing Spondylitis Consortium, Cortes A, Hadler J, Pointon JP, Robinson PC, Karaderi T, et al. Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. (2013) 45:730–8. doi: 10.1038/ng.2667
287. Gracey E, Qaiyum Z, Almaghlouth I, Lawson D, Karki S, Avvaru N, et al. IL-7 primes IL-17 in mucosal-associated invariant T (MAIT) cells, which contribute to the Th17-axis in ankylosing spondylitis. Ann Rheum Dis. (2016) 75:2124–32. doi: 10.1136/annrheumdis-2015-208902
Keywords: spondyloarthritis, anti-TNF therapy, effects of TNF-blockers on the immune system, prediction of responses to anti-TNF therapy, anti-IL-17A therapy, anti-IL-23 therapy
Citation: Menegatti S, Bianchi E and Rogge L (2019) Anti-TNF Therapy in Spondyloarthritis and Related Diseases, Impact on the Immune System and Prediction of Treatment Responses. Front. Immunol. 10:382. doi: 10.3389/fimmu.2019.00382
Received: 16 November 2018; Accepted: 14 February 2019;
Published: 19 March 2019.
Edited by:
Nigil Haroon, University of Toronto, CanadaReviewed by:
Yasser Mohamed El-Sherbiny, Nottingham Trent University, United KingdomYolande Richard, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2019 Menegatti, Bianchi and Rogge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lars Rogge, bGFycy5yb2dnZUBwYXN0ZXVyLmZy
†Present Address: Silvia Menegatti, INSERM U932, Institut Curie, PSL Research University, Paris, France