 Rayoun Ramendra1,2,3
Rayoun Ramendra1,2,3 Stéphane Isnard1,2
Stéphane Isnard1,2 Vikram Mehraj4
Vikram Mehraj4 Jun Chen1,2
Jun Chen1,2 Yonglong Zhang5
Yonglong Zhang5 Malcolm Finkelman5
Malcolm Finkelman5 Jean-Pierre Routy1,2,6*
Jean-Pierre Routy1,2,6*- 1Chronic Viral Illness Service, McGill University Health Centre, Montreal, QC, Canada
- 2Infectious Diseases and Immunity in Global Health Program, Research Institute, McGill University Health Centre, Montreal, QC, Canada
- 3Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
- 4Centre de Recherche du Centre Hospitalier de l'Université de Montréal, Montreal, QC, Canada
- 5Associates of Cape Cod Inc., Falmouth, MA, United States
- 6Division of Hematology, McGill University Health Centre, Montreal, QC, Canada
Immune activation is the driving force behind the occurrence of AIDS and non-AIDS events, and is only partially reduced by antiretroviral therapy (ART). Soon after HIV infection, intestinal CD4+ T cells are depleted leading to epithelial gut damage and subsequent translocation of microbes and/or their products into systemic circulation. Bacteria and fungi are the two most abundant populations of the gut microbiome. Circulating lipopolysaccharide (LPS) and (1→3)-β-D-Glucan (βDG), major components of bacterial and fungal cell walls respectively, are measured as markers of microbial translocation in the context of compromised gut barriers. While LPS is a well-known inducer of innate immune activation, βDG is emerging as a significant source of monocyte and NK cell activation that contributes to immune dysfunction. Herein, we critically evaluated recent literature to untangle the respective roles of LPS and βDG in HIV-associated immune dysfunction. Furthermore, we appraised the relevance of LPS and βDG as biomarkers of disease progression and immune activation on ART. Understanding the consequences of elevated LPS and βDG on immune activation will provide insight into novel therapeutic strategies against the occurrence of AIDS and non-AIDS events.
Introduction
The gastrointestinal tract (GI) is a dynamic setting constantly in contact with nutrients, allergens, commensal microbes, and pathogens. As such, this milieu is equipped with a complex and well balanced system of physical and immunological barriers to allow the absorption of nutrients while preventing the translocation of microbes and their products (1). A physical barrier is formed by firmly linked intestinal epithelial cells (enterocytes) connected via tight junctions. These cells form villi to maximize the absorption of nutrients. The base of each villus forms crypts composed of intestinal stem cells and Paneth cells which secrete growth factors that promote intestinal stem cell proliferation, antimicrobial peptide secretion, and digestive enzyme production (2, 3). In the upper regions of the villus, goblet cells contribute to the physical and chemical barriers by secreting a mucous layer that protects the gut epithelium from the microbiota (4). Patrolling leukocytes in the lamina propria constitute an immunological barrier that ensures any pathogens in the lamina propria are phagocytosed, cleared, and sent to the draining mesenteric lymph nodes (5).
HIV-infection is characterized by the depletion of gut CD4+ T cells, epithelial gut damage, and translocation of microbes and their products into systemic circulation (6). People living with HIV (PLWH) have damage to the gut epithelium which has been shown to precede immune activation in models of SIV-infected rhesus macaques (7, 8). As systemic immune activation is considered the driving force of CD4+ T cell depletion and development of acquired immunodeficiency syndrome (AIDS), it is important to understand the link between epithelial gut damage and systemic immune activation in PLWH.
In 2006, Brenchley et al. were the first to report that increased plasma levels of gram-negative bacterial cell wall antigen lipopolysaccharide (LPS) induces systemic immune activation in both PLWH and SIV-infected rhesus macaques (9). Estes et al. in 2010, demonstrated that elevated plasma levels of LPS in PLWH is a result of HIV-induced epithelial gut damage allowing for the translocation of microbial products from the gut microbiota into systemic circulation (10). Despite the success of antiretroviral therapy (ART), epithelial gut damage, microbial translocation, and to a lesser extent systemic immune activation are not reversed. In parallel, Cani et al. coined the term “metabolic endotoxemia” to describe the phenomenon of obese individuals with high plasma levels of LPS linked to reduced insulin sensitivity and increased risk of metabolic diseases (11). Furthermore, conserved parts of the LPS molecule act as a pathogen-associated molecular patterns (PAMPs) that have been associated in vitro, in animal models, and epidemiologically with increased innate immune activation, inflammation, and risk of developing non-AIDS events in ART-treated PLWH (9, 12).
Increasing awareness about the human gut microbiota reveals that it is a complex community of bacteria, fungi, archaea, viruses, and parasites influencing health and disease (13). However, studies regarding microbial translocation in PLWH have primarily focused on bacterial translocation. (1→3)-β-D-Glucan (βDG), a major component of most fungal cell walls, is commonly used as a biomarker for the diagnosis and management of invasive fungal infections (IFI) and has been recently used as a marker of fungal translocation in people without IFI (14). In 2012, Morris et al. were the first to show elevated plasma levels of βDG in PLWH (15). We and others have found that plasma levels of βDG are associated with epithelial gut damage, immune activation, inflammation, and risk of developing non-AIDS events (16–20). Like epithelial gut damage, plasma levels of βDG do not normalize despite long-term ART (20). These findings show converging evidence that like LPS, βDG also plays a significant role in chronic immune activation and development of non-AIDS events in PLWH (Figure 1).
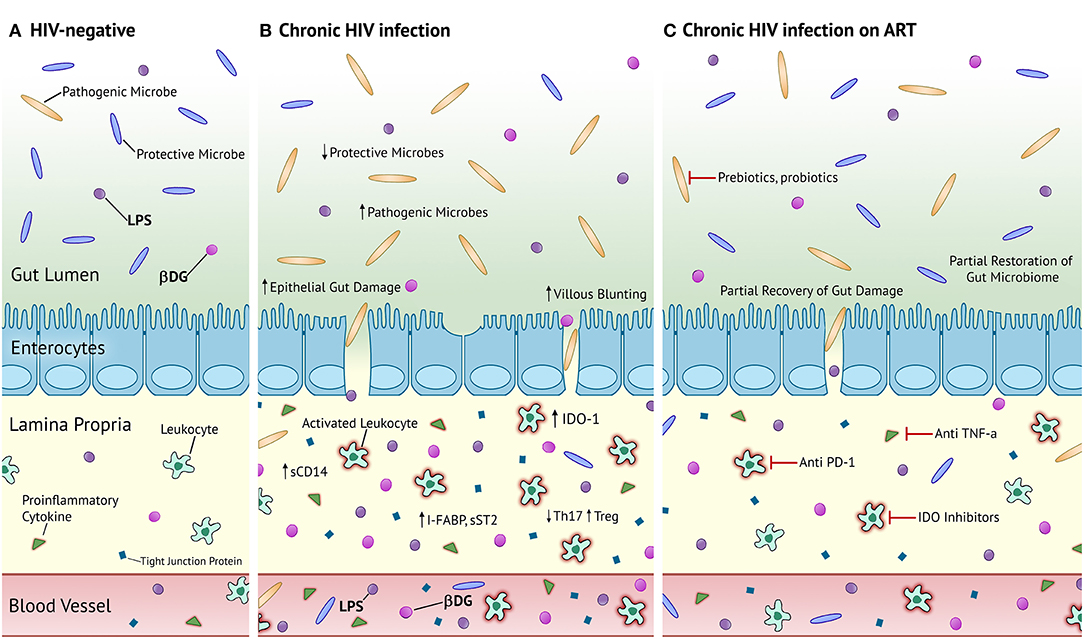
Figure 1. Proposed mechanism of microbial translocation in chronic HIV-infection and chronic HIV-infection on ART compared to HIV- homeostasis. (A) Healthy, HIV-uninfected individuals have an abundance of protective microbes in the gut and tight junctions connecting enterocytes to prevent the translocation of gut microbes and their products. Patrolling leukocytes in the lamina propria are not activated and there is an almost negligible level of inflammation and epithelial damage. (B) PLWH in the chronic stage of infection have increased proportions of pathogenic microbes and decreased proportions of protective microbes in the gut. There is severe villous blunting, epithelial gut damage, and microbial translocation. Patrolling leukocytes are activated and secrete large quantities of pro-inflammatory cytokines. There is elevated circulation of markers of epithelial gut damage, microbial translocation, and inflammation. (C) PLWH on long-term ART have partially restored composition of the gut microbiota, epithelial gut damage, and reduced microbial translocation. Activated leukocytes and systemic inflammation persists. Prebiotics and probiotics are potential therapeutic strategies to fully recover the composition of the gut microbiota. Anti PD-1, anti TNF-α, and IDO inhibitors represent potential therapeutic strategies against persistent immune activation and inflammation. LPS, lipopolysaccharide; βDG, (1→3)-β-D-Glucan; sCD14, soluble cluster of differentiation 14; I-FABP, intestinal fatty acid binding protein; sST2, soluble suppressor of tumorigenicity 2; IDO-1, indoleamine-2,3-dioxygenase 1.
Currently, persistence of systemic immune activation and development of non-AIDS events despite long-term ART represents one of the hurdles in caring for PLWH (21). Herein, we look to comprehensively review the existing English literature regarding the contribution of circulating LPS and βDG in systemic immune activation in PLWH. Understanding the consequences of LPS and βDG antigenemia will help with the development of therapeutic strategies against this “folie à deux.”
Gut Damage
In homeostatic conditions, the microbiota is contained within the gut by the mucous layer, epithelial barrier, and residential leukocytes (22–25).
HIV infection leads to early disruption of the gut epithelial barrier characterized by villous atrophy, crypt hyperplasia, increased gastrointestinal inflammation, and increased intestinal permeability (7, 26–28). Such damage is not completely restored despite long-term ART. Deterioration of the gastrointestinal landscape in PLWH and SIV-infected rhesus macaques has been shown to cause microbial translocation and resultant immune activation (9, 10, 29). Interestingly, SIV-infected sooty mangabeys, which do not progress to AIDS, present without disruption of the GI epithelial layer nor increased microbial translocation, and limited immune activation (30, 31). Thus, understanding the cause(s) and implications of epithelial gut damage in PLWH may help to understand the source(s) of systemic immune activation.
The precise mechanisms responsible for HIV-associated epithelial gut damage remain poorly understood and are now known to precede mucosal immune dysfunction (7). HIV gp120 and Tat proteins have been shown to have detrimental effects on intestinal epithelial cells (32–36). HIV induces inflammasome to produce IL-18, resulting in intestinal epithelial cell death and reduced expression of tight junction proteins, contributing to intestinal permeability and resultant microbial translocation (37, 38). Globally, HIV contributes to epithelial gut damage which is partially improved on ART (39, 40). Markers of epithelial gut damage such as soluble suppressor of tumorigenicity 2 (sST2) and intestinal fatty acid binding protein (I-FABP) have been reported to be elevated in inflammatory bowel diseases, graft vs. host disease, and HIV (41, 42).
As reported by Hensley-McBain et al., alterations to the intestinal epithelial structure precede mucosal immune dysfunction (7). Early mucosal damage in PLWH is partially explained by a substantially high expression of CCR5, a HIV co-receptor, on CD4+ T cells in the gut as compared to peripheral blood (43, 44). This is in line with findings that HIV is 10 times more likely to infect CD4+ T cells in the gut compared to peripheral blood (45). IL-17 producing cells, such as Th17 and Th22 CD4+ T cells, are known to homeostatically maintain the epithelial barrier (7). We and others have reported alterations of the Th17/Treg ratio in PLWH, owing to increased frequency of Tregs and decreased frequency of Th17 CD4+ T cells (46, 47). This in turn impairs the homeostatic response to prevent and restore epithelial gut damage in PLWH. HIV-associated gastrointestinal abnormalities have also been associated with changes in the composition of the gut microbiome (dysbiosis) and translocation of microbial products in PLWH.
Bacterial and Fungal Translocation
In homeostatic conditions, microbial translocation is limited by physical and immunological barriers in the intestine (48). However, when the gut epithelium is damaged and the mucosal immune system is compromised, microbial products translocate out of the gut into systemic circulation via the lamina propria (49, 50). Microbial translocation has been implicated in several conditions including Crohn's disease, ulcerative colitis, graft-vs.-host disease, and HIV (40).
SIV-infected rhesus macaques were shown to have elevated plasma levels of LPS that could be partially reversed with antibiotics. Immunohistochemistry and immunofluorescence of gut biopsies demonstrated that such elevation was a result of bacterial translocation from the gut lumen into systemic circulation via the lamina propria (9, 10). Interestingly, SIV-infected African green monkeys, a natural host of SIV, receiving intravenous injection of LPS had increased viral replication, mucosal CD4+ T cell depletion, and systemic immune activation without inducing epithelial gut damage (51, 52). Altogether, this suggests that translocation of microbial products plays a major role in systemic immune activation and inflammation.
We and others have shown elevation of microbial products in the blood of PLWH (9, 15, 46). Most published studies measured microbial translocation using plasma levels of bacterial DNA fragments, LPS, LPS binding protein (LBP), soluble CD14 (sCD14), and EndoCab (50). Bacterial DNA fragments were quantified using 16S rDNA PCR. Most studies in PLWH use the Limulus Amebocyte Lysate (LAL) assay to measure plasma levels of LPS, a major component of the outer membrane of gram-negative bacteria. LPS binds to LBP, which transfers LPS onto monocytes/macrophages causing the release of soluble CD14 (sCD14) (53). EndoCab is a group of antibodies specific for the core of LPS that are produced by B cells in response to enteric gram-negative bacteria (54). LBP, sCD14, and EndoCab are commonly measured in plasma/serum using solid-phase enzyme-linked immunosorbent assay (ELISA) as circulating biomarkers of LPS-induced innate immune activation (9, 39).
Most research on microbial translocation in PLWH has been focused on bacterial translocation (13). Morris et al. were the first to show elevation of circulating levels of fungal cell wall antigen, βDG, in PLWH (15). Hoenigl et al. have shown that plasma βDG is inversely correlated with the abundance of Lactobacilli in the distal gut (19). Lactobacillus is a protective genus of bacteria that is inversely associated with gut integrity and distal gut permeability. Furthermore, we have shown that plasma levels of βDG is strongly correlated with classical marker of epithelial gut damage, I-FABP, and markers of microbial translocation LPS and sCD14 (20) (Table 1). The origin of circulating βDG was first studied in murine models. As opposed to the human gut microbiome, there is no Candida in the murine gut. Mice fed with live or heat-inactivated Candida had elevated serum levels of βDG. Such elevation induced the production of pro-inflammatory cytokines IL-6 and TNF-α. Administration of Fluconazole, an anti-fungal small molecule, partially inhibited the elevation of serum βDG and systemic inflammation (55). Similar results were found in a murine model of lupus and sepsis (56, 57). As PLWH without invasive fungal infections are highly susceptible to increased proportions of fungal colonization and have high levels of epithelial gut damage, it is highly likely that elevated plasma levels of βDG in PLWH originates from the gut (14). Thus, there is a need to definitely determine whether elevated plasma levels of βDG in PLWH is a result of microbial translocation and involved in systemic immune activation.
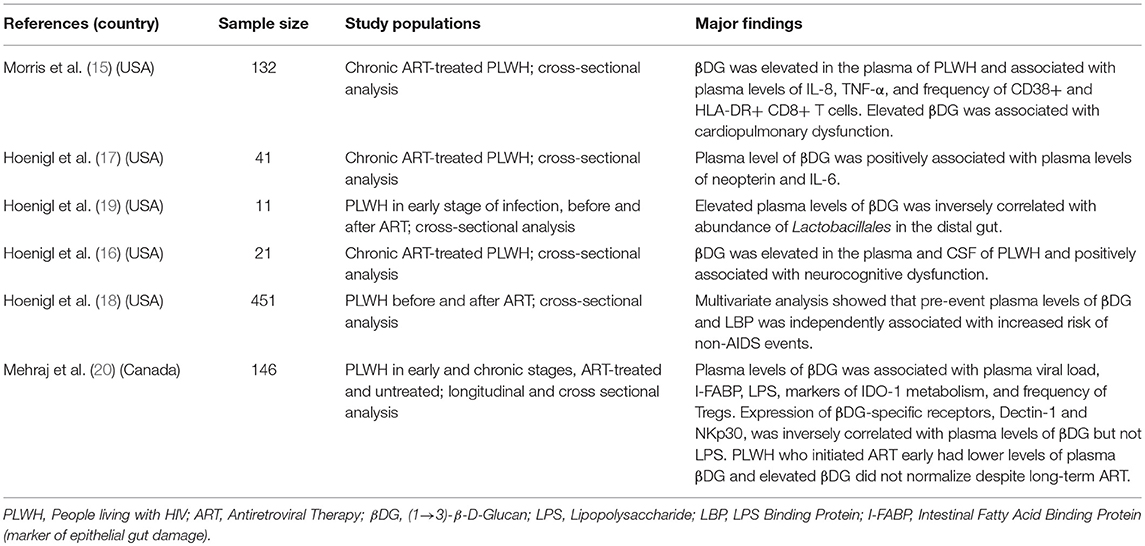
Table 1. Overview of studies associating elevation of plasma levels of βDG with immune activation and immune dysfunction in PLWH.
Microbial Translocation and Systemic Immune Activation
Microbial products such as LPS and βDG represent potent PAMPs that trigger a significant immune response. Several studies have provided convincing evidence that elevated plasma levels of LPS induces immune activation in sepsis, Crohn's disease, ulcerative colitis, obesity, and HIV (9, 11, 58, 59).
LPS is captured by LBP and complexed with CD14, myeloid differentiation 2 protein, and Toll like receptor 4 (TLR4). The formation of this complex is crucial for the immune system to mount a response to LPS. Classical antigen presenting cells (APCs) recognize this complex using TLR4 and subsequently phagocytose LPS while shedding sCD14. B cells also express TLR4 to recognize different parts of the LPS core lipids and secrete EndoCAb to facilitate the phagocytosis of the antigen (60). LPS has been shown to induce the secretion of several pro-inflammatory cytokines including IL-6, IL-8, and TNF-α by APCs (61). Plasma levels of LPS correlated with plasma levels of IFN-α and frequency of activated CD4+ and CD8+ T cells in PLWH (9). Similarly, plasma levels of sCD14 correlated with plasma levels of IL-6 and C reactive protein in PLWH (62).
Meanwhile, βDG is predominantly recognized by complement receptor 3 (CR3), Dectin-1, NKp30, Ephrin type-A receptor 2 (EphA2), and Langerin. CR3 is a ubiquitous heterodimer receptor composed of CD11b and CD18. Recent findings have shown reduced expression of CR3 on both myeloid and plasmacytoid dendritic cells (DC) in PLWH. βDG-specific interactions with CR3 on DC have been shown to increase IL-6 and TNF-α production by activating the Syk-JNK-AP-1 pathway (63). Dectin-1 represents the most prominent myeloid cell receptor for βDG and is expressed on monocytes, macrophages, DCs, and neutrophils (63–65). We have shown that Dectin-1 expression on monocytes is reduced in PLWH and that such expression is inversely correlated with plasma levels of βDG but not LPS (20). βDG-specific binding to Dectin-1 leads to the production of pro-inflammatory cytokines IL-6, IL-8, and TNF-α by myeloid cells (66, 67). βDG is also specifically recognized by NK cells via NKp30, a functional activation receptor (68). In addition, we have shown that NKp30 expression is diminished in PLWH and inversely correlated with plasma levels of βDG but not LPS (20, 69). NKp30-specific binding has been shown to induce activation and the production of pro-inflammatory cytokines such as IL-1β and TNF-α (70, 71). EphA2 is a βDG-specific receptor expressed on epithelial cells, predominantly in the colon and small intestine, that has yet to be measured in PLWH (72). Interestingly, EphA2 has also been identified as a receptor for Kaposi Sarcoma associated herpes virus (also called HHV8), one of the most common HIV-associated co-infections (73). DCs play an important role in maintaining mucosal homeostasis. In the mucosa, they can be distinguished according to their expression of C-type lectins: Langerin [expressed by Langerhans cells (LCs)] and DC-SIGN (expressed by classical DCs). LCs reside in the epithelium of most mucosal surfaces and are thus one of the first APCs to encounter HIV as well as products of microbial translocation. Langerin has been shown to be an important receptor for βDG during Candida and Saccharomyces infections that has yet to be assessed in PLWH without IFI (74). βDG induces the secretion of pro-inflammatory cytokines IL-1β, IL-6, IL-8, IL-23, TNF-α, and chemokine CCL22 that has been shown to increase monocyte recruitment to the colon (66, 75, 76). Indeed, we and others have shown that elevated plasma levels of βDG is correlated with plasma levels of IL-6 and IL-8 in PLWH (15, 20).
Microbial translocation in PLWH is associated with Indoleamine-2,3-deoxygenase-1 (IDO-1) activity and HIV disease progression (77). IDO-1 is expressed in all myeloid cells and activated after PAMPs recognition to metabolize Tryptophan into Kynurenines (78). As such, IDO-1 activity is considered a marker of inflammation and immune activation. We and others have shown that IDO-1 activity is increased in PLWH and does not normalize with early ART. In PLWH, IDO-1 activity is associated with plasma levels of LPS and βDG, increased frequency of Tregs, epithelial gut damage, microbial translocation, immune activation, and HIV reservoir size (20, 46, 79, 80).
Persistent epithelial gut damage and elevated plasma levels of LPS and βDG, despite long-term ART, likely contribute to inflammation and chronic immune activation leading to the development of non-AIDS events in PLWH. In the ART-era, the development of non-AIDS events represents one of the challenges to caring for PLWH. Therefore, both LPS and βDG represent important therapeutic targets to reduce the risk of developing non-AIDS events.
Microbial Translocation, Inflammation, and non-AIDS Events
Despite the significant success of ART, PLWH still present with high rates of non-AIDS events that includes HIV-associated neurocognitive disorders (HANDs), cardiovascular diseases, renal failure, liver steatosis, and cancer (81, 82). Such non-AIDS events have been associated with epithelial gut damage, microbial translocation, and systemic immune activation (50). Hoenigl et al. have observed in a large cohort of ART-treated PLWH that in addition to soluble urokinase-type plasminogen receptor, plasma levels of βDG, and LBP represent two of the best predictors of increased risk of non-AIDS events (18).
Microbial Translocation and HAND
PLWH present with HANDs including asymptomatic neurocognitive impairment, mild neurocognitive disorder, and dementia (83, 84). Previous studies have found strong associations between plasma levels of sCD14, LPS, and βDG with neurocognitive dysfunction (16, 85). Moreover, ART-treated PLWH with severe neurocognitive dysfunction also presented with elevated sCD14 and βDG in their cerebrospinal fluid (CSF) (16). Supporting the concept of the gut-brain axis, increased microbial translocation likely plays a crucial role in the development of HANDs.
Microbial Translocation and Cardiovascular Diseases
A study with more than 27,000 participants showed that PLWH had a two-fold increased risk of developing acute myocardial infarction in every age group compared to matched control participants (86, 87). Elevated circulation of microbial products and resultant inflammation are associated with increased risk of heart disease (88). Plasma levels of LPS have been associated with known risk factors for cardiovascular diseases such as decreased insulin sensitivity and higher total cholesterol (11). Similarly, elevated plasma levels of βDG have also been associated with cardiopulmonary dysfunction (15).
Conclusions and Future Directions
Due to the success of ART, the life expectancy and quality of life of PLWH has dramatically improved over the course of the last decade. While early initiation of ART is associated with lower reservoir size and reduced immune activation, PLWH on ART still suffer from unrecovered epithelial gut damage and chronic immune activation (89). It has been recently reported that epithelial gut damage precedes systemic immune activation in a SIV-infected rhesus macaque model (7). Like epithelial gut damage and systemic immune activation, markers of microbial translocation do not normalize despite long-term ART (20). Thus, understanding the mechanisms via which epithelial gut damage and resultant microbial translocation contribute to chronic immune activation is essential toward improving the prognosis of PLWH.
Circulating microbial polysaccharides LPS and βDG are elevated in PLWH (9, 15). Previous research on microbial translocation in PLWH and SIV-infected rhesus macaques has been primarily measured by plasma levels of LPS using the LAL assay. Of note, it has been initially found that LPS measured by the LAL assay also measures βDG (90). While it has been shown that increased plasma levels of LPS are a result of microbial translocation in PLWH, the source of circulating βDG remains to be clarified. Consumption of certain mushrooms, oat fiber, and seaweed can also increase circulating levels of βDG (91). We and others have shown that plasma levels of βDG are positively associated with marker of epithelial gut damage I-FABP and inversely associated with the abundance of protective bacteria, Lactobacillales, in the distal gut (19, 20). Furthermore, murine models have shown that mice fed with heat-killed or live Candida have increased levels of circulating βDG after cecal ligation and puncture (55). Similarly, people with intestine disorders have increased plasma levels of βDG (56, 92, 93). Hence, there is converging evidence suggesting that elevated plasma levels of βDG in PLWH are a result of increased Candida or other fungal colonization, epithelial gut damage, and subsequent microbial translocation.
Both LPS and βDG have been associated with markers of systemic immune activation, inflammation, and the development of non-AIDS events. As such, the individual and potentially synergistic consequences of elevated plasma levels of LPS and βDG on systemic immune activation must be urgently addressed.
Understanding the respective mechanisms by which these two microbial polysaccharides contribute to chronic immune activation may lead to the development of novel therapeutic strategies against inflammation and the development of non-AIDS events in PLWH. Moreover, genetic factors may play a key role in determining the influence of these PAMPs on systemic immune activation. For example, people with CARD9 deficiencies have been shown to have increased susceptibility to fungal infections (94). Furthermore, Palesch et al. demonstrated that sooty mangabeys, a natural host of SIV, had a frameshift mutation in their TLR4 gene that was associated with a blunted response to TLR4 ligands in vitro (95). Thus, regular genetic variations in receptors for LPS and βDG may also play a pivotal role in determining the consequences of microbial translocation in PLWH.
In 2014, Kristoff et al. gave Sevelamer, a drug known to decrease circulating LPS (96), to SIV-infected pigtailed macaques. A single dose administration led to partially decreased HIV viral replication, decreased circulation of coagulation markers, and decreased immune activation/inflammation (52). However, when Sandler et al. gave Sevelamer to 36 ART-naïve PLWH, they did not observe a decrease in plasma levels of LPS (measured by LAL assay) nor markers of immune activation/inflammation (97). Future clinical trials should aim to reduce the burden of elevated circulation of products of both bacterial and fungal translocation. While therapeutic strategies targeting elevated circulation of PAMPs should be investigated, future studies should also look to target immune signaling molecules (such as TLR4 and Dectin-1) to reduce LPS and βDG induced systemic immune activation.
Overall, elevated plasma levels of LPS and βDG both play an important role in chronic immune activation in PLWH on long-term ART and may represent a “folie à deux” contributing to the development of non-AIDS events. To this end, gaining a comprehensive understanding of the origin and consequences of these circulating microbial polysaccharides is of critical importance to finding therapeutic strategies to restore mucosal homeostasis, and gut dysbiosis in PLWH.
Author Contributions
RR made the first draft, constructed the figure and table, and made revisions to the final draft of the manuscript. RR and SI contributed significantly to conducting the literature review. VM, JC, YZ, and MF critically read and revised the manuscript. J-PR designed the review and critically revised the manuscript.
Funding
This study was supported by the Fonds de recherche du Québec—Santé (FRQ-S): Réseau SIDA/Maladies infectieuses and Thérapie cellulaire, the Canadian Institute of Health Research (CIHR grants MOP 103230 and 154051), the Vaccines & Immunotherapies Core of the CIHR Canadian HIV Trials Network (CTN): grant CTN 257, the Canadian Foundation for AIDS Research (CANFAR) grant 02-512, and the Canadian HIV Cure Enterprise Team Grant (HIG-133050) awarded by the CIHR in partnership with CANFAR. RR is an undergraduate student supported by the H. Grenville Smith Studentship, SI is a post-doctoral fellow supported by the William Turner research fellowship, J-PR is the holder of the Louis Lowenstein Chair in Hematology and Oncology at McGill University.
Conflict of Interest Statement
MF and YZ are employees of Associates of Cape Cod, Inc., the manufacturers of Fungitell, the (1→3)-β-D-Glucan in vitro diagnostic kit.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to study participants for their contribution. We thank Angie Massicotte and Josée Girouard for administrative assistance.
References
1. Perez-Lopez A, Behnsen J, Nuccio SP, Raffatellu M. Mucosal immunity to pathogenic intestinal bacteria. Nat Rev Immunol. (2016) 16:135–48. doi: 10.1038/nri.2015.17
2. Ouellette AJ. Paneth cells and innate mucosal immunity. Curr Opin Gastroenterol. (2010) 26:547–53. doi: 10.1097/MOG.0b013e32833dccde
3. Bry L, Falk P, Huttner K, Ouellette A, Midtvedt T, Gordon JI. Paneth cell differentiation in the developing intestine of normal and transgenic mice. Proc Natl Acad Sci USA. (1994) 91:10335–9. doi: 10.1073/pnas.91.22.10335
4. Birchenough GM, Johansson ME, Gustafsson JK, Bergstrom JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal Immunol. (2015) 8:712–9. doi: 10.1038/mi.2015.32
5. Luissint AC, Parkos CA, Nusrat A. Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology. (2016) 151:616–32. doi: 10.1053/j.gastro.2016.07.008
6. Ghosn J, Taiwo B, Seedat S, Autran B, Katlama C. HIV. Lancet. (2018) 392:685–97. doi: 10.1016/S0140-6736(18)31311-4
7. Hensley-McBain T, Berard AR, Manuzak JA, Miller CJ, Zevin AS, Polacino P, et al. Intestinal damage precedes mucosal immune dysfunction in SIV infection. Mucosal Immunol. (2018) 11:1429–40. doi: 10.1038/s41385-018-0032-5
8. Brenchley JM, Douek DC. Microbial translocation across the GI tract. Annu Rev Immunol. (2012) 30:149–73. doi: 10.1146/annurev-immunol-020711-075001
9. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. (2006) 12:1365–71. doi: 10.1038/nm1511
10. Estes JD, Harris LD, Klatt NR, Tabb B, Pittaluga S, Paiardini M, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. (2010) 6:e1001052. doi: 10.1371/journal.ppat.1001052
11. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
12. Baker JV, Peng G, Rapkin J, Abrams DI, Silverberg MJ, MacArthur RD, et al. Community programs for clinical research on, CD4+ count and risk of non-AIDS diseases following initial treatment for HIV infection. AIDS. (2008) 22:841–8. doi: 10.1097/QAD.0b013e3282f7cb76
13. Mirzaei MK, Maurice CF. Menage a trois in the human gut: interactions between host, bacteria and phages. Nat Rev Microbiol. (2017) 15:397–408. doi: 10.1038/nrmicro.2017.30
14. Farhour Z, Mehraj V, Chen J, Ramendra R, Lu H, Routy JP. Use of (1–>3)-beta-D-Glucan for diagnosis and management of invasive mycoses in HIV-infected patients. Mycoses. (2018) 61:718–22. doi: 10.1111/myc.12797
15. Morris A, Hillenbrand M, Finkelman M, George MP, Singh V, Kessinger C, et al. Serum (1–>3)-beta-D-glucan levels in HIV-infected individuals are associated with immunosuppression, inflammation, and cardiopulmonary function. J Acquir Immune Defic Syndr. (2012) 61:462–8. doi: 10.1097/QAI.0b013e318271799b
16. Hoenigl M, de Oliveira MF, Perez-Santiago J, Zhang Y, Morris S, McCutchan AJ, et al. (1–>3)-beta-D-glucan levels correlate with neurocognitive functioning in HIV-infected persons on suppressive antiretroviral therapy: a cohort study. Medicine. (2016) 95:e3162. doi: 10.1097/MD.0000000000003162
17. Hoenigl M, de Oliveira MF, Perez-Santiago J, Zhang Y, Woods SP, Finkelman M, et al. Correlation of (1–>3)-beta-D-glucan with other inflammation markers in chronically HIV infected persons on suppressive antiretroviral therapy. GMS Infect Dis. (2015) 3:Doc3. doi: 10.3205/id000018
18. Hoenigl M, Moser C, Funderburg N, Bosch R, Kantor A, Zhang Y, et al. Soluble urokinase plasminogen activator receptor (suPAR) is predictive of non-AIDS events during antiretroviral therapy-mediated viral suppression. Clin Infect Dis. (2018). doi: 10.1093/cid/ciy966. [Epub ahead of print].
19. Hoenigl M, Perez-Santiago J, Nakazawa M, de Oliveira MF, Zhang Y, Finkelman MA, et al. (1–>3)-beta-d-Glucan: a biomarker for microbial translocation in individuals with acute or early HIV infection? Front Immunol. (2016) 7:404. doi: 10.3389/fimmu.2016.00404
20. Mehraj V, Ramendra R, Isnard S, Dupuy FP, Ponte R, Chen J, et al. Circulating (1 → 3)-β-D-Glucan is associated with immune activation during HIV infection. Clin Infect Dis.
21. Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. (2013) 39:633–45. doi: 10.1016/j.immuni.2013.10.001
22. Nash AK, Auchtung TA, Wong MC, Smith DP, Gesell JR, Ross MC, et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome. (2017) 5:15. doi: 10.1186/s40168-017-0373-4
23. Waldman AJ, Balskus EP. The human microbiota, infectious disease, and global health: challenges and opportunities. ACS Infect Dis. (2018) 4:14–26. doi: 10.1021/acsinfecdis.7b00232
24. Sheridan BS, Lefrancois L. Intraepithelial lymphocytes: to serve and protect. Curr Gastroenterol Rep. (2010) 12:513–21. doi: 10.1007/s11894-010-0148-6
25. Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. (2009) 124:3–20; quiz 21–2. doi: 10.1016/j.jaci.2009.05.038
26. Batman PA, Miller AR, Forster SM, Harris JR, Pinching AJ, Griffin GE. Jejunal enteropathy associated with human immunodeficiency virus infection: quantitative histology. J Clin Pathol. (1989) 42:275–81. doi: 10.1136/jcp.42.3.275
27. Batman PA, Kotler DP, Kapembwa MS, Booth D, Potten CS, Orenstein JM, et al. HIV enteropathy: crypt stem and transit cell hyperproliferation induces villous atrophy in HIV/Microsporidia-infected jejunal mucosa. AIDS. (2007) 21:433–9. doi: 10.1097/QAD.0b013e3280142ee8
28. Heise C, Dandekar S, Kumar P, Duplantier R, Donovan RM, Halsted CH. Human immunodeficiency virus infection of enterocytes and mononuclear cells in human jejunal mucosa. Gastroenterology. (1991) 100:1521–7. doi: 10.1016/0016-5085(91)90648-5
29. Klatt NR, Harris LD, Vinton CL, Sung H, Briant JA, Tabb B, et al. Compromised gastrointestinal integrity in pigtail macaques is associated with increased microbial translocation, immune activation, and IL-17 production in the absence of SIV infection. Mucosal Immunol. (2010) 3:387–98. doi: 10.1038/mi.2010.14
30. Milush JM, Mir KD, Sundaravaradan V, Gordon SN, Engram J, Cano CA, et al. Lack of clinical AIDS in SIV-infected sooty mangabeys with significant CD4+ T cell loss is associated with double-negative T cells. J Clin Invest. (2011) 121:1102–10. doi: 10.1172/JCI44876
31. Gordon SN, Klatt NR, Bosinger SE, Brenchley JM, Milush JM, Engram JC, et al. Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. J Immunol. (2007) 179:3026–34. doi: 10.4049/jimmunol.179.5.3026
32. Buccigrossi V, Laudiero G, Nicastro E, Miele E, Esposito F, Guarino A. The HIV-1 transactivator factor (Tat) induces enterocyte apoptosis through a redox-mediated mechanism. PLoS ONE. (2011) 6:e29436. doi: 10.1371/journal.pone.0029436
33. Canani RB, De Marco G, Passariello A, Buccigrossi V, Ruotolo S, Bracale I, et al. Inhibitory effect of HIV-1 Tat protein on the sodium-D-glucose symporter of human intestinal epithelial cells. AIDS. (2006) 20:5–10. doi: 10.1097/01.aids.0000198088.85572.68
34. Planes R, Ben Haij N, Leghmari K, Serrero M, BenMohamed L, Bahraoui E. HIV-1 Tat protein activates both the MyD88 and TRIF pathways to induce tumor necrosis factor alpha and interleukin-10 in human monocytes. J Virol. (2016) 90:5886–98. doi: 10.1128/JVI.00262-16
35. Maresca M, Mahfoud R, Garmy N, Kotler DP, Fantini J, Clayton F. The virotoxin model of HIV-1 enteropathy: involvement of GPR15/Bob and galactosylceramide in the cytopathic effects induced by HIV-1 gp120 in the HT-29-D4 intestinal cell line. J Biomed Sci. (2003) 10:156–66. doi: 10.1007/BF02256007
36. Clayton F, Kotler DP, Kuwada SK, Morgan T, Stepan C, Kuang J, et al. Gp120-induced Bob/GPR15 activation: a possible cause of human immunodeficiency virus enteropathy. Am J Pathol. (2001) 159:1933–9. doi: 10.1016/S0002-9440(10)63040-4
37. Allam O, Samarani S, Mehraj V, Jenabian MA, Tremblay C, Routy JP, et al. HIV induces production of IL-18 from intestinal epithelial cells that increases intestinal permeability and microbial translocation. PLoS ONE. (2018) 13:e0194185. doi: 10.1371/journal.pone.0194185
38. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. (2015) 163:1444–56. doi: 10.1016/j.cell.2015.10.072
39. Mehraj V, Jenabian MA, Ponte R, Lebouche B, Costiniuk C, Thomas R, et al. Montreal primary Hiv infection, the plasma levels of soluble ST2 as a marker of gut mucosal damage in early HIV infection. AIDS. (2016) 30:1617–27. doi: 10.1097/QAD.0000000000001105
40. Ponte R, Mehraj V, Ghali P, Couedel-Courteille A, Cheynier R, Routy JP. Reversing gut damage in HIV infection: using non-human primate models to instruct clinical research. EBioMedicine. (2016) 4:40–9. doi: 10.1016/j.ebiom.2016.01.028
41. Mehraj V, Ponte R, Routy JP. The dynamic role of the IL-33/ST2 axis in chronic viral-infections: alarming and adjuvanting the immune response. EBioMedicine. (2016) 9:37–44. doi: 10.1016/j.ebiom.2016.06.047
42. Derikx JP, Luyer MD, Heineman E, Buurman WA. Non-invasive markers of gut wall integrity in health and disease. World J Gastroenterol. (2010) 16:5272–9. doi: 10.3748/wjg.v16.i42.5272
43. Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, et al. CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med. (2004) 200:749–59. doi: 10.1084/jem.20040874
44. Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med. (2004) 200:761–70. doi: 10.1084/jem.20041196
45. Lim SG, Condez A, Lee CA, Johnson MA, Elia C, Poulter LW. Loss of mucosal CD4 lymphocytes is an early feature of HIV infection. Clin Exp Immunol. (1993) 92:448–54. doi: 10.1111/j.1365-2249.1993.tb03419.x
46. Jenabian MA, El-Far M, Vyboh K, Kema I, Costiniuk CT, Thomas R, et al. Montreal primary, and G slow progressor study, immunosuppressive tryptophan catabolism and gut mucosal dysfunction following early HIV infection. J Infect Dis. (2015) 212:355–66. doi: 10.1093/infdis/jiv037
47. Klatt NR, Estes JD, Sun X, Ortiz AM, Barber JS, Harris LD, et al. Loss of mucosal CD103+ DCs and IL-17+ and IL-22+ lymphocytes is associated with mucosal damage in SIV infection. Mucosal Immunol. (2012) 5:646–57. doi: 10.1038/mi.2012.38
48. Macpherson AJ, Smith K. Mesenteric lymph nodes at the center of immune anatomy. J Exp Med. (2006) 203:497–500. doi: 10.1084/jem.20060227
49. Brenchley JM, Douek DC. HIV infection and the gastrointestinal immune system. Mucosal Immunol. (2008) 1:23–30. doi: 10.1038/mi.2007.1
50. Marchetti G, Tincati C, Silvestri G. Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev. (2013) 26:2–18. doi: 10.1128/CMR.00050-12
51. Pandrea I, Gaufin T, Brenchley JM, Gautam R, Monjure C, Gautam A, et al. Cutting edge: experimentally induced immune activation in natural hosts of simian immunodeficiency virus induces significant increases in viral replication and CD4+ T cell depletion. J Immunol. (2008) 181:6687–91. doi: 10.4049/jimmunol.181.10.6687
52. Kristoff J, Haret-Richter G, Ma D, Ribeiro RM, Xu C, Cornell E, et al. Early microbial translocation blockade reduces SIV-mediated inflammation and viral replication. J Clin Invest. (2014) 124:2802–6. doi: 10.1172/JCI75090
53. Triantafilou M, Triantafilou K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. (2002) 23:301–4. doi: 10.1016/S1471-4906(02)02233-0
54. Barclay GR. Endogenous endotoxin-core antibody (EndoCAb) as a marker of endotoxin exposure and a prognostic indicator: a review. Prog Clin Biol Res. (1995) 392:263–72.
55. Panpetch W, Somboonna N, Bulan DE, Issara-Amphorn J, Finkelman M, Worasilchai N, et al. Oral administration of live- or heat-killed Candida albicans worsened cecal ligation and puncture sepsis in a murine model possibly due to an increased serum (1–>3)-beta-D-glucan. PLoS ONE. (2017) 12:e0181439. doi: 10.1371/journal.pone.0181439
56. Issara-Amphorn J, Surawut S, Worasilchai N, Thim-Uam A, Finkelman M, Chindamporn A, et al. The synergy of endotoxin and (1–>3)-beta-D-glucan, from gut translocation, worsens sepsis severity in a lupus model of Fc gamma receptor IIb-deficient mice. J Innate Immun. (2018) 10:189–201. doi: 10.1159/000486321
57. Leelahavanichkul A, Worasilchai N, Wannalerdsakun S, Jutivorakool K, Somparn P, Issara-Amphorn J, et al. Gastrointestinal leakage detected by serum (1–>3)-beta-D-glucan in mouse models and a pilot study in patients with sepsis. Shock. (2016) 46:506–18. doi: 10.1097/SHK.0000000000000645
58. Opal SM. Endotoxins and other sepsis triggers. Contrib Nephrol. (2010) 167:14–24. doi: 10.1159/000315915
59. Caradonna L, Amati L, Magrone T, Pellegrino NM, Jirillo E, Caccavo D. Enteric bacteria, lipopolysaccharides and related cytokines in inflammatory bowel disease: biological and clinical significance. J Endotoxin Res. (2000) 6:205–14. doi: 10.1177/09680519000060030101
60. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. (2014) 5:316. doi: 10.3389/fimmu.2014.00316
61. Freudenberg MA, Tchaptchet S, Keck S, Fejer G, Huber M, Schutze N, et al. Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: benefits and hazards of LPS hypersensitivity. Immunobiology. (2008) 213:193–203. doi: 10.1016/j.imbio.2007.11.008
62. Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. (2011) 203:780–90. doi: 10.1093/infdis/jiq118
63. Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, et al. Dectin-1 is a major β-glucan receptor on macrophages. J Exp Med. (2002) 196:407–12. doi: 10.1084/jem.20020470
64. McDonald JU, Rosas M, Brown GD, Jones SA, Taylor PR. Differential dependencies of monocytes and neutrophils on dectin-1, dectin-2 and complement for the recognition of fungal particles in inflammation. PLoS ONE. (2012) 7:e45781. doi: 10.1371/journal.pone.0045781
65. Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, et al. The B-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol. (2002) 169:3876–82. doi: 10.4049/jimmunol.169.7.3876
66. Elder MJ, Webster SJ, Chee R, Williams DL, Hill Gaston JS, Goodall JC. beta-glucan size controls dectin-1-mediated immune responses in human dendritic cells by regulating IL-1beta production. Front Immunol. (2017) 8:791. doi: 10.3389/fimmu.2017.00791
67. Hefter M, Lother J, Weiss E, Schmitt AL, Fliesser M, Einsele H, et al. Human primary myeloid dendritic cells interact with the opportunistic fungal pathogen Aspergillus fumigatus via the C-type lectin receptor Dectin-1. Med Mycol. (2017) 55:573–8. doi: 10.1093/mmy/myw105
68. Li SS, Kyei SK, Timm-McCann M, Ogbomo H, Jones GJ, Shi M, et al. The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe. (2013) 14:387–97. doi: 10.1016/j.chom.2013.09.007
69. Tremblay-McLean A, Bruneau J, Lebouche B, Lisovsky I, Song R, Bernard NF. Expression profiles of ligands for activating natural killer cell receptors on HIV infected and uninfected CD4(+) T cells. Viruses. (2017) 9:295. doi: 10.3390/v9100295
70. Pandey R, DeStephan CM, Madge LA, May MJ, Orange JS. NKp30 ligation induces rapid activation of the canonical NF-kappaB pathway in NK cells. J Immunol. (2007) 179:7385–96. doi: 10.4049/jimmunol.179.11.7385
71. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
72. Swidergall M, Solis NV, Lionakis MS, Filler SG. EphA2 is an epithelial cell pattern recognition receptor for fungal beta-glucans. Nat Microbiol. (2018) 3:53–61. doi: 10.1038/s41564-017-0059-5
73. Hahn AS, Desrosiers RC. Binding of the Kaposi's sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. J Virol. (2014) 88:8724–34. doi: 10.1128/JVI.01392-14
74. Legentil L, Paris F, Ballet C, Trouvelot S, Daire X, Vetvicka V, et al. Molecular interactions of beta-(1–>3)-glucans with their receptors. Molecules. (2015) 20:9745–66. doi: 10.3390/molecules20069745
75. Noss I, Doekes G, Thorne PS, Heederik DJ, Wouters IM. Comparison of the potency of a variety of beta-glucans to induce cytokine production in human whole blood. Innate Immun. (2013) 19:10–9. doi: 10.1177/1753425912447129
76. Sonnier DI, Bailey SR, Schuster RM, Gangidine MM, Lentsch AB, Pritts TA. Proinflammatory chemokines in the intestinal lumen contribute to intestinal dysfunction during endotoxemia. Shock. (2012) 37:63–9. doi: 10.1097/SHK.0b013e31823cbff1
77. Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. (2013) 5:193ra91. doi: 10.1126/scitranslmed.3006438
78. Favre D, Mold J, Hunt PW, Kanwar B, Loke P, Seu L, et al. Tryptophan catabolism by indoleamine 2,3-dioxygenase 1 alters the balance of TH17 to regulatory T cells in HIV disease. Sci Transl Med. (2010) 2:32ra36. doi: 10.1126/scitranslmed.3000632
79. Jenabian MA, Patel M, Kema I, Kanagaratham C, Radzioch D, Thebault P, et al. Distinct tryptophan catabolism and Th17/Treg balance in HIV progressors and elite controllers. PLoS ONE. (2013) 8:e78146. doi: 10.1371/journal.pone.0078146
80. Chen J, Xun J, Yang J, Ji Y, Liu L, Qi T, et al. Plasma indoleamine 2,3-dioxygenase activity is associated with the size of HIV reservoir in patients receiving antiretroviral therapy. Clin Infect Dis. (2018) 10:676. doi: 10.1093/cid/ciy676
81. Kenneth Castro MDJ, Laurence Slutsker WWG, James Buehler PH, Harold Jaffe W, Ruth Berkelman MD. 1993 Revised Classification System 638 for HIV Infection and Expanded Surveillance Case Definition for AIDS Among Adolescents 639 and Adults. Centre For Disease Control (1992).
82. Hsu DC, Sereti I, Ananworanich J. Serious Non-AIDS events: immunopathogenesis and interventional strategies. AIDS Res Ther. (2013) 10:29. doi: 10.1186/1742-6405-10-29
83. Clifford DB, Ances BM. HIV-associated neurocognitive disorder. Lancet Infect Dis. (2013) 13:976–86. doi: 10.1016/S1473-3099(13)70269-X
84. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE. (2008) 3:e2516. doi: 10.1371/journal.pone.0002516
85. Lyons JL, Uno H, Ancuta P, Kamat A, Moore DJ, Singer EJ, et al. Plasma sCD14 is a biomarker associated with impaired neurocognitive test performance in attention and learning domains in HIV infection. J Acquir Immune Defic Syndr. (2011) 57:371–9. doi: 10.1097/QAI.0b013e3182237e54
86. Justice AC, Dombrowski E, Conigliaro J, Fultz SL, Gibson D, Madenwald T, et al. Veterans aging cohort study (VACS): overview and description. Med Care. (2006) 44:S13–24. doi: 10.1097/01.mlr.0000223741.02074.66
87. Marconi VC, Duncan MS, So-Armah K, Re VL III, Lim JK, Butt AA, et al. Bilirubin is inversely associated with cardiovascular disease among HIV-positive and HIV-negative individuals in VACS (Veterans aging cohort study). J Am Heart Assoc. (2018) 7:7792. doi: 10.1161/JAHA.117.007792
88. Drosatos K, Lymperopoulos A, Kennel PJ, Pollak N, Schulze PC, Goldberg IJ. Pathophysiology of sepsis-related cardiac dysfunction: driven by inflammation, energy mismanagement, or both? Curr Heart Fail Rep. (2015) 12:130–40. doi: 10.1007/s11897-014-0247-z
89. Chen RRJ, Lu H, Routy JP. The early bird gets the worm: benefits and future directions with early antiretroviral therapy initiation in primary HIV infection. Future Virol. (2018) 13:11. doi: 10.2217/fvl-2018-0110
90. Wong J, Zhang Y, Patidar A, Vilar E, Finkelman M, Farrington K. Is endotoxemia in stable hemodialysis patients an artefact? Limitations of the limulus amebocyte lysate assay and role of (1–>3)-beta-D glucan. PLoS ONE. (2016) 11:e0164978. doi: 10.1371/journal.pone.0164978
91. Hashimoto N, Mori T, Hashida R, Sakurai M, Koda Y, Toyama T, et al. False-positive serum (1, 3)-beta-D-glucan elevation due to intake of seaweed in a hematopoietic stem cell transplant recipient. Transpl Infect Dis. (2017) 19. doi: 10.1111/tid.12653
92. Zainab Shahid NSK, Restrepo A, Haider S, Muzaffar J, Grazziutti M, Nucci M, et al. Elevated serum beta-D-glucan (BDG) as a marker for chemotherapy-induced mucosal barrier injury (MBI) in adults with hematologic malignancies: a retrospective analysis. In: IDSA Annual Meeting. Boston, MA (2011).
93. Ellis M, Al-Ramadi B, Finkelman M, Hedstrom U, Kristensen J, Ali-Zadeh H, et al. Assessment of the clinical utility of serial beta-D-glucan concentrations in patients with persistent neutropenic fever. J Med Microbiol. (2008) 57:287–95. doi: 10.1099/jmm.0.47479-0
94. Gavino C, Cotter A, Lichtenstein D, Lejtenyi D, Fortin C, Legault C, et al. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin Infect Dis. (2014) 59:81–4. doi: 10.1093/cid/ciu215
95. Palesch D, Bosinger SE, Tharp GK, Vanderford TH, Paiardini M, Chahroudi A, et al. Sooty mangabey genome sequence provides insight into AIDS resistance in a natural SIV host. Nature. (2018) 553:77–81. doi: 10.1038/nature25140
96. Stinghen AE, Goncalves SM, Bucharles S, Branco FS, Gruber B, Hauser AB, et al. Sevelamer decreases systemic inflammation in parallel to a reduction in endotoxemia. Blood Purif. (2010) 29:352–6. doi: 10.1159/000302723
97. Sandler NG, Zhang X, Bosch RJ, Funderburg NT, Choi AI, Robinson JK, et al. Sevelamer does not decrease lipopolysaccharide or soluble CD14 levels but decreases soluble tissue factor, low-density lipoprotein (LDL) cholesterol, and oxidized LDL cholesterol levels in individuals with untreated HIV infection. J Infect Dis. (2014) 210:1549–54. doi: 10.1093/infdis/jiu305
Keywords: HIV, LPS, (1→3)-β-D-Glucan, immune activation, microbial translocation, antiretroviral therapy, non-AIDS events
Citation: Ramendra R, Isnard S, Mehraj V, Chen J, Zhang Y, Finkelman M and Routy J-P (2019) Circulating LPS and (1→3)-β-D-Glucan: A Folie à Deux Contributing to HIV-Associated Immune Activation. Front. Immunol. 10:465. doi: 10.3389/fimmu.2019.00465
Received: 30 November 2018; Accepted: 21 February 2019;
Published: 18 March 2019.
Edited by:
Sara Gianella Weibel, University of California, San Diego, United StatesReviewed by:
Nicholas Funderburg, The Ohio State University, United StatesCristian Apetrei, University of Pittsburgh, United States
Copyright © 2019 Ramendra, Isnard, Mehraj, Chen, Zhang, Finkelman and Routy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Pierre Routy, amVhbi1waWVycmUucm91dHlAbWNnaWxsLmNh