 Adewunmi Adelaja
Adewunmi Adelaja Alexander Hoffmann
Alexander Hoffmann- 1UCLA-Caltech Medical Scientist Training Program, Department of Microbiology, Immunology, and Molecular Genetics, David Geffen School of Medicine, Molecular Biology Institute, University of California, Los Angeles, Los Angeles, CA, United States
- 2Department of Microbiology, Immunology, and Molecular Genetics, Institute for Quantitative and Computational Biosciences, University of California, Los Angeles, Los Angeles, CA, United States
Precise control of inflammatory gene expression is critical for effective host defense without excessive tissue damage. The principal regulator of inflammatory gene expression is nuclear factor kappa B (NFκB), a transcription factor. Nuclear NFκB activity is controlled by IκB proteins, whose stimulus-responsive degradation and re-synthesis provide for transient or dynamic regulation. The IκB-NFκB signaling module receives input signals from a variety of pathogen sensors, such as toll-like receptors (TLRs). The molecular components and mechanisms of NFκB signaling are well-understood and have been reviewed elsewhere in detail. Here we review the molecular mechanisms that mediate cross-regulation of TLR-IκB-NFκB signal transduction by signaling pathways that do not activate NFκB themselves, such as interferon signaling pathways. We distinguish between potential regulatory crosstalk mechanisms that (i) occur proximal to TLRs and thus may have stimulus-specific effects, (ii) affect the core IκB-NFκB signaling module to modulate NFκB activation in response to several stimuli. We review some well-documented examples of molecular crosstalk mechanisms and indicate other potential mechanisms whose physiological roles require further study.
Introduction
NFκB signaling mediates inflammatory and innate immune responses; the signaling components that comprise the core signaling pathway are well-understood and have been amply reviewed, for example by Mitchell et al. (1), Leifer and Medvedev (2), Pandey et al. (3), and Hayden and Ghosh (4). Here, therefore, is only a brief summary. Of 15 possible NFκB dimers, the predominant mediator of NFκB inflammatory gene expression is the ubiquitous RelA:p50 heterodimer (1). At rest, inhibitors of κB (IκB)s sequester RelA:p50 in the cytoplasm by masking its DNA binding region and nuclear localization signal (5–7). In response to stimuli, IκBs are phosphorylated by IκB kinase (IKK), which triggers their ubiquitination and proteolysis (8, 9). Then, RelA:p50 translocates from the cytoplasm to the nucleus, where it binds and activates promoters and enhancers of target genes, such as nfkbia, which codes for IκBα (10, 11). Since IκBα synthesis is induced by RelA:p50, a tightly coupled negative feedback loop emerges that regulates NFκB activity in a highly dynamic and stimulus-specific fashion (11–13). To tune NFκB signaling, crosstalk mechanisms regulate signal transduction from TLRs to IκBs to NFκB (Figure 1). We describe crosstalk mechanism at four levels: receptors, adaptors, enzymatic complexes, and the IκB-NFκB signaling module (Figure 2). Here, we focus on a few well-established crosstalk mechanisms, and mention others that deserve further study.
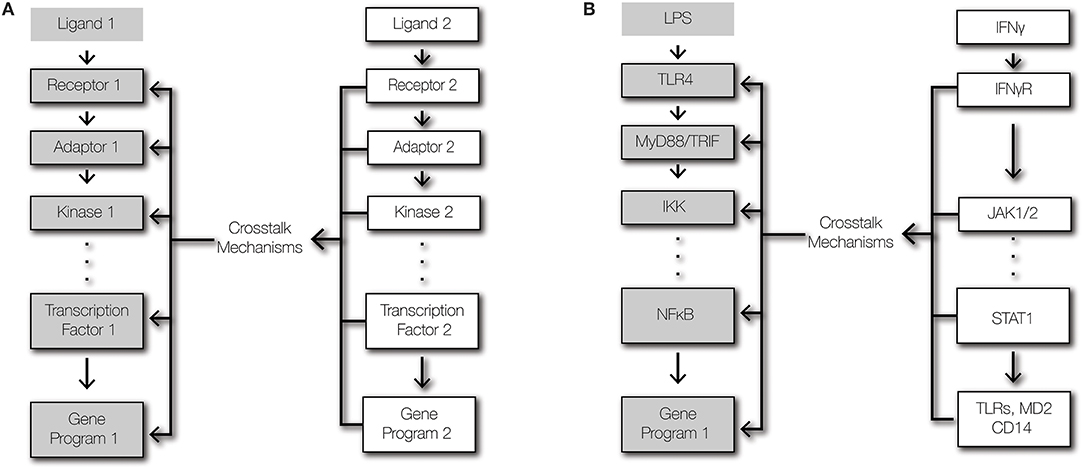
Figure 1. Signaling and crosstalk. (A) Regulatory crosstalk is defined here as the signal transduction within a pathway being altered by a second pathway that affects the abundances or functions of signaling components. (B) Schematic of signaling crosstalk from IFNγ signaling to TLR4-NFκB signaling.
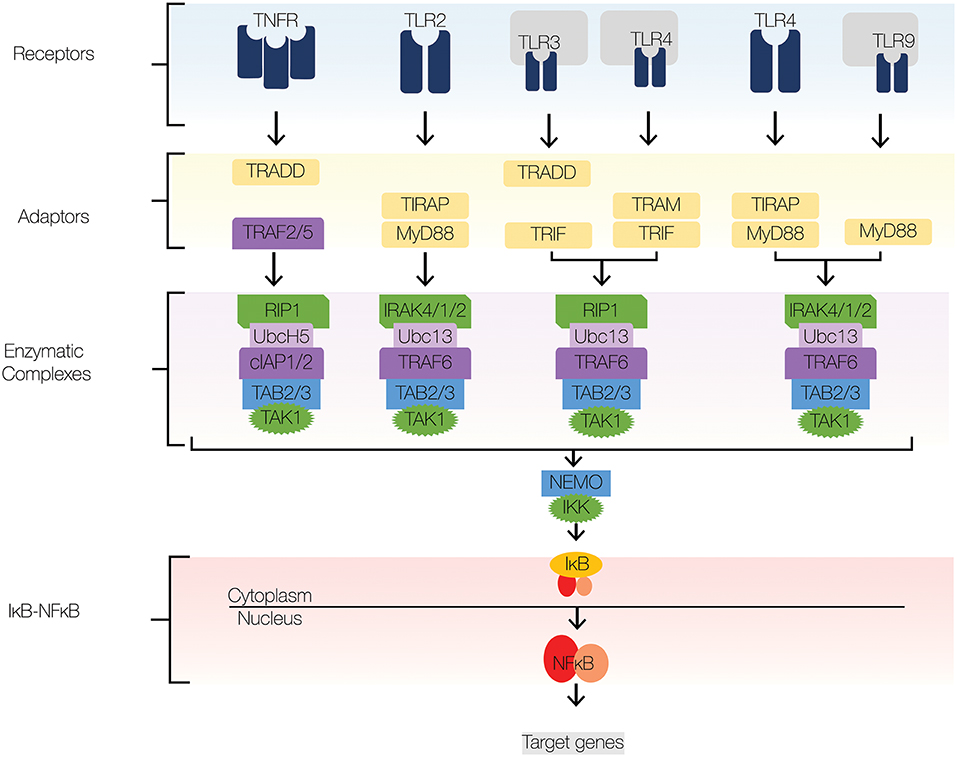
Figure 2. NFκB signaling pathway. The major signaling components of the NFκB signaling pathway include receptors, adaptors, enzymatic complexes, and the IκB-NFκB complex. Upon ligand recognition, cognate receptors engage adaptor proteins that recruit kinases and ubiquitin ligases to the signaling complex. TLR signaling employs adaptor proteins MyD88 and TRIF; both of which contain TIR domains. Sorting adaptor proteins such as TIRAP and TRAM facilitate MyD88 and TRIF association with the signaling complex. MyD88 engages an enzymatic complex that includes IRAK4, IRAK1, IRAK2, TRAF6, Ubc13, TAB2/3, and TAK1. TRIF engages a similar enzymatic complex, which includes RIP1 instead IRAK4,1,2. The enzymatic complexes facilitate the recruitment and activation of IKKβ, which induces the degradation of IκBs and subsequent nuclear translocation of NFκB. Navy blue, TLRs; yellow, adaptors; green, kinases; dark purple, E3 ligases; light purple, E2 conjugases.
To ensure effective host defense against pathogens and to maintain tissue integrity, immune cells must integrate multiple signals to produce appropriate responses (14). Cells of the innate immune system are equipped with pattern recognition-receptors (PRRs) that detect pathogen-derived molecules, such as lipopolysaccharides and dsRNA (3). Once activated, PRRs initiate series of intracellular biochemical events that converge on transcription factors that regulate powerful inflammatory gene expression programs (15). To tune inflammatory responses, pathways that do not trigger inflammatory responses themselves may modulate signal transduction from PRRs to transcription factors through crosstalk mechanisms (Figure 1). Crosstalk allows cells to shape the inflammatory response to the context of their microenvironment and history (16). Crosstalk between two signaling pathways may emerge due shared signaling components, direct interactions between pathway-specific components, and regulation of the expression level of a pathway-specific component by the other pathway (1, 17). Since toll-like receptors (TLRs) are the best characterized PRRs, they provide the most salient examples of crosstalk at the receptor module. Key determinants of tissue microenvironments are type I and II interferons (IFNs), which do not activate NFκB, but regulate NFκB-dependent gene expression (18–21). As such, this review focuses on the cross-regulation of the TLR-NFκB signaling axis by type I and II IFNs.
Whereas, IFNγ is the only type II IFN, the type I IFN family consists of multiple forms of IFNα and IFNβ (22, 23). Type I IFNs ligate interferon-α receptors (IFNAR), which leads to the activation of Janus-activated kinase-1 (JAK1), tyrosine kinase 2 (Tyk2), and IFN-stimulated gene factor 3 (ISGF3) complex, which consists of signal transducer and activator of transcription 1 (STAT1), STAT2, and IFN-regulatory factor (IRF)-9 (23). IFNγ ligates IFNγ-receptor (IFNGR), which leads to the activation of JAK1 and JAK2 and the subsequent STAT1 phosphorylation and homodimerization (22).
Receptor Modules
Receptor Abundance and Localization
IFNγ is a well-described crosstalk mediator that enhances NFκB signaling (Figure 3) (20). By upregulating the expression of TLRs, IFNγ enhances the detection of pathogen-associated molecular patterns (PAMPs) by TLRs in different cellular compartments. At the plasma membrane, TLR2 and TLR4 recognize microbial cell wall components, such as lipopolysaccharides and lipoproteins (24). Similarly, endosomal TLRs, such as TLR3 and TLR9, recognize double stranded RNA and CpG oligonucleotides (24). IFNγ upregulates TLR2, TLR3, TLR4, and TLR9 at the mRNA and protein levels (25–30). Similarly, the inflammatory cytokine, tumor necrosis factor (TNF) upregulates the mRNA expression of TLR2 (31). The significance of TNF-induced and IFNγ-induced upregulation of TLR abundance on NFκB signaling dynamics is unknown. In addition to recognizing PAMPs, TLRs recognize host-derived molecules, such as extracellular matrix proteins, heat-shock proteins, nucleic acids, and high mobility group box 1 (32–37). Whereas, high TLR abundance facilitates detection of pathogens and mobilization host defenses, it may also increase susceptibility to autoimmune diseases and sepsis (24).
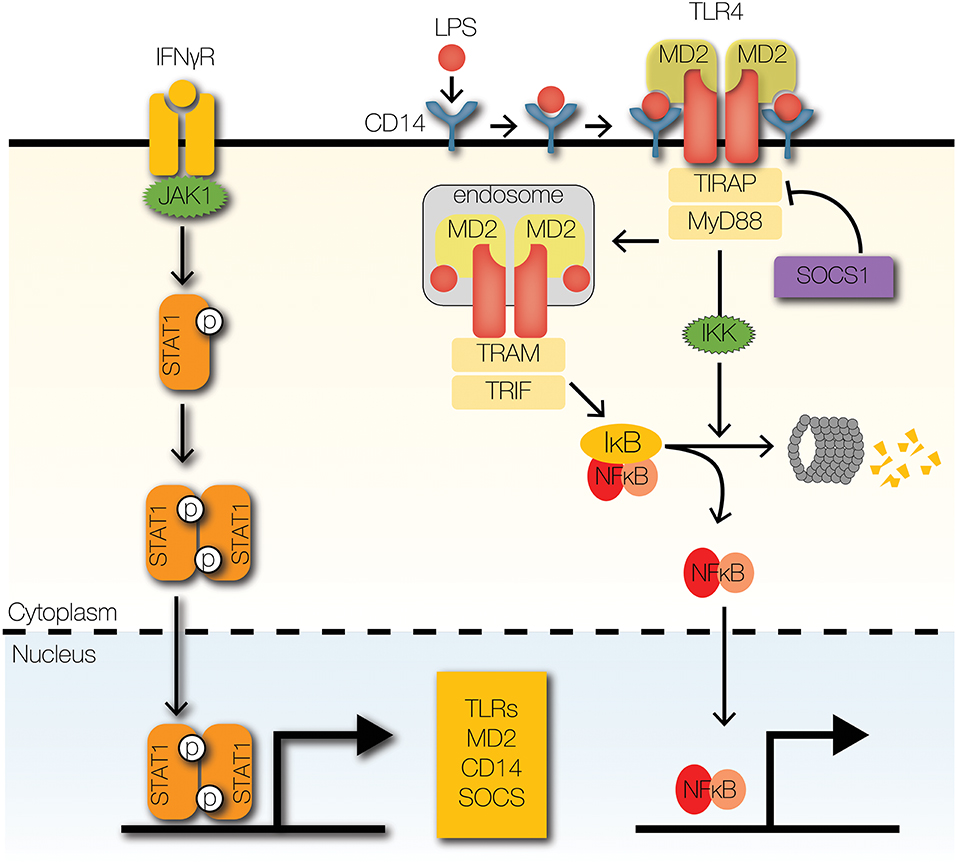
Figure 3. Signaling crosstalk at receptors and adaptors. IFNγ receptor activation leads to the phosphorylation and nuclear translocation of STAT1 homodimers. STAT1 upregulates the expression of several signaling components of the TLR signaling pathway, such asTLRs and co-receptors MD2 and CD14. SOCS1, a STAT1-inducible negative regulator of STAT1 signaling, promotes the degradation of TIRAP by facilitating K48-ubiquitin-mediated proteolysis.
Accessory Protein Abundance
In addition to upregulating TLR expression, IFNγ also upregulates expression of TLR accessory proteins (Figure 3), such as myeloid differentiation factor 2 (MD2) and CD14 (29, 38, 39). Both accessory proteins facilitate the binding of lipopolysaccharide (LPS) to TLR4, in part by regulating localization of TLR4 (40–42). In fact, MD2 is necessary for localization of TLR4 to the plasma membrane, where it can bind LPS and transduce signals to downstream components (41, 43). After activation, TLR4 undergoes dynamin-mediated endocytosis into endosomes, where it continues transmitting signals (44). In the absence of CD14, endocytosis of TLR4 and subsequent signal transmission are attenuated. Further, CD14 and MD2 promote the association of endosomal TLR4 to downstream adaptors, which are critical for signal transduction (41, 42). Although CD14 is primarily associated with TLR4-mediated signaling, it also facilitates TLR2, TLR3, and TLR9 signaling (45–47). Interestingly, accessory proteins may contribute to inflammation in Alzheimer's disease (AD) and atherosclerosis (48). CD36, a scavenger receptor, recognizes amyloid β and oxidized LDL, which contribute to pathogenesis of AD and atherosclerosis, respectively (48). CD36 forms a heterotrimeric complex with TLR4 and TLR6 to induce production of inflammatory mediators (48). Further, IFNγ-activated macrophages significantly upregulate the expression CD36 in disease models of atherosclerosis (49).
Signaling Adapters
While IFNγ upregulates the expression of TLRs and accessory proteins that promote inflammatory responses, it also upregulates negative feedback regulators to maintain homeostasis (Figure 3). To enable negative feedback, IFNγ, TNF, and type I IFNs induce the expression of a family of E3 ubiquitin ligases, aptly named suppressors of cytokine signaling (SOCS) (18, 25, 50). SOCS1 was reported as a negative regulator of TLR4 signaling that is essential for the formation of endotoxin tolerance (51). The putative mechanism by which SOCS1 inhibits TLR signaling is through ubiquitin-mediated degradation of TIR domain containing adaptor (TIRAP), which recruits myeloid differentiation primary response gene 88 (MyD88) to TLR2 and TLR4 by mitigating the effects of electrostatic repulsion (52). The significance of SOCS1 is evident from the fact that SOCS1 deficiency causes neonatal lethality in mice due to overwhelming inflammation (53). However, loss of IFNγ rescues socs1−/− mice, which suggests that the primary role of SOCS1 is to restrain IFNγ-dependent inflammation and pathology.
Since TLRs do not possess the catalytic activity to activate NFκB directly, they engage adaptors such as MyD88 and TIR-domain-containing adapter-inducing interferon-β (TRIF) to propagate signals downstream (54, 55). The expression of MyD88 may be controlled by IFNγ, since myd88 mRNA is IFNγ-inducible (25). Furthermore, MyD88 degradation may also be regulated by the anti-inflammatory cytokine, transforming growth factor (TGF)β, through Smad6-dependent recruitment of Smad ubiquitin regulatory factor (Smurf) 1/2 E3 ubiquitin ligases (56). However, the physiological significance of these crosstalk mechanisms remains to be fully elucidated.
Enzymatic Complexes
Signal transduction from TLRs to NFκB involves recruitment of several enzymes to the TLR signaling complex (3). The recruited kinases and ubiquitin ligases allow for signal amplification while providing pathway specificity (13, 57). The enzymes upstream of the IKK signaling complex provide multiple avenues and nodes for signal integration and crosstalk (57–59). Both the catalytic activity and abundance of these enzymes can be subject to cross-regulation (Figure 4). After engaging TLRs, MyD88 forms an oligomeric complex with IL1R-associated kinases (IRAK) called the Myddosome (60). Formation of the Myddosome complex brings IRAK4 dimers and IRAK1/2 dimers into close proximity for efficient signal transduction (61). In response to IFNγ stimulation, immune cells upregulate the expression of IRAKs and MyD88 (25, 29, 62). In contrast, TNF stimulation upregulates the expression of negative regulators of TLR signaling, such as IRAK-M (63). The expression of IRAK-M in macrophages abrogates signaling downstream of IRAKs, inhibits TLR-induced NFκB activation, and mediates endotoxin tolerance (64). As limiting components in TLR signal transduction, MyD88, and IRAKs form critical junctures for regulatory control of inflammatory responses (60, 65). During endotoxin tolerance, the abundance of IRAKs and the association of TLRs with MyD88 are reduced (62). Therefore, crosstalk at this module can serve a dual purpose: priming and tolerance.
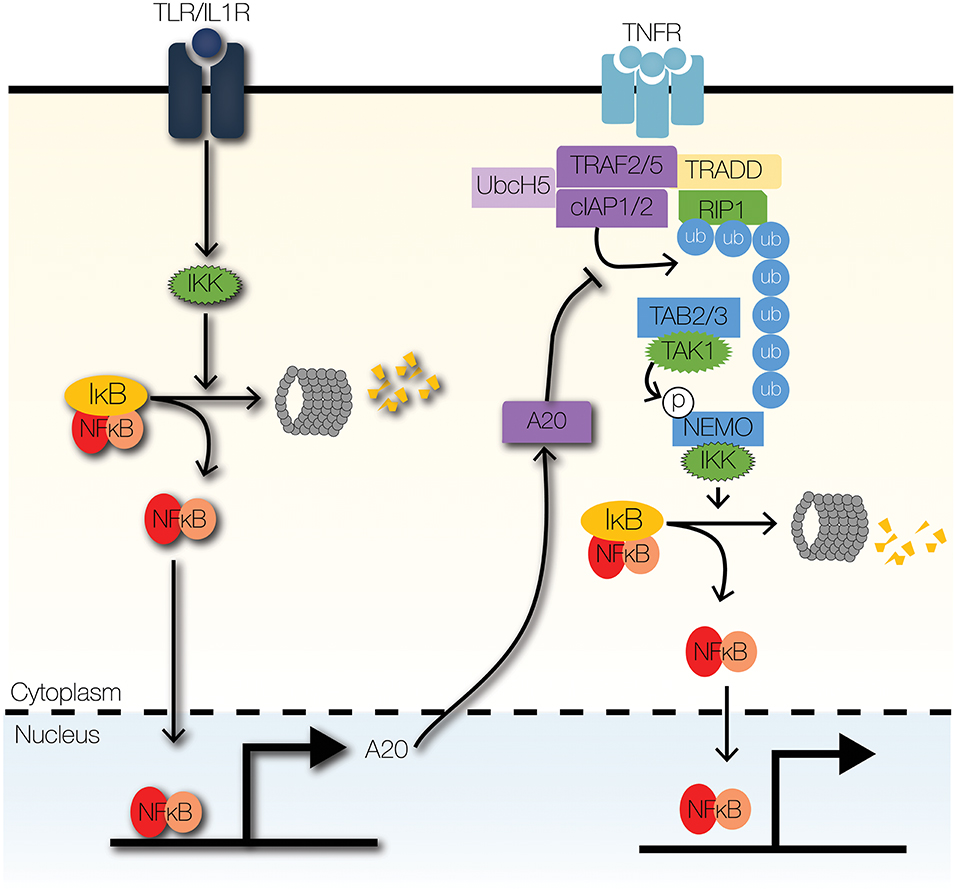
Figure 4. Signaling crosstalk at enzymatic complexes. TLR signaling can modulate TNF signaling through the actions of A20, a ubiquitin-editing enzyme. A20 inhibits the recruitment of IKK to the TNFR signaling complex by inhibiting K63-linked ubiquitination of RIP1. Further, A20 increases the degradation of RIP1 by facilitating K48-linked ubiquitination of RIP1.
Similar to TNF receptor 1 (TNFR1), TRIF engages the adaptor protein tumor necrosis TNFR1-associated death domain protein (TRADD) and the kinase receptor-interacting protein (RIP)1 (66, 67). NFκB activation through TRIF-RIP1 signaling is dependent on Pellino-1, which is an E3 ubiquitin ligase that is essential for the formation of ubiquitin scaffold on RIP1 (68); however, the E3 ubiquitin ligase activity of Pellino-1 may be dispensable for TRIF-dependent activity (69). Whereas, loss of Pellino-1 expression abolishes TRIF-dependent RIP1 ubiquitination, loss of Pellino-1 E3 ubiquitin ligase activity does not affect RIP1 ubiquitination (68, 69). Although the inducible expression of Pellino-1 mRNA (Peli1) is dependent on IFN-regulatory factor 3 (IRF3), evidence suggests Peli1 is also a target gene of ISGF3, which is induced by type I IFNs (70). Whether type I IFNs enhance TRIF-NFκB in a Pellino-1-dependent manner is unknown. Since the loss of Pellino-1 confers resistance to septic shock in response to TLR3 and TLR4 activation, it is possible that type I IFNs cross-regulate TRIF-NFκB through Pellino-1 to regulate septic shock (68). However, direct evidence is lacking.
The primary E3 ubiquitin ligase that transduces signals from MyD88 to IKK is TRAF6 (71–73). Downstream of IRAKs, TRAF6 facilitates the formation of K63-linked ubiquitin scaffold and the recruitment of IKK to the TLR signaling complex (73). TLR-NFκB signaling is regulated by ubiquitin editing enzymes, such as A20 and cylindromatosis (CYLD) (74, 75). We will focus the next section on A20 though it is not IFN-controlled but provides important signaling crosstalk (Figure 4).
A20 is a highly inducible NFκB target gene that attenuates cytokine- and pathogen-mediated inflammatory signaling (76, 77). Loss of A20 is lethal, due to excessive inflammation, cachexia, and organ failure (78, 79). Furthermore, dysregulated A20 signaling contributes to the pathogenesis of atherosclerosis and rheumatoid arthritis (80–82). A20 is an essential negative feedback regulator and terminator of TLR signaling (77). It edits ubiquitin tags on TRAF6 and RIP1 (75, 83). A20 removes K63-linked ubiquitin chains from RIP1 and may add K48-linked ubiquitin chains to target RIPK1 for proteasomal degradation (75). Additionally, A20 disrupts the interactions between TRAF6 and E2 ubiquitin conjugating enzymes, Ubc13 and UbcH5; A20 also enhances proteasomal degradation of Ubc13 and UbcH5c, by catalyzing the deposition of K48-linked ubiquitin chains (83). By mediating signaling crosstalk between TNFR and TLR/IL1R signaling pathways, A20 serves as a memory of recent inflammatory signaling (58, 63).
A20-binding inhibitor of NFκB activation 1(ABIN1; also known TNIP1) is a TNF-inducible binding partner of A20 (84–86). ABIN1 modulates A20-mediated inhibition of IKK-NFκB signaling by enhancing the de-ubiqutination of the IKK regulatory subunit, IKKγ/NEMO (84). The exact mechanism of ABIN1-mediated inhibition of IKK has yet to be elucidated. The observation that ABIN1 has a high affinity for polyubiquitin chains has informed some candidate mechanisms (87). One potential mechanism involves ABIN1 serving as an adaptor that brings A20 and its targets into close proximity (88). Another potential mechanism involves competition with the regulatory subunit of IKK, IKKγ/NEMO for polyubiquitin binding (88). Similar to the loss of A20, the loss of ABIN1 (tnip1−/−) may lead to embryonic lethality (89). Tnip1−/− mice that reach adulthood develop autoimmune disorders spontaneously (87, 90). ABIN3 is another TNF-inducible binding partner of A20 (18, 91). The significance of ABIN3-mediated negative regulation of TLR-NFκB signaling has yet to be established and the mechanism has yet to be elucidated.
Monocyte chemotactic protein [MCP]-induced protein 1 (MCPIP1; also known as Regnase-1a or ZC3H12A) is a TNF-, IL1β-, and IL4-inducible deubiquitinase that negatively regulates NFκB activity (92–94). In the absence of MCPIP1, TLR-induced IKK phosphorylation, and NFκB nuclear translocation are enhanced as a result of elevated TRAF6 ubiquitination (93). The biological importance of MCPIP1 is highlighted by the fact that Zc3h12a−/− mice develop lymphadenopathy, splenomegaly, growth retardation, and chronic autoimmunity and die prematurely (92, 93).
NFκB-IκB Module
IκB Synthesis
Regulation of IκBα synthesis via translational control of nfkbia mRNA, which encodes IκBα, can mediate cross-regulation of NFκB activity (Figure 5B). Type I IFNs, such as IFNβ, enhance TLR-NFκB signaling by repressing the translation of nfkbia (19). Further, stress responses to ultraviolet radiation (UV) and unfolded proteins (UPR) enhance NFκB activity through translation repression of nfkbia (95, 96). Translation of nfkbia is controlled by eukaryotic initiation factor (elF)2α and eIF4E [J. (97, 98)]. Translational repression of nfkbia by eIF2α depends on its phosphorylation by eIF2α kinases, such as PKR (interferon-induced, double-stranded RNA-activated protein kinase), PERK (pancreatic eIF2α kinase/RNA-dependent-protein-kinase-like endoplasmic-reticulum kinase), and GCN2 (general control non-derepressible-2) (96, 97, 99, 101). Whereas, PKR is activated by type I IFNs, GCN2, and PERK are activated by UV and UPR, respectively (100, 101).
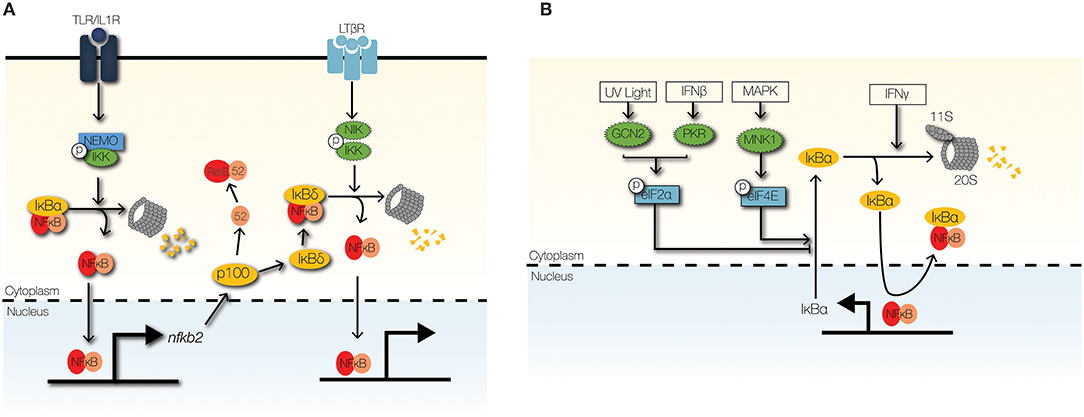
Figure 5. Signaling crosstalk at the IκB module. (A) The non-canonical NFκB signaling pathway can cross-regulate the canonical NFκB through NIK-IKK1-mediated degradation of IκBδ. High-molecular weight complexes of IκBδ trap RelA:p50 dimers in the cytoplasm to limit inflammatory NFκB activity. (B) Stimulus-responsive transcription initiation factors regulate the synthesis of IκBα. GCN2 and PKR phosphorylate eIF2α to inhibit IκBα synthesis in response to UV light and IFNβ, respectively. In contrast, phosphorylation of eIF4E by MNK1 stabilizes the IκBα mRNA. IFNγ promotes the proteolysis of IκBα/ε by upregulating the 11s cap of the immunoproteasome.
IFNγ may also inhibit nfkbia translation and enhance NFκB activity by inhibiting the phosphorylation and activation of eIF4E (102). eIF4E-dependent inhibition of IκBα is controlled by MAPK and mammalian target of rapamycin (mTOR) pathways (98, 102). Interestingly, translation inhibition of IκBα significantly upregulates IFNβ production in response to double-stranded RNA stimulation (98). This observation hints at the possibility of positive feedback regulation of NFκB activity by type I IFNs. Currently, detailed investigations to examine this positive feedback regulation are lacking.
IκB Degradation
Control of IκB degradation can mediate signaling crosstalk to NFκB (Figure 5B). IFNγ enhances NFκB activity by enhancing the degradation of free IκBα, which are unbound to NFκB dimers (19). Free IκBs have short half-lives (<10 min) and can be degraded independently of IKK activity and ubiquitination (99, 103); however, proteolysis of free IκBs is dependent on proteasomal degradation (99, 103). IFNγ enhances proteolysis of free IκBα by the immunoproteasome, which shares the 20S core of the 26S proteasome, but utilizes an 11S cap rather than a 19S cap (19, 104). IFNγ upregulates key components of the IκBα-associated 11S cap: PA28α and PA28β (19). Furthermore, pathological TNF signaling enhances NFκB activity by upregulating the degradation of IκBε by the immunoproteasome in a murine model of inflammatory bowel disease (105). TNF induces the expression PA28γ component of the immunoproteasome cap in colonic epithelial cells, which leads to severe colonic inflammation due to elevated NFκB activity (105).
NFκB Trapping
Cytoplasmic trapping of RelA:p50 dimers by high-molecular weight IκB complexes (IκBsomes) permits multiple layers of inflammatory regulation (106, 107). It provides a gateway for crosstalk through developmental signals and provides a history of recent inflammatory signaling (Figure 5A). Members of the TNF receptor superfamily that transduce developmental signals, such as B-cell activator factor and lymphotoxin-β (LTβ), induce degradation of IκBδ, which is induced in response to inflammatory stimuli such as TLR ligands (108, 109). Although it is induced less rapidly than IκBα, IκBδ possesses a longer half-life and may function as a late brake on NFκB activity (110). Since IκBδ levels are invariant to canonical IKK-degradation, IκBδ functions as regulator of available NFκB dimers that can be activated by inflammatory stimuli (108). Finally, in the absence of IκBδ, priming with TNF or IL1β enhances NFκB signaling rather than inhibiting NFκB signaling (110).
Concluding Remarks
Maintaining a delicate balance between effective host defense and deleterious inflammatory responses requires precise control of NFκB signaling (111). Multiple regulatory circuits have evolved to fine-tune NFκB-mediated inflammation through context-specific crosstalk (112). In this work, we have highlighted specific components of the NFκB signaling pathway for which crosstalk regulation is well-established. Despite decades of research, our current understanding of NFκB signaling remains insufficient to yield effective pharmacological targets (111, 113). Effective and specific pharmacological modulation of NFκB activity requires detailed, quantitative understanding of NFκB signaling dynamics (57). Furthermore, achieving cell-type and context-specific modulation of NFκB would be a panacea for many autoimmune and infectious diseases, as well as malignancies (112–114).
To dissect the dynamic regulation of NFκB signaling, quantitative approaches with single-cell resolution are required (115). By measuring the full distribution of signaling dynamics and gene expression in single cells, rather than simple averages, one can decipher cell-intrinsic properties from tissue-intrinsic properties (116–118). Such single-cell analyses may reveal strategies for targeting pathological cell populations with high specificity, which can mitigate adverse effects of pharmacological therapy (57, 113). Furthermore, with the aid of mathematical and computational modeling, one can conduct experiments in silico that may be prohibitive in vitro or ex vivo (57, 119, 120).
Finally, cross-regulatory pathways may fine-tune NFκB activity in a gene-specific manner. Many studies have identified the molecular components of gene-regulatory networks (GRNs) that control NFκB-dependent gene expression (15, 121). The regulatory mechanisms that define the topology of these GRNs include chromatin remodeling, transcription initiation and elongation, and post-transcriptional processing (15). They allow for combinatorial control by multiple factors and pathways, as well as cross-regulation (15). Further work will be required to delineate them in various physiological contexts.
Author Contributions
AA conducted the literature review, prepared figures, and wrote the manuscript. AH provided supervision, outlined the scope, and edited the manuscript.
Funding
This work was supported by Ruth L. Kirschstein National Research Service Award T32HL69766 and the Medical Scientist Training Program (NIH NIGMS training grant GM008042).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
For the purposes of clarity and brevity, we established a narrow focus for this review. As a result, we did not cite some pertinent contributions. We acknowledge Fay Lin for critical reading of the review.
References
1. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFκB system. Wiley Interdiscip Rev Syst Biol Med. (2016) 8:227–41. doi: 10.1002/wsbm.1331
2. Leifer CA, Medvedev AE. Molecular mechanisms of regulation of toll-like receptor signaling. J Leukocyte Biol. (2016) 100:1–15. doi: 10.1189/jlb.2MR0316-117RR
3. Pandey S, Kawai T, Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harbor Perspect Biol. (2015) 7:a016246. doi: 10.1101/cshperspect.a016246
4. Hayden MS, Ghosh S. Regulation of NF-KB by TNF family cytokines. Semin Immunol. (2014) 26:253–66. doi: 10.1016/j.smim.2014.05.004
5. Zabel U, Henkel T, Silva MS, Baeuerle PA. Nuclear uptake control of NF-κB by MAD-3, an IκB protein present in the nucleus. EMBO J. (1993) 12:201–11. doi: 10.2298/JSC170204032M
6. Ganchi PA, Sun SC, Greene WC, Ballard DW. IκB/MAD-3 masks the nuclear localization signal of NF-κB P65 and requires the transactivation domain to inhibit NF-κB P65 DNA binding. Mol Biol Cell. (1992) 3:1339–52. doi: 10.1091/mbc.3.12.1339
7. Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, Baldwin AS. IκB interacts with the nuclear localization sequences of the subunits of NF-κB: a mechanism for cytoplasmic retention. Genes Dev. (1992) 6:1899–913. doi: 10.1101/gad.6.10.1899
8. Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, et al. Signal-induced site-specific phosphorylation targets IκB α to the ubiquitin-proteasome pathway. Genes Dev. (1995) 9:1586–97. doi: 10.1101/gad.9.13.1586
9. Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent IκB α phosphorylation marks the NF-κB inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Nat Acad Sci USA. (1995) 92:10599–603. doi: 10.1073/pnas.92.23.10599
10. Le Bail O, Schmidt-Ullrich R, Israël A. Promoter analysis of the gene encoding the IκB-α/MAD3 inhibitor of NF-κB: positive regulation by members of the Rel/NF-κB family. EMBO J. (1993) 12:5043–9. doi: 10.1002/j.1460-2075.1993.tb06197.x
11. Chiao PJ, Miyamoto S, Verma IM. Autoregulation of IκB alpha activity. Proc Nat Acad Sci USA. (1994) 91:28–32. doi: 10.1073/pnas.91.1.28
12. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science. (2002) 298:1241–5. doi: 10.1126/science.1071914
13. Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. (2005) 309:1857–61. doi: 10.1126/science.1113319
14. Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. (2012) 12:215–25. doi: 10.1038/nri3167
15. Cheng CS, Behar MS, Suryawanshi GW, Feldman KE, Spreafico R, Hoffmann A. Iterative modeling reveals evidence of sequential transcriptional control mechanisms. Cell Systems. (2017) 4:330–43.e5. doi: 10.1016/j.cels.2017.01.012
16. Rowland MA, Greenbaum JM, Deeds EJ. Crosstalk and the evolvability of intracellular communication. Nat Commun. (2017) 8:1–8. doi: 10.1038/ncomms16009
17. Rowland MA, Fontana W, Deeds EJ. Crosstalk and competition in signaling networks. Biophys J. (2012) 103:2389–98. doi: 10.1016/j.bpj.2012.10.006
18. Park SH, Kang K, Giannopoulou E, Qiao Y, Kang K, Kim G, et al. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat Immunol. (2017) 18:1104–16. doi: 10.1038/ni.3818
19. Mitchell S, Mercado EL, Adelaja A, Ho JQ, Cheng QJ, Ghosh G, et al. An NFκB activity calculator to delineate signaling crosstalk: type I and II interferons enhance NFκB via distinct mechanisms. Front Immunol. 10:1425. doi: 10.3389/fimmu.2019.01425
20. Qiao Y, Giannopoulou EG, Chan CH, Park SH, Gong S, Chen J, et al. Synergistic activation of inflammatory cytokine genes by interferon-γ-induced chromatin remodeling and toll-like receptor signaling. Immunity. (2013) 39:454–69. doi: 10.1016/j.immuni.2013.08.009
21. Cheshire JL, Baldwin AS. Synergistic activation of NF-κB by tumor necrosis factor α and gamma interferon via enhanced IκB α degradation and de novo IκBbeta degradation. Mol Cell Biol. (1997) 17:6746–54. doi: 10.1128/MCB.17.11.6746
22. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
23. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
24. Jiménez-Dalmaroni MJ, Gerswhin ME, Adamopoulos IE. The critical role of toll-like receptors—from microbial recognition to autoimmunity: a comprehensive review. Autoimmun Rev. (2015) 15:1–8. doi: 10.1016/j.autrev.2015.08.009
25. Piccolo V, Curina A, Genua M, Ghisletti S, Simonatto M, Sabò A, et al. Opposing macrophage polarization programs show extensive epigenomic and transcriptional cross-talk. Nat Immunol. (2017) 18:530–40. doi: 10.1038/ni.3710
26. Mita Y, Dobashi K, Shimizu Y, Nakazawa T, Mori M. Toll-like receptor 2 and 4 surface expressions on human monocytes are modulated by interferon-γ and macrophage colony-stimulating factor. Immunol Lett. (2001) 78:97–101. doi: 10.1016/s0165-2478(01)00241-3
27. Kajita AI, Morizane S, Takiguchi T, Yamamoto T, Yamada M, Iwatsuki K. Interferon-gamma enhances TLR3 expression and anti-viral activity in keratinocytes. J Invest Dermatol. (2015) 135:2005–11. doi: 10.1038/jid.2015.125
28. Schroder K, Lichtinger M, Irvine KM, Brion K, Trieu A, Ross IL, et al. PU.1 and ICSBP control constitutive and IFN-gamma-regulated Tlr9 gene expression in mouse macrophages. J Leukoc Biol. (2007) 81:1577–90. doi: 10.1189/jlb.0107036
29. Bosisio D, Polentarutti N, Sironi M, Bernasconi S, Miyake K, Webb GR, et al. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-γ: a molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood. (2002) 99:3427–31. doi: 10.1182/blood.V99.9.3427
30. Ahmad-Nejad P, Häcker H, Rutz M, Bauer S, Vabulas RM, Wagner H. Bacterial CpG-DNA and lipopolysaccharides activate toll-like receptors at distinct cellular compartments. Eur J Immunol. (2002) 32:1958–68. doi: 10.1002/1521-4141(200207)32:7<1958::AID-IMMU1958>3.0.CO;2-U
31. Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, et al. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. (2009) 138:114–28. doi: 10.1016/j.cell.2009.04.020
32. Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. (2006) 177:1272–81. doi: 10.4049/jimmunol.177.2.1272
33. Jana M, Palencia CA, Pahan K. Fibrillar amyloid-peptides activate microglia via TLR2: implications for Alzheimer's disease. J Immunol. (2008) 181:7254–62. doi: 10.4049/jimmunol.181.10.7254
34. Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. J Biol Chem. (2002) 277:15107–12. doi: 10.1074/jbc.M111204200
35. Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Häcker H, et al. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. (2001) 276:31332–9. doi: 10.1074/jbc.M103217200
36. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. (2004) 279:7370–7. doi: 10.1074/jbc.M306793200
37. Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. (2007) 8:487–96. doi: 10.1038/ni1457
38. Tamai R, Sugawara S, Takeuchi O, Akira S, Takada H. Synergistic effects of lipopolysaccharide and interferon-γ in inducing interleukin-8 production in human monocytic THP-1 cells is accompanied by up-regulation of CD14, toll-like receptor 4, MD-2 and MyD88 expression. J Endotoxin Res. (2003) 9:145–53. doi: 10.1179/096805103125001540
39. Mochizuki S, Kobayashi M, Suzuki T, Oikawa A, Koseki T, Nishihara T, et al. γ-interferon enhances expression of CD14/MyD88 and subsequent responsiveness to lipopolysaccharide from actinobacillus actinomycetemcomitans in human gingival fibroblasts. J Periodontal Res. (2004) 39:333–43. doi: 10.1111/j.1600-0765.2004.00749.x
40. Zanoni I, Ostuni R, Marek LR, Barresi S, Barbalat R, Barton GM, et al. CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell. (2011) 147:868–80. doi: 10.1016/j.cell.2011.09.051
41. Nagai Y, Akashi S, Nagafuku M, Ogata M, Iwakura Y, Akira S, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. (2002) 3:667–72. doi: 10.1038/ni809
42. Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem Biophys Res Commun. (2008) 368:94–9. doi: 10.1016/j.bbrc.2008.01.061
43. Meng J, Gong M, Björkbacka H, Golenbock DT. Genome-wide expression profiling and mutagenesis studies reveal that lipopolysaccharide responsiveness appears to be absolutely dependent on TLR4 and MD-2 expression and is dependent upon intermolecular ionic interactions. J Immunol. (2011) 187:3683–93. doi: 10.4049/jimmunol.1101397
44. Husebye H, Halaas Ø, Stenmark H, Tunheim G, Sandanger Ø, Bogen B, et al. Endocytic pathways regulate toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. (2006) 25:683–92. doi: 10.1038/sj.emboj.7600991
45. Baumann CL, Aspalter IM, Sharif O, Pichlmair A, Blüml S, Grebien F, et al. CD14 is a coreceptor of toll-like receptors 7 and 9. J Exp Med. (2010) 207:2689–701. doi: 10.1084/jem.20101111
46. Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity. (2006) 24:153–63. doi: 10.1016/j.immuni.2005.12.012
47. Nakata T, Yasuda M, Fujita M, Kataoka H, Kiura K, Sano H, et al. CD14 directly binds to triacylated lipopeptides and facilitates recognition of the lipopeptides by the receptor complex of toll-like receptors 2 and 1 without binding to the complex. Cell Microbiol. (2006) 8:1899–909. doi: 10.1111/j.1462-5822.2006.00756.x
48. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
49. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
50. Schreiber G. Molecular mechanisms governing differential type I interferons signaling. J Biol Chem. (2017) 292:7285–94. doi: 10.1074/jbc.R116.774562
51. Nakagawa R, Naka T, Tsutsui H, Fujimoto M, Kimura A, Abe T, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. (2002) 17:677–87. doi: 10.1016/S1074-7613(02)00449-1
52. Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, et al. Suppressor of cytokine signaling 1 negatively regulates toll-like receptor signaling by mediating mal degradation. Nat Immunol. (2006) 7:148–55. doi: 10.1038/ni1299
53. Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, et al. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. (1999) 98:597–608. doi: 10.1016/S0092-8674(00)80047-1
54. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
55. Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, et al. MyD88 is an adaptor protein in the HToll/IL-1 receptor family signaling pathways. Mol Cell. (1998) 2:253–8.
56. Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, et al. Smad6-specific recruitment of smurf E3 ligases mediates TGF-B1-induced degradation of MyD88 in TLR4 signalling. Nat Commun. (2011) 2:460. doi: 10.1038/ncomms1469
57. Behar M, Barken D, Werner SL, Hoffmann A. The dynamics of signaling as a pharmacological target. Cell. (2013) 155:448–61. doi: 10.1016/j.cell.2013.09.018
58. Werner SL, Kearns JD, Zadorozhnaya V, Lynch C, O'Dea E, Boldin MP, et al. Encoding NF-κB temporal control in response to TNF: distinct roles for the negative regulators IκBα and A20. Genes Dev. (2008) 22:2093–101. doi: 10.1101/gad.1680708
59. Behar M, Hoffmann A. Tunable signal processing through a kinase control cycle: the IKK signaling node. Biophys J. (2013) 105:231–41. doi: 10.1016/j.bpj.2013.05.013
60. Motshwene PG, Moncrieffe MC, Grossmann JG, Kao C, Ayaluru M, Sandercock AM, et al. An oligomeric signaling platform formed by the toll-like receptor signal transducers MyD88 and IRAK-4. J Biol Chem. (2009) 284:25404–11. doi: 10.1074/jbc.M109.022392
61. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. (2010) 465:885–90. doi: 10.1038/nature09121
62. Adib-Conquy M, Cavaillon JM. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J Biol Chem. (2002) 277:27927–34. doi: 10.1074/jbc.M200705200
63. Park SH, Park-Min KH, Chen J, Hu X, Ivashkiv LB. Tumor necrosis factor induces GSK3 kinase-mediated cross-tolerance to endotoxin in macrophages. Nat Immunol. (2011) 12:607–15. doi: 10.1038/ni.2043
64. Kobayashi K, Hernandez LD, Galán JE, Janeway CA, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of toll-like receptor signaling. Cell. (2002) 110:191–202. doi: 10.1016/S0092-8674(02)00827-9
65. Latty SL, Sakai J, Hopkins L, Verstak B, Paramo T, Berglund NA, et al. Activation of toll-like receptors nucleates assembly of the MyDDosome signaling hub. ELife. (2018) 7:1–15. doi: 10.7554/eLife.31377
66. Ermolaeva MA, Michallet MC, Papadopoulou N, Utermöhlen O, Kranidioti K, Kollias G, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. (2008) 9:1037–46. doi: 10.1038/ni.1638
67. Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of toll-like receptor 3-induced NFκB activation. Nat Immunol. (2004) 5:503–7. doi: 10.1038/ni1061
68. Chang M, Jin W, Sun SC. Peli1 facilitates TRIF-dependent toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol. (2009) 10:1089–95. doi: 10.1038/ni.1777
69. Enesa K, Ordureau A, Smith H, Barford D, Cheung PC, Patterson-Kane J, et al. Pellino1 is required for interferon production by viral double-stranded RNA. J Biol Chem. (2012) 287:34825–35. doi: 10.1074/jbc.M112.367557
70. Cheng Z, Taylor B, Ourthiague DR, Hoffmann A. Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci Signal. (2015) 8:ra69. doi: 10.1126/scisignal.aaa5208
71. Gohda J, Matsumura T, Inoue J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not Toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signaling. J Immunol. (2004) 173:2913–7. doi: 10.4049/jimmunol.173.5.2913
72. Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, et al. Specificity in toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature. (2006) 439:204–7. doi: 10.1038/nature04369
73. Deng L, Wang C, Spencer E, Yang L, Braun A, You J, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. (2000) 103:351–61. doi: 10.1016/S0092-8674(00)00126-4
74. Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. (2003) 424:801–5. doi: 10.1038/nature01802
75. Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kB signalling. Nature. (2004) 430:694–9. doi: 10.1038/nature02794
76. Song HY, Rothe M, Goeddel DV. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-κB activation. Proc Nat Acad Sci USA. (1996) 93:6721–5.
77. Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, et al. The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat Immunol. (2004) 5:1052–60. doi: 10.1038/ni1110
78. Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J Exp Med. (2008) 205:451–64. doi: 10.1084/jem.20071108
79. Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science. (2000) 289:2350–4. doi: 10.1126/science.289.5488.2350
80. Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-κB target genes. Proc Nat Acad Sci USA. (2007) 104:18601–6. doi: 10.1073/pnas.0709011104
81. Matmati M, Jacques P, Maelfait J, Verheugen E, Kool M, Sze M, et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet. (2011) 43:908–12. doi: 10.1038/ng.874
82. Walle LV, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. A20 protects against arthritis. Nature. (2014) 512:69–73. doi: 10.1038/nature13322
83. Shembade N, Ma A, Harhaj EW. Inhibition of NF-B signaling by A20 through disruption of ubiquitin enzyme complexes. Science. (2010) 327:1135–9. doi: 10.1126/science.1182364
84. Mauro C, Pacifico F, Lavorgna A, Mellone S, Iannetti A, Acquaviva R, et al. ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J Biol Chem. (2006) 281:18482–8. doi: 10.1074/jbc.M601502200
85. Tian B, Nowak DE, Brasier AR. A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics. (2005) 6:137. doi: 10.1186/1471-2164-6-137
86. Adamson A, Boddington C, Downton P, Rowe W, Bagnall J, Lam C, et al. Signal transduction controls heterogeneous NF-κB dynamics and target gene expression through cytokine-specific refractory states. Nat Commun. (2016) 7:12057. doi: 10.1038/ncomms12057
87. Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, et al. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med. (2011) 208:1215–28. doi: 10.1084/jem.20102177
88. Ma A, Malynn BA. A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol. (2012) 12:774–85. doi: 10.1038/nri3313
89. Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. (2009) 457:906–9. doi: 10.1038/nature07575
90. Zhou J, Wu R, High AA, Slaughter CA, Finkelstein D, Rehg JE. A20-binding inhibitor of NF-κB (ABIN1) controls toll-like receptor-mediated CCAAT/enhancer-binding protein β activation and protects from inflammatory disease. Proc Nat Acad Sci USA. (2011) 108:E998–1006. doi: 10.1073/pnas.1106232108
91. Wullaert A, Verstrepen L, Van Huffel S, Adib-Conquy M, Cornelis S, Kreike M, et al. LIND/ABIN-3 is a novel lipopolysaccharide-inducible inhibitor of NF-κB activation. J Biol Chem. (2007) 282:81–90. doi: 10.1074/jbc.M607481200
92. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating MRNA decay. Nature. (2009) 458:1185–90. doi: 10.1038/nature07924
93. Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J Exp Med. (2010) 207:2959–73. doi: 10.1084/jem.20092641
94. Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattukudy PE, et al. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. (2008) 283:6337–46. doi: 10.1074/jbc.M707861200
95. O'Dea EL, Kearns JD, Hoffmann A. UV as an amplifier rather than inducer of NF-κB activity. Mol Cell. (2008) 30:632–41. doi: 10.1016/j.molcel.2008.03.017
96. Tam AB, Mercado EL, Hoffmann A, Niwa M. ER stress activates NF-κB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE. (2012) 7:e45078. doi: 10.1371/journal.pone.0045078
97. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, et al. Translational repression mediates activation of nuclear factor κB by phosphorylated translation initiation factor 2. Mol Cell Biol. (2004) 24:10161–8. doi: 10.1128/MCB.24.23.10161
98. Herdy B, Jaramillo M, Svitkin YV, Rosenfeld AB, Kobayashi M, Walsh D, et al. Translational control of the activation of transcription factor NF-κB and production of type I interferon by phosphorylation of the translation factor EIF4E. Nat Immunol. (2012) 13:543–50. doi: 10.1038/ni.2291
99. Mathes E, O'Dea EL, Hoffmann A, Ghosh G. NF-κB dictates the degradation pathway of IκBα. EMBO J. (2008) 27:1357–67. doi: 10.1038/emboj.2008.73
100. de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukocyte Biol. (2001) 69:912–20. doi: 10.1016/j.coviro.2011.10.008
101. Jiang H-Y, Wek RC. GCN2 Phosphorylation of EIF2α activates NF-κB in response to UV irradiation. Biochem J. (2005) 385 (Pt 2):371–80. doi: 10.1042/BJ20041164
102. Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, et al. Interferon-γ regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. (2015) 16:838–49. doi: 10.1038/ni.3205
103. O'Dea EL, Barken D, Peralta RQ, Tran KT, Werner SL, Kearns JD, et al. A homeostatic model of IκB metabolism to control constitutive NF-κB activity. Mol Systems Biol. (2007) 3:111. doi: 10.1038/msb4100148
104. Fortmann KT, Lewis RD, Ngo KA, Fagerlund R, Hoffmann A. A regulated, ubiquitin-independent degron in IκBα. J Mol Biol. (2015) 427:2748–56. doi: 10.1016/j.jmb.2015.07.008
105. Xu J, Zhou L, Ji L, Chen F, Fortmann K, Zhang K, et al. The REGγ-proteasome forms a regulatory circuit with IκBε and NFκB in experimental colitis. Nat Commun. (2016) 7:10761. doi: 10.1038/ncomms10761
106. Savinova OV, Hoffmann A, Ghosh G. The Nfκb1 and Nfκb2 proteins P105 and P100 function as the core of high-molecular-weight heterogeneous complexes. Mol Cell. (2009) 34:591–602. doi: 10.1016/j.molcel.2009.04.033
107. Tao Z, Fusco A, Huang DB, Gupta K, Young Kim D, Ware CF, et al. P100/IκBδ sequesters and inhibits NF-κB through κB some formation. Proc Nat Acad Sci USA. (2014) 111:15946–51. doi: 10.1073/pnas.1408552111
108. Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, et al. A fourth IκB protein within the NF-κB signaling module. Cell. (2007) 128:369–81. doi: 10.1016/j.cell.2006.12.033
109. Almaden JV, Tsui R, Liu YC, Birnbaum H, Shokhirev MN, Ngo KA, et al. A pathway switch directs BAFF signaling to distinct NFκB transcription factors in maturing and proliferating B cells. Cell Rep. (2014) 9:2098–111. doi: 10.1016/j.celrep.2014.11.024
110. Shih VF, Kearns JD, Basak S, Savinova OV, Ghosh G, Hoffmann A. Kinetic control of negative feedback regulators of NF-κB/RelA determines their pathogen- and cytokine-receptor signaling specificity. Proc Nat Acad Sci USA. (2009) 106:9619–24. doi: 10.1073/pnas.0812367106
111. Herrington FD, Carmody RJ, Goodyear CS. Modulation of NF-κB signaling as a therapeutic target in autoimmunity. J Biomol Screen. (2016) 21:223–42. doi: 10.1177/1087057115617456
112. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. (2018) 18:309–24. doi: 10.1038/nri.2017.142
113. Begalli F, Bennett J, Capece D, Verzella D, D'Andrea D, Tornatore L, et al. Unlocking the NF-κB conundrum: embracing complexity to achieve specificity. Biomedicines. (2017) 5:50. doi: 10.3390/biomedicines5030050
114. de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Ann Rev Immunol. (2015) 33:823–74. doi: 10.1146/annurev-immunol-032414-112227
115. Junkin M, Kaestli AJ, Cheng Z, Jordi C, Albayrak C, Hoffmann A, et al. High-content quantification of single-cell immune dynamics. Cell Rep. (2016) 15:411–22. doi: 10.1016/j.celrep.2016.03.033
116. Paszek P, Ryan S, Ashall L, Sillitoe K, Harper CV, Spiller DG, et al. Population robustness arising from cellular heterogeneity. Proc Nat Acad Sci. (2010) 107:11644–9. doi: 10.1073/pnas.0913798107
117. Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single-cell NF-κB dynamics reveal digital activation and analogue information processing. Nature. (2010) 466:267–71. doi: 10.1038/nature09145
118. Lane K, Van Valen D, DeFelice MM, Macklin DN, Kudo T, Jaimovich A, et al. Measuring signaling and RNA-seq in the same cell links gene expression to dynamic patterns of NF-κB activation. Cell Systems. (2017) 4:458–69.e5. doi: 10.1016/j.cels.2017.03.010
119. Brodland GW. How computational models can help unlock biological systems. Semin Cell Dev Biol. (2015) 47–48:62–73. doi: 10.1016/j.semcdb.2015.07.001
120. Zhang ZB, Wang QY, Ke YX, Liu SY, Ju JQ, Lim WA, et al. Design of tunable oscillatory dynamics in a synthetic NF-κB signaling circuit. Cell Systems. (2017) 5:460–70.e5. doi: 10.1016/j.cels.2017.09.016
Keywords: NFκB, PAMPs (pathogen-associated molecular patterns), interferon-beta (IFNβ), signaling crosstalk, immunoproteasome, TRIF, A20 (TNFAIP3), IκBs
Citation: Adelaja A and Hoffmann A (2019) Signaling Crosstalk Mechanisms That May Fine-Tune Pathogen-Responsive NFκB. Front. Immunol. 10:433. doi: 10.3389/fimmu.2019.00433
Received: 15 September 2018; Accepted: 19 February 2019;
Published: 02 July 2019.
Edited by:
Myong-Hee Sung, National Institutes of Health (NIH), United StatesReviewed by:
Iain Fraser, National Institutes of Health (NIH), United StatesKoichi S. Kobayashi, Texas A&M University Baylor College of Dentistry, United States
Kathryn Miller-Jensen, Yale University, United States
Copyright © 2019 Adelaja and Hoffmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander Hoffmann, YWhvZmZtYW5uQHVjbGEuZWR1