 Duaa O. Khair1
Duaa O. Khair1 Heather J. Bax1,2Silvia Mele1
Heather J. Bax1,2Silvia Mele1 Silvia Crescioli1Giulia Pellizzari1Atousa Khiabany1Mano Nakamura1Robert J. HarrisElise French1Ricarda M. Hoffmann1,2Iwan P. Williams1
Silvia Crescioli1Giulia Pellizzari1Atousa Khiabany1Mano Nakamura1Robert J. HarrisElise French1Ricarda M. Hoffmann1,2Iwan P. Williams1 Anthony Cheung1,3
Anthony Cheung1,3 Benjamin Thair1
Benjamin Thair1 Charlie T. Beales1
Charlie T. Beales1 Emma Touizer1Adrian W. Signell1
Emma Touizer1Adrian W. Signell1 Nahrin L. Tasnova1James F. Spicer2
Nahrin L. Tasnova1James F. Spicer2 Debra H. Josephs1,2Jenny L. Geh4Alastair MacKenzie Ross4Ciaran Healy4Sophie Papa2
Debra H. Josephs1,2Jenny L. Geh4Alastair MacKenzie Ross4Ciaran Healy4Sophie Papa2 Katie E. Lacy1
Katie E. Lacy1 Sophia N. Karagiannis1*
Sophia N. Karagiannis1*- 1St. John's Institute of Dermatology, School of Basic & Medical Biosciences, Guy's Hospital, King's College London, London, United Kingdom
- 2School of Cancer & Pharmaceutical Sciences, Guy's Hospital, King's College London, London, United Kingdom
- 3Breast Cancer Now Research Unit, School of Cancer & Pharmaceutical Sciences, Guy's Cancer Centre, King's College London, London, United Kingdom
- 4Department of Plastic Surgery at Guy's, King's, and St. Thomas' Hospitals, London, United Kingdom
The immune system employs several checkpoint pathways to regulate responses, maintain homeostasis and prevent self-reactivity and autoimmunity. Tumor cells can hijack these protective mechanisms to enable immune escape, cancer survival and proliferation. Blocking antibodies, designed to interfere with checkpoint molecules CTLA-4 and PD-1/PD-L1 and counteract these immune suppressive mechanisms, have shown significant success in promoting immune responses against cancer and can result in tumor regression in many patients. While inhibitors to CTLA-4 and the PD-1/PD-L1 axis are well-established for the clinical management of melanoma, many patients do not respond or develop resistance to these interventions. Concerted efforts have focused on combinations of approved therapies aiming to further augment positive outcomes and survival. While CTLA-4 and PD-1 are the most-extensively researched targets, results from pre-clinical studies and clinical trials indicate that novel agents, specific for checkpoints such as A2AR, LAG-3, IDO and others, may further contribute to the improvement of patient outcomes, most likely in combinations with anti-CTLA-4 or anti-PD-1 blockade. This review discusses the rationale for, and results to date of, the development of inhibitory immune checkpoint blockade combination therapies in melanoma. The clinical potential of new pipeline therapeutics, and possible future therapy design and directions that hold promise to significantly improve clinical prognosis compared with monotherapy, are discussed.
Introduction
Immune-mediated destruction of tumors has long been considered a potential route of therapeutic intervention. Partial spontaneous regression of melanoma lesions has previously been associated with the presence of endogenous tumor infiltrating lymphocytes (TILs) and the presence of TILs in patient samples has been shown to correlate with improved clinical outcomes and better prognosis (1). Infusion with CD8+ TILs has been reported to induce some responses in patients when combined with other treatments including IL-2 (2). Immunotherapy via cytokine infusion has also been extensively trialed, with IL-2, IL-12, and IFNα2b to activate T cells, showing anti-tumor effects in pre-clinical models and clinical trials, with IL-2 and IFNα2b approved for clinical use (3, 4). Cytokine treatments have however been associated with severe adverse effects resembling severe systemic infections and sometimes resulting in toxic shock or capillary leak syndrome as reported in randomized clinical trials (5, 6). Though not without challenges, these trials confirmed the possibility of reigniting components of the immune system as a cancer therapy.
Increased understanding of tumor evolution and the complex interactions in the tumor microenvironment (TME) has revealed numerous mechanisms by which tumors may escape immune destruction and actively suppress immune activity (7). Immunosuppression by tumor cells may partially be mediated through FoxP3+ regulatory T cell (T-reg) recruitment via tumor-secreted chemokines as shown in an ex vivo study (8, 9). Critically, tumor resident T-reg can highly express cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), an important checkpoint that acts as a negative regulator of effector T cell (T-eff) activity in vivo, studied in different models including CTLA-4-deficient mice (10) (Figure 1). Suppression may also be mediated by tumor expression of the Programmed-death ligand 1 (PD-L1; B7-H1; CD274), known to trigger T cell apoptosis in vivo in mouse tumors (11) and to promote formation of FoxP3+ T-regs upon interaction with the T cell-associated checkpoint receptor Programmed-death 1 (PD-1, also known as CD279) (12) (Figure 1). These checkpoints, have become therapeutic targets in immune checkpoint blockade therapy, with the aim of overcoming TME-mediated immunosuppression and restoring anti-tumor immune activity (13). Monoclonal antibodies targeting CTLA-4 and PD-1 have now been approved for the treatment of melanoma. These new therapeutic modalities were developed in parallel with targeted MAPK pathway inhibitor therapies, such as vemurafenib and dabrafenib, approved for a subset of melanomas bearing point mutations in the kinase BRAF (e.g., BRAFV600E), and the MEK inhibitors trametinib and cobimetinib, all designed to cause cancer cell death via interruption of the MAPK pathway (Table 1). Together, these agents have led to an increase in medial survival for advanced melanoma from 9 months in 2010 to over 3.5 years.
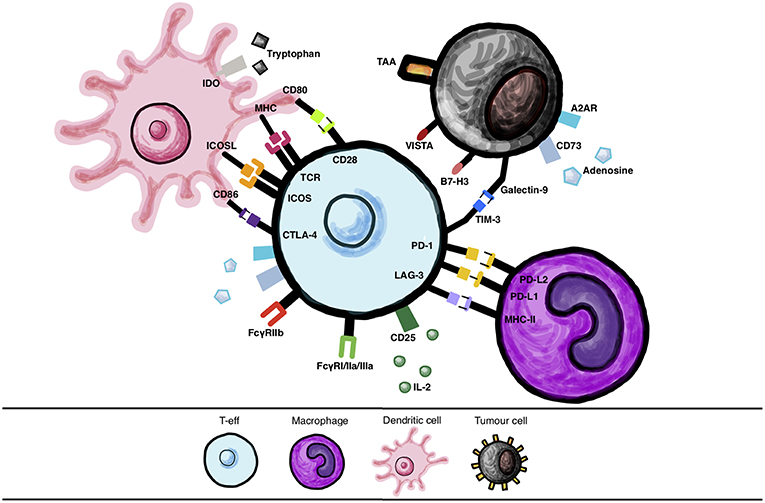
Figure 1. Immune cell interactions via checkpoint molecules and their ligands. Various interactions between checkpoint molecules and their ligands expressed by different cells, such as immune cells (dendritic cells (DC)s, T-effector cells (T-eff), macrophages) and between T-eff and tumor cells, that may be targeted with therapy.
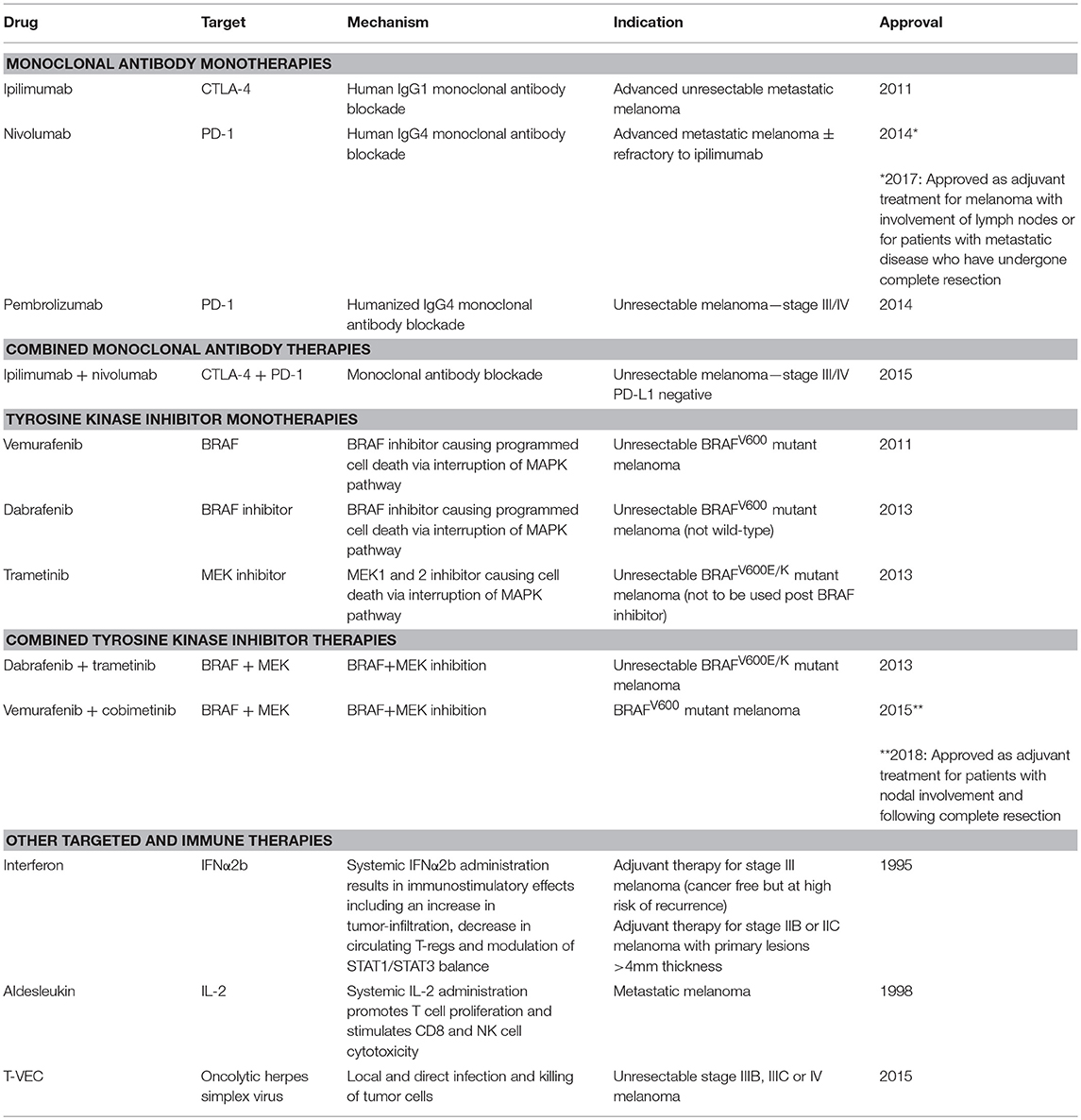
Table 1. Approved targeted, antibody and other immunotherapies and combination treatments for malignant melanoma.
While CTLA-4 and PD-1 blockade has proved successful in improving survival rates, many patients do not respond or develop resistance to these interventions. Alongside combinations of checkpoint inhibitors already in clinical use, research into new checkpoints as therapeutic targets has shown promise in pre-clinical and clinical studies, either alone or combined with established agents. Focusing on malignant melanoma as the tumor type for which the first pivotal immunotherapy breakthroughs were demonstrated, in this review, we discuss current and future checkpoint blockade and other immunooncology combination therapies, and the rationale for potential synergistic effects (Table 2).
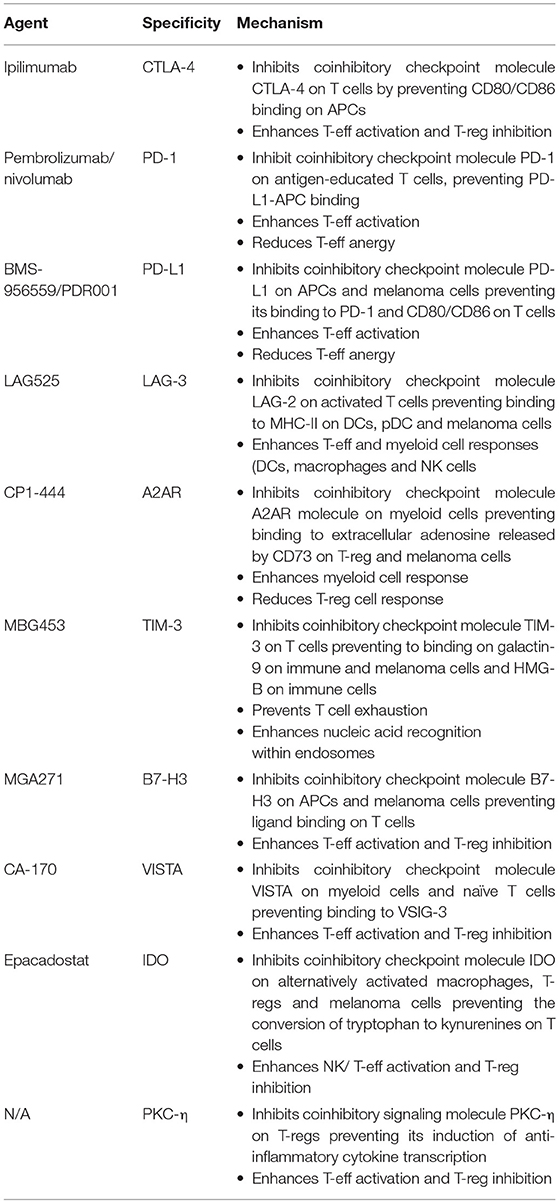
Table 2. Proposed mechanisms of action of selected immune checkpoint blocking agents.
Therapies Targeting CTLA-4 and PD-1
Anti-CTLA-4 Monotherapy
CTLA-4 is a CD28 homolog expressed constitutively on the surface of both T-reg cells and activated T cells (14). CTLA-4 binds to CD28 co-receptors CD80/60 with a higher affinity and avidity than CD28, thus superseding positive CD28 signaling and thus allowing for inhibition of T cell activation (15, 16). In order to function effectively as an immune checkpoint via endocytosis CTLA-4 is not only able to competitively inhibit T cell co-stimulation but can also clear CD28 ligands CD80/CD86 from the surrounding cells including APCs by trans-endocytosis in vivo (17). Physiologically, CTLA-4 has been shown in vitro and in mouse models in vivo, to suppress T cell responses including activation, proliferation, and pro-inflammatory cytokine production (IFN-γ and IL-2) by antigen-presenting cells (APCs) such as dendritic cells (DCs) and macrophages (18) (Figure 1).
Studies on cells expressing human CTLA-4 in murine models of melanoma, that investigated antibodies aimed at blocking CTLA-4 checkpoints, have documented effects such as enhanced T-eff function, inhibition of T-reg activity and selective depletion of T-reg cells via antibody Fc binding of Fcγ-receptors on atypical macrophages in tumor lesions (19, 20). Hypotheses that CTLA-4 blockade could enhance anti-tumor response were tested by a pre- clinical study using transplantable murine melanoma cell lines, demonstrating that CTLA-4 inhibitors induced rejection of melanoma (21). Ex vivo studies of peripheral blood mononuclear cells (PBMCs) and matched melanoma metastases from patients with melanoma treated with ipilimumab have shown evidence that ipilimumab also works by depleting T-reg cell populations by antibody-dependent cell-mediated cytotoxicity (ADCC) mediated by CD16 (FcγRIIIA)-expressing, nonclassical monocytes. In the same study, patients who responded to ipilimumab treatment had higher ratios of intratumoral CD68-expressing vs. CD163-expressing macrophages before treatment and lower T-reg infiltration after treatment (22). Clinical trials involving ipilimumab have demonstrated a dose-dependent response to the antibody in late-stage melanoma patients, with pooled analysis consistently showing improved survival in patients with metastatic disease above historical controls (23, 24). By blocking this key immune escape mechanism, overall survival rates for ipilimumab were significantly improved, alone or in combination with a glycoprotein 100 peptide (GP-100) vaccine when compared to vaccine alone (15, 25). Ipilimumab, a fully humanized IgG1 antibody, was the first anti-CTLA-4 treatment approved by FDA in 2011 (Table 1).
Anti-PD-1 Monotherapy
Another immune checkpoint, the programmed death 1 (PD-1) immunoglobulin-based receptor predominantly expressed on activated, antigen-educated T cells can recognize two ligands, PD-L1 and PD–L2 (B7-DC; CD273). PD-L1 is expressed broadly across many cell types, including leukocytes and tissue cells, whereas PD-L2 expression is limited and specific to expression on immune cells: antigen presenting and stromal cells. Ligation of PD-1 to PD-L1 causes phosphorylation and activation of SHP-2, a phosphatase that can inactivate many downstream molecules in TCR signaling (26). In vitro and in vivo studies in mouse models of cancer showed that PD-L1 can also enhance the generation of peripherally induced T-regs, (iT-reg), increasing Foxp3 expression and sustaining their immunoregulatory actions such as suppression of CD4+ T-eff cells (27). The co-stimulatory molecule CD28 of which CTLA-4 is a homolog, is also preferentially targeted by PD-1-mediated dephosphorylation (28). By this mechanism, PD-1 mediates two immune checkpoints, by reducing immune hyperstimulation via PD-L1 and maintaining tolerance in lymphoid tissues via PD-L2. Both ligands PD-L1 and PD-L2 can also be induced by cytokine signaling during inflammation (29).
PD-L1 expression on tumor cells is often upregulated, resulting in inhibition of T cell responses (15). In melanoma, the expression of PD-L1 may be prognostic, and could correlate with Breslow thickness (30). Mouse melanoma metastasis to the liver was shown to be impaired in PD-1-deficient mice and anti-PD-1 monoclonal antibody administration could inhibit the spread of tumor cells via recruitment of T-eff (31) by blocking the interaction of PD-1 with its ligands (14). Anti-PD-1 and anti-PD-L1 blocking strategies produce different immunologic effects as anti-PD-L1 has effects on more than one pathway. PD-L1 signals negatively to T cells by interacting with both CD80/CD86 and PD-1 (32) preventing both pathways, without interacting with PD-L2, which activates T cell response by producing co-stimulatory signals (32). Anti-PD-L1 studies demonstrated temporary arrest of the growth of melanoma cells in mouse models (33). Following the approval of ipilimumab, the anti-PD-1 monoclonal antibodies nivolumab and pembrolizumab gained FDA approval in 2014 (34) and EMA approval in 2015, following trials showing significantly improved patient outcomes (Table 1) (35). Nivolumab is a fully human IgG4 monoclonal antibody which was shown to improve median overall survival to 8.9 months compared to 6.8 months in patients treated with dacarbazine in a phase III study involving patients with previously untreated melanoma (36). Studies into metastatic melanoma have shown superior overall survival of 1 year (72.9 to 42.1%) and better objective response rate (40 to 13.9%) with nivolumab plus dacarbazine compared to dacarbazine plus placebo (36). Pembrolizumab, a humanized IgG4 monoclonal antibody has also shown similar efficacy to nivolumab, with one phase III study reporting increased long-term survival rates when compared to ipilimumab in patients with unresectable melanoma (37).
Comparing Anti-CTLA-4 and Anti-PD-1 Therapies and Toxicity in the Clinic
Both anti-CTLA-4 and anti-PD-1 therapies aim to restore T cell effector function in the TME and establish immune dominance over tumors. Overall, these immune checkpoint blockade monotherapies have generated significant improvements in patient outcomes against traditional dacarbazine therapy, with anti-PD-1 blockade seemingly more effective.
In 173 patients with advanced melanoma unresponsive to ipilimumab treatment, the overall response rate (ORR) to pembrolizumab was 26%, with the most severe adverse event (AE) reported as grade 3 fatigue in 5 patients (38). In comparison, CTLA-4-blockade treatment-associated toxicity has not been insignificant; 60% of patients treated with ipilimumab experienced adverse immune effects, which were severe (grade 3 or 4) in 10–15% of cases (25). Head-to-head trials of pembrolizumab vs. ipilimumab have shown 47.3 and 26.5% 6-month progression-free-survival (PFS) rates respectively, with AEs grade 3 or higher at rates of 19.9 and 13.3% (39). Nivolumab was shown to be effective in a range of cancers, producing a 28% ORR in melanoma and grade 3 or 4 drug- related AEs occurred in 14% of 296 patients across all groups (including three deaths from pulmonary toxicity) (40). Research in one study demonstrated 1-year and 2-year survival rates of 62 and 43%, respectively (35). Although both therapies have been successful in treating many cases, anti-PD-1 have thus far been more efficacious than anti-CTLA-4 monoclonal antibodies and have fewer adverse drug reactions. This difference may be because PD-1 is expressed on mature T cells; PD-L1 is expressed on antigen-presenting cells such as DCs and macrophages, and other immune cells as well as on tumor cells, while CTLA-4 is widely expressed on T cells across the body including those circulating in lymph nodes and skin. CTLA-4 inhibitor-mediated anti-tumor activity may therefore extend to secondary lymphoid organs rather than only within the TME (13). This wide expression distribution may potentially result in the disruption of other immune-regulating mechanisms and triggering of autoimmune-like events, consistent with toxicities observed in the clinic with anti-CTLA-4 antibody treatment.
Rationale and Pre-clinical Evidence for Checkpoint Blockade Combination Therapy
While checkpoint inhibitor monotherapy provides significant benefits, this is generally only the case in subsets of patients. CTLA-4 and PD-1 are not functionally redundant, acting at different locations and times in the generation of T-eff (41). This may mean that combination therapies may act in a complementary or even synergistic fashion.
Checkpoint blockade therapies are also known to be subject to various forms of resistance mechanisms. Anti-CTLA-4 blockade primary resistance has been shown to correlate with a loss of IFN-γ signaling genes in vitro and clinically in patients who had poor clinical responses to ipilimumab therapy (42). Furthermore, in patients, PD-L1 expression on circulating CD4+ T CD8+ T cells may be predictive of resistance to anti-CTLA-4 treatment, providing a potential rationale for combination with PD-1/PD-L1 blockade therapy (43). Studies in murine models and patients receiving anti-PD-1 treatment also point to key roles of infiltrating myeloid cells and their signaling pathways, as well as upregulation of alternative immune checkpoints such as T cell immunoglobulin mucin-3 (TIM-3), all associated with resistance to checkpoint inhibition (44, 45). Checkpoint inhibitor monotherapy has been shown to trigger activation of compensatory T cell-associated checkpoints. Pre-clinical evidence supports a rationale for combination therapy as, by blocking more than one of these pathways, including PD-1, LAG-3, and CTLA-4, may reduce tumor growth (46, 47). Murine studies demonstrated that B16 melanoma cell rejection in mice was improved by combined anti-CTLA-4 and anti-PD-1 antibody therapies (47). The results indicated that combination therapy was more than twice as effective as monotherapy in terms of B16 melanoma rejection by increasing T cell infiltration and the presence of T-eff in the TME; IFN-γ and other pro-inflammatory cytokines were observed to be upregulated, producing an inflammatory rather than immunosuppressive TME (47). Further pre-clinical studies suggested that anti-CTLA-4 and anti-PD-1 therapies may have synergistic effects, increasing the numbers of TILs, reducing T-reg and retarding tumor growth (48). Certain subgroups of patients, such as elderly patients, tend to respond better to anti-PD-1 agents, a phenomenon which may be attributed to a depletion of the number of T-regs in older patients (49). These findings, taken together with studies suggesting that anti-CTLA-4 treatment can reduce CTLA-4-expressing T-regs, may lend further merit to stratifying appropriate patient groups to receive combination therapies (20, 22).
Therefore, since CTLA-4 and PD-1 inhibitors exert anti-tumor effects through different mechanisms of action, combining these agents could potentially lead to more efficacious treatments. Furthermore, blockade of one pathway resulted in increased activity and an upregulation of other inhibitory pathways, an effect that could perhaps be mitigated by combined therapy. Emerging evidence provided a rationale for the development of combined blockade regimens as well as acceleration of research into further blockade targets. Although toxicity profiles especially those associated with CTLA-4 blockade use may be an important concern in combined therapies, pre-clinical and clinical findings into the efficacy of combining checkpoint blockade antibodies showed encouraging results in melanoma.
Combined CTLA-4 and PD-1 mAb Therapies in the Clinic
In the first clinical trial investigating the efficacy and safety of combined checkpoint blockade antibodies published in 2013 (50), 53 patients with melanoma were treated concurrently with nivolumab and ipilimumab, while 33 patients received only ipilimumab. In the double therapy group the ORR was 40% for combination treatment and the ORR was 20% in the monotherapy group. However, drug-related toxicity was higher in the combined therapy group; 53% of patients experienced grade 3 or 4 AEs compared to 18% in the monotherapy group. These drug related reactions were managed with medications.
Prolonged PFS was also reported in a phase II dose-escalation study of combined nivolumab and ipilimumab in 142 patients (51). ORRs in the combination and ipilimumab alone groups were 61 and 11%, respectively with drug-related AEs of grade 3 or 4 were exhibited by 54 and 24% of the patients, respectively (51). These drug-related AEs were also managed by immune modulation drug intervention. Follow up on these patients (median 24.5 months) showed a 63.8% 2-year overall survival for the combination group compared to 53.6% for ipilimumab alone (51). This clearly indicated the benefit of combined therapy and its longevity.
The registration phase III Checkmate 067 trial randomized 945 previously-untreated patients to nivolumab, ipilimumab and the two combined drugs, showing a median PFS of 6.9 months, 2.9 months and 11.5 months and 3-year overall survival (OS) rates of 52, 34, and 58%, respectively (52). This large well powered trial confirmed the superior efficacy of combination therapy and nivolumab monotherapy when compared to ipilimumab monotherapy as PFS was consistently longer for patients taking preparations that included nivolumab; this included subgroups categorized by PD-L1 or BRAF mutation status and metastasis stage (53). For patients with BRAF mutations the PFS was 11.7 months (11.2 months for those with wild-type BRAF). Positive PD-L1 status patients fared better with a median PFS of 14 months in both the nivolumab mono and dual therapy groups compared to only 3.9 months for patients taking ipilimumab alone. Checkmate 067 also reported higher rates of complete response in patients on a combined regimen (11.5% compared with 8.9% for nivolumab alone and only 2.2% for ipilimumab alone). Tumor burden change (a parameter which can be used for predicting treatment response) was also significantly higher in the combination group;−51.9% compared with−34.5% and 5.9% for nivolumab alone and ipilimumab alone, respectively.
Furthermore, treatment-related adverse events, as reported in the Checkmate 067 trial comparing ipilimumab, nivolumab and the two combined, revealed higher rates of toxicity associated with the combination therapy (96% compared with 86% in both monotherapy regimens) and this also more frequently led to discontinuation of treatment in the combined group than either monotherapy group. Grade 3 or 4 AEs occurred in 59% of patients given dual therapy compared with 21 and 28% of patients on nivolumab or ipilimumab alone, respectively, and these grade 3 or 4 adverse reactions were most frequently gastrointestinal in nature. Side effects were managed with established safety guidelines and usually resolved within 3–4 weeks and the 3-year survival rate for patients who discontinued treatment was 67% (52). These indicate that combined treatment elicited higher rates of toxicity than either monotherapy and that benefit from dual therapy was conferred despite discontinuation of treatment. Median survival in patients with PD-L1-positive tumors was the same in both the combination and nivolumab alone groups. This may reflect T cell infiltration enhanced by ipilimumab, thus favoring a TME that may be amenable to anti-PD-1 agent action (54).
Previous studies of anti-PD-1 monotherapies have suggested that efficacy was higher in patients whose tumors expressed PD-L1 at levels ≥5%, compared with those whose tumors showed lower expression (54). Where the patients were PD-L1-negative however, another study demonstrated that PFS was longer in the combination group (11.2 months) than in the nivolumab alone group (5.3 months) (53). Overall studies to-date support these combination therapies, which appear to benefit patients with low PD-L1 tumor expression.
Studies have also successfully treated melanoma with combined checkpoint blockade regimens where the two antibodies were administered sequentially. A phase III study of two groups of patients with unresectable stage III or IV melanoma investigated patients treated with nivolumab then ipilimumab (n = 68), or vice versa (n = 70); with nivolumab used as maintenance therapy for both groups until toxicity or disease progression (55). Toxicity was comparable between the groups, however, the nivolumab/ipilimumab exhibited a greater 12-month survival (76%) compared with its counterpart cohort of patients who received the treatments in reverse.
Reducing the dose of ipilimumab in combination with PD-1 directed therapy has resulted in lower combined therapy toxicity rates. Nivolumab with low-dose ipilimumab has been approved in some jurisdictions for the treatment of renal cell carcinoma where toxicity rates have reportedly been reduced compared with combined therapies with higher doses of ipilimumab (56). Research has suggested pembrolizumab as an alternative to nivolumab in a combined regimen with ipilimumab (57). A phase I study, investigating the safety of combining standard-dose (2 mg/kg) pembrolizumab with low-dose (1 mg/kg reduced from 3 mg/kg) ipilimumab, was not powered to examine efficacy (although it suggested comparable level of efficacy to a nivolumab/ipilimumab combination) (57). However, the study demonstrated a more manageable toxicity profile with the pembrolizumab preparation (57).
In summary, the first phase I trials demonstrated higher efficacy than previous monotherapy regimens in small patient cohorts, with a concurrent increase in drug-related toxicity (58). Phase III trials have confirmed these findings showing improved outcomes for advanced-stage melanoma patients when treated with both anti-CTLA-4 and anti-PD-1 therapeutics, which showed benefits for patients with PD-L1 negative tumors (53). Significant toxicity associated with dual therapy limits its usage in patients with comorbidities. However, improved clinical outcomes led to the FDA approval of the combination of nivolumab and ipilimumab for the treatment of unresectable stage III/IV PD-L1 negative melanoma (Table 1). In 2018 the EMA adopted a positive opinion recommending that nivolumab in combination with ipilimumab is indicated for the treatment of unresectable or metastatic melanoma in adults.
Other Potential Immune Checkpoint Therapy Targets and Combinations With Established Checkpoint Inhibitors
As research into therapies utilizing CTLA-4 and PD-1/PD-L1 blockade to treat melanoma has advanced, further targets have been sought out in an effort to overcome issues such as incomplete tumor regression or relapse following treatment. A concerted effort is underway focusing on inhibitory molecules whose mechanisms may operate within the TME and could have complementary functions to those of approved immunotherapies. Studies in the circulation and tumors of patients with melanoma reveal that the TME promotes T cell, exhaustion demonstrated by upregulation of markers of immunosenescence, thereby allowing T cell impairment and immune escape (59). These markers represent targets for immunotherapy to counteract the immune escape of cancer cells. Targeted treatments combining anti-CTLA-4 or anti-PD-1/PD-L1 alongside blockade of novel checkpoints have the potential to produce comparable effects, perhaps with fewer adverse drug reactions than those of the dual anti-CTLA-4/anti-PD-1 regimens.
LAG-3/PD-L1 Blockade
The lymphocyte-activation gene 3 (LAG-3) is a co-inhibitory receptor known primarily to be expressed on exhausted TILs which have less potent effector functions (60, 61). LAG-3 may downregulate T cell responses via interaction with major histocompatibility complex class-II (MHC-II) on DCs (61) (Figure 1). Preclinical studies have shown that, as a result of persistent melanoma antigen expression, LAG-3 expression on TILs is increased, thereby inhibiting T cell action and reducing IFN-γ production within the TME under the influence of PD-1 co-stimulation (61). It has been hypothesized that LAG-3 blockade might produce milder side effects than those observed with checkpoint inhibitors currently in clinical use. Autoimmunity developed by Lag3−/−Pdcd1−/− mice (deficient in LAG-3 and PD-1) was slower: approximately 10 weeks compared with 3–4 weeks in CTLA-4 deficient mice, and less penetrant (80% vs. 100%) than the phenotype observed in Ctla4−/− mice (60). Indeed, in one phase I trial of LAG3/PD-1 targeting combined therapy, similar safety profiles to nivolumab monotherapy were reported (62). Moreover, in vivo studies in murine cancer models have shown that when expressed at high levels, concomitant LAG-3/PD-1 expression is mostly restricted to infiltrating TILs (60). This may signify that a combination immunotherapy targeting these two molecules may encourage tumor-specific responses, avoiding non-specific or self-antigen specific immune responses, perhaps rendering such treatment less toxic than CTLA-4 blockade. Indeed, preclinical evidence that LAG-3 is synergistically efficacious in combination with anti-CTLA or anti-PD-1 therapies is driving clinical development (46).
In addition, PD-L1 overexpression in melanoma tumors has been associated with increased LAG-3 expression. This may point to potential synergistic treatment effects. Pre-clinical studies demonstrated that combination blockade of PD-1 and LAG-3 can induce immune activation and associated tumor rejection in fibrosarcoma and colorectal cancer models in mice (60). LAG-3 has been shown to be expressed on a subset of alternatively-activated human plasmacytoid DCs (pDC). These cells may be enriched in human melanoma tumor sites and produce anti-inflammatory cytokines in response to interactions with MHC class II (61, 63). This may indicate that LAG-3 blockade may promote innate immunity host defense mechanisms. Additionally, melanoma resistance to FAS-mediated apoptosis has been proposed as a mechanism of immune escape mediated by tumor cells expressing MHC class II through engagement with LAG-3 (CD223) expressed on TILs (64). Murine studies have indicated that combined use of anti-LAG-3 and anti-PD-L1 in melanoma treatment overcame the requirement for tumor specific T-reg depletion (65). Because LAG-3 engages with DCs, it is possible that LAG-3 blockade can promote innate immunity, a first host defense mechanism, hence stopping tumor growth at an early stage.
The human IgG4 monoclonal LAG-3 antibody relatlimab is in late phase clinical trials in combination with nivolumab versus nivolumab monotherapy for first line advanced melanoma treatment (Table 3). This regimen holds promise of both efficacy and de-escalation of toxicity.

Table 3. Selection of current anti-LAG-3 (relatlimab) clinical trials. Information sourced from ClinicalTrials.gov.
TIM-3/PD-1 Blockade
T cell immunoglobulin and mucin 3 (TIM-3), a co-inhibitory receptor expressed on T cells, has both inhibitory and activating properties (Figure 1). It has been shown to induce T cell apoptosis, anergy and exhaustion via interaction with galectin-9 on immune cells (66). TIM- 3 expressed on TILs has also been found to bind to galectin-9 expressed on tumor cells in vitro, promoting immune escape (66). In addition, TIM-3 interactions with the high mobility group box 1 (HMGB1) protein, which is involved in the recruitment of nucleic acids into endosomes to be sensed by the innate immunity, impairs this mechanism promoting tumor escape (67). Since TIM-3 has been established as an exhaustion marker in cancer, it is an attractive immunotherapy target (68). Compared with single agent PD-1 blockade in murine cancer models, it has been shown that combined TIM-3/PD-1 blockade led to superior tumor regression (68). In vivo and ex vivo research into the properties of TIM-3 has shown that a melanoma peptide vaccine induced CD8+ T cells to upregulate PD-1 and to an extent TIM-3 in immunized patients. Simultaneous TIM-3 and PD-1 blockade enhanced the proliferation of CD8+ T cells (69). Dual anti-TIM-3 (MBG453) plus anti-PD-1 (PDR001) blockade is currently being analyzed in phase I/II trials (NCT02817633, NCT02608268, NCT03099109, NCT03066648).
B7-H3/CTLA-4 Blockade
B7-H3 (CD276) is a receptor of the CD28 (a co-stimulatory molecule) and B7 (a co-inhibitory molecule) family molecules found on APCs (Figure 1). B7-H3 has been found to be over-expressed in melanoma, favoring tumor growth and conferring anti-apoptotic processes (70). When targeted by an Fc-optimized anti-B7-H3 (MGA271) humanized IgG1 antibody, potent antibody-dependent cellular cytotoxicity (ADCC) against melanoma in vitro and in vivo was observed (71). Studies have suggested than B7-H3 blockade could suppress its co-stimulatory properties. Anti-sense oligonucleotides shown to inhibit B7-H3 expression on DCs resulted in inhibition of IFN-γ production by DC-activated T cells and the proliferation of T cells in vitro (72). Subsequently, ipilimumab plus enoblituzumab, a first in class mAb targeting B7-H3, have been the subject of a phase I trial (NCT02381314), and also enoblituzumab in combination with pembrolizumab is being tested in refractory cancers (NCT02475213) (73).
VISTA/PD-1 Blockade
V-domain Ig suppressor of T cell activation (VISTA) is a PD-L1 homolog and co-inhibitory receptor of the B7 family predominantly expressed on various hematopoietic cells (myeloid derived suppressor cells (MDSCs), tumor associated macrophages (TAMS) and DCs) (74) and on leukocytes such as naïve T cells (Figure 1). Particularly high levels of VISTA were found on tumor infiltrating myeloid cells in murine models (75). VISTA may participate in the suppression of T-eff responses and T-reg induction via interaction with its putative ligand VSIG-3 (75, 76). VSIG-3 is thought to inhibit T cell function and, in the presence of TCR signaling, it may impair T cell proliferation via the VSIG-3/VISTA pathway (77). As well as expression on T cells, it has also been noted to be upregulated in tumors such as colorectal cancer or hepatocellular carcinoma (77). A murine study using B16-OVA melanoma cell lines demonstrated that VISTA blockade with a monoclonal antibody (13F3) enhanced T-eff response within the TME (78). Therefore, blockade of VISTA could enhance innate immunity since it is expressed on myeloid cells thereby promoting early melanoma eradication. VISTA is also expressed on naïve T cells, and inhibition of VISTA could promote early T cell reaction in response to tumor cells (79). VISTA can control T cell activation through nonredundant functions distinct from the PD-1/PD-L1 pathway in controlling T cell activation and antibodies targeting both checkpoints have shown efficacy in preclinical models (80). Therefore, concurrent targeting VISTA and PD-1 pathways has been proposed as a therapeutic approach. The small molecule antagonist CA-170 electively targets PD-L1/2 and VISTA and is presently tested in a phase I trial (NCT02812875) in advanced solid tumors and lymphomas.
A2AR/PD-L1 Blockade
The adenosine-adenosine A2A receptor (A2AR) pathway is of interest as a target for immunotherapy, since the extracellular adenosine, often found in TME due to hypoxia, can inhibit T cell proliferation and cytotoxicity via the A2AR receptor (CD73) on myeloid cells such as macrophages (81) (Figure 1). CD73 is a checkpoint molecule expressed on T-reg which converts AMP to adenosine in this pathway and this checkpoint is also expressed on melanoma cells. Studies of patient samples ex vivo have shown that via adenosine interaction, tumor cells are able to inhibit immune responses and to simultaneously enhance neovascularization and cancer cell growth via vascular endothelial growth factor (VEGF) and IL-6 expression (82, 83). Synergistic effects of combined CTLA-4 and CD73 as well as combinations of PD-1 and CD73 blockade immunotherapies in breast cancer and colon carcinoma pre-clinical models (84, 85) have been observed. Furthermore, murine studies have demonstrated suppressed melanoma growth and enhanced lymphocytic infiltration in the TME in A2AR-deficient mice. Early phase trials investigating the merits of a combined anti-CD73/anti-PD-L1/anti-PD1 therapies in patients with solid tumors are underway, to ascertain whether or not targeting multiple sites in the pathway can show enhanced anti-tumor effects (e.g. NCT02655822, NCT02503774) (Table 4).

Table 4. Selection of current A2AR/CD73 inhibitor clinical trials. Information sourced from ClinicalTrials.gov.
IDO/CTLA-4, PD-1 and PD-L1 Blockade
Indoleamine 2'3' dioxygenase (IDO, IDO1 and IDO2) is a catabolic enzyme produced by macrophages and T-regs to convert tryptophan, which is needed for T cell effector function, to kynurines (86) (Figure 1). Immune tolerance is promoted by IDO-mediated tryptophan deficiency, with naïve T cells observed to differentiate into T-reg (87). This protein can also be expressed by tumors alongside prostaglandin E2, thereby aiding in immune escape of these cancers via NK cell inhibition (88) and T-reg recruitment resulting in IL-10 mediated induction of myeloid-derived suppressor cells (MDSC) (89). Ex vivo-derived and tumor-associated MDSC have been shown to express PD-L1 and MHC-II, and correlated with expression of their receptors, PD-1 and LAG- 3, on T cells, known to be associated with immunosuppression of T cell functions. PD-L1-expressing MDSCs could trigger immunosuppressive effects via IDO (90). Importantly, a pre-clinical study in a melanoma model in mice demonstrated IDO overexpression post-treatment with anti-CTLA-4 and anti-PD-1 (14). This conferred resistance and tumor growth, a property found to be reversible by combination treatment with anti-CTLA-4 and IDO inhibitors (14). Studies using the murine B16.SIY melanoma mouse model have shown that combinations of CTLA-4 or PD-1/PD-L1 with IDO blockade restored both IL-2 production and CD8+ T cell proliferation within the TME (48), pointing to the potential merits of a combinational targeting approach.
IDO and galectin-3 expression are known to promote T-reg upregulation, while suppressing T-eff production (91, 92). Blockade of these two molecules reversed these effects (91), and although further investigations into combined anti-PD-1/IDO (epacadostat) inhibitors underway in a phase I and II trial (NCT02318277) were promising, a phase III trial studying an epacodostat-pembrolizumab combination was abandoned due to a lack of significant improvement in both primary and secondary endpoints—PFS and overall survival (OS) (93). Epacodostat has had to be discontinued in several trials assessing its efficacy as a monotherapy including KEYNOTE-006, KEYNOTE-010, KEYNOTE-087 and KEYNOTE-052, due to adverse reactions (94). These disappointing results may influence the design of future studies aiming to assess IDO as a potential immunotherapy target. However, different approaches to direct IDO inhibition, including an IDO peptide vaccine, have been promising and have shown significant delay in tumor growth and prolonged survival in a B16 murine model (95). Early phase trials of combinations such as a PD-L1/IDO peptide vaccine with nivolumab (NCT03047928) in advanced melanoma are underway.
PKC-η/PD-1 Blockade
A protein which may synergize with CTLA-4 to mediate immune tolerance is PKC-η, an intrinsic downstream signaling checkpoint molecule (96) (Figure 1). PKC-η induces anti-inflammatory cytokine transcription by activating NFkB downstream of the cascade (97). Downregulation of PKC-η in murine models reduced the tumor-suppressive activity of T-reg but did not enhance autoimmune colitis (96). It is possible that inhibition of PKC-η could potentially produce effective T-reg suppression and with fewer adverse drug reactions if used in combination with anti-CTLA-4 blockade. Pre-clinical studies on Rag1 mice have shown that a PKC-η deficiency reduced tumor growth (98) and there are currently studies investigating the efficacy of midostaurin, a tyrosine kinase inhibitor, for the treatment of leukemia as well as studies on the pan-PKC inhibitor AEB071 sotrastaurin which is being tested in uveal melanoma (NCT02273219, NCT02601378, and NCT01430416).
Antibody Engineering and Novel Combinations to Improve Checkpoint Blockade
Combining checkpoint blockade antibodies may be an effective strategy for treating melanoma (Figure 1). However, there remain numerous avenues available for the refinement of existing therapies. With advances in protein engineering, antibodies can be manipulated to introduce novel functionality (99). For combined checkpoint blockade therapies, there is evidence that such engineering could further improve clinical efficacy. Anti-CTLA-4 antibodies have been observed in mouse models of cancer to deplete T-reg cells by ADCC, a function that relies on specific antibody isotypes engaging with FcγR on effector cells (monocytes, macrophages, NK cells) that are cytotoxic to T-regs within the TME (20, 100) (Figure 2A).
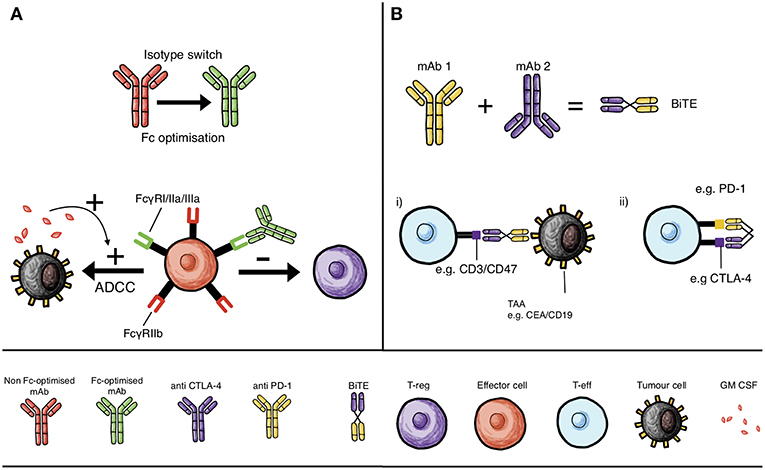
Figure 2. Monoclonal antibody mechanisms of action and interactions with immune cells that can influence checkpoint inhibition: isotype switching, Fc domain optimization and BiTEs. (A) Isotype switching and Fc optimization of mAbs to increase binding to activatory Fc receptors on effector cells, such as monocytes, macrophages and NK cells, to enhance T-reg depletion by ADCC, and in the presence of GM-CSF, to enhance ADCC of tumor cells. (B) Development of bispecific T cell engaging antibody structures (BiTE) that can either simultaneously engage tumor-associated antigen (TAA, e.g., CEA or CD19) and T cell specific molecules (e.g., CD3 or CD47) to promote cytolysis of tumor cells (B,i) or simultaneously inhibit two T cell checkpoint molecules; such as CTLA-4 and PD-1, to circumvent mechanisms of resistance (B,ii).
Until recently, there has been an assumption that Fc receptors would not contribute to the anti-tumor activity of antibodies recognizing checkpoint molecules (101). FcγRs expressed mainly on hematopoietic cells such as NK cells, DCs and macrophages (102) can activate or inhibit; in particular, FcγRI, FcγRIIa, and FcγRIIIa are activating receptors while FcγRIIb is inhibitory (101) (Figure 2A). Importantly, FcγRs are co-expressed on the same cells such as monocytes and macrophages, allowing thresholds for activation/inhibition to be fine-tuned (101). Ipilimumab, for instance, exhibits T-reg depletion properties specifically in the presence of FcγR-expressing monocytes and natural killer (NK) cells, consistent with clinical studies that have demonstrated correlations between specific checkpoint inhibitor antibody subtypes of activating hFcγRs and clinical responses (100) (Figure 2A). An example of this includes variants in the gene FCGR (FCGR2A and FCGR3A) which bind more avidly to human IgG1 and IgG2 subtypes, thereby increasing ADCC-mediated cell death and thus have been associated with improved outcomes (100). Whether these single nucleotide polymorphisms (SNPs) can affect the response of other immunomodulatory monoclonal antibodies requires further investigation.
In vitro studies of anti-CTLA-4 monoclonal antibodies with the same IgG1 and IgG2 Fc variants such as ipilimumab and tremelimumab exhibited superior tumor killing with antibodies able to maximize T-reg depletion by ADCC (20, 100) (Figure 2A). Additionally, preclinical studies have shown that targeting CTLA-4 on T-reg cells conferred minimal tumor protection in comparison to when antibody bound to CTLA-4 on both T-eff and T-reg compartments in vivo (101). Preclinical studies demonstrated a requirement for enriching the TME with effector cells such as myeloid cells expressing high levels of FcγRs (103). One phase II trial showed that granulocyte–macrophage colony-stimulating factor (GM-CSF) plus ipilimumab improved overall survival in patients with metastatic melanoma compared with ipilimumab alone, possibly as a result of a macrophage enriched TME. These findings suggest that macrophages, may be key effector cells in mediating ADCC (101, 103) (Figure 2A).
One molecule of note is CD25, highly expressed on T-reg cells in the TME in mouse models and in human tumors, but with minimal expression in the effector cell compartment (104). Anti-CD25 antibody-induced ADCC can be enhanced by optimizing the antibody isotype to engage activating FcγRs in mouse models of cancer (104). Experiments on mouse models have also shown that anti-CD25 therapies can work concurrently with other immunomodulatory drugs such as anti-PD-1 monoclonal antibodies in the TME. Fc-optimized anti-CD25 drug combinations may prove promising through improving the drug therapeutic window (104). However, it is important to note that CD25 is also expressed at lower levels on activated memory T cell populations (105) potentially highlighting a risk for unwelcome T-eff depletion with anti-CD25 treatment.
Bispecific T cell engaging antibodies (BiTE) constitute an extensive class of agents able to recognize a range of tumor antigens expressed on cancer cells on one arm, while simultaneously engaging a T cell-specific molecule such as CD3 through the other arm (Figure 2B). BiTEs are able to link the two, promoting cytolysis of tumor cells. A bispecific T cell engaging (BiTE) antibody which recognizes the tumor antigen carcinoembryonic antigen (CEA) with one arm and CD3 with the other has been suggested for improving checkpoint blockade therapies (106) (Figure 2B). Following BiTE-mediated cytolysis, upregulation of PD-1 impaired T cell functions (107). If the BiTEs are delivered in conjunction with PD-1 checkpoint blockers, these T cell modulating effects may be reversed (107). It is therefore possible that checkpoint blockers could synergize with such cancer immunotherapies when used in combination.
Bispecific antibodies have also been generated which target checkpoint molecules directly. CD47 is an immune-regulatory molecule expressed on the surface of many cells and all human cancers. It can prevent phagocytosis of cancer cells by macrophages and dendritic cells by binding to SIRPα on their surface (108). However, the ubiquity of CD47 makes it difficult to specifically target with antibodies. Considering this, a bispecific antibody recognizing both CD47 and a cancer-specific antigen, in this case CD19 for B cell lymphoma, has been generated. In vitro, this antibody could promote effective phagocytosis of the cancerous cells (109). It is possible to imagine that this targeted checkpoint blockade approach may be exploited for use melanoma, provided an appropriate targeting antigen can be identified. Furthermore, a BiTE designed to simultaneously target two immune checkpoints on T-eff cells, such as CTLA-4 and PD-1, may improve tumor killing and increase efficacy since dual therapies of anti-PD-1 and anti-CTLA-4 have proven effective (Figure 2B). This could circumvent mechanisms of resistance and eschew the need for two mAbs to be administered concurrently.
T cell activation upregulates ICOS expression and targeting the ICOS/ICOSL pathway improved the potency of anti-CTLA-4 therapy in preclinical models (110, 111). The cellular vaccine IVAX (irradiated ICOSL-positive tumor cells) has been shown to function synergistically in the context of CTLA-4 blockade. The combination has been shown to lead to effector cell migration to and survival in the TME, and consequent enhanced tumor elimination in murine models (110) (Figure 3A).
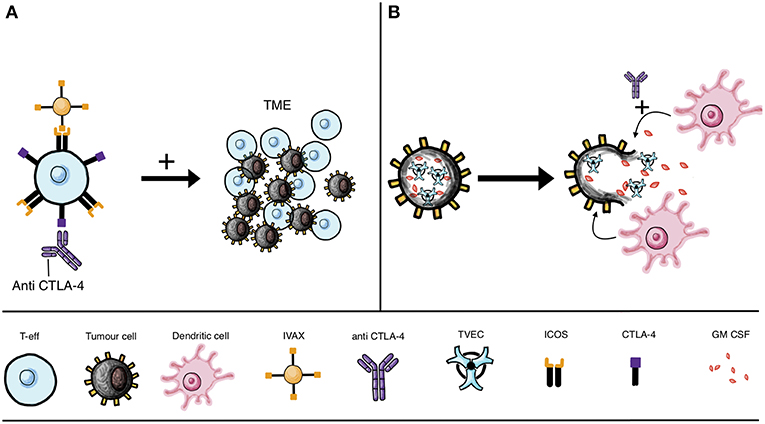
Figure 3. IVAX vaccine and oncolytic viruses combined with checkpoint blockade. (A) Combinatorial therapy may include CTLA-4 blockade with anti-CTLA-4 mAb (leading to T cell activation and subsequent ICOS upregulation) together with the IVAX vaccine [which engages ICOS to increase T-eff migration to the tumor microenvironment (TME)]. (B) Approaches beyond antibody engineering include attenuated oncolytic virus, such as T-VEC unable to replicate in healthy cells, instead preferentially invading cancer cells. The replicating virus lyses the tumor cells and is armed, for instance with GM-CSF, which when released can recruit DCs to prime T cells to identify and destroy tumor cells. Efficacy of oncolytic virus may be improved by combination with checkpoint inhibitors, such as anti-CTLA-4 treatment to boost T cell activation.
Beyond Antibody Engineering: Oncolytic Viruses as Immunotherapies
Oncolytic viruses (OVs) are being developed as cancer immunotherapies, with the modified herpes simplex virus type 1 talimogene laherparepvec (T-VEC, or OncoVEXGM−CSF) being approved by the FDA for malignant melanoma (112), and the ClinicalTrials.gov database currently listing 22 trials studying oncolytic herpes simplex virus following the success of T-VEC for metastatic melanoma (113) (Figure 3B). Many tumor cells cannot adequately protect themselves against viral infection, making the development of oncolytic agents an attractive therapy option that may selectively infect tumor cells (113–115). Among the properties of OVs is their ability to improve the immune system response to tumors, and in combination with checkpoint inhibitors they also enhance checkpoint blockade (116–118). By lysing tumor cells, OV treatment can lead to the release of tumor-associated antigens by the dying cells and efficient cross-presentation of antigens to DCs, thus enhancing anti-tumor responses (113, 116) (Figure 3B).
In clinical practice, OVs are made cancer specific by attenuating the virus so that preferential infection of tumor cells occurs (119). In addition to this, cytokine expression of OVs may increase the anti-cancer properties of these therapies avoiding systemic toxicity due to the preferential action of OVs on tumor cells (119). IL-12 in particular is an important cancer immune response modulator and oncolytic herpes viruses (oHSVs) expressing IL-12 have shown efficacy in preclinical studies on glioblastoma multiforme (120). ‘Arming’ these OVs with an expression of a particular cytokine may be key for achieving anti-tumor efficacy. The clinically-licensed T-VEC for instance is armed with GM-CSF, an APC activator (121). A murine study also found that an IL-12 armed oHSVs delayed tumor proliferation and reduced tumor growth more effectively than its unarmed counterpart (119) (Figure 3B).
Although viral-mediated tumor lysis can be enhanced by tropism targeting and viral arming with immune stimulatory cytokines, clinical efficacy has not been consistent across all patient groups (122). For instance, the cytidine deaminase Apolipoprotein B Editing Complex 3 (APOBEC3), normally involved in the host response to retrovirus infection, is also reported to be associated with virus-resistant tumors. Expression of APOBEC3 been shown to be upregulated in B16 murine melanoma cells infected with an oncolytic vesicular stomatitis virus (VSV) (122). Tumor cell resistance to VSV both in vivo and in vitro was demonstrated where APOBEC3 was upregulated and knockdown of this enzyme reduced VSV escape from immune clearance (122). This highlights the need for further research to clarify methods for mitigating and preventing tumor resistance to immunotherapies and other novel therapies that often limit efficacy.
Studies have suggested the potential merit of combining OVs with checkpoint inhibitors. In a Phase II study, CD8-expressing T cells, immune activation markers and PD-L1 expression were increased with intralesional administration of coxsackievirus A21 (CVA21), which has a propensity for tumor cells expressing ICAM-1 (123). Phase Ib trials have shown that this combination has minimal additional toxicity or adverse drug reactions, with one study recording an ORR of 73% and a disease control rate (DCR) of 91% of patients with advanced melanoma (124, 125). In the case of T-VEC, long term protection and significant effects on untreated tumors was seen only when the OV was combined with anti-CTLA-4 (121). Finally, several studies are exploring novel OV combinations. One is an oncolytic vaccinia virus armed with a superagonist IL-15 (IL-15-IL-15Rα fusion) given alone or in combination with anti-PD-1: combined treatment demonstrated T cell-driven anti-tumor efficacy in in vivo mouse models of cancer (126). Furthermore, TNFα armed viruses have seen success in inducing tumor regression and vascular collapse in solid masses when administered in tumor-bearing mice alongside therapies that inhibit anti-apoptosis proteins in vitro (127).
The administration of OVs has often been limited to intratumoral delivery in patients; T-VEC is currently only licensed as a locally-administered treatment (128). This is due to concerns regarding neutralizing antibodies (NAbs) which are often present in human populations in response to common viruses such as HSV and reovirus and may impair the efficacy of systemic delivery of OVs (128). This may be a limitation since systemic delivery of OV therapy would in theory be more effective for targeting metastatic lesions. However, one report (128) found that by loading neutralized reovirus with antibodies in immune complexes onto human monocytes, resulted in the transfer of the virus to melanoma cells, cancer cell lysis and in restriction of cancer growth in a murine model of melanoma (128). This indicates that monocytes and antibodies recognizing viral proteins may be important components in maintaining the potency of OV therapy and may lead to new strategies for OV treatment.
In summary, oncolytic viruses may provide an alternative avenue for cancer immunotherapy due to their properties in targeting cancer cells with some specificity and due to some promising results in combination with immune checkpoint blockade. It appears that these therapies are most efficient when armed with specific cytokine expression and attenuation of the virus is important for both reducing toxicity and allowing preferential infection of cancer cells.
Challenges and Future Directions
Anti-CTLA-1 and anti-PD-1 therapies have been the most extensively researched due to documented benefits for melanoma treatment as evidenced by higher rates of tumor clearance and, crucially, long term disease eradication in some patients compared with traditional treatments (129). Importantly, further research has emphasized improved response and survival rates when combining anti-CTLA-4 and anti-PD-1 therapy (130), leading to the approval of ipilimumab plus nivolumab in BRAF wild-type melanoma in 2015 and regardless of BRAF tumor subtype in 2016 (131). As more and more immune checkpoints are identified, and their mechanisms of action elucidated, new options for single and combined therapy regimens can be derived (Table 5). Combining different checkpoint blockade agents may produce potent effects, due in part to targeting more than one immune pathways. The benefits of combination therapies are not limited to the use of multiple checkpoint inhibitors but can extend to combining anti-CTLA or anti-PD-1 drugs with other targeted agents such as MEK and BRAF inhibitors. One such example is a Phase III trial combining PD-L1 and MEK inhibition (atezolizumab and cobimetinib) vs. pembrolizumab in advanced BRAFV600 wild-type melanoma (NCT03273153). Combinations of checkpoint inhibitors with chemotherapy, radiotherapy and other immunotherapies such as oncolytic viruses and cell-based therapy approaches are also being investigated. Many challenges, including selecting optimal combinations, predicting and managing toxic effects for different combinations and development of clinically-useful biomarkers to help support the use of such treatments are and will continue to be the subject of intense study.
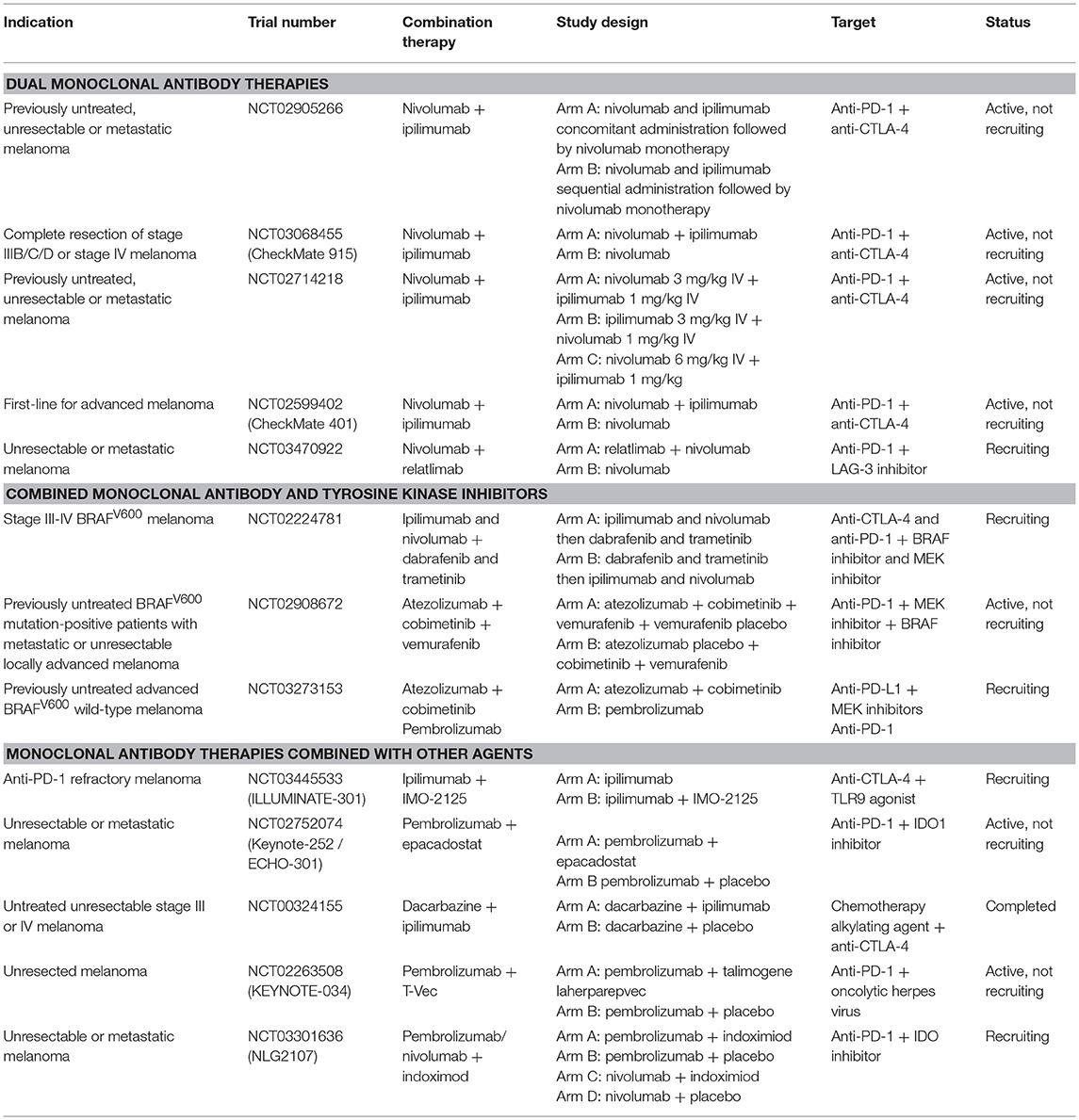
Table 5. Examples of combination phase III monoclonal antibody immunotherapy trials in melanoma. Information sourced from ClinicalTrials.gov.
Toxicities of immunotherapies remains an important challenge. Adverse effects (50, 62), can be managed according to information from resources such as the European Society for Medical Oncology (ESMO) Clinical Practice Guidelines (132, 133). It has been shown that recurrence of immune-related adverse events (irAEs) in patients in whom it was necessary to discontinue treatment as a result of irAEs can occur upon re-challenge with immunotherapies such as anti-PD-1. For instance, a retrospective analysis showed that colitis was less likely to recur in comparison to hepatitis, pancreatitis, pneumonitis and nephritis, and re-challenge with PD-1 blockade has been reported to be tolerated better than other agents (134). Studies have also suggested various biomarkers such as IL-17 or eosinophilia to help predict toxicity in patients, something that could allow early recognition of pathology and thus prompt intervention (133, 135). Investigation into new immunotherapies and combinations may also reveal treatment algorithms with more favorable toxicity profiles than the current regimens.
Improved efficacy has often been associated with increased irAEs, as reported in trials such as Checkmate 067, where the ipilimumab/nivolumab combination had a 10% higher rate of toxicity than either drug administered alone (52). However, immune-related toxic effects may potentially be abrogated in future therapeutic regimens by various approaches. These may include the increasing specificity of targeted immune therapies, to help reduce systemic effects on non-target healthy cells, as well as the implementation of combinatorial therapies with non-overlapping targets. For instance LAG-3 may promote more tumor-specific responses than currently licensed immunotherapies: concomitant LAG-3/PD-1 expression is mostly restricted to infiltrating TILs, expressed at high levels in murine models (60), and mouse studies into other combinations such as a PKC-η/PD-1 blockade therapy has shown a reduction in T-reg immunosuppressive activity without the concomitant increase in autoimmunity (96). Thus, although data from current licensed therapies have suggested a correlation between combinatorial therapies and increased toxicity, this may not necessarily be the case with different checkpoint blockade combinations. In the future, ideally patients would be able to access fully personalized medicine in which treatment such as immunotherapies would be specific to the patient immune response as well as the specific genetic characteristics of their cancers—this would reduce unwanted side effects and maximize the antitumor effect in each individual patient.
The mechanisms behind the lack of clinical responses to single or combination treatments in many patients, how tumors develop resistance to novel immunotherapies and the best criteria to select patients for maximum treatment effect are insufficiently elucidated. A focus on functional evaluations in the context of patient immunity for novel regimens alongside clinical testing may shed some light on the most effective combinations and the best strategies to reduce adverse effects. This may reveal immune or genomic biomarkers linked with better treatment outcomes. For instance, cancer development and therapeutic response may relate to host factors such as the gut microbiome. Approximately 20% of malignancies are linked with microorganism infection and a patient's gastrointestinal microbiota can both positively and negatively influence cancer susceptibility (136). The effects of the microbiome in cancer susceptibility, progression and response to treatment are far reaching insofar as the microbiome is known to guide the immune system's response, host metabolism of medication and endogenously produced chemicals, and can also influence the balance of cell growth and death (136). A recent study of patients with melanoma exploring the role of the microbiome in influencing clinical response to anti-PD-1 therapy found that “favorable” gut microbiota (characterized by higher gut microbe diversity and levels of Ruminococcaceae/Faecalibacterium) mediated higher levels of antigen presentation and T-eff function both in the periphery and within the TME. This promoted better systemic and anti-tumor immune responses compared with patients with “unfavorable” microbiomes (137). Bacteroides has been linked to a poorer anti-tumor response in patients treated with ipilimumab and patient microbiomes enriched with Faecalibacterium and Firmicutes have also been correlated with more efficacious clinical response to ipilimumab (138).
Research exploring predictive markers for checkpoint treatment response has pointed to mutational burden, PD-1 ligand (PD-L1) expression, circulating tumor cells (CTCs) and miRNA signatures. The challenge of identifying biomarkers should also be explored in combination therapies. For melanoma, mutational burden and PD-L1 studied in pretreatment biopsies have been evaluated as predictive markers for guiding therapy. However, these do not always correlate with response (139). A study exploring a noninvasive blood-based monitoring method of tumor burden, aimed at improving prediction of response for melanoma patients undergoing immune checkpoint inhibition therapy, found longitudinal digital measurements of circulating tumor cells (CTCs) to be predictive of clinical outcomes in patients with metastatic melanoma (139). Other research studies have reported falling levels of circulating free DNA (ctDNA) for tumor survival-promoting mutant kinase BRAF, NRAS, and KIT alleles in melanoma patients who are undergoing immunotherapy (140, 141). Alongside their use as biomarkers of response, perhaps an optimal immune and mutant kinase targeted therapy combination could be derived to further improve clinical outcomes. Tumor lymphocyte and NK cell infiltration and IFNγ upregulation, have been proposed as potential predictors of response alongside mutational burden, however these need to be standardized and widely evaluated in clinical practice (142–144). Tumor mutational load and PD-L1 expression are often clinically available to physicians. PD-L1 expression has been reported to have a good negative predictive value in lung cancer; however, the same results have not been shown in melanoma (142). Finally, new technologies such as analyzing T cell clonality are promising but require specialist equipment not yet widely available as clinical tools (142). Reliable predictive markers of long term outcome are also particularly important for combined therapies with their high toxicity profiles. Identifying signatures to guide selection of patients who do not need to be exposed to the increased risk of toxicity currently inherent in combination strategies would greatly aid clinical decision making.
Conclusion
Combinations of existing and novel immune checkpoint inhibitors and discovering predictive biomarkers promise to further build on the success of immunotherapy. A comprehensive approach will be required to produce the most efficacious combination immunotherapies able to circumvent mechanisms of resistance. Antibody engineering may help capitalize on tumor killing effector mechanisms through knowledge of SNPs and FcR-expressing immune effector cells known to influence patient responses to other immunomodulatory monoclonal antibodies. These in addition to considering host factors such as the microbiome, which can affect medication metabolism and uptake, and tumor-associated molecular and immunological characteristics such as mutational burden, expression of checkpoint molecules and immune cell infiltration in the TME, may ultimately determine patient response to treatment and help optimize immunotherapy for melanoma.
Author Contributions
DK, SK, KL, and SP conceived and designed the study; DK, HB, SK, BT, CB, ET, AS, NT, KL, and SP searched and studied the literature; DK, SK, BT, CB, ET, AS, NT, KL, and SP wrote the manuscript; DK, SK, HB, KL, and SP discussed and interpreted the literature findings; SM, SC, GP, AK, MN, RJH, EF, RMH, AC, IW, JS, DJ, JG, CH, and AM commented and helped to edit the manuscript; SK supervised the study.
Funding
The authors acknowledge support by the Medical Research Council (MR/L023091/1); Cancer Research UK (C30122/A11527; C30122/A15774); The Academy of Medical Sciences; Breast Cancer Now (147) working in partnership with Walk the Walk; CRUK/NIHR in England/DoH for Scotland, Wales and Northern Ireland Experimental Cancer Medicine Centre (C10355/A15587). The research was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre (BRC) based at Guy's and St Thomas' NHS Foundation Trust and King's College London (IS-BRC-1215-20006). The authors are solely responsible for study design, data collection, analysis, decision to publish, and preparation of the manuscript. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. James FR, Jiminez-Linan M, Alsop J, Mack M, Song H, Brenton JD, et al. Association between tumour infiltrating lymphocytes, histotype and clinical outcome in epithelial ovarian cancer. BMC Cancer. (2017) 17:657. doi: 10.1186/s12885-017-3585-x
2. Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched “young” tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. (2010) 16:6122–31. doi: 10.1158/1078-0432.CCR-10-1297
3. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. (2004) 4:11–22. doi: 10.1038/nrc1252
4. Litvin O, Schwartz S, Wan Z, Schild T, Rocco M, Oh NL, et al. Interferon alpha/beta enhances the cytotoxic response of MEK inhibition in melanoma. Mol Cell. (2015) 57:784–96. doi: 10.1016/j.molcel.2014.12.030
5. Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. (1997) 3:409–17.
6. Rosenberg SA, Lotze MT, Yang JC, Topalian SL, Chang AE, Schwartzentruber DJ, et al. Prospective randomized trial of high-dose interleukin-2 alone or in conjunction with lymphokine-activated killer cells for the treatment of patients with advanced cancer. J Natl Cancer Inst. (1993) 85:622–32. doi: 10.1093/jnci/85.8.622
7. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
8. Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. (2010) 127:759–67. doi: 10.1002/ijc.25429
9. Ishida T, Ishii T, Inagaki A, Yano H, Komatsu H, Iida S, et al. Specific recruitment of CC chemokine receptor 4–positive regulatory T cells in hodgkin lymphoma fosters immune privilege. Cancer Res. (2006) 66:5716–22. doi: 10.1158/0008-5472.CAN-06-0261
10. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
11. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
12. Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci USA. (2008) 105:9331–6. doi: 10.1073/pnas.0710441105
13. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
14. Aris M, Mordoh J, Barrio MM. Immunomodulatory monoclonal antibodies in combined immunotherapy trials for cutaneous melanoma. Front Immunol. (2017) 8:1024. doi: 10.3389/fimmu.2017.01024
15. Swart M, Verbrugge I, Beltman JB. Combination approaches with immune-checkpoint blockade in cancer therapy. Front Oncol. (2016) 6:233. doi: 10.3389/fonc.2016.00233
16. Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. (2013) 94:25–39. doi: 10.1189/jlb.1212621
17. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
18. Yokosuka T, Kobayashi W, Takamatsu M, Sakata-Sogawa K, Zeng H, Hashimoto-Tane A, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. (2010) 33:326–39. doi: 10.1016/j.immuni.2010.09.006
19. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. (2009) 206:1717–25. doi: 10.1084/jem.20082492
20. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
21. Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. (1999) 11:483–93. doi: 10.1016/S1074-7613(00)80123-5
22. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
23. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. (2010) 11:155–64. doi: 10.1016/S1470-2045(09)70334-1
24. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. (2015) 33:1889–94. doi: 10.1200/JCO.2014.56.2736
25. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
26. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
27. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
28. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292
29. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
30. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. (2010) 116:1757–66. doi: 10.1002/cncr.24899
31. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. (2005) 17:133–44. doi: 10.1093/intimm/dxh194
32. Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. (2007) 27:111–22. doi: 10.1016/j.immuni.2007.05.016
33. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
34. Johnson DB, Peng C, Sosman JA. Nivolumab in melanoma: latest evidence and clinical potential. Ther Adv Med Oncol. (2015) 7:97–106. doi: 10.1177/1758834014567469
35. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
36. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
37. Schachter J, Ribas A, Long GV, Arance A, Grob J, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. (2017) 390:1853–62. doi: 10.1016/S0140-6736(17)31601-X
38. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. (2014) 384:1109–17. doi: 10.1016/S0140-6736(14)60958-2
39. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
40. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
41. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. (2017) 170:1133.e17. doi: 10.1016/j.cell.2017.07.024
42. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. (2016) 167:404.e9. doi: 10.1016/j.cell.2016.08.069
43. Jacquelot N, Roberti MP, Enot DP, Rusakiewicz S, Ternes N, Jegou S, et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun. (2017) 8:2. doi: 10.1038/s41467-017-00608-2
44. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
45. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. (2016) 539:443–7. doi: 10.1038/nature20554
46. Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. (2016) 6:e1249561. doi: 10.1080/2162402X.2016.1249561
47. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. (2010) 107:4275–80. doi: 10.1073/pnas.0915174107
48. Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. (2014) 2:3. doi: 10.1186/2051-1426-2-3
49. Kugel CH III, Douglass SM, Webster MR, Kaur A, Liu Q, Yin X, et al. Age correlates with response to anti-PD1, reflecting age-related differences in intratumoral effector and regulatory T-cell populations. Clin Cancer Res. (2018) 24:5347–56. doi: 10.1158/1078-0432.CCR-18-1116
50. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus Ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
51. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
52. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
53. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:1270–1. doi: 10.1056/NEJMoa1504030
54. Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. (2016) 17:1558–68. doi: 10.1016/S1470-2045(16)30366-7
55. Weber JS, Gibney G, Sullivan RJ, Sosman JA, Slingluff CL Jr, Lawrence DP, et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): an open-label, randomised, phase 2 trial. Lancet Oncol. (2016) 17:943–55. doi: 10.1016/S1470-2045(16)30126-7
56. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
57. Long GV, Atkinson V, Cebon JS, Jameson MB, Fitzharris BM, McNeil CM, et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): an open-label, phase 1b trial. Lancet Oncol. (2017) 18:1202–10. doi: 10.1016/S1470-2045(17)30428-X
58. Abdel-Wahab N, Shah M, Suarez-Almazor ME. Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS ONE. (2016) 11:e0160221. doi: 10.1371/journal.pone.0160221
59. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
60. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
61. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. (2016) 107:1193–7. doi: 10.1111/cas.12986
62. Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipet EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti-LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti-PD-1/PD-L1 therapy. J Clin Oncol. (2017) 35:9520. doi: 10.1200/JCO.2017.35.15_suppl.9520
63. Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol. (2014) 134:1893–902. doi: 10.1038/jid.2014.29
64. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. (2011) 186:5173–83. doi: 10.4049/jimmunol.1002050
65. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. (2013) 190:4899–909. doi: 10.4049/jimmunol.1300271
66. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
67. Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. (2009) 462:99–103. doi: 10.1038/nature08512
68. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
69. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res. (2014) 74:1045–55. doi: 10.1158/0008-5472.CAN-13-2908
70. Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. (2016) 22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428
71. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. (2012) 18:3834–45. doi: 10.1158/1078-0432.CCR-12-0715
72. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. (2001) 2:269–74. doi: 10.1038/85339
73. Urba W, Chmielowski B, Loo D, Baughman J, Chen F, Moore P, et al. A Phase I, open-label, dose escalation study of MGA271 in combination with ipilimumab in patients with B7-H3-expressing melanoma, squamous cell cancer of the head and neck or non-small cell lung cancer. J Immunother Cancer. (2015) 3(Suppl. 2):P176. doi: 10.1186/2051-1426-3-S2-P176
74. Lines JL, Sempere LF, Wang L, Pantazi E, Mak J, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
75. Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. (2014) 2:510–7. doi: 10.1158/2326-6066.CIR-14-0072
76. Prodeus A, Abdul-Wahid A, Sparkes A, Fischer NW, Cydzik M, Chiang N, et al. VISTA.COMP — an engineered checkpoint receptor agonist that potently suppresses T cell–mediated immune responses. JCI Insight. (2017) 2:e94308. doi: 10.1172/jci.insight.94308
77. Wang J, Wu G, Manick B, Hernandez V, Renelt M, Bi M, et al. VSIG-3/IGSF11 is a ligand of VISTA/PD-1H and inhibits human T cell function. J Immunol. (2017) 198(Suppl. 1):154.1.
78. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev. (2017) 276:66–79. doi: 10.1111/imr.12525
79. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
80. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA. (2015) 112:6682–7. doi: 10.1073/pnas.1420370112
81. Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, et al. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. (2009) 183:5487–93. doi: 10.4049/jimmunol.0901247
82. Szajnik-Szczepanski M, Derbis M, Lach M, Patalas P, Michalak M, Drzewiecka H, et al. Abstract A79: the adenosine pathway in ovarian carcinoma: tumor cells and tumor-derived exosomes express CD39 and CD73 ectonucleotidases, produce adenosine and mediate immune suppression. Clin Cancer Res. (2013) 19:A79. doi: 10.1158/1078-0432.OVCA13-A79
83. Coombs MRP, Belderbos ME, Gallington LC, Bont L, Levy O. The adenosine system modulates Toll-like receptor function: basic mechanisms, clinical correlates and translational opportunities. Exp Rev Anti Infect Ther. (2011) 9:261–9. doi: 10.1586/eri.10.158
84. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
85. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
86. Prendergast GC, Smith C, Thomas S, Mandik-Nayak L, Laury-Kleintop L, Metz R, et al. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol Immunother. (2014) 63:721–35. doi: 10.1007/s00262-014-1549-4
87. Sucher R, Kurz K, Weiss G, Margreiter R, Fuchs D, Brandacher G. IDO-mediated tryptophan degradation in the pathogenesis of malignant tumor disease. Int J Tryptophan Res. (2010) 3:113–20. doi: 10.4137/IJTR.S4157
88. Pietra G, Manzini C, Rivara S, Vitale M, Cantoni C, Petretto A, et al. Melanoma cells inhibit natural killer cell function by modulating the expression of activating receptors and cytolytic activity. Cancer Res. (2012) 72:1407–15. doi: 10.1158/0008-5472.CAN-11-2544
89. Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. (2015) 13:412–24. doi: 10.1016/j.celrep.2015.08.077
90. Pinton L, Solito S, Damuzzo V, Francescato S, Pozzuoli A, Berizzi A, et al. Activated T cells sustain myeloid-derived suppressor cell-mediated immune suppression. Oncotarget. (2016) 7:1168–84. doi: 10.18632/oncotarget.6662
91. Melief SM, Visser M, van der Burg SH, Verdegaal EME. IDO and galectin-3 hamper the ex vivo generation of clinical grade tumor-specific T cells for adoptive cell therapy in metastatic melanoma. Cancer Immunol Immunother. (2017) 66:913–26. doi: 10.1007/s00262-017-1995-x
92. Hsu DK, Chen Huan-Yuan, Liu Fu-Tong. Galectin-3 regulates T-cell functions. Immunol Rev. (2009) 230:114–27. doi: 10.1111/j.1600-065X.2009.00798.x
93. Carroll J. Keytruda/Epacadostat Combo Crashes in PhIII Melanoma Study, Raising Questions About the Future of IDO for Incyte. (2018) Available online at: https://endpts.com/keytruda-epacadostat-combo-crashes-in-phiii-melanoma-study-raising-questions-about-the-future-of-ido-for-incyte/ (Accessed August 14, 2018).
94. Incyte and Merck Provide Update on Phase 3 Study of Epacadostat in Combination with KEYTRUDA® (pembrolizumab) in Patients with Unresectable or Metastatic Melanoma. (2018) Available online at: https://www.businesswire.com/news/home/20180406005141/en/Incyte-Merck-Provide-Update-Phase-3-Study (Accessed August 14, 2018).
95. Nandre R, Verma V, Gaur P, Vir P, Wang H, Kang B, et al. IDO vaccine enhances antigen-specific anti-tumor effects by reducing IDO-expressing antigen presenting cells and MDSCs [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018 Apr 14-18 Chicago, IL; Philadelphia, PA: AACR; Cancer Res. (2018).
96. Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol. (2014) 15:465–72. doi: 10.1038/ni.2866
97. Diaz-Meco M, Moscat J. The atypical PKCs in inflammation: NF-κB and beyond. Immunol Rev. (2012) 246:154–67. doi: 10.1111/j.1600-065X.2012.01093.x
98. Pedros C, Canonigo-Balancio A, Kong K, Altman A. Requirement of Treg-intrinsic CTLA4/PKCη signaling pathway for suppressing tumor immunity. JCI Insight. (2017) 2:e95692. doi: 10.1172/jci.insight.95692
99. Ayyar BV, Arora S, O'Kennedy R. Coming-of-Age of antibodies in cancer therapeutics. Trends Pharmacol Sci. (2016) 37:1009–28. doi: 10.1016/j.tips.2016.09.005
100. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:663.e4. doi: 10.1016/j.ccell.2018.02.010
101. Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol. (2014) 35:290–8. doi: 10.1016/j.it.2014.05.002
102. Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. (2007) 19:239–45. doi: 10.1016/j.coi.2007.01.005
103. Hodi FS, Lee SJ, McDermott DF, Rao UNM, Butterfield LH, Tarhini AA, et al. Multicenter, randomized phase II trial of GM-CSF (GM) plus ipilimumab (Ipi) versus Ipi alone in metastatic melanoma: E1608. JCO. (2013) 31:CRA9007. doi: 10.1200/jco.2013.31.18_suppl.cra9007
104. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. (2017) 46:577–86. doi: 10.1016/j.immuni.2017.03.013
105. Kmieciak M, Gowda M, Graham L, Godder K, Bear HD, Marincola FM, et al. Human T cells express CD25 and Foxp3 upon activation and exhibit effector/memory phenotypes without any regulatory/suppressor function. J Transl Med. (2009) 7:89. doi: 10.1186/1479-5876-7-89
106. Huehls AM, Coupet TA, Sentman CL. Bispecific T cell engagers for cancer immunotherapy. Immunol Cell Biol. (2016) 93:290–6. doi: 10.1038/icb.2014.93
107. Osada T, Patel SP, Hammond SA, Osada K, Morse MA, Lyerly HK. CEA/CD3-bispecific T cell-engaging (BiTE) antibody-mediated T lymphocyte cytotoxicity maximized by inhibition of both PD1 and PD-L1. Cancer Immunol Immunother. (2015) 64:677–88. doi: 10.1007/s00262-015-1671-y
108. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. (2012) 109:6662–7. doi: 10.1073/pnas.1121623109
109. Johnson Z, Papaioannou A, Bernard L, Cosimo E, Daubeuf B, Richard F, et al. Bispecific antibody targeting of CD47/CD19 to promote enhanced phagocytosis of patient B lymphoma cells. J Clin Oncol. (2015) 33:e14016. doi: 10.1200/jco.2015.33.15_suppl.e14016
110. Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. (2014) 211:715–25. doi: 10.1084/jem.20130590
111. Amatore F, Gorvel L, Olive D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin Ther Targets. (2018) 22:343–51. doi: 10.1080/14728222.2018.1444753
112. Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, et al. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. JCO. (2013) 31:LBA9008. doi: 10.1200/jco.2013.31.18_suppl.lba9008
113. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
114. Samson A, Scott KJ, Taggart D, West EJ, Wilson E, Nuovo GJ, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. (2018) 10:eaam7577. doi: 10.1126/scitranslmed.aam7577
115. Marin-Acevedo J, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. (2017) 11:8. doi: 10.1186/s13045-017-0552-6
116. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. (2013) 12:103. doi: 10.1186/1476-4598-12-103
117. Jiang H, Rivera-Molina Y, Gomez-Manzano C, Clise-Dwyer K, Bover L, Vence LM, et al. Oncolytic adenovirus and tumor-targeting immune modulatory therapy improve autologous cancer vaccination. Cancer Res. (2017) 77:3894–907. doi: 10.1158/0008-5472.CAN-17-0468
118. Sivendran S, Pan M, Kaufman HL, Saenger Y. Herpes simplex virus oncolytic vaccine therapy in melanoma. Expert Opin Biol Ther. (2010) 10:1145–53. doi: 10.1517/14712598.2010.495383
119. Leoni V, Vannini A, Gatta V, Rambaldi J, Sanapo M, Barboni C, et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. (2018) 14:e1007209. doi: 10.1371/journal.ppat.1007209
120. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a phase I clinical trial to evaluate M032, a genetically engineered HSV-1 expressing IL-12, in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma. Hum Gene Ther Clin Dev. (2016) 27:69–78. doi: 10.1089/humc.2016.031
121. Moesta AK, Cooke K, Piasecki J, Mitchell P, Rottman JB, Fitzgerald K, et al. Local delivery of OncoVEX(mGM-CSF) generates systemic antitumor immune responses enhanced by cytotoxic T-lymphocyte-associated protein blockade. Clin Cancer Res. (2017) 23:6190–202. doi: 10.1158/1078-0432.CCR-17-0681
122. Huff AL, Wongthida P, Kottke T, Thompson JM, Driscoll CB, Schuelke M, et al. APOBEC3 mediates resistance to oncolytic viral therapy. Mol Ther Oncolytics. (2018) 11:1–13. doi: 10.1016/j.omto.2018.08.003
123. Andtbacka RHI, Curti B, Hallmeyer S, Feng Z, Paustian C, Bifulco C, et al. (3336) Phase II CALM extension study: enhanced immune-cell infiltration within the tumour micro-environment of patients with advanced melanoma following intralesional delivery of Coxsackievirus A21. Eur J Cancer. (2015) 51:S677. doi: 10.1016/S0959-8049(16)31854-8
124. Silk AW, Kaufman H, Gabrail N, Mehnert J, Bryan J, Norrell J, et al. Abstract CT026: phase 1b study of intratumoral Coxsackievirus A21 (CVA21) and systemic pembrolizumab in advanced melanoma patients: interim results of the CAPRA clinical trial. Cancer Res. (2017) 77:CT026. doi: 10.1158/1538-7445.AM2017-CT026
125. Pandha HS, Ralph C, Harrington K, Curti BD, Sanborn RE, Akerley WL, et al. Keynote-200 phase 1b: a novel combination study of intravenously delivered coxsackievirus A21 and pembrolizumab in advanced cancer patients. JCO. (2017) 35:TPS3108. doi: 10.1200/JCO.2017.35.15_suppl.TPS3108
126. Kowalsky SJ, Liu Z, Feist M, Berkey SE, Ma C, Ravindranathan R, et al. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol Ther. (2018) 26:2476–86. doi: 10.1016/j.ymthe.2018.07.013
127. Beug ST, Pichette SJ, St-Jean M, Holbrook J, Walker DE, LaCasse EC, et al. Combination of IAP antagonists and TNF-alpha-armed oncolytic viruses induce tumor vascular shutdown and tumor regression. Mol Ther Oncolytics. (2018) 10:28–39. doi: 10.1016/j.omto.2018.06.002
128. Berkeley RA, Steele LP, Mulder AA, van dW, Kottke TJ, Thompson J, et al. Antibody-neutralized reovirus is effective in oncolytic virotherapy. Cancer Immunol Res. (2018) 6:1161–73. doi: 10.1158/2326-6066.CIR-18-0309
129. Medina PJ, Adams VR. PD-1 Pathway inhibitors: immuno-oncology agents for restoring antitumor immune responses. Pharmacotherapy. (2016) 36:317–34. doi: 10.1002/phar.1714
130. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:86. doi: 10.3389/fonc.2018.00086
131. Rozeman EA, Dekker TJA, Haanen JBAG, Blank CU. Advanced melanoma: current treatment options, biomarkers, and future perspectives. Am J Clin Dermatol. (2018) 19:303–17. doi: 10.1007/s40257-017-0325-6
132. Haanen JBAG, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. (2017) 28(Suppl. 4):iv119–iv142. doi: 10.1093/annonc/mdx225
133. Linardou H, Gogas H. Toxicity management of immunotherapy for patients with metastatic melanoma. Ann Transl Med. (2016) 4:272. doi: 10.21037/atm.2016.07.10
134. Nagai H, Muto M. Optimal management of immune-related adverse events resulting from treatment with immune checkpoint inhibitors: a review and update. Int J Clin Oncol. (2018) 23:410–20. doi: 10.1007/s10147-018-1259-6
135. Callahan MK, Yang A, Tandon S, Xu Y, Subudhi SK, Roman RA, et al. Evaluation of serum IL-17 levels during ipilimumab therapy: correlation with colitis. JCO. (2011) 29:2505. doi: 10.1200/jco.2011.29.15_suppl.2505
137. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. (2018) 359:97–103. doi: 10.1126/science.aan4236
138. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. (2017) 28:1368–79. doi: 10.1093/annonc/mdx108
139. Hong X, Sullivan RJ, Kalinich M, Kwan TT, Giobbie-Hurder A, Pan S, et al. Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. Proc Natl Acad Sci USA. (2018) 115:2467–72. doi: 10.1073/pnas.1719264115
140. Ashida A, Sakaizawa K, Uhara H, Okuyama R. Circulating tumour DNA for monitoring treatment response to anti-PD-1 immunotherapy in melanoma patients. Acta Derm Venereol. (2017) 97:1212–8. doi: 10.2340/00015555-2748
141. Lee JH, Long GV, Boyd S, Lo S, Menzies AM, Tembe V, et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann Oncol. (2017) 28:1130–6. doi: 10.1093/annonc/mdx026
142. Espinosa E, Márquez-Rodas I, Soria A, Berrocal A, Manzano JL, Gonzalez-Cao M, et al. Predictive factors of response to immunotherapy—a review from the Spanish Melanoma Group (GEM). Ann Transl Med. (2017) 5:389. doi: 10.21037/atm.2017.08.10
143. Alavi S, Stewart AJ, Kefford RF, Lim SY, Shklovskaya E, Rizos H. Interferon signaling is frequently downregulated in melanoma. Front Immunol. (2018) 9:1414. doi: 10.3389/fimmu.2018.01414
Keywords: checkpoint inhibitors, combination immunotherapy, immunooncology therapeutics, melanoma, CTLA-4, PD-1, PD-L1, antibody engineering
Citation: Khair DO, Bax HJ, Mele S, Crescioli S, Pellizzari G, Khiabany A, Nakamura M, Harris RJ, French E, Hoffmann RM, Williams IP, Cheung A, Thair B, Beales CT, Touizer E, Signell AW, Tasnova NL, Spicer JF, Josephs DH, Geh JL, MacKenzie Ross A, Healy C, Papa S, Lacy KE and Karagiannis SN (2019) Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 10:453. doi: 10.3389/fimmu.2019.00453
Received: 30 August 2018; Accepted: 20 February 2019;
Published: 19 March 2019.
Edited by:
Christian Ostheimer, Martin Luther University of Halle-Wittenberg, GermanyReviewed by:
Nicole Joller, University of Zurich, SwitzerlandDaniel Olive, Aix Marseille Université, France
Copyright © 2019 Khair, Bax, Mele, Crescioli, Pellizzari, Khiabany, Nakamura, Harris, French, Hoffmann, Williams, Cheung, Thair, Beales, Touizer, Signell, Tasnova, Spicer, Josephs, Geh, MacKenzie Ross, Healy, Papa, Lacy and Karagiannis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophia N. Karagiannis, c29waGlhLmthcmFnaWFubmlzQGtjbC5hYy51aw==