 Franziska Eckert1,2*
Franziska Eckert1,2* Kerstin Zwirner1
Kerstin Zwirner1 Simon Boeke1,2,3Daniela Thorwarth2,3
Simon Boeke1,2,3Daniela Thorwarth2,3 Daniel Zips1,2
Daniel Zips1,2 Stephan M. Huber1
Stephan M. Huber1- 1Department of Radiation Oncology, University Hospital Tuebingen, Tuebingen, Germany
- 2German Cancer Consortium (DKTK) Partnersite Tuebingen, German Cancer Research Center (DKFZ), Heidelberg, Germany
- 3Section for Biomedical Physics, Department of Radiation Oncology, University Hospital Tuebingen, Tuebingen, Germany
In order to compensate for the increased oxygen consumption in growing tumors, tumors need angiogenesis and vasculogenesis to increase the supply. Insufficiency in this process or in the microcirculation leads to hypoxic tumor areas with a significantly reduced pO2, which in turn leads to alterations in the biology of cancer cells as well as in the tumor microenvironment. Cancer cells develop more aggressive phenotypes, stem cell features and are more prone to metastasis formation and migration. In addition, intratumoral hypoxia confers therapy resistance, specifically radioresistance. Reactive oxygen species are crucial in fixing DNA breaks after ionizing radiation. Thus, hypoxic tumor cells show a two- to threefold increase in radioresistance. The microenvironment is enriched with chemokines (e.g., SDF-1) and growth factors (e.g., TGFβ) additionally reducing radiosensitivity. During recent years hypoxia has also been identified as a major factor for immune suppression in the tumor microenvironment. Hypoxic tumors show increased numbers of myeloid derived suppressor cells (MDSCs) as well as regulatory T cells (Tregs) and decreased infiltration and activation of cytotoxic T cells. The combination of radiotherapy with immune checkpoint inhibition is on the rise in the treatment of metastatic cancer patients, but is also tested in multiple curative treatment settings. There is a strong rationale for synergistic effects, such as increased T cell infiltration in irradiated tumors and mitigation of radiation-induced immunosuppressive mechanisms such as PD-L1 upregulation by immune checkpoint inhibition. Given the worse prognosis of patients with hypoxic tumors due to local therapy resistance but also increased rate of distant metastases and the strong immune suppression induced by hypoxia, we hypothesize that the subgroup of patients with hypoxic tumors might be of special interest for combining immune checkpoint inhibition with radiotherapy.
Introduction
Solid tumors are prone to encounter chronic or intermittent hypoxic microenvironment. Hypoxia results from an imbalance of O2 consumption by the tumor and O2 delivery by perfused tumor vessels. The latter is limited since tumor vasculogenesis and angiogenesis usually lags behind expansion of tumor mass. In addition, tumor vessels often show aberrant architecture, may have dilated or blind-ending lumina, and lack normal vessel walls (1). As a consequence, increasing intra-tumoral pressure may compress the vessel lumen accentuating malperfusion of the tumor. Concomitant to insufficient O2 and nutrient supply, this malperfusion restricts delivery of systemically administered drugs such as chemotherapeutics or immunomodulating antibodies limiting the efficacy of these therapies in hypoxic tumor areas (2). Beyond that, hypoxia attenuates DNA damages conferred by ionizing radiation.
Oxygen tensions vary considerable in areas of diffusion-limited chronic hypoxia or perfusion-limited cycles of intermittent hypoxia and reperfusion, hence, triggering a plethora of different cellular adaptation processes (3). Oxygen-sensing processes comprise stabilization of hypoxia-inducible factor (HIF), nutrient depletion-induced down-regulation of the mTOR (mammalian target of rapamycin) pathway (4), impairment of oxidative folding of proteins in the endoplasmic reticulum and unfolded protein response (5), DNA replication stress (6), or oxygen-dependent remodeling of chromatin (7–9). Adaptations to hypoxia include metabolic reprogramming that maintains structural integrity (10), as well as energy (4), redox (11, 12), pH (13), and lipid (14) homeostasis of the hypoxic tumor cell. These complex adaptations, however, induce tumor heterogeneity and may be accompanied by adoption of more malignant phenotypes (15).
Therefore, intratumoral hypoxia has major implications in cancer biology and treatment resistance. Based on the knowledge of an increased radioresistance of hypoxic cancer cells and impaired prognosis for patients with hypoxic tumors, imaging modalities for hypoxia and treatment strategies to overcome the disadvantages of hypoxia have been developed in radiation oncology. With the rise of immunotherapy in cancer over the recent years and the establishment of immune checkpoint inhibition as a standard treatment for several cancer entities, well-known concepts in cancer and radiobiology have been evaluated for their effects on immune responses to cancer. For hypoxia, pronounced immunosuppressive properties have been described by several groups. This article aims at giving an overview and converging the knowledge about tumor hypoxia in the context of radiotherapy and immunotherapy of cancer patients, hypothesizing that patients with hypoxic cancers might benefit most from combination treatments in curative treatment settings.
Hypoxia-Associated Malignant Progression of Tumor Cells
Master regulators of metabolic reprogramming under hypoxia are the O2-sensitive hypoxia-inducible transcription factors (HIFs), the cellular nutrient sensing mTOR and the energy-sensing AMP kinase, as well as the unfolded protein response. They induce downregulation of anabolic metabolism, up-regulation of nutrient import and glycolysis, a switch from oxidative phosphorylation to lactic acid fermentation, up-regulation of acid extrusion pathways such as monocarboxylate transport, adaptation of glutamine metabolisms to maintain fuelling of the citrate pool, alteration of lipid metabolism, attenuation of mitochondrial reactive oxygen species (ROS) formation and/or up-regulation of oxidative defense [for recent reviews (4, 16, 17)].
Metabolic reprogramming may be paralleled by a HIF-regulated phenotypic switch leading to cellular plasticity of tumor and stroma cells which drives tumor heterogeneity. In particular, a hypoxic microenvironment may stimulate in a subset of tumor cells neuroendocrine differentiation, epithelial-mesenchymal transition (EMT) (or neural/glial-mesenchymal transition in brain tumors) or induction of cancer stem (-like)/tumor initiating cells (CSCs) (11). Signaling cascades that induce CSC phenotypes in distinct hypoxic niches are probably triggered by ROS that are formed during the metabolic adaptation to hypoxia (Figure 1). Notably, EMT and CSC induction seems to be highly interrelated and involve HIF signaling [for review see (18, 19)]. Importantly, EMT and upregulation of CSC properties are accompanied by a change from a “grow” to a “go” phenotype. As a consequence, hypoxic tumors are at higher risk of tissue infiltration and metastasis (18, 19).
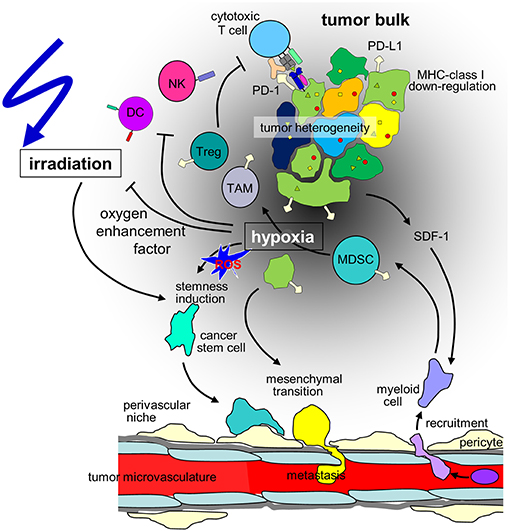
Figure 1. Hypothesis of the influence of hypoxia on cancer cells and the immune microenvironment in the context of radiotherapy of solid tumors. Hypoxia may stimulate in a subset of tumor cells mesenchymal transition and metastasis or induction of cancer stem(-like) cells. The radioresistant phenotype of the latter together with the decline in radiation-induced DNA damage with decrease in oxygen tension (oxygen enhancement factor) contribute to the radioresistance of hypoxic tumors. Moreover, hypoxia/radiation-induced migration may lower locoregional tumor control by radiotherapy. In addition, tumor hypoxia recruits immunosuppressive cell types such as regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) that mature to M2-polarized tumor associated macrophages (TAMs) via stromal cell-derived factor-1 (SDF-1) chemokine signaling. Dendritic cell (DC) function is modulated to TH2 polarized immune responses which suppress anti-tumor immunity. Finally, hypoxia may induce downregulation of MHC class-I molecules and Natural Killer (NK) cell-activating ligands and upregulation of programmed death-ligand-1 (PD-L1) on tumor cells. (ROS: reactive oxygen species).
Moreover, hypoxia and in particular ROS formation during reoxygenation have been shown to favor genetic instability and to increase mutagenesis in tumors by induction of DNA damage and/or deregulation of DNA damage response and apoptotic pathways fostering malignant progression of tumor cells (10, 11). Notably, genetic instability has been associated with response to immune checkpoint inhibition on the one hand and decreased tumor immunogenicity by formation of immune-evasive subclones on the other hand (20, 21). Beyond malignant progression and immune evasion, hypoxia confers resistance to chemo- (2) and radiation therapy as described in the next paragraphs.
Radioresistance of Hypoxic Tumor Cells
About half of all cancer patients undergo radiation therapy often applied in fractionated regimens. Conceptually, a radiation dose of 1 Gy with high energy photons causes about 20 DNA double strand breaks (DSBs) per nucleus on average in normoxic tissue (22). Nuclear DNA DSBs have been proposed to be most hazardous for the cell since when left unrepaired they inevitably provoke chromosome aberrations in mitosis. Tumors are thought to become eradicated if the quantity of radiation induced DSBs exceeds the capacity of DNA DSB repair by non-homologous end joining in G1 phase of cell cycles and additional homologous recombination in S and G2 phase (23). Hypoxia has turned out to be a negative predictive factor for the response to radiation therapy (24) due to lowering the efficacy of ionizing radiation by a factor of 2–3. Mechanistically, this so-called oxygen enhancement ratio (OER) most probably reflects three processes in irradiated cells: O2 fixation of DNA damages, O2-dependent formation of ROS by the mitochondria, as well as hypoxia-induced acquisition of a radioresistant phenotype.
O2 Fixation of DNA Damages
Radiation therapy damages cells by ionization of molecules. Among those, H2O with the far highest concentration (more than 50 M) of all molecules in a cell absorbs the largest fraction of the radiation energy. Energy transfer to H2O leads to formation of hydrogen (•H) and hydroxyl radicals (•OH) in a process referred to as radiolysis of H2O. Formation of •H radicals has been proposed to confer reductive stress to the irradiated cells (25) while the high reactivity and low lifetime of •OH radicals may remove hydrogen atoms from neighboring macromolecules resulting in formation of macromolecule radicals. With a lower stochastic probability formation of macromolecule radicals also occurs upon direct absorption of radiation energy by the macromolecules. Now, the O2 tension comes into the play. Under normoxia, at high O2 partial pressure in the cell, the radical atom within the macromolecule has been suggested to become oxidized which may be associated with the cleavage of molecular bonds of the macromolecule. Under hypoxia, however, at low cellular O2 tension and reductive cellular redox state (which comprises a high ratio between reduced and oxidized glutathione and a high capacity of oxidative defense), macromolecule radicals have been proposed to become “repaired” chemically (Figure 1).
Thus, a high O2 tension may evoke DNA strand breaks whenever radiation-induced radical formation occurs within the phosphate deoxyribose backbone of the DNA. If radical formation concurs in close vicinity in both anti-parallel DNA strands, high oxygen pressure promotes formation of DNA DSBs. This so-called oxygen fixation hypothesis which was developed in the late 1950's, however, explains only insufficiently the oxygen enhancement ratio in radiation therapy. It neither considers hypoxia-mediated effects on DNA repair (26) nor radiation-induced secondary cell damages by mitochondrial ROS formation. The latter are also highly O2-dependent as discussed in the following paragraphs.
Mitochondrial ROS Formation
Early microbeam technologies which allow irradiation of cellular substructures provided strong evidence for a much higher efficacy of ionizing radiation when the nucleus was targeted as compared to selective irradiation of the cytoplasm (27). Therefore, as central dogma of radiation therapy, the genotoxic effects of radiation has been attributed for many years to an interaction between ionizing radiation and the nucleus as primary mechanism (25). Notwithstanding, more recent work, however, suggests that nuclear DNA damage does not exclusively require irradiation of the nucleus and even can be observed in unirradiated bystander cells [for review see (28)]. Notably, inhibiting ROS formation reportedly prevents nuclear DNA damage of the beam-targeted and the bystander cells (29) indicating ROS mediated spreading of the absorbed radiation energy. Furthermore, experiments comparing cells with mitochondrial DNA-proficient (ρ+) and -deficient (ρ0) mitochondria strongly suggest the involvement of mitochondrial electron transport chain in genotoxic damage mediated by radiation (29–33). Most importantly, the fraction of mitochondrial ROS formation-dependent DNA damage has been proposed to increase with O2 tension (34).
Mechanistically, ionizing radiation reportedly increase intracellular free Ca2+ concentration in several tumor entities such as lymphoma (35), leukemia (36, 37), or glioblastoma (38). Intracellular Ca2+ buffering experiments demonstrated that Ca2+, in turn, stimulates in the presence of O2 mitochondrial ROS formation (30) probably in concert with the transient energy crises observed in irradiated cells (39, 40). Both, low ATP/ADP ratios and high Ca2+ concentrations disinhibit mitochondrial electron transport chain, leading to hyperpolarization of the inner mitochondrial membrane potential ΔΨm which is directly linked to superoxide anion (•) formation by slippage of single electrons to O2 [for review see (41)]. Ca2+-mediated • formation by the electron transport chain, in turn, provokes mitochondrial membrane permeability transition and eventually dissipation of ΔΨm and mitochondrial disintegration (42). Of note, radiation-stimulated permeability transition of few affected mitochondria and consequent local release of mitochondrial Ca2+ has been proposed to stimulate Ca2+-overflow, ROS formation, and Ca2+ re-release of adjacent mitochondria, thereby propagating radiation-induced mitochondrial ROS formation through the mitochondrial network in a spatial-temporal manner (30).
As a matter of fact, inhibitors of mitochondrial permeability transition blocked radiation-induced mitochondrial ROS formation (30) and in some but not all cell lines O2-dependent radiosensitivity (43). Combined, these observations strongly suggest that O2 tension-dependent mitochondrial ROS formation and adjunct DNA damage contribute significantly to the OER phenomenon. Beyond stimulation of mitochondrial ROS formation, radiation has been reported to up-regulate activity of uncoupling proteins (UCPs) in the inner mitochondrial membrane (34). UCPs shortcircuit ΔΨm thereby directly counteracting radiation-stimulated mitochondrial ROS formation [for review see (41)]. As described in the next paragraph, adaptation to hypoxia may also involve up-regulation of mitochondrial uncoupling.
Radioresistant Phenotypes Induced by Hypoxia
Adaptation of cells to hypoxia has been described for highly oxidative phosphorylation-dependent normal proximal tubule cells. By repeatedly subjecting these cells to hypoxia and re-oxygenation cycles over weeks strong up-regulation of oxidative defense and mitochondrial uncoupling was induced. Besides diminishing reoxygenation-induced ΔΨm hyperpolarization, • formation, and consecutive cell damage, mitochondrial uncoupling confers cross-resistance to ionizing radiation (44). Importantly, tumors such as proximal tubule-derived renal clear cell carcinoma show high upregulation of mitochondrial uncoupling proteins (44) pointing to hypoxia-induced mitochondrial uncoupling as one potential mechanism of induced resistance in vivo. Similarly, cyclic hypoxia and reoxygenation reportedly upregulates in vitro the mitochondrial citrate carrier SLC25A1 in cancer cell lines that contributes to an increased radioresistance-conferring oxidative defense (11). Beyond that, further metabolic pathways up-regulated in hypoxic cells such as glutamine-dependent glutathione formation (12) or glycolysis-associated pyruvate accumulation [for review see (4)] result in increased capacity of radical scavenging that may confer radioresistance.
Moreover, the above mentioned hypoxia-triggered induction/selection of CSCs reportedly associates with an increased intrinsic radioresistance (Figure 1). CSCs have been supposed to express higher oxidative defense, pre-activated and highly efficient DNA repair and anti-apoptotic pathways rendering them less vulnerable to ionizing radiation [for review see (18)]. Beyond that, CSCs may overexpress certain Ca2+ and electrosignaling pathways that improve stress response upon irradiation (45, 46) as demonstrated for the mesenchymal subpopulation of glioblastoma stem cells (47).
Finally, at least in theory, the above mentioned hypoxia-induced migratory phenotype of tumor cells might limit efficacy of radiotherapy in fractionated regimens. One might speculate that highly migratory cells evade from the target volume covered by the radiation beam. In glioblastoma, stabilization of HIF-1α stimulates auto/paracrine SDF-1 (CXCL12)/CXCR4-mediated chemotaxis the programming of which strongly depends on electrosignaling as one key regulator of chemotaxis (48). Likewise, ionizing radiation stimulates the same pathways also by activating the HIF-1α/SDF-1/CXCR4 axis (48). It is, therefore, tempting to speculate that hypoxia and radiation cooperate in stimulating hypermigration during fractionated radiotherapy. Evidence, however, that hypermigration indeed has any relevance for local tumor control by radiation therapy in the clinical setting is missing. Nevertheless, tumor hypoxia is a severe obstacle of radiation therapy. The next section deals with concepts of visualization and effective treatment of hypoxic tumors for radiation therapy.
Treatment Modifications Targeting Hypoxia in Radiation Oncology
Cellular effects on radiation-response under hypoxia in vitro (49, 50) cannot be directly transferred to xenografts in vivo and tumors in patients. The OER (determined to be 2–3 in vitro (51), as described above) seems to be lower in vivo. This is on the one hand due to the fact that parts of the tumor volume are sufficiently oxygenated since oxygen tension is decreasing only gradually around perfused blood vessels (52–54). On the other hand, depending on the tumor entity, decrease of the bulk tumor mass during fractionated radiation may lead to tumor reoxygenation (55, 56). Extensive research on the tumor microenvironment (hypoxia, vasculature, necrosis and metabolism) and its impact on radioresistance has been done in xenograft models for head and neck squamous cell carcinoma (HNSCC), glioblastoma, non-small cell lung cancer (NSLCL) and colorectal carcinoma and sarcoma cell lines (51, 57–61). In vivo models were also used to show the predictive value of functional tumor imaging with hypoxia sensitive tracers for positron emission tomography (PET) imaging (62–64). Based on hypoxia imaging, different approaches including dose escalation, HIF1α-inhibitors, hypoxia activated prodrugs and hyperbaric oxygen (HBO) or carbogen breathing were studied to overcome treatment resistance with promising results (65–67).
In a clinical setting of HNSCC and cervix cancer, an association between oxygen tension and radioresistance could be shown. For 35 patients with locally advanced HNSCC invasive pO2-measurement with oxygen sensitive electrodes with >15% of pO2 values below 2.5 mm HG, was associated with reduced local control at 2 years (68). In a prognostic validation study as well as in a multicenter study with more than 390 patients, the results could be confirmed (69). There are matching results of worse prognosis for patients with cervical cancer with decreased pO2 values before radiotherapy (70, 71). With advances in imaging methods, non-invasive measurement of hypoxia, based on positron emission tomography (PET) with different hypoxia specific tracers, e.g., [18F]fluoromisonidazole (FMISO), [18F]fluoroazomycin arabinoside (F-AZA), [18F]fluortanidazole (HX4) and [64Cu]diacetyl-bis(N4-methylthiosemicarbazone (Cu-ATSM), and magnetic resonance imaging (MRI) were established and could be correlated to outcome in HNSCC, cervical cancer and NSCLC (72–81). Hypoxia imaging is also closely related to other functional imaging modalities such as FDG-PET or functional MRI (82–84). Based on this evidence, there were major efforts to target hypoxia in the curative setting of radiotherapy during the last decades.
In parallel to the findings of hypoxia as a common phenomenon in solid tumors in the fifties, efforts were started to increase tumor oxygenation by HBO treatment under 2 to 4 atmospheres (85). Due to small numbers of patients in these trials and difficulties of irradiation in pressure chambers, the promising results could not advance into clinical use. Inhalation of carbogen with nicotinamide was the topic of a large phase III trial, which showed decreased regional failure (86). Another approach is the use of hypoxia specific agents like nitroimidazoles. In a trial of The Danish Head and Neck Cancer group (DAHANCA 5) the addition of nimorazole to standard treatment showed an increase in locoregional control (LRC) as well as disease-free survival (DFS) for patients with increased osteopontin levels (87) or a specific gene expression profile (88), both linked to hypoxia. Since then nimorazole is standard of care in Denmark during radiotherapy of HNSCC. To evaluate this combined approach, a large European Organization for Research and Treatment of Cancer (EORTC) phase III trial was conducted with results pending (NCT01880359). With the possibilities of modern radiotherapy techniques like intensity modulated radiotherapy (IMRT) and image-guided radiotherapy (IGRT), first trials with dose escalation based on [18F]fluorodeoxyglucose (FDG) or FMISO are conducted with conflicting results for toxicity and local control data pending (89, 90). A large meta-analysis of all studies with hypoxic modification in HNSCC of 32 trials with more than 4,800 patients included, showed a significant survival benefit of the intervention vs. the control group (91). In a phase II trial an increased radiation dose could not overcome the worse prognosis of hypoxic NSCLC (92). In summary, the big hopes of targeting hypoxia could not be translated directly into the clinic (93).
Immunosuppression in the Hypoxic Tumor Microenvironment
Hypoxia in the tumor microenvironment influences the interaction between cancers and the immune system on all levels. Cancer cells regulate the interaction surface with immune cells, the cytokine microenvironment is altered, and immune cell function is reshaped.
Immune-Relevant Changes in Cancer Cells Under Hypoxia
Cancer cells under hypoxic conditions show a downregulation of MHC class-I molecules (94) (Figure 1), which are crucial for the immune recognition and immune mediated lysis of tumor cells (95). Several immune checkpoints are upregulated in hypoxic conditions. HIF-1α mediates the upregulation of HLA-G (96), which has been described as immunosuppressive (97, 98). In pancreatic cancer HLA-G is a negative prognostic marker, and downregulation of ILT-2 (the receptor of HLA-G) in immune cells activates anti-tumor immunity (99). In addition, hypoxia induces upregulation of CTLA-4 and PD-L1 on tumor cells via HIF-1α in several different mouse and human tumor cell lines (Figure 1). Enhanced PD-L1 abundance could be linked to a HIF-1α binding site in the PD-L1 promotor (100). In renal cell carcinoma elevated PD-L1 levels were correlated with HIF1α levels linked to impaired function of the Von-Hippel-Lindau (VHL) protein (101). In patient samples, HIF1α genes and expression also correlated with PD-L1 expression. The functional link of PD-L1 expression and HIF1α was established by knock-down experiments (101, 102). In hepatocellular carcinoma patient samples PD-L1 expression also was linked to hypoxia and showed prognostic value (103).
Hypoxia has also been linked to downregulation of DNA damage response proteins such as RAD51 in prostate cancer (104), and RAD51 and BRCA1 in breast cancer (105), respectively. BRCA1 downregulation has been shown to be epigenetically regulated in different cancer cell lines (106). Impaired DNA-double-strand-break repair under hypoxic condition might lead to a higher mutation rates and more malignant phenotypes (104). On the other hand, more mutations might also lead to more neoantigens possibly supporting tumor-immune responses. Intriguingly, mutational burden is one of the most promising predictive factor for treatment with immune-checkpoint-inhibition (107). In concordance, the antigenic landscape of prostate cancer is modified by the applied oxygen tension (108) in vitro.
Hypoxic Immune Microenvironment
The immune microenvironment of tumors also undergoes profound changes with the development of intratumoral hypoxia. Hypoxia induced downregulation of ADAM-10 (109) and upregulation of CCL28 (110, 111) and IL-10 (112) all lead to immunosuppression via shedding of MHC class I chain-related molecule A (MICA) and hampering cytolytic action of immune cells, Treg recruitment and enhancing suppressor MDSc, respectively. Hampered anti-tumor immunity in hypoxic tumors is mainly mediated by adenosine receptor signaling (113). Adenosine is formed by hydrolysis of tumor cell-derived ATP in the extracellular space (114). Adenosine receptors are a direct target of HIF1α and have been reported to enable stem (like) cell enrichment in breast cancer (115). Clinical data as well as in vivo data in an autochthonous mouse model linked adenosine A2A receptor with carcinogenesis and immune resistance of HNSCC (116). Tumor reactive CD8+ cells express A2A receptors and show enhanced activity upon downregulation or blockade thereof (117). Oral A2A receptor inhibitors have been developed and tested preclinically (118). Ex vivo testing suggests synergistic effects with immune checkpoint blockade (119).
Consequently, several cell subsets required for efficient anti-cancer immune responses have been described to be impaired or inhibited by hypoxia. Mechanisms of the innate immune system, such as NK cell-mediated killing of cancer cells is disturbed due to downregulation of the respective activating ligands on tumor cells (120). Concerning adaptive immunity, several critical steps are hampered under hypoxic conditions. Dendritic cell function is modulated to TH2 polarized immune responses, consequently, T cells primed under hypoxia preferably are TH2-polarized and thus suppress anti-tumor immunity (121) (Figure 1). At the same time, the development of anti-cancer TH1 cells is inhibited (122) and CD8+ effector T cells are inhibited in their proliferative activity under hypoxia, possibly via IL-10 (112).
Regulatory T Cells
In addition, major immunosuppressive cell types in the tumor microenvironment are upregulated under hypoxic conditions, such as regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) and tumor associated macrophages (TAMs) (Figure 1). Tregs have been described as major players in cancer immunosuppression by inhibiting effector T cells and fostering angiogenesis (123) and have been described to be increased in hypoxic tumors (124). Several mechanisms for this phenomenon have been proposed. In gastric cancer, FoxP3 (as a marker for Tregs) is strongly associated with HIF-1α and TGFβ and acts as negative prognostic factor. In vitro, TGFβ blockade diminished the Treg induction under hypoxic conditions (125). This has been linked to hypoxia-induced NANOG expression (126). SDF-1/CXCR4 signaling induced by hypoxia also has been linked to Treg recruitment (127). Another major mechanism described for ovarian as well as for liver cancer is the induction of CCL28. In ovarian cancer CCL28 recruits Tregs and leads to accelerated tumor growth in vitro as well as in orthotopic models of intraperitoneal tumors (110). These findings have been confirmed for hepatocellular carcinoma (111). The interplay of these different factors for Treg accumulation has not been clarified yet.
Myeloid-Derived Suppressor Cells (MDSCs) and Tumor Associated Macrophages (TAMs)
Hypoxia leads to the recruitment of MDSCs (128) as well as their accumulation (129) in a hepatocellular carcinoma model as well as in gliomas (130). In the tumor microenvironment MDSCs differentiate to macrophages (131). In hypoxia, macrophages are preferably polarized to the immunosuppressive M2 phenotype (132, 133). M2 macrophages support tumor growth directly (134–136) and simultaneously prevent immune destruction (137, 138). Interestingly, myeloid cells have also been described to be involved in the formation of pre-metastatic niches in secondary organs (139, 140).
Rationale for Combining Radiotherapy and Immunotherapy
Immune Checkpoint Inhibition for Cancer Therapy
Immune checkpoint inhibition (ICI) gained increasing interest as a new paradigm in cancer treatment as several encouraging clinical trials were published (141–143). However, in some other studies, ICI showed less promising results (144, 145). There is still a considerable number of patients who do not response at all, solely achieve a partial response or relapse in spite of notable initial response, yet. Several other immunotherapy approaches are being developed (146) [such as cytokine based therapy (147–149) or vaccines (150, 151)], however, the clinical development is most advanced for CTLA-4 and PD-1/PD-L1 blockade.
As reviewed in Wolchok et al. (152) CTLA-4 has been identified as a negative regulator of T-cell activation binding to the B7 protein on antigen presenting cells. This interaction prevents the binding of CD28 to B7, a necessary costimulatory signal for T cell activation following the recognition of respective antigens by the T-cell-receptor representing a very early step in the immune cascade (153). CTLA-4 deficient mice show massive lymphoproliferation, multi-organ tissue destruction and early letality (154). Blockade of CTLA-4 has been shown to induce T cell activation (155, 156) and anti-tumor immunity in preclinical models (157). These findings translated into clinical benefits and long-term cancer control first in patients with malignant melanoma (158, 159). A recent compilation of finished and ongoing clinical trial shows the application of CTLA-4 blockade in numerous cancer entities, therapeutic settings and combinatorial approaches (160).
In clinical cancer therapy, blockade of the PD-1/PD-L1 axis has become even more prominent as indicated by the numbers of ongoing clinical trials (160). The inhibitory effect of PD-1/PD-L1 interaction is predominant during the inflammatory phase in peripheral tissues (161). Similar to CTLA-4, mice deficient for PD-1 developed severe autoimmune symptoms indicating an inhibitory function of PD-1 on immune activation (162). It was soon linked to immune-evasion of tumors as cancer cells show a high expression of PD-L1 and thus directly inhibit T-cell activation in the tumor microenvironment (163). PD-1 also plays a major role in T-cell exhaustion in chronic inflammatory processes and cancer (164). After initial signs of safety and activity of blocking PD-1 for cancer treatment (165), numerous randomized trials have shown clinical benefit of single-agent or combined treatment using PD-1 or PD-L1 antibodies (166).
Immune Effects of Radiation
Rare abscopal effects (response of distant, non-irradiated lesions) in irradiated patients have been described many years ago [reviewed in (167)], but the interaction of radiation and tumor specific immune responses was increasingly understood later on (168).
In addition to direct cytotoxic effects of radiotherapy and reoxygenation in solid tumors during fractionated radiation, local irradiation also affects the tumor immune microenvironment. In contrast to the predominant perception of radiotherapy being basically immunosuppressive, several mechanisms have been identified how irradiation might lead to better anti-tumor immune responses as summarized by Demaria and Formenti (169). Radiation influences every step of the “cancer immunity cycle” (170). The cancer cell death induced by irradiation does not only lead to antigen release, but has been characterized as immunogenic cell death characterized by the release of danger signals (171, 172) such as membranous calreticulin exposure and release of HMGB1 and ATP into the extracellular space leading to activation of the innate immune system (173, 174) (Figure 2). Radiation induces upregulation of MHC-I complexes on cancer cells (175) and priming and maturation of antigen-presenting cells (176, 177). After traveling to draining lymph nodes, these antigen-presenting cells are able to prime T cells specific for tumor associated antigens (178). The primed and activated effector T cells show increased infiltration into irradiated tumors (179–181). In addition to the effects on T cell based anti-tumor immune responses, irradiation is able to repolarize macrophages to a tumor inhibiting M1-subtype (182) and activate natural killer cells (183) (Figure 2).
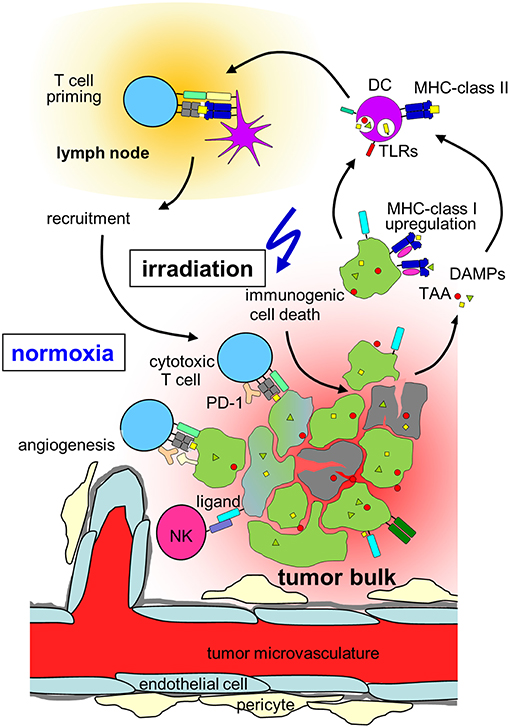
Figure 2. Hypothesis on radiation-induced immunogenic cell death in normoxic tumors. In a normoxic tumor microenvironment, irradiation may lead to effective anti-tumor immune responses by induction of upregulation of MHC class-I on the tumor, immunogenic cell death, release of danger associated molecular patterns (DAMPs) activating toll-like receptors (TLRs) and induction of new tumor associated antigens (TAAs). Maturation of dendritic cells (DCs) and upregulation of MHC-class II is followed by T cell priming in the draining lymph node, cytotoxic T cells and natural killer (NK) cells travel back to the tumor and lead to lysis of tumor cells. Please note, that radiation also induces immunosuppressive processes in normoxic tumors (which are not depicted) such as up-regulation of programmed death-ligand-1 (PD-L1) or Tregs (for details, see chapter Immune effects of radiation).
On the other hand (and explaining the scarce clinical evidence for anti-tumor immune induction by radiotherapy alone) irradiation induces immunosuppressive mechanisms in solid tumors (184). One major mechanism is the upregulation of PD-L1 in irradiated tumors (185–187). Even combined treatment of CTLA-4 blockade with irradiation led to upregulated PD-L1 level and treatment resistance, which could be overcome by adding PD-1/PD-L1 blockade to the regimen in a preclinical model (188). In addition, radiation leads to the accumulation of Tregs (189, 190) as well as the release of immunosuppressive molecules such as TGFβ (191, 192). Curative, normofractionated radiotherapy leads to significant changes in the peripheral immune status of the patients with a decrease of naïve CD4+ lymphocytes and an increase in Tregs (193–195). These findings led to the rationale of combining cancer radiotherapy with immune checkpoint inhibition (196).
Combined Radiation and Immune Checkpoint Inhibition
The rationale of combining immunotherapy and radiotherapy has been discussed intensely in several review articles [e.g., (197, 198)]. Initial clinical signs of synergistic and abscopal effects after combination therapy of radiotherapy and immune checkpoint inhibition were reported in a patient with malignant melanoma who had progressed on Ipilimumab but showed a second systemic response after palliative radiotherapy for a paraspinal lesion (199). Initial phase II studies in melanoma showed an abscopal response rate of 18% (200). Immune checkpoint inhibition has been combined with palliative radiotherapy (201) as well as with ablative stereotactic irradiation (202). Furthermore, a recent trial in stage III non-small cell lung cancer encourages efforts of combining both therapeutic strategies in curative settings as well (203). Here, Durvalumab (a monoclonal PD-L1-antibody) consolidation after definitive radiochemotherapy showed significantly prolonged progression-free survival rates and increased overall survival compared to the placebo group with short time between end of radiochemotherapy and start of checkpoint-blockade showing an even larger effect in a subgroup analysis (203, 204).
However, in spite of first efforts (205), the optimal regimen of timing, target organ, dosage and fractionation remains elusive and future trials and translational research need to address these important questions to maximize the potentially beneficial combination effects of radiotherapy and immunotherapy (206). The underlying molecular mechanisms are being investigated intensely and might lead to more promising designs for future clinical trials. PD-1 signaling has been linked to abscopal responses by knock-out and inhibition in in vivo models of stereotactic radiotherapy (207). The identification of radiation fractionation schedules leading to abscopal effects in combination with CTLA-4 blockade in an in vivo model of breast cancer was linked to the induction of cytosolic double-stranded DNA. With high radiation doses, the induction of the exonuclease TREX-1 degrading the DNA fragments, no abscopal effects were observed (208).
Rationale for Selecting Patients With Hypoxic Tumors for Combination Treatment
To the best of our knowledge, there are no data on combined radiotherapy and immune checkpoint inhibition focusing on hypoxic tumors. However, as hypoxic tumors are intrinsically more radioresistant than normoxic counterparts and show reduced local control and higher rates of distant metastases, there is a specific clinical need in this subgroup of patients for more effective therapies. As hypoxia also leads to dramatically impaired anti-tumor immune responses, enhancing immune-mediated tumor control mechanisms might be a promising strategy, especially because the combination of immune checkpoint inhibition and radiotherapy has been described to improve local control as well as to induce abscopal effects leading to better systemic tumor control. The here described effects of hypoxia with increased mutational load and upregulation of immune checkpoints such as PD-L1 might even hint at improved responsiveness of hypoxic tumors to immune checkpoint inhibition, further strengthening the hypothesis that patients with hypoxic tumors might be a subgroup of specific interest for combination concepts of radiotherapy with immune checkpoint inhibition (Figure 3).
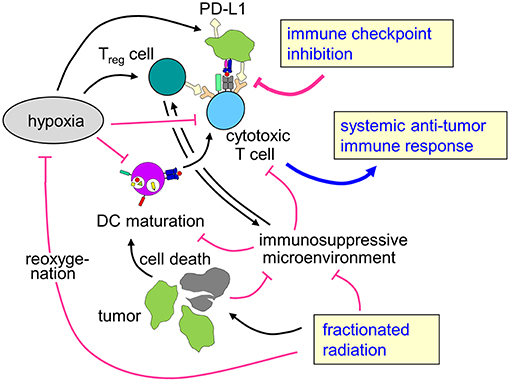
Figure 3. Rationale for combining radiotherapy and immune checkpoint inhibition to overcome therapy resistance of hypoxic tumors. Tumor hypoxia is a key player for the prognosis of cancer patients and resistance to radiotherapy and possibly also for anti-tumor immune response. Fractionated radiotherapy may lead to reoxygenation. The profound immune suppressive microenvironment (see chapter Immunosuppression in the hypoxic tumor microenvironment) predominantly in hypoxic tumors as well as upregulation of immune checkpoint molecules might hint at a rationale to combine fractionated radiotherapy with immune checkpoint inhibition in patients with hypoxic tumors to enhance local control and systemic anti-tumor immune effects.
Author Contributions
FE and SH designed the concept and wrote the manuscript. KZ wrote the chapter Rationale for combining radiotherapy and immunotherapy. SB wrote the chapter Treatment modifications targeting hypoxia in radiation oncology. DT, DZ, and all authors read and approved the manuscript.
Funding
FE was partly funded by the Else-Kroener-Fresenius Research Foundation under Grant 2015_Kolleg.14. SH was partly funded by grants from the German Cancer Aid (70112872, 70113144).
Conflict of Interest Statement
FE has a research collaboration with Merck KgAa. DZ, DT, KZ, SB, and FE have research and educational grants from Elekta, Philips, Siemens, Sennewald. SH has a research collaboration with Novocure.
Acknowledgments
We acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of University of Tübingen.
References
1. Nagy JA, Chang SH, Dvora AM, Dvorak F. Why are tumour blood vessels abnormal and why is it important to know? Br J Cancer. (2009) 100:865–9. doi: 10.1038/sj.bjc.6604929
2. Graha K. Unger: overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J Nanomed. (2018) 13:6049–58. doi: 10.2147/ijn.s140462
3. Vaupe P. Mayer: hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol. (2014) 812:19–24. doi: 10.1007/978–1-4939–0620-8_3
4. Rey S, Schito L, Wouters BG, Eliaso S, Kerbel S. Targeting hypoxia-inducible factors for antiangiogenic cancer therapy. Trends Cancer. (2017) 3:529–41. doi: 10.1016/j.trecan.2017.05.002
5. Wouter BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. (2008) 8:851–64. doi: 10.1038/nrc2501
6. Ng N, Purshouse K, Foskolou IP, Olcin MM, Hammond M. Challenges to DNA replication in hypoxic conditions. FEBS J. (2018) 285:1563–71. doi: 10.1111/febs.14377
7. Adriaens ME, Prickaerts PM, van den Beucken T, Dahlmans VEH, Eijssen LM, Beck T, et al. Quantitative analysis of ChIP-seq data uncovers dynamic and sustained H3K4me3 and H3K27me3 modulation in cancer cells under hypoxia. Epigene Chromatin. (2016) 9:48. doi: 10.1186/s13072–016-0090–4
8. Lu Y, Liu Y, Oec S, Glazer M. Hypoxia promotes resistance to EGFR inhibition in SCLC cells via the histone demethylases, LSD1 and PLU-1. Mol Cancer Res. (2018) 16:1458–69. doi: 10.1158/1541–7786.mcr-17–0637
9. Prickaerts P, Adriaens ME, Beucken TVD, Koch E, Dubois L, Dahlmans VEH, et al. Voncken: hypoxia increases genome-wide bivalent epigenetic marking by specific gain of H3K27me3. Epigene Chromatin. (2016) 9:46. doi: 10.1186/s13072–016-0086–0
10. Koritzinsk M, Wouters G. The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin Radiat Oncol. (2013) 23:252–61. doi: 10.1016/j.semradonc.2013.05.006
11. Hlouschek J, Hansel C, Jendrosse V, Matschke J. The mitochondrial citrate carrier (SLC25A1) sustains redox homeostasi, itochondrial metabolism supporting radioresistance of cancer cells with tolerance to cycling severe hypoxia. Front Oncol. (2018) 8:170. doi: 10.3389/fonc.2018.00170
12. Matschke J, Riffkin H, Klein D, Handrick R, Ludemann L, Metzen E, et al. Targeted inhibition of glutamine-dependent glutathione metabolism overcomes death resistance induced by chronic cycling hypoxia. Antioxid Redox Signal. (2016) 25:89–107. doi: 10.1089/ars.2015.6589
13. van den Beucken T, Koritzinsky M, Niessen H, Dubois L, Savelkouls K, Mujcic H, et al. Hypoxia-induced expression of carbonic anhydrase 9 is dependent on the unfolded protein response. J Biol Chem. (2009) 284:24204–12. doi: 10.1074/jbc.M109.006510
14. Matschke J, Wiebeck E, Hurst S, Rudne J, Jendrossek V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat Oncol. (2016) 11:75. doi: 10.1186/s13014–016-0647–1
15. van den Beucken T, Koch E, Chu K, Rupaimoole R, Prickaerts P, Adriaens M, et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICE. Nat Commun. (2014) 5:5203. doi: 10.1038/ncomms6203
16. Xi H, Simon C. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem. (2017) 292:16825–32. doi: 10.1074/jbc.R117.799973
17. Tyszka-Czochara M, Konieczny P, Majka M. Recent advances in the role of AMP-activated protein kinase in metabolic reprogramming of metastatic cancer cells: targeting cellular bioenergetics and biosynthetic pathways for anti-tumor treatment. J Physiol Pharmacol. (2018) 69:337–49. doi: 10.26402/jpp.2018.3.07
18. Peitzsch C, Perrin R, Hill RP, Dubrovska A, Kurth I. Hypoxia as a biomarker for radioresistant cancer stem cells. Int J Radiat Biol. (2014) 90:636–52. doi: 10.3109/09553002.2014.916841
19. Terry S, Faouzi Zaarour R, Hassan Venkatesh G, Francis A, El-Sayed W, Buart S, et al. Role of hypoxic stress in regulating tumor immunogenicity, resistance and plasticity. Int J Mol. Sci. (2018) 19:3044. doi: 10.3390/ijms19103044
20. Scanlon SE, Glazer PM. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair. (2015) 32:180–9. doi: 10.1016/j.dnarep.2015.04.030
21. Terry S, Buart S, Chouaib S. Hypoxic stress-induced tumor and immune plasticity, suppression, and impact on tumor heterogeneity. Front Immunol. (2017) 8:1625. doi: 10.3389/fimmu.2017.01625
22. Matsuya Y, Ohtsubo Y, Tsutsumi K, Sasaki K, Yamazaki R, Date H. Quantitative estimation of DNA damage by photon irradiation based on the microdosimetric-kinetic model. J Radiat Res. (2014) 55:484–93. doi: 10.1093/jrr/rrt222
23. Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. (2003) 22:5792–812. doi: 10.1038/sj.onc.1206679
24. Thoday JM, Read J. Effect of oxygen on the frequency of chromosome aberrations produced by X-rays. Nature. (1947) 160:608.
25. Reisz JA, Bansal N, Qian J, Zhao W, Furdui CM. Effects of ionizing radiation on biological molecules–mechanisms of damage and emerging methods of detection. Antioxid Redox Signal. (2014) 21:260–92. doi: 10.1089/ars.2013.5489
27. Munro TR. The site of the target region for radiation-induced mitotic delay in cultured mammalian cells. Radiat Res. (1970) 44:747–57.
28. Zhou H, Hong M, Chai Y, Hei TK. Consequences of cytoplasmic irradiation: studies from microbeam. J Radiat Res. (2009) 50(Suppl. A):A59–65. doi: 10.1269/jrr.08120S
29. Tartier L, Gilchrist S, Burdak-Rothkamm S, Folkard M, Prise KM. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. (2007) 67:5872–9. doi: 10.1158/0008-5472.CAN-07-0188
30. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. (2001) 61:3894–901.
31. Yamazaki H, Yoshida K, Yoshioka Y, Isohashi F, Ozeki S, Koizumi M, et al. Impact of mitochondrial DNA on hypoxic radiation sensitivity in human fibroblast cells and osteosarcoma cell lines. Oncol Rep. (2008) 19:1545–9. doi: 10.3892/or.19.6.1545
32. Cloos CR, Daniels DH, Kalen A, Matthews K, Du J, Goswami PC, et al. Mitochondrial DNA depletion induces radioresistance by suppressing G2 checkpoint activation in human pancreatic cancer cells. Radiat Res. (2009) 171:581–7. doi: 10.1667/RR1395.1
33. Zhang B, Davidson MM, Hei TK. Mitochondria regulate DNA damage and genomic instability induced by high LET radiation. Life Sci Space Res. (2014) 1:80–8. doi: 10.1016/j.lssr.2014.02.006
34. Richardson RB, Harper ME. Mitochondrial stress controls the radiosensitivity of the oxygen effect: Implications for radiotherapy. Oncotarget. (2016) 7:21469–83. doi: 10.18632/oncotarget.7412
35. Klumpp D, Misovic M, Szteyn K, Shumilina E, Rudner J, Huber SM. Targeting TRPM2 channels impairs radiation-induced cell cycle arrest and fosters cell death of T cell leukemia cells in a Bcl-2-dependent manner. Oxid Med Cell Longev. (2016) 2016:8026702. doi: 10.1155/2016/8026702
36. Heise N, Palme D, Misovic M, Koka S, Rudner J, Lang F, et al. Non-selective cation channel-mediated Ca2+-entry and activation of Ca2+/calmodulin-dependent kinase II contribute to G2/M cell cycle arrest and survival of irradiated leukemia cells. Cell Physiol Biochem. (2010) 26:597–608. doi: 10.1159/000322327
37. Palme D, Misovic M, Schmid E, Klumpp D, Salih HR, Rudner J, et al. Kv3.4 potassium channel-mediated electrosignaling controls cell cycle and survival of irradiated leukemia cells. Pflugers Arch. (2013) 465:1209–21. doi: 10.1007/s00424-013-1249-5
38. Stegen B, Klumpp L, Misovic M, Edalat L, Eckert M, Klumpp D, et al. K+ channel signaling in irradiated tumor cells. Eur Biophys J. (2016) 45:585–98. doi: 10.1007/s00249-016-1136-z
39. Dittmann K, Mayer C, Rodemann HP, Huber SM. EGFR cooperates with glucose transporter SGLT1 to enable chromatin remodeling in response to ionizing radiation. Radiother Oncol. (2013) 107:247–51. doi: 10.1016/j.radonc.2013.03.016
40. Huber SM, Misovic M, Mayer C, Rodemann HP, Dittmann K. EGFR-mediated stimulation of sodium/glucose cotransport promotes survival of irradiated human A549 lung adenocarcinoma cells. Radiother Oncol. (2012) 103:373–9. doi: 10.1016/j.radonc.2012.03.008
41. Huber SM, Butz L, Stegen B, Klumpp D, Braun N, Ruth P, et al. Ionizing radiation, ion transports, and radioresistance of cancer cells. Front Physiol. (2013) 4:212. doi: 10.3389/fphys.2013.00212
42. Vercesi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. (1997) 17:43–52.
43. Anoopkumar-Dukie S, Conere T, Sisk GD, Allshire A. Mitochondrial modulation of oxygen-dependent radiosensitivity in some human tumour cell lines. Br J Radiol. (2009) 82:847–54. doi: 10.1259/bjr/35746067
44. Braun N, Klumpp D, Hennenlotter J, Bedke J, Duranton C, Bleif M, et al. UCP-3 uncoupling protein confers hypoxia resistance to renal epithelial cells and is upregulated in renal cell carcinoma. Sci Rep. (2015) 5:13450. doi: 10.1038/srep13450
45. Stegen B, Butz L, Klumpp L, Zips D, Dittmann K, Ruth P, et al. Ca2+-Activated IK K+ channel blockade radiosensitizes glioblastoma cells. Mol Cancer Res. (2015) 13:1283–95. doi: 10.1158/1541-7786.MCR-15-0075
46. Klumpp D, Frank SC, Klumpp L, Sezgin EC, Eckert M, Edalat L, et al. TRPM8 is required for survival and radioresistance of glioblastoma cells. Oncotarget. (2017) 8:95896–913. doi: 10.18632/oncotarget.21436
47. Klumpp L, Sezgin EC, Skardelly M, Eckert F, Huber SM. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr Neuropharmacol. (2018) 16:627–35. doi: 10.2174/1570159X15666170808115821
48. Edalat L, Stegen B, Klumpp L, Haehl E, Schilbach K, Lukowski R, et al. BK K+ channel blockade inhibits radiation-induced migration/brain infiltration of glioblastoma cells. Oncotarget. (2016) 7:14259–78. doi: 10.18632/oncotarget.7423
49. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OCA. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. (1953) 26:638–48. doi: 10.1259/0007–1285-26–312-638
50. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. (1955) 9:539–49.
51. Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. (2009) 9:442–58. doi: 10.2174/156652409788167087
52. Horsman MR. Measurement of tumor oxygenation. Int J Radiat Oncol Biol Phys. (1998) 42:701–4. doi: 10.1016/S0360–3016(98)00332–0
53. Maftei CA, Bayer C, Shi K, Vaupel P. Intra- and intertumor heterogeneities in total, chronic, and acute hypoxia in xenografted squamous cell carcinomas. detection and quantification using (immuno-)fluorescence techniques. Strahlenther Onkol. (2012) 188:606–15. doi: 10.1007/s00066–012-0105–4
54. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: a review. Cancer Res. (1989) 49:6449–65.
55. Harriss W, Bezak E, Yeoh E, Hermans M. Measurement of reoxygenation during fractionated radiotherapy in head and neck squamous cell carcinoma xenografts. Austr Phys Eng Sci Med. (2010) 33:251–63. doi: 10.1007/s13246–010-0032–6
56. Kallman RF. The phenomenon of reoxygenation and its implications for fractionated radiotherapy. Radiology. (1972) 105:135–42. doi: 10.1148/105.1.135
57. Gulliksrud K, Vestvik IK, Galappathi K, Mathiesen B, Rofstad EK. Detection of different hypoxic cell subpopulations in human melanoma xenografts by pimonidazole immunohistochemistry. Radiat Res. (2008) 170:638–50. doi: 10.1667/rr1400.1
58. Moulder JE, Rockwell S. Hypoxic fractions of solid tumors: experimental techniques, methods of analysis, and a survey of existing data. Int J Radiat Oncol Biol Phys. (1984) 10:695–712.
59. Yaromina A, Kroeber T, Meinzer A, Boeke S, Thames H, Baumann M, et al. Exploratory study of the prognostic value of microenvironmental parameters during fractionated irradiation in human squamous cell carcinoma xenografts. Int J Radiat Oncol Biol Phys. (2011) 80:1205–13. doi: 10.1016/j.ijrobp.2011.02.015
60. Yaromina A, Thames H, Zhou X, Hering S, Eicheler W, Dorfler A, et al. Radiobiological hypoxia, histological parameters of tumour microenvironment and local tumour control after fractionated irradiation. Radiother Oncol. (2010) 96:116–22. doi: 10.1016/j.radonc.2010.04.020
61. Zips D, Boke S, Kroeber T, Meinzer A, Bruchner K, Thames HD, et al. Prognostic value of radiobiological hypoxia during fractionated irradiation for local tumor control. Strahlenther Onkol. (2011) 187:306–10. doi: 10.1007/s00066–011-2210–1
62. Ali R, Apte S, Vilalta M, Subbarayan M, Miao Z, Chin FT, et al. 18F-EF5 PET is predictive of response to fractionated radiotherapy in preclinical tumor models. PLoS ONE. (2015) 10:e0139425. doi: 10.1371/journal.pone.0139425
63. Peeters SG, Zegers CM, Biemans R, Lieuwes NG, van Stiphout RG, Yaromina A, et al. TH-302 in combination with radiotherapy enhances the therapeutic outcome and is associated with pretreatment [18F]HX4 hypoxia PET imaging. Clin Cancer Res. (2015) 21:2984–92. doi: 10.1158/1078–0432.ccr-15–0018
64. Tran LB, Bol A, Labar D, Cao-Pham TT, Jordan B, Gregoire V, et al. Predictive value of (18)F-FAZA PET imaging for guiding the association of radiotherapy with nimorazole: a preclinical study. Radiother Oncol. (2015) 114:189–94. doi: 10.1016/j.radonc.2014.12.015
65. Brizel DM, Lin S, Johnson JL, Brooks J, Dewhirst MW, Piantadosi CA. The mechanisms by which hyperbaric oxygen and carbogen improve tumour oxygenation. Br J Cancer (1995)72:1120–4.
66. Helbig L, Koi L, Bruchner K, Gurtner K, Hess-Stumpp H, Unterschemmann K, et al. BAY 87–2243, a novel inhibitor of hypoxia-induced gene activation, improves local tumor control after fractionated irradiation in a schedule-dependent manner in head and neck human xenografts. Radiat Oncol. (2014) 9:207. doi: 10.1186/1748–717x-9–207
67. Yaromina A, Granzier M, Biemans R, Lieuwes N, van Elmpt W, Shakirin G, et al. A novel concept for tumour targeting with radiation: Inverse dose-painting or targeting the “Low Drug Uptake Volume”. Radiother Oncol. (2017) 124:513–20. doi: 10.1016/j.radonc.2017.04.020
68. Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol. (1996) 41:31–9.
69. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. an international multi-center study. Radiother Oncol. (2005) 77:18–24. doi: 10.1016/j.radonc.2005.06.038
70. Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, et al. Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol. (1998) 48:149–56.
71. Hockel M, Schlenger K, Aral B, Mitze M, Schaffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res. (1996) 56:4509–15.
72. Dirix P, Vandecaveye V, De Keyzer F, Stroobants S, Hermans R, Nuyts S. Dose painting in radiotherapy for head and neck squamous cell carcinoma: value of repeated functional imaging with (18)F-FDG PET, (18)F-fluoromisonidazole PET, diffusion-weighted MRI, and dynamic contrast-enhanced MRI. J Nucl Med. (2009) 50:1020–7. doi: 10.2967/jnumed.109.062638
73. Hatakenaka M, Shioyama Y, Nakamura K, Yabuuchi H, Matsuo Y, Sunami S, et al. Apparent diffusion coefficient calculated with relatively high b-values correlates with local failure of head and neck squamous cell carcinoma treated with radiotherapy. Am J Neuroradiol. (2011) 32:1904–10. doi: 10.3174/ajnr.A2610
74. Kinoshita T, Fujii H, Hayashi Y, Kamiyama I, Ohtsuka T, Asamura H. Prognostic significance of hypoxic PET using 18F-FAZA and 62Cu-ATSM in non-small-cell lung cancer. Lung Cancer. (2016) 91:56–66. doi: 10.1016/j.lungcan.2015.11.020
75. Lambrecht M, Van Calster B, Vandecaveye V, De Keyzer F, Roebben I, Hermans R, et al. Integrating pretreatment diffusion weighted MRI into a multivariable prognostic model for head and neck squamous cell carcinoma. Radiother Oncol. (2014) 110:429–34. doi: 10.1016/j.radonc.2014.01.004
76. Rischin D, Hicks RJ, Fisher R, Binns D, Corry J, Porceddu S, et al. Prognostic significance of [18F]-misonidazole positron emission tomography-detected tumor hypoxia in patients with advanced head and neck cancer randomly assigned to chemoradiation with or without tirapazamine: a substudy of Trans-Tasman Radiation Oncology Group Study 98.02. J Clin Oncol. (2006) 24:2098–104. doi: 10.1200/jco.2005.05.2878
77. Thorwarth D, Eschmann SM, Paulsen F, Alber M. A model of reoxygenation dynamics of head-and-neck tumors based on serial 18F-fluoromisonidazole positron emission tomography investigations. Int J Radiat Oncol Biol Phys. (2007) 68:515–21. doi: 10.1016/j.ijrobp.2006.12.037
78. Wiedenmann NE, Bucher S, Hentschel M, Mix M, Vach W, Bittner MI, et al. Serial [18F]-fluoromisonidazole PET during radiochemotherapy for locally advanced head and neck cancer and its correlation with outcome. Radiother Oncol. (2015) 117:113–7. doi: 10.1016/j.radonc.2015.09.015
79. Zips D, Zophel K, Abolmaali N, Perrin R, Abramyuk A, Haase R, et al. Exploratory prospective trial of hypoxia-specific PET imaging during radiochemotherapy in patients with locally advanced head-and-neck cancer. Radiother Oncol. (2012) 105:21–8. doi: 10.1016/j.radonc.2012.08.019
80. Eschmann SM, Paulsen F, Reimold M, Dittmann H, Welz S, Reischl G, et al. Prognostic impact of hypoxia imaging with 18F-misonidazole PET in non-small cell lung cancer and head and neck cancer before radiotherapy. J Nucl Med. (2005) 46:253–60.
81. Boeke S, Thorwarth D, Monnich D, Pfannenberg C, Reischl G, La Fougere C, et al. Geometric analysis of loco-regional recurrences in relation to pre-treatment hypoxia in patients with head and neck cancer. Acta Oncol. (2017) 56:1571–6. doi: 10.1080/0284186x.2017.1372626
82. Leibfarth S, Simoncic U, Monnich D, Welz S, Schmidt H, Schwenzer N, et al. Analysis of pairwise correlations in multi-parametric PET/MR data for biological tumor characterization and treatment individualization strategies. Eur J Nucl Med Mol Imag. (2016) 43:1199–208. doi: 10.1007/s00259–016-3307–7
83. Monnich D, Thorwarth D, Leibfarth S, Pfannenberg C, Reischl G, Mauz PS, et al. Overlap of highly FDG-avid and FMISO hypoxic tumor subvolumes in patients with head and neck cancer. Acta Oncol. (2017) 56:1577–82. doi: 10.1080/0284186x.2017.1363910
84. Simoncic U, Leibfarth S, Welz S, Schwenzer N, Schmidt H, Reischl G, et al. Thorwarth: Comparison of DCE-MRI kinetic parameters and FMISO-PET uptake parameters in head and neck cancer patients. Med Phys. (2017) 44:2358–68. doi: 10.1002/mp.12228
85. Overgaard J, Horsman MR. Modification of hypoxia-induced radioresistance in tumors by the use of oxygen and sensitizers. Semin Radiat Oncol. (1996) 6:10–21. doi: 10.1053/srao0060010
86. Janssens GO, Rademakers SE, Terhaard CH, Doornaert PA, Bijl HP, van den Ende P, et al. Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: results of a phase III randomized trial. J Clin Oncol. (2012) 30:1777–83. doi: 10.1200/jco.2011.35.9315
87. Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR. Plasma osteopontin, hypoxia, and response to the hypoxia sensitiser nimorazole in radiotherapy of head and neck cancer: results from the DAHANCA 5 randomised double-blind placebo-controlled trial. Lancet Oncol. (2005) 6:757–64. doi: 10.1016/s1470–2045(05)70292–8
88. Toustrup K, Sorensen BS, Lassen P, Wiuf C, Alsner J, Overgaard J. Gene expression classifier predicts for hypoxic modification of radiotherapy with nimorazole in squamous cell carcinomas of the head and neck. Radiother Oncol. (2012) 102:122–9. doi: 10.1016/j.radonc.2011.09.010
89. Berwouts D, Madani I, Duprez F, Olteanu AL, Vercauteren T, Boterberg T, et al. Long-term outcome of (18) F-fluorodeoxyglucose-positron emission tomography-guided dose painting for head and neck cancer: Matched case-control study. Head Neck. (2017) 39:2264–75. doi: 10.1002/hed.24892
90. Welz S, Monnich D, Pfannenberg C, Nikolaou K, Reimold M, La Fougere C, et al. Prognostic value of dynamic hypoxia PET in head and neck cancer: Results from a planned interim analysis of a randomized phase II hypoxia-image guided dose escalation trial. Radiother Oncol. (2017) 124:526–32. doi: 10.1016/j.radonc.2017.04.004
91. Overgaard J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck–a systematic review and meta-analysis. Radiother Oncol. (2011) 100:22–32. doi: 10.1016/j.radonc.2011.03.004
92. Vera P, Thureau S, Chaumet-Riffaud P, Modzelewski R, Bohn P, Vermandel M, et al. Phase II study of a radiotherapy total dose increase in hypoxic lesions identified by (18)F-Misonidazole PET/CT in patients with non-small cell lung carcinoma (RTEP5 Study). J Nucl Med. (2017) 58:1045–53. doi: 10.2967/jnumed.116.188367
93. Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. (2007) 25:4066–74. doi: 10.1200/jco.2007.12.7878
94. Sethumadhavan S, Silva M, Philbrook P, Nguyen T, Hatfield SM, Ohta A, et al. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE. (2017) 12:e0187314. doi: 10.1371/journal.pone.0187314
95. Blander JM. The comings and goings of MHC class I molecules herald a new dawn in cross-presentation. Immunol Rev. (2016) 272:65–79. doi: 10.1111/imr.12428
96. Yaghi L, Poras I, Simoes RT, Donadi EA, Tost J, Daunay A, et al. Hypoxia inducible factor-1 mediates the expression of the immune checkpoint HLA-G in glioma cells through hypoxia response element located in exon 2. Oncotarget. (2016) 7:63690–707. doi: 10.18632/oncotarget.11628
97. Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: An Immune Checkpoint Molecule. Adv Immunol. (2015) 127:33–144. doi: 10.1016/bs.ai.2015.04.001
98. Gonzalez A, Rebmann V, LeMaoult J, Horn PA, Carosella ED, Alegre E. The immunosuppressive molecule HLA-G and its clinical implications. Crit Rev Clin Lab Sci. (2012) 49:63–84. doi: 10.3109/10408363.2012.677947
99. Wu D, Kuiaste I, Moreau P, Carosella E, Yotnda P. Rescuing lymphocytes from HLA-G immunosuppressive effects mediated by the tumor microenvironment. Oncotarget. (2015) 6:37385–97. doi: 10.18632/oncotarget.6044
100. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
101. Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. (2016) 139:396–403. doi: 10.1002/ijc.30077
102. Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal cell carcinoma programmed death-ligand 1, a new direct target of hypoxia-inducible factor-2 alpha, is regulated by von hippel-lindau gene mutation status. Eur Urol. (2016) 70:623–32. doi: 10.1016/j.eururo.2015.11.029
103. Semaan A, Dietrich D, Bergheim D, Dietrich J, Kalff JC, Branchi V, et al. CXCL12 expression and PD-L1 expression serve as prognostic biomarkers in HCC and are induced by hypoxia. Virchows Arch. (2017) 470:185–96. doi: 10.1007/s00428–016-2051–5
104. Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, et al. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiother Oncol. (2005) 76:168–76. doi: 10.1016/j.radonc.2005.06.025
105. Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene. (2007) 26:2048–57. doi: 10.1038/sj.onc.1210001
106. Lu Y, Chu A, Turker MS, Glazer PM. Hypoxia-induced epigenetic regulation and silencing of the BRCA1 promoter. Mol Cell Biol. (2011) 31:3339–50. doi: 10.1128/mcb.01121–10
107. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 Inhibition. N Engl J Med. (2017) 377:2500–1. doi: 10.1056/NEJMc1713444
108. Ma T, Schreiber CA, Knutson GJ, Khattouti AE, Sakiyama MJ, Hassan M, et al. Effects of oxygen on the antigenic landscape of prostate cancer cells. BMC Res Notes. (2015) 8:687. doi: 10.1186/s13104-015-1633-7
109. Barsoum IB, Hamilton TK, Li X, Cotechini T, Miles EA, Siemens DR, et al. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: role of nitric oxide. Cancer Res. (2011) 71:7433–41. doi: 10.1158/0008–5472.can-11–2104
110. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. (2011) 475:226–30. doi: 10.1038/nature10169
111. Ren L, Yu Y, Wang L, Zhu Z, Lu R, Yao Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget. (2016) 7:75763–73. doi: 10.18632/oncotarget.12409
112. Vuillefroy de Silly R, Ducimetiere L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR. Phenotypic switch of CD8+ T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol. (2015) 45:2263–75. doi: 10.1002/eji.201445284
113. Sitkovsky M, Ohta A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J Mol Med. (2013) 91:147–55. doi: 10.1007/s00109–013-1001–9
114. Vaupel P, Multhoff G. Adenosine can thwart antitumor immune responses elicited by radiotherapy: Therapeutic strategies alleviating protumor ADO activities. Strahlenther Onkol. (2016) 192:279–87. doi: 10.1007/s00066–016-0948–1
115. Lan J, Lu H, Samanta D, Salman S, Lu Y, Semenza GL. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci USA. (2018)115:E9640–8. doi: 10.1073/pnas.1809695115
116. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer. (2017) 16:99. doi: 10.1186/s12943–017-0665–0
117. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A2A adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor-reactive CD8+ T Cells from Tumor-Induced Immunosuppression. J Immunol. (2018) 201:782–91. doi: 10.4049/jimmunol.1700850
118. Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, et al. A2AR Antagonism with CPI-444 induces antitumor responses and augments efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in Preclinical Models. Cancer Immunol Res. (2018) 6:1136–49. doi: 10.1158/2326–6066.cir-18–0056
119. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia. (2017) 19:530–6. doi: 10.1016/j.neo.2017.02.004
120. Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, et al. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. (2013) 43:2756–64. doi: 10.1002/eji.201343448
121. Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology. (2009) 128(Suppl. 1):e237–49. doi: 10.1111/j.1365–2567.2008.02954.x
122. Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, et al. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ effector T cell function and promoting treg activity. Cell Physiol Biochem. (2017) 41:1271–84. doi: 10.1159/000464429
123. Facciabene A, Santoro S, Coukos G. Know thy enemy: Why are tumor-infiltrating regulatory T cells so deleterious? Oncoimmunology. (2012)1:575–7.
124. Noman MZ, Hasmim M, Messai Y, Terry S, Kieda C, Janji B, et al. Hypoxia: a key player in antitumor immune response. a review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol. (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
125. Deng B, Zhu JM, Wang Y, Liu TT, Ding YB, Xiao WM, et al. Intratumor hypoxia promotes immune tolerance by inducing regulatory T cells via TGF-beta1 in gastric cancer. PLoS ONE. (2013) 8:e63777. doi: 10.1371/journal.pone.0063777
126. Hasmim M, Noman MZ, Messai Y, Bordereaux D, Gros G, Baud V, et al. Cutting edge: Hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-beta1. J Immunol. (2013) 191:5802–6. doi: 10.4049/jimmunol.1302140
127. Yan M, Jene N, Byrne D, Millar EK, O'Toole SA, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Res. (2011) 13:R47. doi: 10.1186/bcr2869
128. Chiu DK, Xu IM, Lai RK, Tse AP, Wei LL, Koh HY, et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology. (2016) 64:797–813. doi: 10.1002/hep.28655
129. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467–017-00530–7
130. Guo X, Xue H, Shao Q, Wang J, Guo X, Chen X, et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSF. Oncotarget. (2016) 7:80521–42. doi: 10.18632/oncotarget.11825
131. Corzo CA, Condamine T, Lu L, Cotter MJ, Youn JI, Cheng P, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. (2010) 207:2439–53. doi: 10.1084/jem.20100587
132. Shrivastava R, Asif M, Singh V, Dubey P, Ahmad Malik S, Lone MU, et al. M2 polarization of macrophages by Oncostatin M in hypoxic tumor microenvironment is mediated by mTORC2 and promotes tumor growth and metastasis. Cytokine. (2018). doi: 10.1016/j.cyto.2018.03.032. [Epub ahead of print].
133. Tripathi C, Tewari BN, Kanchan RK, Baghel KS, Nautiyal N, Shrivastava R, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. (2014) 5:5350–68. doi: 10.18632/oncotarget.2110
134. Meng F, Li C, Li W, Gao Z, Guo K, Song S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life. (2014) 66:835–46. doi: 10.1002/iub.1336
135. Partecke LI, Gunther C, Hagemann S, Jacobi C, Merkel M, Sendler M, et al. Induction of M2-macrophages by tumour cells and tumour growth promotion by M2-macrophages: a quid pro quo in pancreatic cancer. Pancreatology. (2013) 13:508–16. doi: 10.1016/j.pan.2013.06.010
136. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. (2015) 62:607–16. doi: 10.1016/j.jhep.2014.10.029
137. Han Q, Shi H, Liu F. CD163+ M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int Immunopharmacol. (2016) 34:101–6. doi: 10.1016/j.intimp.2016.01.023
138. Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood. (2006) 108:525–35. doi: 10.1182/blood-2005–09-3777
139. Sceneay J, Parker BS, Smyth MJ, Moller A. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology. (2013) 2:e22355. doi: 10.4161/onci.22355
140. Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, McDonald PC, et al. Carbonic anhydrase IX promotes myeloid-derived suppressor cell mobilization and establishment of a metastatic niche by stimulating G-CSF production. Cancer Res. (2015) 75:996–1008. doi: 10.1158/0008–5472.can-14–3000
141. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
142. Bellmunt J, de Wit R, Vaughn DJ, Fraldet Y, Lee JL, Fong L, et al. Pembrolizumab as Second-line therapy for advanced urothelial carcinoma. N Engl J Med. (2017) 376:1015–26. doi: 10.1056/NEJMoa1613683
143. Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab vs investigator's choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol. (2018) 81:45–51. doi: 10.1016/j.oraloncology.2018.04.008
144. Shitara K, Ozguroglu M, Bang YJ, Di Bartolomeo M, Mandala M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet. (2018) 392:123–33. doi: 10.1016/S0140–6736(18)31257–1
145. Reardona DA, Omuroa A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: checkmate 143. Neuro-Oncology. (2017) 19:21. doi: 10.1093/neuonc/nox036.071
146. Eckert F, Gaipl US, Niedermann G, Hettich M, Schilbach K, Huber SM, et al. Beyond checkpoint inhibition - Immunotherapeutical strategies in combination with radiation. Clin Transl Radiat Oncol. (2017) 2:29–35. doi: 10.1016/j.ctro.2016.12.006
147. Eckert F, Jelas I, Oehme M, Huber SM, Sonntag K, Welker C, et al. Tumor-targeted IL-12 combined with local irradiation leads to systemic tumor control via abscopal effects in vivo. Oncoimmunology. (2017) 6:e1323161. doi: 10.1080/2162402x.2017.1323161
148. Eckert F, Schmitt J, Zips D, Krueger MA, Pichler BJ, Gillies SD, et al. Enhanced binding of necrosis-targeting immunocytokine NHS-IL12 after local tumour irradiation in murine xenograft models. Cancer Immunol Immunother. (2016) 65:1003–13. doi: 10.1007/s00262–016-1863–0
149. Schilbach K, Alkhaled M, Welker C, Eckert F, Blank G, Ziegler H, et al. Cancer-targeted IL-12 controls human rhabdomyosarcoma by senescence induction and myogenic differentiation. Oncoimmunology. (2015) 4:e1014760. doi: 10.1080/2162402x.2015.1014760
150. Basler L, Kowalczyk A, Heidenreich R, Fotin-Mleczek M, Tsitsekidis S, Zips D, et al. Abscopal effects of radiotherapy and combined mRNA-based immunotherapy in a syngeneic, OVA-expressing thymoma mouse model. Cancer Immunol Immunother. (2018) 67:653–62. doi: 10.1007/s00262–018-2117–0
151. Fotin-Mleczek M, Zanzinger K, Heidenreich R, Lorenz C, Kowalczyk A, Kallen KJ, et al. mRNA-based vaccines synergize with radiation therapy to eradicate established tumors. Radiat Oncol. (2014) 9:180. doi: 10.1186/1748–717x-9–180
152. Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. (2013) 1291:1–13. doi: 10.1111/nyas.12180
153. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 Costimulation: from mechanism to therapy. Immunity. (2016) 44:973–88. doi: 10.1016/j.immuni.2016.04.020
154. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. (1995) 270:985–8.
155. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65.
156. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. (1994) 1:405–13.
157. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6.
158. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
159. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. (2015) 33:1889–94. doi: 10.1200/jco.2014.56.2736
160. Vanpouille-Box C, Lhuillier C, Bezu L, Aranda F, Yamazaki T, Kepp O, et al. Trial watch: Immune checkpoint blockers for cancer therapy. Oncoimmunology. (2017) 6:e1373237. doi: 10.1080/2162402x.2017.1373237
161. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
162. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51.
163. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
164. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. (2006) 439:682–7. doi: 10.1038/nature04444
165. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. (2010) 28:3167–75. doi: 10.1200/jco.2009.26.7609
166. Liu B, Song Y, Liu D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J Hematol Oncol. (2017) 10:174. doi: 10.1186/s13045–017-0541–9
167. Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. (2016) 40:25–37. doi: 10.1016/j.currproblcancer.2015.10.001
168. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. (2004) 58:862–70. doi: 10.1016/j.ijrobp.2003.09.012
169. Demaria S, Formenti SC. Role of T lymphocytes in tumor response to radiotherapy. Front Oncol. (2012) 2:95. doi: 10.3389/fonc.2012.00095
170. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
171. Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. (2015) 25:11–7. doi: 10.1016/j.semradonc.2014.07.005
172. Song KH, Jung SY, Kang SM, Kim MH, Ahn J, Hwang SG, et al. Induction of immunogenic cell death by radiation-upregulated karyopherin alpha 2 in vitro. Eur J Cell Biol. (2016) 95:219–27. doi: 10.1016/j.ejcb.2016.04.002
173. Galluzzi L, Kepp O, Kroemer G. Immunogenic cell death in radiation therapy. Oncoimmunology. (2013) 2:e26536. doi: 10.4161/onci.26536
174. Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. (2014) 3:e955691. doi: 10.4161/21624011.2014.955691
175. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. (2006) 203:1259–71. doi: 10.1084/jem.20052494
176. Jochems C, Fantini M, Fernando RI, Kwilas AR, Donahue RN, Lepone LM, et al. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget. (2016) 7:37762–72. doi: 10.18632/oncotarget.9326
177. Mazorra Z, Lavastida A, Concha-Benavente F, Valdes A, Srivastava RM, Garcia-Bates TM, et al. Nimotuzumab Induces NK Cell Activation, Cytotoxicity, dendritic cell maturation and expansion of egfr-specific t cells in head and neck cancer patients. Front Pharmacol. (2017) 8:382. doi: 10.3389/fphar.2017.00382
178. Sanchez-Paulete AR, Teijeira A, Cueto FJ, Garasa S, Perez-Gracia JL, Sanchez-Arraez A, et al. Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol. (2017) 28(Suppl. 12):44–55. doi: 10.1093/annonc/mdx237
179. Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. (2016) 2:286–94. doi: 10.1016/j.trecan.2016.05.002
180. Dovedi S J, Cheadle EJ, Popple AL, Poon E, Morrow M, Stewart R, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal t-cell populations when combined with PD-1 blockade. Clin Cancer Res. (2017) 23:5514–26. doi: 10.1158/1078–0432.ccr-16–1673
181. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. (2005) 174:7516–23. doi: 10.4049/jimmunol.174.12.7516
182. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS+/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. (2013) 24:589–602. doi: 10.1016/j.ccr.2013.09.014
183. Canter RJ, Grossenbacher SK, Foltz JA, Sturgill IR, Park JS, Luna JI, et al. Radiotherapy enhances natural killer cell cytotoxicity and localization in pre-clinical canine sarcomas and first-in-dog clinical trial. J Immunother Cancer. (2017) 5:98. doi: 10.1186/s40425–017-0305–7
184. Wennerberg E, Vanpouille-Box C, Bornstein S, Yamazaki T, Demaria S, Galluzzi L. Immune recognition of irradiated cancer cells. Immunol Rev. (2017) 280:220–30. doi: 10.1111/imr.12568
185. Derer A, Spiljar M, Baumler M, Hecht M, Fietkau R, Frey B, et al. Chemoradiation Increases PD-L1 expression in certain melanoma and glioblastoma cells. Front Immunol. (2016) 7:610. doi: 10.3389/fimmu.2016.00610
186. Fujimoto D, Uehara K, Sato Y, Sakanoue I, Ito M, Teraoka S, et al. Alteration of PD-L1 expression and its prognostic impact after concurrent chemoradiation therapy in non-small cell lung cancer patients. Sci Rep. (2017) 7:11373. doi: 10.1038/s41598–017-11949–9
187. Shen MJ, Xu LJ, Yang L, Tsai Y, Keng PC, Chen Y, et al. Radiation alters PD-L1/NKG2D ligand levels in lung cancer cells and leads to immune escape from NK cell cytotoxicity via IL-6-MEK/Erk signaling pathway. Oncotarget. (2017) 8:80506–20. doi: 10.18632/oncotarget.19193
188. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. (2014) 124:687–95. doi: 10.1172/jci67313
189. Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, et al. Stereotactic radiotherapy increases functionally suppressive regulatory T Cells in the tumor microenvironment. Cancer Immunol Res. (2017) 5:992–1004. doi: 10.1158/2326–6066.cir-17–0040
190. Persa E, Balogh A, Safrany G, Lumniczky K. The effect of ionizing radiation on regulatory T cells in health and disease. Cancer Lett. (2015) 368:252–61. doi: 10.1016/j.canlet.2015.03.003
191. Carl C, Flindt A, Hartmann J, Dahlke M, Rades D, Dunst J, et al. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell Mol Life Sci. (2016) 73:427–43. doi: 10.1007/s00018–015-2003–2
192. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
193. Eckert F, Schaedle P, Zips D, Schmid-Horch B, Rammensee HG, Gani C, et al. Impact of curative radiotherapy on the immune status of patients with localized prostate cancer. Oncoimmunology. (2018) 7:e1496881. doi: 10.1080/2162402x.2018.1496881
194. Schaue D, Comin-Anduix B, Ribas A, Zhang L, Goodglick L, Sayre JW, et al. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin Cancer Res. (2008) 14:4883–90. doi: 10.1158/1078-0432.CCR-07-4462
195. van Meir H, Nout RA, Welters MJ, Loof NM, de Kam ML, van Ham JJ, et al. Impact of (chemo)radiotherapy on immune cell composition and function in cervical cancer patients. Oncoimmunology. (2016) 6:e1267095. doi: 10.1080/2162402X.2016.1267095
196. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. (2015) 16:e498–509. doi: 10.1016/S1470-2045(15)00007-8
197. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. (2013) 105:256–65. doi: 10.1093/jnci/djs629
198. Shahabi V, Postow MA, Tuck D, Wolchok JD. Immune-priming of the tumor microenvironment by radiotherapy: rationale for combination with immunotherapy to improve anticancer efficacy. Am J Clin Oncol. (2015) 38:90–7. doi: 10.1097/COC.0b013e3182868ec8
199. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. (2012) 366:925–31. doi: 10.1056/NEJMoa1112824
200. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. (2015) 520:373–7. doi: 10.1038/nature14292
201. Hiniker SM, Reddy SA, Maecker HT, Subrahmanyam PB, Rosenberg-Hasson Y, Swetter SM, et al. A prospective clinical trial combining radiation therapy with systemic immunotherapy in metastatic melanoma. Int J Radiat Oncol Biol Phys. (2016) 96:578–88. doi: 10.1016/j.ijrobp.2016.07.005
202. Luke JJ, Lemons JM, Karrison TG, Pitroda SP, Melotek JM, Zha Y, et al. Safety and clinical activity of pembrolizumab and multisite stereotactic body radiotherapy in patients with advanced solid tumors. J Clin Oncol. (2018) 36:1611–8. doi: 10.1200/jco.2017.76.2229
203. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in stage iii non-small-cell lung cancer. N Engl J Med. (2017) 377:1919–29. doi: 10.1056/NEJMoa1709937
204. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after chemoradiotherapy in Stage III NSCLC. N Engl J Med. (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
205. Tang C, Welsh JW, de Groot P, Massarelli E, Chang JY, Hess KR, et al. Ipilimumab with stereotactic ablative radiation therapy: phase i results and immunologic correlates from peripheral t cells. Clin Cancer Res. (2017) 23:1388–96. doi: 10.1158/1078–0432.CCR-16–1432
206. Gandhi SJ, Minn AJ, Vonderheide RH, Wherry EJ, Hahn SM, Maity A. Awakening the immune system with radiation: optimal dose and fractionation. Cancer Lett. (2015) 368:185–90. doi: 10.1016/j.canlet.2015.03.024
207. Park SS, Dong H, Liu X, Harrington SM, Krco CJ, Grams MP, et al. PD-1 restrains radiotherapy-induced abscopal effect. Cancer Immunol Res. (2015) 3:610–9. doi: 10.1158/2326–6066.cir-14–0138
Keywords: immunotherapy, radiotherapy, hypoxia, T cells, cancer, Tregs, immune checkpoint inhibition
Citation: Eckert F, Zwirner K, Boeke S, Thorwarth D, Zips D and Huber SM (2019) Rationale for Combining Radiotherapy and Immune Checkpoint Inhibition for Patients With Hypoxic Tumors. Front. Immunol. 10:407. doi: 10.3389/fimmu.2019.00407
Received: 29 October 2018; Accepted: 15 February 2019;
Published: 12 March 2019.
Edited by:
Franz Rödel, Universitätsklinikum Frankfurt, GermanyReviewed by:
Verena Jendrossek, University of Duisburg-Essen, GermanyHaidong Dong, Mayo Clinic, United States
Copyright © 2019 Eckert, Zwirner, Boeke, Thorwarth, Zips and Huber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franziska Eckert, ZnJhbnppc2thLmVja2VydEBtZWQudW5pLXR1ZWJpbmdlbi5kZQ==