 David G. Jackson
David G. Jackson- MRC Human Immunology Unit, Radcliffe Department of Medicine, MRC Weatherall Institute of Molecular Medicine, University of Oxford, Oxford, United Kingdom
The lymphatics fulfill a vital physiological function as the conduits through which leucocytes traffic between the tissues and draining lymph nodes for the initiation and modulation of immune responses. However, until recently many of the molecular mechanisms controlling such migration have been unclear. As a result of careful research, it is now apparent that the process is regulated at multiple stages from initial leucocyte entry and intraluminal crawling in peripheral tissue lymphatics, through to leucocyte exit in draining lymph nodes where the migrating cells either participate in immune responses or return to the circulation via efferent lymph. Furthermore, it is increasingly evident that most if not all leucocyte populations migrate in lymph and that such migration is not only important for immune modulation, but also for the timely repair and resolution of tissue inflammation. In this article, I review the latest research findings in these areas, arising from new insights into the distinctive ultrastructure of lymphatic capillaries and lymph node sinuses. Accordingly, I highlight the emerging importance of the leucocyte glycocalyx and its novel interactions with the endothelial receptor LYVE-1, the intricacies of endothelial chemokine secretion and sequestration that direct leucocyte trafficking and the significance of the process for normal immune function and pathology.
Introduction
The lymphatics form an extensive network that facilitates the drainage of plasma leaked from the peripheral vasculature and its re-uptake by the venous circulation for maintenance of fluid homeostasis (1, 2). Moreover, they constitute an essential compartment of the immune system, providing conduits for the trafficking of antigen loaded dendritic cells (DCs), memory and regulatory T cells (TMEM and TREG) and neutrophils to draining lymph nodes (dLNs) in the process of immune activation, modulation and peripheral tolerance (3–8). In addition, they mediate the clearance of macrophages that remove pathogens and tissue debris during resolution of tissue inflammation and infection, and are exploited by microbial pathogens such as Group A streptococci, Salmonella, Brucella and M. tuberculosis, and parasitic nematodes that use the lymphatics for host colonization and systemic dissemination (9–13). Understanding the mechanisms by which cells enter and exit lymphatics in tissues and lymph nodes and their detailed choreography will therefore be essential to understanding how such processes help regulate immunity, and how they might be manipulated for therapeutic intervention.
The lymphatic network is quite distinct from the blood vasculature in terms of both structure and physiology. Notably, the lymphatics start as blind-ended capillaries that are freely permeable to fluids, and have discontinuous overlapping junctions pre-adapted to cell transit, unlike the conventional tight junctions that seal most blood vessels (8, 14, 15). In addition, unlike the blood circulation, cell trafficking in most afferent lymphatics involves intravasation rather than extravasation, in keeping with their role in accommodating the passage to dLNs of tissue resident leucocytes and transient immune cell populations recruited from the circulation (3–5, 8, 16–18). Moreover, during entry to lymphatic capillaries, leucocytes are exposed to the very low shear rates associated with interstitial fluid flow, as distinct from the high shear rates experienced during extravasation from blood capillaries (19). Reflecting such different environments, some of the molecular mechanisms for leucocyte entry and trafficking in the lymphatics are quite different to those in blood vessels. Nevertheless, as will be apparent from this present review, some are broadly similar. Just as extravasation of leucocytes from blood is triggered by inflammation and the induced expression of dedicated chemokines and adhesion molecules in the vascular endothelium, so too is the entry of most leucocytes to afferent lymph vessels. Indeed, as discussed later, certain key adhesion molecules are shared by both vasculatures.
In the following sections, I describe the latest findings on how leucocytes exploit chemotactic and adhesive mechanisms to enter and migrate within lymphatic vessels, as well as exit the lymphatic sinuses in dLNs to fulfill their various immune functions. I begin with an outline of the major leucocyte populations that migrate in lymph, and the characteristic architecture of lymphatic endothelial junctions. Based largely on knowledge gained from studies on DCs, I go on to provide a detailed account of the key steps in lymphatic trafficking from interstitial migration, lymphatic entry, and intraluminal crawling, to transit within downstream dLNs (Figure 1). In my discourse, I highlight the newly discovered role of the lymphatic endothelial HA receptor LYVE-1 in lymphatic entry and its functional relationship with other more ubiquitous adhesion receptors in endothelial transit, and the possibility of their co-operation in a “lymphatic synapse.” I also describe the co-ordinated triggering of chemokine release by transmigrating DCs, and some of the additional mechanisms employed by neutrophils and certain T cell populations during lymphatic transit. Lastly, I speculate on how knowledge of lymphatic trafficking mechanisms might be exploited in the future to develop new therapies for immune and inflammatory disorders.
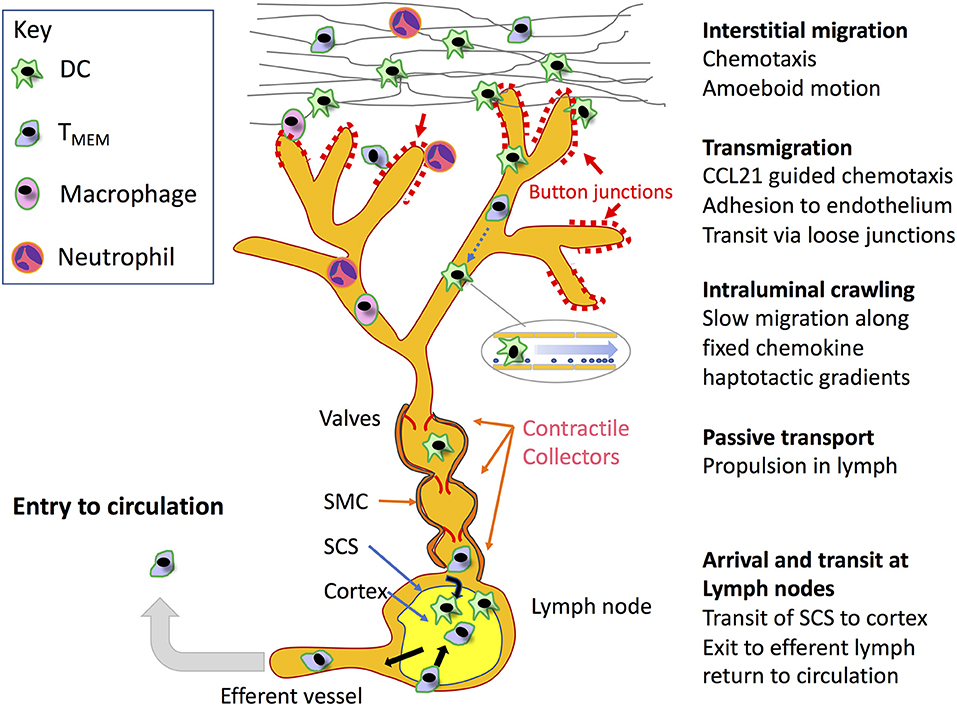
Figure 1. Key steps in leucocyte migration through the lymphatic system. Beginning in the interstitium, motile leucocytes including recirculating (TRCM) cells and neutrophils recruited from the blood, in addition to antigen loaded tissue resident DCs and macrophages are mobilized through inflammation to migrate toward initial lymphatic capillaries for access to dLNs by means of chemokine-directed amoeboid movement and in some cases integrin-mediated adhesion to fibronectin. The leucocytes then transit the blind-ended lymphatic capillaries at discrete sites within their discontinuous button-like endothelial junctions through receptor-mediated adhesion and chemokine directed squeezing, or in the case of neutrophils via lipoxin mediated chemorepulsion (detailed in Figures 2,4, respectively). Although not depicted, it is also possible that leucocytes can enter downstream vessels through conventional zipper-like junctions, and exit at intermediate stages to the surrounding tissues. On entering the capillary lumen, the transmigrated cells crawl in a semi-directional manner along the endothelial surface, guided by gradients of immobilized CCL21 established under low shear flow, using adhesion to ICAM-1 for traction. On entering downstream valved collectors, the leucocytes are now conveyed by passive transport, propelled by lymph flow generated by smooth muscle contraction. Ultimately, the migrating cells arrive within the SCS of dLNs, where they immediately transmigrate to the underlying cortex to initiate or modify immune responses in the case of DCs and likely neutrophils and macrophages, or continue to the medullary sinuses where they transit to the cortex to either remain there or pass to the efferent lymphatics to re-enter the blood circulation in the case of TMEMs. Although not shown in the Figure, naïve T and B cells also recirculate directly from the blood through lymph nodes at specialized high-endothelial venules to enter the cortex and re-exit through efferent lymph.
Leucocyte Populations that Traffic via Lymph
Classic cannulation studies carried out in domestic animals and applicable also to mice and humans showed the major cell populations migrating in normal afferent lymph are T cells (80–90%), followed by antigen presenting DCs and very small numbers of B cells which together account for most of the remaining 10–15%. Most of the T cells are antigen-experienced CD4+ CD45RO+ effector memory (TEM) cells, recently re-defined as the recirculating memory (TRCM) subset (20, 21), which, having entered the extra-lymphoid tissues from blood, engage in immune surveillance for cognate antigens before exiting via the afferent lymphatics to dLNs where they modulate recall immune responses (22–25). Notably, lymphocytes of the CD4 subset in afferent lymph outnumber those of the cytotoxic CD8 subset by some 5× fold (26–28), which mostly remain immotile as tissue-resident (TRM) cells. Furthermore, more recent cell tracking studies using photoconvertible Kaede mice have revealed that a significant proportion (25%) of the CD4 population are FOXP3+ TREGs, thus uncovering a previously unrecognized role for the lymphatics in conveying these important immunoregulatory cells. By comparison, only low numbers of naïve T cells are usually present in afferent lymph, and despite the fact these can be shown to enter lymphatic capillaries after adoptive transfer in mice, their normally low frequency in tissue means they rarely do so in vivo. The likely functional significance of TEM cell migration in afferent lymph may be to allow the amplification and polarization of immune responses in the dLNs and maintenance of the T cell memory pool, as well as enabling the re-entry of these lymphocytes to the circulation to target pathogen dissemination in further tissue sites (7, 21).
The second most numerous leucocyte population in afferent lymph are DCs, which ferry endocytosed antigens from the tissues, primarily for immune priming in dLNs. Although small numbers of DCs migrate in lymph under steady state conditions to maintain peripheral tolerance to self-antigens (29–33), the majority are mobilized by inflammation, which induces a program of differentiation and the expression of appropriate chemokine receptors for vessel entry (34–36) (see below). DCs, more than any other cell type, have been the subject of studies into the mechanisms of lymphatic trafficking, not least because they are normal tissue residents whose migration can be readily monitored by dye uptake in experimental mice (37). Moreover, in laboratory animals the lymph migrating DCs are almost as numerous as T cells, owing to the fact the pathogen-low environment in which they are bred and maintained generates a smaller pool of circulating memory cells (38).
During inflammation, the numbers of T cells and DCs in afferent lymph increase several fold along with an increase in lymph vessel permeability and the rate of lymph flow (8, 34). Furthermore, the afferent lymph can contain type I and II macrophages which are recruited to inflamed tissues for the clearance of debris and remodeling of the extracellular matrix (ECM) and which utilize the lymphatics for subsequent exit in a process that is becoming increasingly appreciated as critical for resolution and the return to normal homeostasis (39–41). In addition, this lymph contains subsets of neutrophils that are rapidly recruited to the tissues during sepsis and trauma and subsequently exit via the inflamed lymphatics to dLNs (42, 43). Although the majority of neutrophils in tissues are short-lived (T1/2 6–12 h) and undergo early apoptosis before removal by macrophage efferocytosis (44–46), the lymph-migrating cells have an extended lifespan (47). Most notably they can transport phagocytosed pathogens such as Leishmania, H. pylori and M. bovis BCG to dLNs where they can influence the polarity of protective T cell responses through cytokine release and crosstalk with DCs, thus bridging the gap between innate and adaptive immunity (48–51). Indeed, neutrophils can migrate via lymph more rapidly than any other leucocyte populations, reportedly arriving in the ipsilateral dLNs some 12–72 h earlier than either DCs or macrophages (52–56).
In contrast to afferent lymph, the leucocyte population present in the efferent lymphatics that exit from the lymph node hilum are mostly naïve T and B cells. Having entered the lymph nodes through high endothelial blood venules in a separate circuit to probe for antigens presented by DCs in the cortex and paracortex, these are ultimately returned to the circulation through the subclavian vein (36). Notably, during the onset of infection or inflammation, the efflux of this recirculating population from the lymph node is halted transiently (3–4 days) so as to prolong their residence time and thus increase the efficiency of immune recognition (57).
The Distinctive Architecture of Initial Lymphatic Vessels
As already mentioned, the afferent lymphatics initiate as blind-ended capillaries that branch and merge with larger collecting vessels, emptying their contents into dLNs before exiting as efferent vessels that reconnect either directly, or via other intervening nodes to the venous blood (Figure 1). In keeping with their fluid draining function, the blind-ended capillaries have only a rudimentary basement membrane (BM), and lack any investment by actin-containing smooth muscle cells. Moreover, the endothelial cells that make up the first few millimeters of these initial capillaries have a distinctive oakleaf shape that allows them to interdigitate and form loose discontinuous junctions (Figure 2), quite unlike the continuous junctions of endothelia in most blood vessels (14, 58–61). As revealed by electron microscopy (EM) and confocal imaging studies of such capillaries in mice, the alternating membrane flaps making up these structures are pinned at their sides by discrete assemblages some 3 μm wide and spaced 3 μm apart, that contain the adherens-junction protein VE-cadherin and the tight junction proteins Claudin-5, ZO-1 (zonula occludens-1), ESAM (endothelial selective adhesion molecule) and JAM-A (14). In contrast, the flaps remain free at their tips, where they guard openings of ~0.5–1 μm, that are decorated by CD31 and the lymphatic endothelial HA receptor LYVE-1 as detailed below (14). It is through these openings that DCs appear to enter the lymphatic capillaries. Importantly, the process is far from passive, as the dimensions of the DCs are many times greater than the gaps through which they must enter, and hence transit requires pushing and squeezing via intimate contact with the endothelium (14, 62–65). While the rationale for such an elaborate arrangement of buttons and flaps is not fully clear, it likely represents a compromise between the conflicting requirements of high vessel permeability for fluid uptake and a more restrictive barrier for regulating leucocyte entry. Indeed, for the experimenter, such architecture poses problems for establishing in vitro models to study vessel entry, as primary cultured lymphatic endothelial cell (LEC) monolayers form only partial surrogates of discontinuous junctions (66), and full authentication requires the application of whole animal models or crawl-in assays with tissue explants.
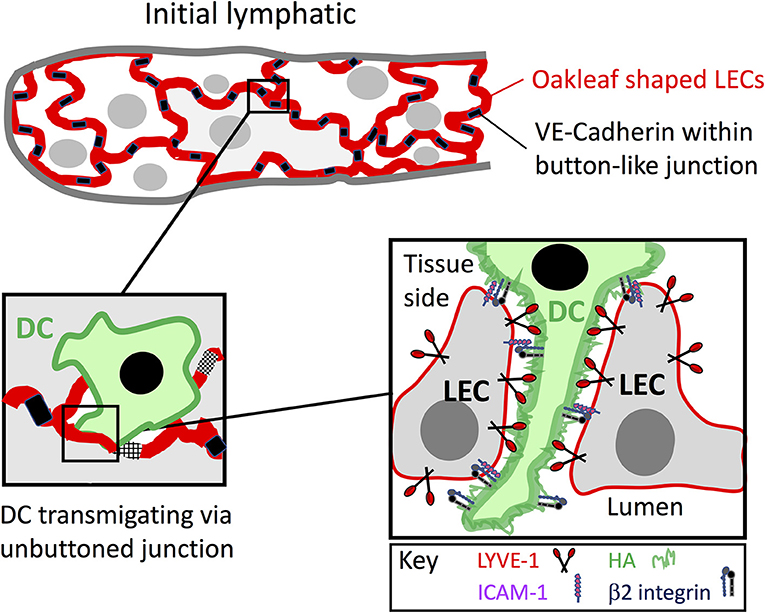
Figure 2. Leucocyte entry to initial lymphatic capillaries through formation of LYVE-1 endothelial transmigratory cups. Entry to the afferent lymphatics proceeds within the discontinuous junctions between the oakleaf shaped lymphatic endothelial cells of initial capillaries that are tightly buttoned at their sides by VE-cadherin and lined at their tips by the HA receptor LYVE-1 (red). As depicted in the Figure, migrating leucocytes such as DCs, macrophages and likely T cells that assemble a surface HA glycocalyx engage with LYVE-1 and transduce signals for VE-cadherin disassembly and junctional unbuttoning that lead to formation of a continuous LYVE-1 lined interface at portals termed transmigratory cups (see text). The weak avidity dependent nature of HA interactions with the LYVE-1 homodimers, combined with the multivalent nature of the long HA polymers are thought to enable low friction migration of the DCs through the endothelial openings. In addition, ICAM-1 and VCAM-1 expressed within the cups bind to integrin ligands on the DC surface activated by local secretion of CCL21 and likely provide the necessary traction for diapedesis. The precise details of the interplay between LYVE-1 and these receptors are not yet known.
In contrast to the initial capillaries, downstream pre-collector and valved collector vessels lack buttons and instead adopt conventional tight or “zipper” like junctions that allow the formation of a more fully sealed vasculature (14, 15). The collectors are also covered by smooth muscle cells that promote contractile pumping and which convey migrating leucocytes to the dLNs with minimal leakage. Of note, these zippered junctions constitute the default state of capillaries in the developing embryo and early neonate, and button-like junctions emerge only during the later neonatal period, co-incident with the establishment of full immune function (67). Curiously, the nascent lymphatics generated by lymphangiogenesis in chronically inflamed tissues also have conventional zippers, raising the possibility that vessel entry via zippers rather than buttons may be permissible in certain contexts (15).
Migration Through Interstitial Matrix and the Peri-Lymphatic Basement Membrane
In order to access the initial lymphatic capillaries for exit from the tissues and onward trafficking to lymph nodes, leucocytes must first migrate distances of several hundred microns through the surrounding interstitial matrix, a variably dense complex of type I collagen fibrils, fibronectin, hyaluronan (HA), and heparan sulfate proteoglycans (HSPGs) (68). The rate of such interstitial migration is thought to be comparable with that of subsequent migration through afferent lymph capillaries [4.4 vs. 5.7 μm/min in the case of DCs (69) and see below] and the time taken for DCs to navigate to a lymphatic capillary from the point of initial mobilization has been estimated as ~1 h (70). In mice, under conditions of normal homeostasis, the numbers of migrating cells in sites such as skin are small and the populations consist mostly of immature dermal DCs and effector TMEM cells en route to lymph nodes during immune surveillance. However, in response to inflammation, as deduced from in vivo studies using skin contact sensitizing agents and complete Freund's adjuvant (CFA), this traffic increases significantly. For practical reasons, most studies of leucocyte interstitial migration in the context of lymphatic trafficking have focused on DCs in the mouse dermis and epidermis. Although sessile in the steady state, these cells are subsequently induced to differentiate and crawl toward LVs, in response to inflammatory signals received from cytokines, prostaglandins and leukotrienes released from keratinocytes activated by microbial products and other Toll-like receptor (TLR) ligands (71). The movement of activated DCs is further stimulated by the increase in interstitial fluid flow that is characteristic of tissue inflammation. Nevertheless, the major driving force for leucocyte interstitial migration under resting and inflamed conditions is chemotaxis, directed primarily by the chemokine CCL21 secreted radially from lymphatic endothelium and its G-protein coupled receptor CCR7 on both migrating DCs and T cells (32, 36, 57, 72). Elegant studies by Weber and Sixt using single cell tracking in mouse ear skin revealed DC migration begins in a random fashion until the cells reach a distance of ~90 μm from an initial capillary, at which point their migration becomes directional and persistent (73). This directionality of motion is imposed by fixed, haptotactic gradients of CCL21 secreted constitutively from lymphatic vessel endothelial cells and sequestered by collagen and HSPGs in the surrounding interstitial matrix via the positively charged chemokine N-terminal domain (73). Moreover, the gradients fall away with distance from the vessel periphery in such a way as to provide a polarity that can be sensed by DCs in the typical size range of 15–50 μm. Importantly, DC migration under both resting and inflamed conditions is independent of β2 integrin/ICAM-1 adhesion, as demonstrated by seminal studies in mice that compared intact and pan-integrin knockout cells (74). Migration proceeds instead by “amoeboid movement” whereby CCL21 directs the pushing and squeezing of DCs through the 3D collagen matrix, primarily by triggering actin polymerization and actomyosin contraction at the leading and trailing edges via the small GTPases Rac1 and Rac2 and nuclear contraction via the Rho associated kinase ROCK (69, 74, 75). To date few studies have addressed the mechanisms of interstitial migration employed by leucocyte populations other than DCs. Although in the case of CD4+ effector TMEM, such migration in CFA inflamed ear dermis was also found to be independent of β2 integrin/ICAM-1 adhesion (76), it was nevertheless reliant on guidance via β1and β3 integrin-mediated adhesion to matrix fibronectin, laid down in association with parallel oriented collagen fibers (77). In the looser fibronectin-rich ECM network that is characteristic of inflamed tissues, such adhesion may be a greater requirement for the chemokine driven migration of T cells due to their smaller size and inability to extend dendritic processes.
Lastly, before engaging with the external surface of initial lymphatic capillaries, migrating leucocytes must traverse the surrounding BM, a rudimentary structure comprised mainly of type IV collagen and the non-network forming laminin isoform α4, before they can transmigrate the endothelium to enter the vessel lumen. In contrast to the more close-knit BM of blood vessels (78), the BM of lymphatic capillaries is sparse and highly perforated by gaps of ~1 μm diameter that are completely devoid of any ECM components. Making up 30% of the vessel surface, these gaps offer points through which migrating cells can access the underlying vessel endothelium with the aid of physical expansion (to ~2 μm). Studies of inflamed mouse ear skin indicate that those gaps which overlie sites of vessel entry are marked by discrete deposits of sequestered CCL21 which direct the transit of DCs by chemotaxis/haptotaxis rather than simply chemokinesis. Indeed, migrating DCs have been observed to extend cellular processes toward these CCL21 puncta and apparently make physical contact with them (71). Furthermore, detailed imaging of such events in ex vivo crawl-in assays with dermal tissue explants (79) suggested these invadopodia-like protrusions enable DCs to transiently expand the BM portals by physical squeezing without the parallel requirement for proteolytic re-modeling observed during leucocyte transit of blood vascular BMs. On traversing the BM the migrating leucocytes then encounter the lymphatic endothelium next to their interdigitating flaps, which bend inwards to accommodate entry to the vessel lumen (79).
Transmigration of the Vessel Endothelium and Entry to the Lymphatics
Integrin-Mediated Adhesion via Endothelial Microvilli
Similar to leucocyte transmigration of blood vessel endothelium, transmigration of lymphatic endothelium involves prior adhesive interactions between β2 integrins and their Ig superfamily counter receptors. Of note, early in vivo studies using mouse models of skin inflammation and contact hypersensitivity showed that the endothelial leucocyte adhesion receptor ICAM-1 is involved in the migration of epidermal Langerhans cells to skin dLNs (80, 81). Nevertheless, the requirement for adhesion receptors in lymphatic transmigration has been disputed, and one particularly prominent study using pan-integrin (Int−/−) deficient mice reported that under normal resting conditions, DCs can enter lymphatic vessels and migrate to dLNs independently of all integrins, just as documented for interstitial migration, by means of chemokine-directed amoeboid motion (74). Importantly however, these studies tracked the migration of exogenous DCs that had been adoptively transferred to a non-inflamed dermis and hence reported a mode of trafficking that proceeds only quite rarely in normal tissues (74). Indeed, other studies that tracked endogenous DC trafficking in the uninflamed skin of CD11c YFP+/VE-Cadherin Cre/Rosa26 Fl RFP+ chimeric mice detected few if any cells entering dermal lymphatic capillaries, and such entry was observed only after adoptive transfer into tissue inflamed by either contact hypersensitization, exposure to adjuvants (CFA) or treatment with bacterial LPS (71). Hence, an integrin-independent mode of lymphatic vessel transmigration likely applies only to the relatively small minority of immature DCs that traffic constitutively in immune surveillance.
In contrast, in inflamed tissues where DCs migrate more extensively via lymph, transmigration is indeed dependent on integrin-mediated adhesion mechanisms. As evidenced by studies with cultured primary LECs, and confirmed by transcriptional profiling of lymph vessel endothelium isolated from inflamed mouse skin, exposure to inflammatory cytokines and contact sensitizing agents results in rapid (T1/2 ~3 h) upregulation of the key integrin counter-receptors ICAM-1 and VCAM-1 together with various other adhesion molecules associated with leucocyte endothelial transit such as E-selectin, and a range of different chemokines that are attractants for DCs, monocytes, lymphocytes and neutrophils (82, 83). Furthermore, ICAM-1 and VCAM-1 blocking mAbs have been demonstrated to impair adhesion and transmigration of bone marrow DCs (BMDC) across inflamed LEC monolayers in vitro, as well as entry and trafficking of endogenous DCs to dLNs in vivo, both in murine models of skin hypersensitivity and dermal vaccine-induced T-cell immunity (82, 84). Likewise, in CD45.2 mice, LFA-1 blocking mAbs were shown to impair trafficking of injected CD45.1 DCs to dLNs from TNFα treated footpads (85). Most notably, it has been observed using in vitro confocal imaging of murine BMDCs engaged in transit across inflamed lymphatic endothelium, that ICAM-1 and VCAM-1 are concentrated within finger-like projections somewhat analogous to the transmigratory cups described originally in blood vascular endothelium (86–89) (Figure 2 and see below). These extend around the transmigrating cells, facilitating their adhesion via activated (mAb 24+ve) forms of β2 integrin (LFA-1) present in complementary projections on the DC surface (85). Reportedly, similar ICAM-1 lined membrane protrusions were also observed around transmigrating DCs in vivo both in mouse ear and human dermal tissues.
The transmigration of T cells across lymphatic endothelium also involves integrin-mediated adhesion. Accordingly, in studies employing antibody blockade, ICAM-1 and its β2 integrin ligand LFA-1 were shown to be functionally required for T cell adhesion and transmigration of TNFα treated murine LECs in vitro, as well as entry to lymphatic vessels and trafficking to dLNs in the inflamed skin of oxazolone and adjuvant treated mice in vivo (76). Indeed, in an analogous manner to DCs, ICAM-1 was observed to be distributed in microvillar projections around CD4+ T cells and ionomycin/PMA-activated peripheral blood mononuclear cells adhering to a lymphatic endothelium (85). Hence it appears that similar mechanisms are employed by both CD4 T cells and DCs for lymphatic vessel entry through extension of ICAM/VCAM enriched endothelial microvilli (19, 90). Furthermore, as detailed later in this review, the initial stages of neutrophil entry involves adhesion through the β2 integrins LFA-1 and Mac-I and most likely their main lymphatic endothelial counter-receptor ICAM-1, as evidenced from experiments using adoptive transfer of Int−/− neutrophils and receptor blocking mAbs (42, 43, 56, 91). Indeed, it was clearly shown that administration of either β2 integrin or ICAM-1 blocking mAbs impaired the entry of GFPlysM labeled neutrophils to the initial lymphatic capillaries in mice, causing the cells to logjam at the vessel periphery (56), in a manner reminiscent of DCs given similar blockade in oxazolone-treated mouse skin (17, 82). Such harnessing of integrins, ICAM-1 and VCAM-1 for leucocyte adhesion to the basolateral surface of lymphatic vessel endothelium and subsequent transendothelial migration is in many ways analogous to their involvement in the abluminal crawling of newly extravasated leucocytes on the pericyte surface of blood vessels (92) and stands as an example of how comparable mechanisms can operate in the two vasculatures albeit in reverse orientation.
Transit via Hyaluronan and LYVE-1 Transmigratory Cups
In addition to integrins and their counter-receptors, it has recently been demonstrated that transmigration of DCs requires critical involvement of LYVE-1 (93) within the button-like junctions of initial lymphatic capillaries (14, 15) (Figure 2), and its engagement with the large mucopolysaccharide ligand HA present on the DC surface (19, 90). Closely related to the leucocyte receptor CD44 (94, 95), LYVE-1 contains a conserved lectin-like HA-binding domain, termed the Link module, at its N-terminus (19, 96). Although HA is a ubiquitous component of perivascular ECMs (97), it can also be synthesized by DCs and other leucocytes including macrophages and T cells as a surface glycocalyx (98–100), and it is the selective interaction of LYVE-1 with this latter structure in preference to ambient HA that facilitates DC transmigration (90, 101). Such specificity is possible because of the strict avidity-dependent nature of LYVE-1: HA interactions. Because the receptor binds only a short 8–20 saccharide region of HA with low affinity (KD 125 μM), it therefore relies on homo-dimerization and clustering, as well as a high ligand density to achieve the multiplicity of co-ordinate binding interactions required for tethering of the polymer chains (96, 101, 102), a phenomenon that has been termed superselectivity (103). Intriguingly, in vitro confocal imaging studies using primary murine LEC monolayers have shown that LYVE-1 is recruited along with ICAM-1 and VCAM-1 to the transmigratory cups that form upon initial DC contact with endothelium, and furthermore that engagement of LYVE-1 with the HA glycocalyx is actually critical for their formation (90). Consistent with a functional role for the receptor in DC transmigration, LYVE-1 HA blocking mAbs also impaired both the adhesion and transit of DCs across LEC monolayers in vitro. Similar features of LYVE-1 transmigratory cups have been observed in vivo, during DC transit of lymphatic capillaries in oxazolone sensitized mouse skin. Moreover, interference with LYVE-1 mediated DC interactions in such studies by LYVE-1 gene deletion, antibody blockade or DC HA glycocalyx depletion resulted in the characteristic logjamming of endogenous and adoptively transferred DCs on the basolateral surface of dermal lymphatics, and impaired their capacity to prime antigen specific T cells in dLNs, mirroring the effects seen with β2 integrin blockade (90). Notably, besides DCs, LYVE-1 also mediates adhesion and transmigration of macrophages across lymphatic endothelium through similar mechanisms (102). Indeed, in a murine model of myocardial infarction where damage-inducing M1 macrophages infiltrate the ischaemic myocardium and are subsequently cleared via cardiac lymphatics (104, 105), the process is blocked by Lyve1 deletion, which delays resolution and leads to fibrotic scarring (41). Whether or not T cells also engage LYVE-1 in such structures has yet to be determined. However, in common with DCs and macrophages these also have a capacity for HA biosynthesis, and hence may well assemble a similar HA surface glycocalyx (98). Curiously, another receptor, CLEVER-1, containing an HA-binding “Link” domain related to LYVE-1 has been reported to mediate CD4 and CD8 T cell trafficking through afferent murine skin lymphatics (see Table 1) as well as transmigration across monolayers of lymphatic endothelium in vitro (115–117). However, the mode of action of CLEVER-1 is distinctly different to LYVE-1, as the Link module was shown to be non-functional and the site mediating transmigration instead found to reside within a distant EGF repeat region (116, 118).

Table 1. Adhesion receptors in lymphatic endothelium involved in regulating leucocyte entry and trafficking.
Importantly, the role of LYVE-1 in lymphatic transmigration extends beyond merely supporting cell adhesion. For example, it has been demonstrated that engagement of the receptor can transduce signals for endothelial junctional relaxation, in particular the phosphorylation and detachment of VE-cadherin located within the button-like foci of initial capillaries from which LYVE-1 is selectively excluded (14, 106). Hence, engagement with the DC glycocalyx and un-buttoning of the VE-cadherin lined junctions at vessel entry sites may well promote coalescence of the alternating endothelial flaps and redistribution of LYVE-1 to form a single continuous interface for leucocyte diapedesis (2, 19). In addition, the large contour lengths of HA polymers, which can extend to several microns, likely mask access to the more compact integrins [extracellular domains ~20 nm (107)] on the underlying DC surface, and allow the polysaccharide to make primary contact with the capillary endothelium. It has also been postulated that the low affinity of LYVE-1 HA-binding supports crawling of DCs along the vessel surface toward junctional portals through the inherently low friction of the interaction (19). This role as lubricant is supported by physicochemical studies of LYVE-1 HA binding mechanics at the single molecule level using atomic force microscopy, which indicate the individual interactions are weak and that they rupture collectively under the low forces experienced in interstitial flow [see (19)]. It contrasts markedly with the behavior of CD44, a receptor tuned for leucocyte capture in post-capillary venules, which forms bonds that are stronger and detach sequentially in a Velcro (hook and loop) like fashion in response to the higher forces experienced in blood flow (108, 109). Nevertheless, the transit of cells through lymphatic endothelium must involve traction, and if this is not provided by LYVE-1, then it is likely that DCs use both HA and integrin-based adhesion either on different faces of the cell, or in sequential fashion during diapedesis.
Undoubtedly, many other adhesion molecules located in and around the buttoned junctions of lymphatic endothelium contribute to leucocyte transmigration, and may even specify the entry of discrete leucocyte populations. A number of such receptors including Mannose receptor (110–114), ALCAM (CD166, Activated Leucocyte Cell Adhesion Molecule) (119), L1CAM (CD171) (120), 4-1BB (CD137) (121), CD99, and CD31 (PECAM-1) (122) have already been implicated in the process from various in vitro and in vivo studies (Table 1). However, the precise functional roles played by each of these receptors, and how they are individually choreographed during lymphatic trafficking have yet to be elucidated.
Directional Guidance of Leucocyte Transmigration by Chemokines
The Key Roles of CCL21 and CCR7
In concert with adhesion receptors, critical cues for the guidance of leucocytes during the process of lymphatic vessel entry are provided by chemokines synthesized and secreted in the main by underlying LECs. However, the emerging view is that these may operate as much by inducing the transient arrest of migrating leucocytes at endothelial junctions as by guiding their migration along conventional chemotactic gradients. CCL21 released from the endothelium has been identified as the primary chemokine controlling the entry of DCs to afferent lymph, based initially on the findings from elegant studies in mice showing that CCL21 neutralizing mAbs or CCR7 gene deletion decreased or delayed DC migration from the dermis to dLNs (32, 72, 123, 124). Curiously, mice express two separate genes for CCL21 that encode a lymph node isoform CCL21ser and an afferent vessel isoform CCL21leu (125, 126), and it has logically been assumed (though not formally proven) that the latter (CCL21leu) controls the lymphatic entry step (32, 72, 123, 124, 126). Although a naturally occurring genetic deletion of CCL21ser in the plt/plt mouse line compromises DC trafficking via lymph (125–127), this likely reflects a more distal defect in either entry or retention in downstream dLNs.
In addition to DCs, CCL21 is also the primary chemokine driving the entry of T cells to afferent lymph vessels in the periphery, in particular the antigen-experienced CD4+ and CD8+ TEM population that exits from the circulation to patrol the inflamed tissues. Approximately 50% of this population in the skin of humans and mice are CCR7+, and as confirmed from cannulation studies in sheep, almost all the T cells that migrate in afferent lymph express CCR7 and respond chemotactically to CCL21 (23, 128). Furthermore, parallel studies with CFSE labeled T cells in gene deficient mice have indicated their entry to the afferent lymphatics in both dermis and lung and trafficking to dLNs is almost entirely dependent on expression of CCR7 (20, 23). These migratory CCR7+ T cells which can ultimately re-enter the blood via the thoracic duct are clearly distinct from the CCR7− T effector (TEM) population that remains resident within the tissues as sentinels, and have since been defined phenotypically as a (CCR7int/+ CD62Lint CD69− CD103+/− E-selectin ligand+) recirculating memory (TRCM) cell subset, based on their tracking in photoconvertible fluorescent Kaede mice (21). Indeed, they also include the important immunosuppressive CD4+ T regulatory cell (TREG) as well as inflammation-associated Th1 and Th17 cell subsets (129, 130). Curiously however, the importance of CCL21/CCR7 for T cell entry and trafficking is diminished in chronic as compared to acute inflammation (130), and hence it is likely that other inflammation-induced chemokines become involved at later time points (see below).
It has also been reported that CCL21 chemotaxis helps direct the entry of neutrophils to afferent lymphatics. Accordingly, in a study of skin inflammation evoked by topical CFA administration in mice, the lymph migrating neutrophil population was identified exclusively as CCR7+ and trafficking to dLNs was decreased almost 4 × fold in CCR7−/− animals (55). Likewise, the entry of neutrophils to the cremaster muscle lymphatics following TNFα treatment, which induces CCL21 release, was reported to be almost completely (>97%) inhibited in CCR7−/− mice (43). Whether CCL21 is the primary chemokine in every context is however open to question, as another study of neutrophil migration in S. aureus treated mice found that neutrophil entry to the dermal lymphatics was directed by CXCL12, on the basis of inhibition by the CXCR4 receptor antagonist AMD3100 (91).
Leucocyte-Induced Chemokine Release
Synthesis of CCL21 is markedly upregulated in murine and human lymphatic endothelium in response to inflammation (35, 66, 131) whereby the chemokine accumulates in intracellular storage vesicles in readiness for secretion, notably at the basolateral surface of the endothelium where leucocytes transmigrate (132). Intriguingly, in a recent seminal study of DCs by Vaahtomeri et al it was reported that leucocytes themselves can trigger such secretion from lymphatic vessels through a contact dependent mechanism in which the transmigrating cells extend filopodia toward the endothelium, provoking a Ca2+ flux that triggers disassembly of cortical actin and the exocytosis of pre-stored CCL21 from trans Golgi vesicles along linear microtubule tracks, for fusion with the plasma membrane (Figure 3) (65). As visualized using EM, the exocytosed CCL21 is then retained focally in the form of minute puncta at the basolateral surface of the endothelium close to intercellular junctions where they are thought to induce local arrest of DCs through β2 integrin activation. Such confined release may well avoid the desensitization of CCR7 on the migrating cells that might otherwise be evoked by formation of a conventional transendothelial CCL21 gradient (133). Diapedesis—the actual transit process, then proceeds by CCL21-driven re-arrangement of the DC actomyosin cytoskeleton that allows the cell to push and squeeze through the flap-like protrusions between adjacent oakleaf shaped endothelial cells in the initial capillary junctions (65), aided by Semaphorin 3A induced signaling via RhoA and ROCK for contraction of the uropod (134). The CCL21 secreted focally in response to DC contact is thus functionally distinct from the CCL21 that is secreted homeostatically for interstitial migration. Interestingly, the DC adhesion-induced CCL21 exocytotic mechanism described by Vaahtomeri et al. does not appear to be initiated by integrins. Indeed, the identities of the receptors on DCs and endothelium responsible have yet to be determined. Moreover, it is likely that the process of DC-induced CCL21 secretion orchestrates the assembly of LYVE-1, ICAM-1, and VCAM-1 enriched endothelial transmigratory cups, as independent studies reported that corresponding structures consistently formed close to CCL21 puncta in LEC monolayers in vitro, and their assembly was blocked by CCL21 neutralizing antibody (76, 85, 90, 135). It remains to be determined whether other leucocyte populations such as CD4+ T cells can trigger local CCL21 secretion in a similar manner to DCs and whether the coupling of chemokine release and transmigratory cup formation is a general phenomenon for vessel entry.
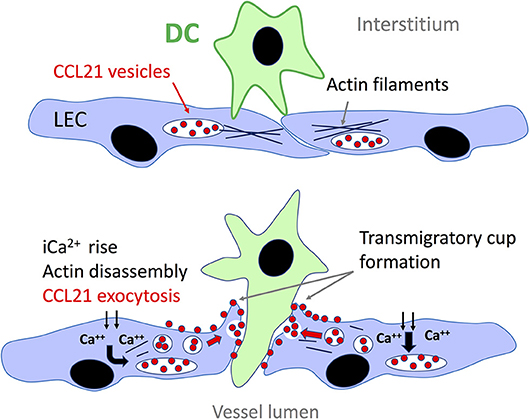
Figure 3. Local secretion of CCL21 from lymphatic endothelium triggered by DC contact. Contacts with the basolateral surface of lymphatic endothelium via filopodia extended from the migrating DCs trigger an influx of Ca2+ that leads to disassembly of the adjacent cortical actin network and enables translocation of endothelial trans-Golgi vesicles containing pre-stored CCL21 to the plasma membrane along parallel microtubule tracks. Fusion of the vesicles with the plasma membrane, most likely orchestrated by Ca2+ binding synaptotagmins, leads to the local release of CCL21 which is retained on the plasma membrane surface close to the endothelium:DC interface, where it directs DC transit through β2 integrin activation and actomyosin-mediated pushing and squeezing, upon the formation of LYVE-1 and ICAM-1/VCAM-1 lined transmigratory cups (see text and Figure 2). The identity of the adhesion molecules involved in making initial contact between DC and lymphatic endothelium are not currently known.
Additional Inflammatory Chemokines and Exosome—Mediated Secretion
Besides CCL21, LECs synthesize a variety of other chemokines including CCL1, CCL2, CCL5, CCL20, CXCL12, and CX3CL1 that are chemotactic for T cells, DCs and monocytes expressing the G-protein coupled receptors CCR8, CCR2, CCR5, CCR4, and CX3CR, respectively, and CXCL1, CXCL2, CXCL5, and CXCL8 that are chemotactic for neutrophils that express CXCR1 and CXCR2. In common with CCL21 these are all upregulated by exposure of the endothelium to inflammatory cytokines or other inflammatory stimuli (82, 83). In most cases however, the mechanisms underlying their release and extracellular localization in relation to junctional entry sites are not so well-understood. Amongst those chemokines that have been studied in any significant detail, CXCL12 (SDF-1) has been reported to direct the entry of DCs and epidermal Langerhans cells to lymphatic vessels in mice and migration to dLNs as assessed by FITC skin painting (136, 137). More specifically, its receptor CXCR4 was shown to be highly expressed by lymph-migrating MHC class II+ DC in skin and co-administration of a synthetic CXCR4 antagonist (4-F-benzoyl-TN14003) impaired their migration to dLNs and capacity to promote T cell proliferation after contact hypersensitization with the hapten DNBS (136). Nevertheless, the authors concluded that the role of CXCL12/CXCR4 axis, although operating in parallel for DC migration was subordinate to that of CCL21 and CCR7. Furthermore, CXCL12 does not appear to control the exit of T cells to afferent lymphatic vessels in inflamed skin, despite the fact they express CXCR4 and exhibit responsiveness to the chemokine in vitro (138). Additionally, CCL1, which regulates DC and tumor cell transit across the LN SCS via its primary receptor CCR8, has also been implicated in directing transmigration of monocytes and monocyte derived DCs across inflamed peripheral lymphatics (139–141).
More recently, CX3CL1 was shown to promote both in vitro transmigration and in vivo lymphatic entry of CX3CR+ monocyte-derived DCs in the skin of oxazolone-hypersensitised mice in parallel with CCL21 (142). Unique amongst chemokines, CX3CL1 is synthesized as a membrane-anchored molecule that is subsequently cleaved by proteases including the disintegrin and metalloproteases ADAM10 and ADAM17 to generate a conventional soluble chemoattractant, and it is this form that is released basolaterally from cytokine-activated endothelium (142). Moreover, as reported within the last few months (143), CX3CL1 is also secreted from lymphatic vessels in CD9+ and CD63+ exosomes that form halos around the periphery of lymphatic vessels in inflamed mouse and human tissues. Intriguingly, these exosomes which carry the membrane-anchored form of CX3CL1 on their surface can elicit cellular protrusions in monocyte-derived DCs and promote their transmigration across human dermal LEC (HDLEC) monolayers, in co-operation with CCL21, as well as entry to intact lymphatic vessels in ex vivo exposed skin (143). Why lymphatic vessels should employ two such different modes of CX3CL1 release is unclear. However, as exosomes act as vehicles for the release of chemokines other than CX3CL1 it is possible they direct the entry of multiple different inflammatory leucocyte populations.
Interestingly, most of the remaining chemokines such as CCL2, CCL5, CCL20, and the neutrophil chemokines CXCL2, CXCL5, and CXCL8 are preferentially secreted from the luminal rather than the basolateral face of lymphatic endothelium (17, 56, 66, 132, 142), unlike CCL21 and CX3CL1, and hence it is currently unclear how they might regulate leucocyte entry to afferent lymphatic vessels. Indeed, the reduced trafficking of epidermal Langerhans cells and CD8+ dermal DCs to dLNs observed in CCR2−/− and to a lesser extent in CCR5−/− mice appeared to result from their accumulation inside rather than outside dermal lymphatics and hence it is more likely that such chemokines direct intraluminal crawling rather than initial vessel entry (144).
Finally, the steady-state levels of secreted chemokines on and around lymphatic capillaries are regulated by a group of “atypical” chemokine receptors present in lymphatic endothelium that lack signaling capacity and act primarily as chemokine scavengers. This group which includes ACKR1 (Duffy antigen), ACKR2, formerly known as D6, ACKR3 (CXCR7), and ACKR4 (CCRL1) bind and internalize inflammatory CC chemokines and prevent their inappropriate accumulation on the surface of lymphatic capillaries while also helping to establish the polarity of their gradients (145). Accordingly, the action of ACKR2 which scavenges the inflammatory CC chemokines CCL2, CCL3, CCL4, and CCL5 is thought to aid in the preferential entry of mature activated CCR7+ DCs via CCL21, largely by preventing an accumulation of CCR2/CCR5 macrophages at the vessel surface that might otherwise block their access (146, 147). It has also been posited that ACKR4 helps preserve the responsiveness of DCs to CCL21 during entry to dermal lymphatics in inflamed tissues by scavenging CCL19 released from stromal cells and preventing a build-up of the chemokine that might otherwise de-sensitize CCR7 and lead to DC stasis (148).
Chemoattractive Guidance for T Cells via Sphingosine-1-Phosphate and Lymphotoxins
Besides conventional chemokines, T cells also engage two other chemoattractive pathways for vessel entry, that function co-operatively with CCL21 and may impinge at least partly on the integrin: ICAM/VCAM mediated transmigratory mechanism described above. The first involves the chemotactic lipid sphingosine 1 phosphate (S1P), best known as a regulator of lymphocyte exit from lymph nodes but which appears also to be a negative regulator of CD4+ T cell entry to initial lymphatics in inflamed peripheral tissues. Normally present at high concentrations in lymph and low concentrations in lymph nodes, the resulting gradients of S1P direct transit of T cells bearing the G-protein coupled receptor S1PR to efferent lymph (149, 150). However, in a murine alloantigen-induced model of inflammation, increased synthesis of S1P in peripheral tissue or administration of the S1PR1 functional antagonist FTY720 (fingolimod) signals retention and arrest of CD4+ T cells, which logjam around the basolateral surface of initial lymphatic capillaries (151). Moreover, as shown using in vitro transmigration assays with monolayers of the lymphatic endothelial like cell line SVEC4-10, S1P treatment of T cells blocked their transit and caused their arrest through β2 integrin-mediated adhesion to ICAM-1 and VCAM-1 (151). Whether this S1P/S1PR driven chemotactic mechanism also operates in conjunction with CCL21/CCR7 to positively regulate T cell transmigration and whether ICAM-1 and VCAM-1 are recruited to structures similar to or distinct from DC transmigratory cups during S1P mediated transmigration/arrest remains uncertain.
Secondly, the main immunoregulatory TREG population that migrates between tissues and lymph nodes to maintain peripheral tolerance and immune suppression have been shown to employ lymphotoxin (LT), a lymphokine member of the TNF superfamily, to transit inflamed lymphatic vessels by engaging its signal transducing receptor LTβR in lymphatic endothelium (152, 153). Best known for its role in inducing lymph node neogenesis via lymphoid tissue inducer (LTi) cells, the trimeric lymphotoxin molecule is expressed on the surface of TREG as a membrane-bound LTα1β2 heterotrimer. Moreover, genetic deletion of LTα in murine TREGs or treatment with soluble LTβR Ig fusion protein was shown to disrupt their entry to lymphatic capillaries in mouse skin, while leaving the transit of naïve CD4 and CD8 cells unaffected (153). More specifically, the interaction between LT and LTβR in vitro induces the rapid extension of VCAM-1 enriched lamellipodia-like protrusions akin to transmigratory cups in lymphatic endothelium, that appear to facilitate TREG transmigration (153). Curiously however, the VCAM counter-receptor involved in LT-mediated TREG transit has not been identified. While RNA array data suggest that TREGs express much higher levels of LTα than other T cell types (154), it remains unclear whether this mechanism is used by other T or B cell populations for lymphatic transmigration, or indeed whether the LTβR is redistributed to TREG transmigratory structures in lymphatic endothelium together with LYVE-1 and VCAM-1 must await further investigation.
Transmigration via Chemorepulsion—the Unusual Mechanism used by Neutrophils
In comparison to other leucocyte populations, neutrophils deploy a particularly unique and complex mechanism to enter lymphatic capillaries. While studies in mice harboring bacterial infections have shown that entry involves adhesion via β2 integrins, like that of DCs and T cells, more detailed in vitro studies using inflamed human LEC monolayers and mouse tissue explants revealed that such adhesion is just the first in a co-ordinated series of events that induce release of neutrophil elastase and the matrix metalloproteinases MMP8 and MMP9 and focal secretion of the arachidonate-derived chemorepellant lipid 12-hydroxyeicosatetraenoate (12(S)HETE), that together evoke local endothelial junctional retraction (Figure 4) (56, 155). These studies indicated the openings created by such retraction were transient and resolved spontaneously without any attendant cell death. Moreover, they served as portals for enhanced transit of successive waves of neutrophils which formed loosely attached swarms over the LEC monolayers resembling those previously described in lymph nodes of infected mice. Notably, the rate of in vitro neutrophil transmigration was some 10 × fold higher than that of DCs, even in neutrophil pre-exposed LEC monolayers, suggesting they exclusively target such portals for entry without inducing significant endothelial damage (56). The contact dependent nature of this neutrophil-induced activation process is curiously reminiscent of the contact-induced secretion of CCL21 by transmigrating DCs. Indeed, neutrophil adhesion to human LEC monolayers triggered the secretion of several chemokines including CXCL2, CXCL5, and CXCL8 from the basolateral surface that could potentially direct in vitro neutrophil transit, although experiments with neutralizing mAbs revealed only CXCL8 (IL-8) fulfilled this role (56). Whether a murine IL-8 ortholog guides neutrophil transmigration similarly in mice along with the recently reported actions of CCL21 and CXCL12 (43, 55, 91, 136, 137) remains to be determined. Importantly, neutrophils as distinct from other lymph migrating leucocyte populations do not synthesize an HA glycocalyx, and hence cannot engage LYVE-1 transmigratory cups for vessel entry. The likely significance of the alternative transmigration mechanism is that it offers a far more rapid mode of lymphatic entry than through the button junctions of initial capillaries, in keeping with the primary function of these cells in the rapid response to tissue injury. It is also noteworthy that an analogous lipoxin-mediated process of endothelial retraction has also been described for lymphatic metastasis of tumors in mice, and certain human cancers (156).
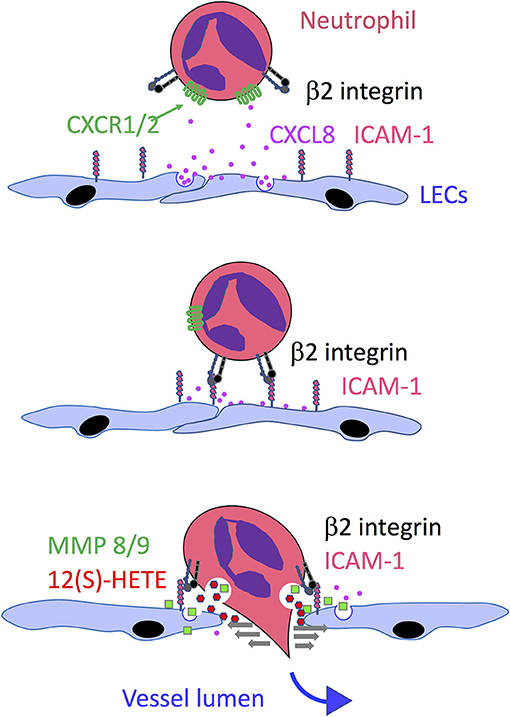
Figure 4. Model of neutrophil transmigration across lymphatic endothelium, Based on a combination of in vitro studies with HDLECs and in vivo studies in mice, the model depicts the guidance of neutrophils toward the basolateral surface of endothelium following inflammation-induced release of the chemokine CXCL8 (IL-8) which transmits signals through its G protein coupled receptors CXCR1 and CXCR2 to activate the neutrophil β2 integrin LFA-1 and engage its endothelial counter-receptor ICAM-1. This in turn triggers local secretion of the matrix metalloproteinases MMP8 and MMP9 and neutrophil elastase (which also bind to the endothelial surface) and exocytosis of the chemorepellent 12(S)-HETE that together promote transient junctional retraction and neutrophil transmigration.
Intraluminal Crawling
Having transited the lymphatic endothelium, migrating leucocytes enter the vessel lumen and begin their onward journey to the dLNs. In initial capillaries, which are non-contractile and lack smooth muscle investment, the rate of lymph flow has been variably estimated as up to 200–300 μm/min (157, 158), some 2–3 orders of magnitude slower than in blood capillaries and sinusoids, and only marginally exceeding that in the interstitium (69). It is not until these capillaries merge into downstream contractile collectors that the flow rates even approach those of blood vessels. Surprisingly, intravital imaging studies revealed that DCs migrate within initial capillaries (6–8 μm/min) at an even slower rate than lymph itself, and that the majority of newly transmigrated cells are not conveyed by passive flow, but rather crawl along the luminal surface of the lymphatic capillaries until they enter downstream collectors (69–71, 159). Moreover, the crawling leucocytes exhibit semi-random patterns of migration, frequently changing direction before moving downstream (160). Indeed, using time-lapsed microscopy of YFP tagged DCs in the ear tissue of lymphatic reporter mice, it was confirmed that the rate and directionality of intraluminal crawling is almost completely unaffected by changes in lymph flow and even proceeds in its absence (160). Instead, crawling was shown to be driven by chemotaxis/haptotaxis, guided by physical gradients of CCL21 sequestered on the luminal surface of the vessel, as evidenced by confocal and immune EM imaging of mouse dermis, and by the demonstration that its downstream directionality in mouse dermal lymphatics was abrogated by CCL21 blocking mAbs or CCR7 gene deletion in the case of both endogenous and adoptively transferred DCs (160). The spontaneous establishment of such intraluminal gradients was elegantly demonstrated in vitro by flow chamber experiments with transfected LECs which affirmed that under levels of shear close to those of afferent lymph (0.015 dynes/cm2), fluorescent CCL21 secreted from the luminal surface of the endothelium underwent re-binding downstream to form authentic, directionally oriented gradients for DC migration (160) (see also Figure 1). The macromolecules responsible for sequestering the CCL21 gradient on the endothelium likely include the lymphatic marker podoplanin (161) that reportedly binds the chemokine with high affinity (KD 70 nM) in Biacore analyses (162), and HSPGs, whose digestion or specific knockdown has been shown to disrupt CCL21-dependent DC adhesion to LEC monolayers under flow (163–165). Thus, lymph flow generates the chemotactic gradients which drive intraluminal crawling, rather than conveying cells through physical propulsion. The necessary traction for crawling is provided by β2 integrin-mediated adhesion to ICAM-1 on the luminal surface of the endothelium, whose expression is upregulated in inflammation and whose blockade by mAbs was shown to reduce DC crawling velocity in vitro (69). Like DC migration in the interstitium, efficient intraluminal crawling of DCs in inflamed lymphatics also depends on signaling via the Rho-associated protein kinase ROCK for dissociation of β2 integrin from ICAM-1 and uropod retraction in a continual process of adhesion and detachment from the endothelium (69).
As regards other leucocyte populations, semi-directional crawling behavior has also been observed for CD4+ T cells migrating in the lumen of initial dermal lymphatic capillaries, as evidenced in a recent study that used time-lapsed imaging to track CD2 DsRed fluorescent lymphocytes in Prox1 GFP mice reporter mice (76). Furthermore, the speed of T cell intraluminal crawling and the degree of motility were both markedly increased in the inflamed dermal lymphatics of skin contact hypersensitised mice, supported again by integrin-mediated adhesive interactions with ICAM-1 on the inner surface of the endothelium (76). Just as described for DCs, these T cells became detached from the luminal endothelium as the initial capillaries merged with downstream collectors where they were passively drawn into lymph flow by vessel pumping. Neutrophils also crawl within the lumen of initial lymphatic capillaries in a mostly downstream direction toward lymphatic collectors, and at broadly comparable velocities (mean 6–13 μm/min) to DCs and T cells. In common with these other leucocyte populations, migration was shown to be directed by haptotactic gradients of CCL21 sequestered on the capillary floor in the direction of lymph flow, with traction provided by β2 integrin mediated ICAM-1 adhesion (91). The faster recruitment of neutrophils to dLNs compared to DCs and T cells via afferent lymph may therefore be the product of more rapid mobilization in the tissues, the ability to translocate stored CCR7 from intracellular vesicles rather than relying on de novo synthesis (43), and a more efficient mode of endothelial transmigration.
On passing from initial capillaries to the smooth muscle invested pre-collectors and collectors, migrating leucocytes encounter a large increase in lymph flow rate (>1 mm/min) that likely renders intraluminal crawling redundant, and hence it is thought the cells are conveyed toward downstream lymph nodes by passive lymph flow.
Arrival and Transit at LNs
The major destination for leucocytes migrating through tissue lymphatics is the lymph node, an organ the size of a small bean in most peripheral tissues. With the exception of recirculating TRCM cells which reside transiently before returning to the blood circulation via efferent lymphatics, most leucocytes reaching the nodes proceed no further and having fulfilled their immune function, ultimately die there. In either case the cells arrive into the subcapsular sinus (SCS), a labyrinthine compartment continuous with the afferent lymphatics that is situated just beneath the outer capsule of the node. From there, they transit across the SCS endothelium to access the T and B cell-rich cortex, in the case of DCs and neutrophils to prime or re-activate T cell immune responses, and in the case of TMEM and TREGs to influence or downregulate such responses, before they egress and circulate back to the tissues (see Figure 1). While exit from the nodes is known to be directed by the sphingosine 1-phosphate (S1P) receptor S1PR on recirculating cells and S1P in efferent lymph (150, 166, 167), it has often been assumed that cell entry from afferent lymph is a more passive process. However, it is becoming increasingly clear that the SCS endothelium represents a checkpoint for nodal entry that is regulated both by chemokines and adhesion receptors.
In one particularly informative study using mouse eGFP tagged leucocytes microinjected into pre-nodal (popliteal) lymphatics, it was found that DCs and CD4+ T cells used separate routes to transit across the SCS (168). Whereas, DCs invariably crossed directly through the floor of the SCS to the cortical zones, T cells instead continued to the adjoining medullary sinuses, previously considered as exit routes from the nodes, before transmigrating to the underlying parenchyma (168). Moreover, DC transmigration across the SCS was shown to be directed by CCR7/CCL21 dependent chemotaxis, whereas CD4+ T cells required this chemokine receptor pair only for subsequent haptotactic crawling within the underlying parenchyma (168). Curiously however, when CD4+ T cells were co-injected into mice, the former then switched to the SCS route for transmigration, suggesting that DCs in some way remodel the endothelium during transit. More recent work has revealed that the role of CCL21 in directing DC transit can also be aided or augmented by other chemokines. Notably CCL1 released from the floor of the SCS was reported to induce the entry of monocyte-derived DCs to the LN parenchyma via its cognate receptor CCR8 (139). More recently, in Th2 immunized mice, the alternative CCR8 ligand CCL8 released from CD169+ SCS macrophages was shown to potentiate transit of DCs across the SCS by enhancing CCR7 signal transduction (141). Significantly, polarized expression of the alternative CCL19/CCL21 scavenging receptor CCRL1 (ACKR4) in the ceiling of the SCS was reported to maintain the gradient of CCL21 inside the SCS that drives DC transit, as its deletion led to trapping of DCs within the SCS lumen (169).
Knowledge of the contribution made by adhesion receptors in leucocyte transmigration across the SCS floor is still rather sketchy in comparison with peripheral lymphatics. Curiously, a key role has been identified for the transmembrane protein PLVAP (aka Pal-E, MECA 32) that forms diaphragm-like structures in the SCS and certain blood vessels and which primarily regulates the size selective entry of macromolecules to the underlying cortical fibrillar conduit network for presentation by LN HEVs (170). Notably, deletion of PLVAP was found to result not only in the uncontrolled entry of small macromolecules to the cortex, but also the enhanced transmigration of injected splenic T cells. Based on in vitro studies with isolated SCS endothelium, it was concluded that PLVAP diaphragms guard entry portals remote from VE-cadherin buttoned junctions, through which T cells transmigrate by extension of their leading edges (170). Whether PLVAP plays a direct or indirect role in the process and also mediates transit of DCs and other leucocytes remains unknown. More recently, transcriptional profiling has identified further candidate receptors that might regulate the differential transit of leucocytes across SCS and medullary sinuses. In particular, the macrophage scavenger receptor MSR1 (CD203) was found to be selectively expressed in SCS, where it was shown to mediate T cell adhesion in ex vivo assays with frozen sections. Like PLVAP however, the receptor appears to act as a regulator of SCS transit rather than a gatekeeper, as the process was enhanced not retarded in MSR−/− mice (171). Studies have yet to determine whether transit through SCS or other LN sinuses involves leucocyte adhesion via β2 integrins in resting or inflammatory conditions, or interactions between the leucocyte HA glycocalyx and LYVE-1 as in the case of initial lymphatic capillaries. Nevertheless, the observation that transit of DCs is accompanied by marked morphological changes in the floor of the SCS endothelium, including modulation of LYVE-1 and realignment of the SCS-lining CD169+ macrophages is indicative that the process is complex and that these and other adhesion receptors may well play contributory roles (36). Finally, it should be stressed that dLN sinuses and the surrounding afferent lymphatic network undergo considerable expansion following antigen challenge or inflammation in peripheral tissues, through a process of lymphangiogenesis driven by VEGF-A released primarily by B cells and macrophages arriving through afferent lymph (172–174). This is accompanied by a transient increase in LN size, cellularity and lymph flow that induces DC mobilization and migration, and markedly enhances DC transit into the deep underlying LN cortex and paracortex for lymphocyte activation (174–176). It is highly likely that such changes also enhance the entry of other migrant leucocyte populations to dLNs and it is hoped that future research in this area will yield much needed insights into the underlying mechanisms.
Summary and Conclusions
Over the past decade, research using new techniques and animal models for tracking and imaging cell migration, combined with the efforts of a wide interdisciplinary community of interested scientists, has led to huge advances in our understanding of leucocyte trafficking in the lymphatic system and its immune significance. As this pace of advance seems set to continue in the immediate future, we can anticipate that the resulting mechanistic insights will translate into new targets and therapies for immune disorders and even the treatment of lymph metastasising cancers.
As outlined in this text, we now have detailed insight into the first key step in such trafficking, the entry of cells to the lymphatic vessels. In the case of DCs, this has revealed an intricate and closely co-ordinated mechanism in which physical contact of migrating leucocytes with lymphatic endothelium triggers the local exocytosis of CCL21 and formation of LYVE-1+ transmigratory cups which envelop the migrating cells and promote their transit into the vessel lumen. Moreover, parallel observations that transmigrating T cells and macrophages elicit the formation of similar endothelial protrusions containing ICAM-1, VCAM-1 and/or LYVE-1, and reliance on an HA glycocalyx or β2 integrin adhesion, raise the possibility that lymph-migrating leucocytes exploit a common mechanism for vessel entry (76, 85, 101). Indeed, this could be considered as a form of “lymphatic synapse,” through which appropriate input from other chemokine receptors or signaling components such as lymphotoxin/LTβR and S1P/S1PR1 might direct selective entry of TREGs or other lymph migrating cell populations including Natural Killer (NK) cells and innate lymphoid cells (ILCs). However, the notion of a synapse may not apply to neutrophils which use a unique mechanism of integrin-dependent proteolysis and lipoxin-mediated endothelial retraction to “invade” lymphatic vessels. This unusually specialized process may have evolved to enable these professional phagocytes to exit almost instantaneously from sites of infection and reach the dLNs well-ahead of slower migrating DCs. In comparison, we know little about how the docking and adhesion of transmigrating leucocytes is choreographed and how the many “accessory” adhesion molecules including the Mannose receptor, ALCAM, CLEVER-1, CD31, CD99, and others (Table 1) integrate with key cup-forming components such as ICAM-1, VCAM-1, and LYVE-1. Though most leucocytes that employ such cups have been observed to enter the afferent lymphatics at button junctions in initial vessels, it seems unlikely that these are the only junctional types to allow entry. During inflammation-induced lymphangiogenesis for example, newly sprouting vessels assemble zippered rather than buttoned junctions, and in chronic inflammation, when leucocyte traffic via lymph is markedly increased, zippers replace buttons. It will be interesting to determine whether leucocytes have the ability to enter through zipper junctions and whether the process involves different molecular mechanisms to buttons and transit via a transcellular or paracellular route. A further priority will be to ascertain whether these mechanisms of lymphatic entry are universally applicable or vary between tissue beds such as the intestines, brain and central nervous system (177), given that most of our current insight has been gained from studies on mouse dermis, due to its greater accessibility.
Recent research has also provided surprising insight into how leucocytes, having entered the initial lymphatics, migrate within the vessel lumen toward downstream lymph nodes. Rather than being conveyed by passive lymph flow, it is now apparent that DCs, CD4+ T cells and neutrophils actively crawl along the internal surface of initial vessels using guidance from CCL21 and transient integrin-mediated adhesion. Yet why such mechanisms should have evolved to deliberately slow the downstream progress of antigen presenting and immune effector cells is unclear. Might the intimate contact with endothelium imposed by intraluminal crawling enable for example en route uptake of foreign antigens or maturation signals by DCs, or MHC-mediated antigen presentation to recirculating T cells or TREGs for immune tolerance? Could it provide a platform for interactions between different leucocyte populations themselves? Or might intraluminal crawling provide DCs or T cells with the option to exit and re-enter lymphatic vessels prior to reaching dLNs in order to sample or respond to antigen in the surrounding tissues? Though not yet reported in initial lymphatics vessels, there is evidence that migrating leucocytes exit collectors in adipose tissue during bacterial infection, and that DCs which interdigitate the vessel endothelium to regulate vessel permeability and sample the surrounding tissue can subsequently detach and migrate to dLNs (178).
With the many insights into lymphatic trafficking and its immune consequences that have been gained from basic research, there is increasing scope for clinical translation and the development of new immune based therapies based on migration blockade. Judicious targeting of integrins, ICAM-1, chemokine receptors, S1P and particularly lymph-specific adhesion molecules such as LYVE-1 by appropriate blocking mAbs may be envisaged as therapeutic strategies for transplant rejection, to prevent DC migration from engrafted tissues and consequently activation of alloimmune responses in host dLNs. This is particularly applicable in the case of corneal allografts, where such therapies could be applied locally, thus avoiding off-target effects associated with systemic antibody administration. In the case of TREGs, the inclusion of lymphotoxin blockade to impair their migration to lymph nodes for immune suppression might also provide an adjunct to checkpoint inhibitors for tumor immunotherapy. As a corollary, our understanding of the main factors regulating DC migration from inflamed tissues could be exploited to optimize vaccine delivery. For many years the notion of using DCs as adjuvants for adoptive cancer immunotherapy has fuelled efforts to enhance their maturation ex vivo using TLR ligands and inflammatory cytokines and to optimize antigen loading and presentation. Combining these approaches with pre-conditioning of vaccination sites to boost lymphatic vessel density, the efficiency of DCs for vessel entry and nodal transport should lead to much greater clinical efficacy. Likewise, boosting the exit of macrophages via lymphatics could aid in the resolution of inflammation and tissue recovery in conditions such as myocardial infarction where their delayed removal results in a failure of cardiomyocyte replenishment, and an increase in tissue scarring and fibrosis (41, 179–181).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
DJ gratefully acknowledges financial support in the form of Unit Funding (Grant MC_UU_00008/2) from the UK Medical Research Council.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author thanks all his previous and present research team members for their seminal contributions to this work, and colleagues including Prof. Paul Riley, Dr. Cornelia Halin, Dr. Peter Baluk, and Prof. Ralf Richter for continued collaborations and helpful discussions.
References
1. Swartz MA, Hubbell JA, Reddy ST. Lymphatic drainage function and its immunological implications: from dendritic cell homing to vaccine design. Semin Immunol. (2008) 20:147–56. doi: 10.1016/j.smim.2007.11.007
2. Jackson DG. Lymphatic regulation of cell trafficking. J Clin Cell Immunol. (2014) 5:258–67. doi: 10.4172/2155-9899.1000258
3. Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. (2005) 5:617–28. doi: 10.1038/nri1670
4. Platt AM, Randolph GJ. Dendritic cell migration through the lymphatic vasculature to lymph nodes. Adv Immunol. (2013) 120:51–68. doi: 10.1016/B978-0-12-417028-5.00002-8
5. Teijeira A, Russo E, Halin C. Taking the lymphatic route: dendritic cell migration to draining lymph nodes. Semin Immunopathol. (2014). doi: 10.1007/s00281-013-0410-8
6. Tewalt EF, Cohen JN, Rouhani SJ, Engelhard VH. Lymphatic endothelial cells - key players in regulation of tolerance and immunity. Front Immunol. (2012) 3:305. doi: 10.3389/fimmu.2012.00305
7. Hunter MC, Teijeira A, Halin C. T cell trafficking through lymphatic vessels. Front Immunol. (2016) 7:613. doi: 10.3389/fimmu.2016.00613
8. Randolph GJ, Ivanov S, Zinselmeyer BH, Scallan JP. The Lymphatic system: integral roles in immunity. Annu Rev Immunol. (2017) 35:31–52. doi: 10.1146/annurev-immunol-041015-055354
9. Lynskey NN, Banerji S, Johnson LA, Holder KA, Reglinski M, Wing PA, et al. Rapid lymphatic dissemination of encapsulated group A streptococci via lymphatic vessel endothelial receptor-1 interaction. PLoS Pathog. (2015) 11:e1005137. doi: 10.1371/journal.ppat.1005137
10. Lerner TR, de Souza Carvalho-Wodarz C, Repnik U, Russell MR, Borel S, Diedrich CR, et al. Lymphatic endothelial cells are a replicative niche for Mycobacterium tuberculosis. J Clin Invest. (2016) 126:1093–108. doi: 10.1172/JCI83379
11. Babu S, Nutman TB. Immunology of lymphatic filariasis. Parasite Immunol. (2014) 36:338–46. doi: 10.1111/pim.12081
12. Corthesy-Theulaz IE, Hopkins S, Bachmann D, Saldinger PF, Porta N, Haas R, et al. Mice are protected from Helicobacter pylori infection by nasal immunization with attenuated Salmonella typhimurium phoPc expressing urease A and B subunits. Infect Immun. (1998) 66:581–6.
13. von Bargen K, Gagnaire A, Arce-Gorvel V, de Bovis B, Baudimont F, Chasson L, et al. Cervical lymph nodes as a selective niche for brucella during oral infections. PLoS ONE. (2014) 10:e0121790. doi: 10.1371/journal.pone.0121790
14. Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, et al. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. (2007) 204:2349–62. doi: 10.1084/jem.20062596
15. Yao LC, Baluk P, Srinivasan RS, Oliver G, McDonald DM. Plasticity of button-like junctions in the endothelium of airway lymphatics in development and inflammation. Am J Pathol. (2012) 180:2561–75. doi: 10.1016/j.ajpath.2012.02.019
16. Schineis P, Runge P, Halin C. Cellular traffic through afferent lymphatic vessels. Vascul Pharmacol. (2018) 112:31–41. doi: 10.1016/j.vph.2018.08.001
17. Johnson LA, Jackson DG. Cell traffic and the lymphatic endothelium. Ann N Y Acad Sci. (2008) 1131:119–33. doi: 10.1196/annals.1413.011
18. Johnson LA, Jackson DG. Control of dendritic cell trafficking in lymphatics by chemokines. Angiogenesis. (2014) 17:335–45. doi: 10.1007/s10456-013-9407-0
19. Jackson DG. Hyaluronan in the lymphatics: the key role of the hyaluronan receptor LYVE-1 in leucocyte trafficking. Matrix Biol. (in press). doi: 10.1016/j.matbio.2018.02.001
20. Bromley SK, Thomas SY, Luster AD. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol. (2005) 6:895–901. doi: 10.1038/ni1240
21. Bromley SK, Yan S, Tomura M, Kanagawa O, Luster AD. Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J Immunol. (2013) 190:970–6. doi: 10.4049/jimmunol.1202805
22. Mackay CR, Kimpton WG, Brandon MR, Cahill RN. Lymphocyte subsets show marked differences in their distribution between blood and the afferent and efferent lymph of peripheral lymph nodes. J Exp Med. (1988) 167:1755–65. doi: 10.1084/jem.167.6.1755
23. Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, et al. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. (2005) 6:889–94. doi: 10.1038/ni1238
24. Cahill RN, Frost H, Trnka Z. The effects of antigen on the migration of recirculating lymphocytes through single lymph nodes. J Exp Med. (1976) 143:870–88. doi: 10.1084/jem.143.4.870
25. Cahill RN, Poskitt DC, Frost DC, Trnka Z. Two distinct pools of recirculating T lymphocytes: migratory characteristics of nodal and intestinal T lymphocytes. J Exp Med. (1977) 145:420–8. doi: 10.1084/jem.145.2.420
26. Mackay CR. T-cell memory: the connection between function, phenotype and migration pathways. Immunol Today. (1991) 12:189–92. doi: 10.1016/0167-5699(91)90051-T
27. Mackay CR, Marston WL, Dudler L. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J Exp Med. (1990) 171:801–17. doi: 10.1084/jem.171.3.801
28. Yawalkar N, Hunger RE, Pichler WJ, Braathen LR, Brand CU. Human afferent lymph from normal skin contains an increased number of mainly memory/effector CD4+ T cells expressing activation, adhesion and co-stimulatory molecules. Eur J Immunol. (2000) 30:491–7. doi: 10.1002/1521-4141(200002)30:2<491::AID-IMMU491>3.0.CO;2-H
29. Huang FP, Platt N, Wykes M, Major JR, Powell TJ, Jenkins CD, et al. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J Exp Med. (2000) 191:435–44. doi: 10.1084/jem.191.3.435
30. Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. (2002) 196:1079–90. doi: 10.1084/jem.20020991
31. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
32. Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. (2004) 21:279–88. doi: 10.1016/j.immuni.2004.06.014
33. Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ. Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med. (2008) 205:2839–50. doi: 10.1084/jem.20081430
34. MacPherson GG, Jenkins CD, Stein MJ, Edwards C. Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J Immunol. (1995) 154:1317–22.
35. MartIn-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. (2003) 198:615–21. doi: 10.1084/jem.20030448
36. Forster R, Braun A, Worbs T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. (2012) 33:271–80. doi: 10.1016/j.it.2012.02.007
37. Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, et al. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci USA. (2008) 105:10871–6. doi: 10.1073/pnas.0802278105
38. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. (2016) 532:512–6. doi: 10.1038/nature17655
39. Bellingan GJ, Caldwell H, Howie SEM, Dransfield I, Haslett C. In vivo fate of the inflammatory macrophage during the resolution of inflammation. J Immunol. (1996) 157:2577–85.
40. Bellingan GJ, Laurent GJ. Fate of macrophages once having ingested apoptotic cells: lymphatic clearance or in situ apoptosis? In: Rossi AG, Sawatzky DA, editors. The resolution of inflammation. Basel: Birkhauser Verlag (2008) 75–91. doi: 10.1007/978-3-7643-7506-5_5
41. Vieira JM, Norman S, Villa Del Campo C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest. (2018) 128:3402–12. doi: 10.1172/JCI97192
42. Gorlino CV, Ranocchia RP, Harman MF, Garcia IA, Crespo MI, Moron G, et al. Neutrophils exhibit differential requirements for homing molecules in their lymphatic and blood trafficking into draining lymph nodes. J Immunol. (2014) 193:1966–74. doi: 10.4049/jimmunol.1301791
43. Arokiasamy S, Zakian C, Dilliway J, Wang W, Nourshargh S, Voisin MB. Endogenous TNFalpha orchestrates the trafficking of neutrophils into and within lymphatic vessels during acute inflammation. Sci Rep. (2017) 7:44189. doi: 10.1038/srep44189
44. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
45. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
46. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
47. Kim MH, Granick JL, Kwok C, Walker NJ, Borjesson DL, Curry FR, et al. Neutrophil survival and c-kit+-progenitor proliferation in Staphylococcus aureus-infected skin wounds promote resolution. Blood. (2011) 117:3343–52. doi: 10.1182/blood-2010-07-296970
48. Bennouna S, Bliss SK, Curiel TJ, Denkers EY. Crosstalk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J Immunol. (2003) 171:6052–8. doi: 10.4049/jimmunol.171.11.6052
49. Megiovanni AM, Sanchez F, Robledo-Sarmiento M, Morel C, Gluckman JC, Boudaly S. Polymorphonuclear neutrophils deliver activation signals and antigenic molecules to dendritic cells: a new link between leukocytes upstream of T lymphocytes. J Leukoc Biol. (2006) 79:977–88. doi: 10.1189/jlb.0905526
50. Beauvillain C, Delneste Y, Scotet M, Peres A, Gascan H, Guermonprez P, et al. Neutrophils efficiently cross-prime naive T cells in vivo. Blood. (2007) 110:2965–73. doi: 10.1182/blood-2006-12-063826
51. Culshaw S, Millington OR, Brewer JM, McInnes IB. Murine neutrophils present Class II restricted antigen. Immunol Lett. (2008) 118:49–54. doi: 10.1016/j.imlet.2008.02.008
52. Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, et al. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. (2005) 106:1843–50. doi: 10.1182/blood-2005-03-1281
53. Maletto BA, Ropolo AS, Alignani DO, Liscovsky MV, Ranocchia RP, Moron VG, et al. Presence of neutrophil-bearing antigen in lymphoid organs of immune mice. Blood. (2006) 108:3094–102. doi: 10.1182/blood-2006-04-016659
54. Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. (2008) 29:487–96. doi: 10.1016/j.immuni.2008.07.012
55. Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, et al. CCR7 is involved in the migration of neutrophils to lymph nodes. Blood. (2011) 117:1196–204. doi: 10.1182/blood-2009-11-254490
56. Rigby DA, Ferguson DJ, Johnson LA, Jackson DG. Neutrophils rapidly transit inflamed lymphatic vessel endothelium via integrin-dependent proteolysis and lipoxin-induced junctional retraction. J Leukoc Biol. (2015) 98:897–912. doi: 10.1189/jlb.1HI0415-149R
57. Permanyer M, Bosnjak B, Forster R. Dendritic cells, T cells and lymphatics: dialogues in migration and beyond. Curr Opin Immunol. (2018) 53:173–9. doi: 10.1016/j.coi.2018.05.004
58. Leak LV. The structure of lymphatic capillaries in lymph formation. Fed Proc. (1976) 35:1863–71.
59. Leak LV, Burke JF. Fine structure of the lymphatic capillary and the adjoining connective tissue area. Am J Anat. (1966) 118:785–809. doi: 10.1002/aja.1001180308
60. Leak LV, Jamuar MP. Ultrastructure of pulmonary lymphatic vessels. Am Rev Respir Dis. (1983) 128(2 Pt 2):S59–65.
61. Zoltzer H. Initial lymphatics–morphology and function of the endothelial cells. Lymphology. (2003) 36:7–25.
62. Romani N, Ratzinger G, Pfaller K, Salvenmoser W, Stossel H, Koch F, et al. Migration of dendritic cells into lymphatics-the Langerhans cell example: routes, regulation, and relevance. Int Rev Cytol. (2001) 207:237–70. doi: 10.1016/S0074-7696(01)07007-3
63. Stoitzner P, Holzmann S, McLellan AD, Ivarsson L, Stossel H, Kapp M, et al. Visualization and characterization of migratory Langerhans cells in murine skin and lymph nodes by antibodies against Langerin/CD207. J Invest Dermatol. (2003) 120:266–74. doi: 10.1046/j.1523-1747.2003.12042.x
64. Stoitzner P, Pfaller K, Stossel H, Romani N. A close-up view of migrating Langerhans cells in the skin. J Invest Dermatol. (2002) 118:117–25. doi: 10.1046/j.0022-202x.2001.01631.x
65. Vaahtomeri K, Brown M, Hauschild R, De Vries I, Leithner AF, Mehling M, et al. Locally triggered release of the chemokine CCL21 promotes dendritic cell transmigration across lymphatic endothelia. Cell Rep. (2017) 19:902–9. doi: 10.1016/j.celrep.2017.04.027
66. Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, et al. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med. (2001) 194:797–808. doi: 10.1084/jem.194.6.797
67. Ni A, Lashnits E, Yao LC, Baluk P, McDonald DM. Rapid remodeling of airway vascular architecture at birth. Dev Dyn. (2010) 239:2354–66. doi: 10.1002/dvdy.22379
68. Lammermann T, Germain RN. The multiple faces of leukocyte interstitial migration. Semin Immunopathol. (2014) 36:227–51. doi: 10.1007/s00281-014-0418-8
69. Nitschke M, Aebischer D, Abadier M, Haener S, Lucic M, Vigl B, et al. Differential requirement for ROCK in dendritic cell migration within lymphatic capillaries in steady-state and inflammation. Blood. (2012) 120:2249–58. doi: 10.1182/blood-2012-03-417923
70. Sen D, Forrest L, Kepler TB, Parker I, Cahalan MD. Selective and site-specific mobilization of dermal dendritic cells and Langerhans cells by Th1- and Th2-polarizing adjuvants. Proc Natl Acad Sci USA. (2010) 107:8334–9. doi: 10.1073/pnas.0912817107
71. Tal O, Lim HY, Gurevich I, Milo I, Shipony Z, Ng LG, et al. DC mobilization from the skin requires docking to immobilized CCL21 on lymphatic endothelium and intralymphatic crawling. J Exp Med. (2011) 208:2141–53. doi: 10.1084/jem.20102392
72. Saeki H, Moore AM, Brown MJ, Hwang ST. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J Immunol. (1999) 162:2472–5.
73. Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. (2013) 339:328–32. doi: 10.1126/science.1228456
74. Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. (2008) 453:51–5. doi: 10.1038/nature06887
75. Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, et al. Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science. (2004) 305:1150–3. doi: 10.1126/science.1099159
76. Teijeira A, Hunter MC, Russo E, Proulx ST, Frei T, Debes GF, et al. T cell migration from inflamed skin to draining lymph nodes requires intralymphatic crawling supported by ICAM-1/LFA-1 interactions. Cell Rep. (2017) 18:857–65. doi: 10.1016/j.celrep.2016.12.078
77. Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, Lambert K, et al. Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin alphaV. Nat Immunol. (2013) 14:949–58. doi: 10.1038/ni.2682
78. Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, et al. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. (2006) 203:1519–32. doi: 10.1084/jem.20051210
79. Pflicke H, Sixt M. Preformed portals facilitate dendritic cell entry into afferent lymphatic vessels. J Exp Med. (2009) 206:2925–35. doi: 10.1084/jem.20091739
80. Ma J, Wang JH, Guo YJ, Sy MS, Bigby M. In vivo treatment with anti-ICAM-1 and anti-LFA-1 antibodies inhibits contact sensitization-induced migration of epidermal Langerhans cells to regional lymph nodes. Cell Immunol. (1994) 158:389–99. doi: 10.1006/cimm.1994.1285
81. Xu H, Guan H, Zu G, Bullard D, Hanson J, Slater M, et al. The role of ICAM-1 molecule in the migration of Langerhans cells in the skin and regional lymph node. Eur J Immunol. (2001) 31:3085–93. doi: 10.1002/1521-4141(2001010)31:10<3085::AID-IMMU3085>3.0.CO;2-B
82. Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. (2006) 203:2763–77. doi: 10.1084/jem.20051759
83. Vigl B, Aebischer D, Nitschke M, Iolyeva M, Rothlin T, Antsiferova O, et al. Tissue inflammation modulates gene expression of lymphatic endothelial cells and dendritic cell migration in a stimulus-dependent manner. Blood. (2011) 118:205–15. doi: 10.1182/blood-2010-12-326447
84. Teoh D, Johnson LA, Hanke T, McMichael AJ, Jackson DG. Blocking development of a CD8+ T cell response by targeting lymphatic recruitment of APC. J Immunol. (2009) 182:2425–31. doi: 10.4049/jimmunol.0803661
85. Teijeira A, Garasa S, Pelaez R, Azpilikueta A, Ochoa C, Marre D, et al. Lymphatic endothelium forms integrin-engaging 3D structures during DC transit across inflamed lymphatic vessels. J Invest Dermatol. (2013) 133:2276–85. doi: 10.1038/jid.2013.152
86. Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. (2004) 167:377–88. doi: 10.1083/jcb.200404129
87. Nieminen M, Henttinen T, Merinen M, Marttila-Ichihara F, Eriksson JE, Jalkanen S. Vimentin function in lymphocyte adhesion and transcellular migration. Nat Cell Biol. (2006) 8:156–62. doi: 10.1038/ncb1355
88. Millan J, Hewlett L, Glyn M, Toomre D, Clark P, Ridley AJ. Lymphocyte transcellular migration occurs through recruitment of endothelial ICAM-1 to caveola- and F-actin-rich domains. Nat Cell Biol. (2006) 8:113–23. doi: 10.1038/ncb1356
89. Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. (2009) 30:384–96. doi: 10.1016/j.immuni.2008.12.020
90. Johnson LA, Lawrance W, Roshorn YM, Hanke T, Banerji S, Jackson DG. The lymphatic vessel endothelial receptor LYVE-1 mediates dendritic cell entry to the afferent lymphatics via transmigratory cups that engage the leukocyte hyaluronan glycocalyx. Nat Immunol. (2017) 18:762–70. doi: 10.1038/ni.3750
91. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun. (2015) 6:7139. doi: 10.1038/ncomms8139
92. Weninger W, Biro M, Jain R. Leukocyte migration in the interstitial space of non-lymphoid organs. Nat Rev Immunol. (2014) 14:232–46. doi: 10.1038/nri3641
93. Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, et al. LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J Cell Biol. (1999) 144:789–801. doi: 10.1083/jcb.144.4.789
94. Teriete P, Banerji S, Noble M, Blundell CD, Wright AJ, Pickford AR, et al. Structure of the regulatory hyaluronan binding domain in the inflammatory leukocyte homing receptor CD44. Mol Cell. (2004) 13:483–96. doi: 10.1016/S1097-2765(04)00080-2
95. Banerji S, Wright AJ, Noble M, Mahoney DJ, Campbell ID, Day AJ, et al. Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat Struct Mol Biol. (2007) 14:234–9. doi: 10.1038/nsmb1201
96. Banerji S, Hide BR, James JR, Noble ME, Jackson DG. Distinctive properties of the hyaluronan-binding domain in the lymphatic endothelial receptor Lyve-1 and their implications for receptor function. J Biol Chem. (2010) 285:10724–35. doi: 10.1074/jbc.M109.047647
97. Jackson DG. Immunological functions of hyaluronan and its receptors in the lymphatics. Immunol Rev. (2009) 230:216–31. doi: 10.1111/j.1600-065X.2009.00803.x
98. Mahaffey CL, Mummert ME. Hyaluronan synthesis is required for IL-2-mediated T cell proliferation. J Immunol. (2007) 179:8191–9. doi: 10.4049/jimmunol.179.12.8191
99. Mummert ME. Immunologic roles of hyaluronan. Immunol Res. (2005) 31:189–206. doi: 10.1385/IR:31:3:189
100. Mummert ME, Mummert D, Edelbaum D, Hui F, Matsue H, Takashima A. Synthesis and surface expression of hyaluronan by dendritic cells and its potential role in antigen presentation. J Immunol. (2002) 169:4322–31. doi: 10.4049/jimmunol.169.8.4322
101. Lawrance W, Banerji S, Day AJ, Bhattacharjee S, Jackson DG. Binding of hyaluronan to the native lymphatic vessel endothelial receptor LYVE-1 is critically dependent on Receptor Clustering and Hyaluronan Organization. J Biol Chem. (2016) 291:8014–30. doi: 10.1074/jbc.M115.708305
102. Banerji S, Lawrance W, Metcalfe C, Briggs DC, Yamauchi A, Dushek O, et al. Homodimerization of the lymph vessel endothelial receptor LYVE-1 through a redox-labile disulfide is critical for hyaluronan binding in lymphatic endothelium. J Biol Chem. (2016) 291:25004–18. doi: 10.1074/jbc.M116.736926
103. Dubacheva GV, Curk T, Mognetti BM, Auzely-Velty R, Frenkel D, Richter RP. Superselective targeting using multivalent polymers. J Am Chem Soc. (2014) 136:1722–5. doi: 10.1021/ja411138s
104. Randolph GJ. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. (2008) 19:462–8. doi: 10.1097/MOL.0b013e32830d5f09
105. Randolph GJ. Proliferating macrophages prevail in atherosclerosis. Nat Med. (2013) 19:1094–5. doi: 10.1038/nm.3316
106. Hou WH, Liu IH, Tsai CC, Johnson FE, Huang SS, Huang JS. CRSBP-1/LYVE-1 ligands disrupt lymphatic intercellular adhesion by inducing tyrosine phosphorylation and internalization of VE-cadherin. J Cell Sci. (2011) 124(Pt 8):1231–44. doi: 10.1242/jcs.078154
107. Nermut MV, Green NM, Eason P, Yamada SS, Yamada KM. Electron microscopy and structural model of human fibronectin receptor. EMBO J. (1988) 7:4093–9. doi: 10.1002/j.1460-2075.1988.tb03303.x
108. Bano F, Banerji S, Howarth M, Jackson D, Richter R. A Single molecule assay to probe monovalent and multivalent bonds between hyaluronan and its key leukocyte receptor CD44 under force. Sci Rep. (2016) 6:34176. doi: 10.1038/srep34176
109. Bano F, Tammi MI, Kang DW, Harris EN, Richter RP. Single-molecule unbinding forces between the polysaccharide hyaluronan and its binding proteins. Biophys J. (2018) 114:2910–22. doi: 10.1016/j.bpj.2018.05.014
110. Linehan SA, Martinez-Pomares L, da Silva RP, Gordon S. Endogenous ligands of carbohydrate recognition domains of the mannose receptor in murine macrophages, endothelial cells and secretory cells; potential relevance to inflammation and immunity. Eur J Immunol. (2001) 31:1857–66. doi: 10.1002/1521-4141(200106)31:6<1857::AID-IMMU1857>3.0.CO;2-D
111. Linehan SA, Martinez-Pomares L, Stahl PD, Gordon S. Mannose receptor and its putative ligands in normal murine lymphoid and nonlymphoid organs: in situ expression of mannose receptor by selected macrophages, endothelial cells, perivascular microglia, and mesangial cells, but not dendritic cells. J Exp Med. (1999) 189:1961–72. doi: 10.1084/jem.189.12.1961
112. Irjala H, Alanen K, Grenman R, Heikkila P, Joensuu H, Jalkanen S. Mannose receptor (MR) and common lymphatic endothelial and vascular endothelial receptor (CLEVER)-1 direct the binding of cancer cells to the lymph vessel endothelium. Cancer Res. (2003) 63:4671–6.
113. Marttila-Ichihara F, Turja R, Miiluniemi M, Karikoski M, Maksimow M, Niemela J, et al. Macrophage mannose receptor on lymphatics controls cell trafficking. Blood. (2008) 112:64–72. doi: 10.1182/blood-2007-10-118984
114. Salmi M, Karikoski M, Elima K, Rantakari P, Jalkanen S. CD44 binds to macrophage mannose receptor on lymphatic endothelium and supports lymphocyte migration via afferent lymphatics. Circ Res. (2013) 112:1577–82. doi: 10.1161/CIRCRESAHA.111.300476
115. Irjala H, Elima K, Johansson EL, Merinen M, Kontula K, Alanen K, et al. The same endothelial receptor controls lymphocyte traffic both in vascular and lymphatic vessels. Eur J Immunol. (2003) 33:815–24. doi: 10.1002/eji.200323859
116. Salmi M, Koskinen K, Henttinen T, Elima K, Jalkanen S. CLEVER-1 mediates lymphocyte transmigration through vascular and lymphatic endothelium. Blood. (2004) 104:3849–57. doi: 10.1182/blood-2004-01-0222
117. Karikoski M, Irjala H, Maksimow M, Miiluniemi M, Granfors K, Hernesniemi S, et al. Clever-1/Stabilin-1 regulates lymphocyte migration within lymphatics and leukocyte entrance to sites of inflammation. Eur J Immunol. (2009) 39:3477–87. doi: 10.1002/eji.200939896
118. Prevo R, Banerji S, Ni J, Jackson DG. Rapid plasma membrane-endosomal trafficking of the lymph node sinus and high endothelial venule scavenger receptor/homing receptor stabilin-1 (FEEL-1/CLEVER-1). J Biol Chem. (2004) 279:52580–92. doi: 10.1074/jbc.M406897200
119. Iolyeva M, Karaman S, Willrodt AH, Weingartner S, Vigl B, Halin C. Novel role for ALCAM in lymphatic network formation and function. FASEB J. (2013) 27:978–90. doi: 10.1096/fj.12-217844
120. Maddaluno L, Verbrugge SE, Martinoli C, Matteoli G, Chiavelli A, Zeng Y, et al. The adhesion molecule L1 regulates transendothelial migration and trafficking of dendritic cells. J Exp Med. (2009) 206:623–35. doi: 10.1084/jem.20081211
121. Teijeira A, Palazon A, Garasa S, Marre D, Auba C, Rogel A, et al. CD137 on inflamed lymphatic endothelial cells enhances CCL21-guided migration of dendritic cells. FASEB J. (2012) 26:3380–92. doi: 10.1096/fj.11-201061
122. Torzicky M, Viznerova P, Richter S, Strobl H, Scheinecker C, Foedinger D, et al. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) and CD99 are critical in lymphatic transmigration of human dendritic cells. J Invest Dermatol. (2012) 132:1149–57. doi: 10.1038/jid.2011.420
123. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. (1999) 189:451–60. doi: 10.1084/jem.189.3.451
124. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. (1999) 99:23–33. doi: 10.1016/S0092-8674(00)80059-8
125. Vassileva G, Soto H, Zlotnik A, Nakano H, Kakiuchi T, Hedrick JA, et al. The reduced expression of 6Ckine in the plt mouse results from the deletion of one of two 6Ckine genes. J Exp Med. (1999) 190:1183–8. doi: 10.1084/jem.190.8.1183
126. Mori S, Nakano H, Aritomi K, Wang CR, Gunn MD, Kakiuchi T. Mice lacking expression of the chemokines CCL21-ser and CCL19 (plt mice) demonstrate delayed but enhanced T cell immune responses. J Exp Med. (2001) 193:207–18. doi: 10.1084/jem.193.2.207
127. Nakano H, Gunn MD. Gene duplications at the chemokine locus on mouse chromosome 4: multiple strain-specific haplotypes and the deletion of secondary lymphoid-organ chemokine and EBI-1 ligand chemokine genes in the plt mutation. J Immunol. (2001) 166:361–9. doi: 10.4049/jimmunol.166.1.361
128. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. (2006) 176:4431–9. doi: 10.4049/jimmunol.176.7.4431
129. Menning A, Hopken UE, Siegmund K, Lipp M, Hamann A, Huehn J. Distinctive role of CCR7 in migration and functional activity of naive- and effector/memory-like Treg subsets. Eur J Immunol. (2007) 37:1575–83. doi: 10.1002/eji.200737201
130. Brown MN, Fintushel SR, Lee MH, Jennrich S, Geherin SA, Hay JB, et al. Chemoattractant receptors and lymphocyte egress from extralymphoid tissue: changing requirements during the course of inflammation. J Immunol. (2010) 185:4873–82. doi: 10.4049/jimmunol.1000676
131. Eberhard Y, Ortiz S, Ruiz Lascano A, Kuznitzky R, Serra HM. Up-regulation of the chemokine CCL21 in the skin of subjects exposed to irritants. BMC Immunol. (2004) 5:7. doi: 10.1186/1471-2172-5-7
132. Johnson L, Jackson D. Inflammation-induced secretion of CCL21 in lymphatic endothelium is a key regulator of integrin-mediated dendritic cell transmigration. Int Immunol. (2010) 22:639–49. doi: 10.1093/intimm/dxq435
133. Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol. (2011) 13:67–76. doi: 10.1038/ni.2173
134. Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol. (2010) 11:594–600. doi: 10.1038/ni.1885
135. Rouzaut A, Garasa S, Teijeira A, Gonzalez I, Martinez-Forero I, Suarez N, et al. Dendritic cells adhere to and transmigrate across lymphatic endothelium in response to IFN-alpha. Eur J Immunol. (2010) 40:3054–63. doi: 10.1002/eji.201040523
136. Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. (2007) 171:1249–57. doi: 10.2353/ajpath.2007.070225
137. Kabashima K, Sugita K, Shiraishi N, Tamamura H, Fujii N, Tokura Y. CXCR4 engagement promotes dendritic cell survival and maturation. Biochem Biophys Res Commun. (2007) 361:1012–6. doi: 10.1016/j.bbrc.2007.07.128
138. Geherin SA, Wilson RP, Jennrich S, Debes GF. CXCR4 is dispensable for T cell egress from chronically inflamed skin via the afferent lymph. PLoS ONE. (2014) 9:e95626. doi: 10.1371/journal.pone.0095626
139. Qu C, Edwards EW, Tacke F, Angeli V, Llodra J, Sanchez-Schmitz G, et al. Role of CCR8 and other chemokine pathways in the migration of monocyte-derived dendritic cells to lymph nodes. J Exp Med. (2004) 200:1231–41. doi: 10.1084/jem.20032152
140. Das S, Sarrou E, Podgrabinska S, Cassella M, Mungamuri SK, Feirt N, et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J Exp Med. (2013) 210:1509–28. doi: 10.1084/jem.20111627
141. Sokol CL, Camire RB, Jones MC, Luster AD. The chemokine receptor CCR8 promotes the migration of dendritic cells into the lymph node parenchyma to initiate the allergic immune response. Immunity. (2018) 49:449–63 e6. doi: 10.1016/j.immuni.2018.07.012
142. Johnson LA, Jackson DG. The chemokine CX3CL1 promotes trafficking of dendritic cells through inflamed lymphatics. J Cell Sci. (2013) 126(Pt 22):5259–70. doi: 10.1242/jcs.135343
143. Brown M, Johnson LA, Leone DA, Majek P, Vaahtomeri K, Senfter D, et al. Lymphatic exosomes promote dendritic cell migration along guidance cues. J Cell Biol. (2018) 217:2205–21. doi: 10.1083/jcb.201612051
144. Sato N, Ahuja SK, Quinones M, Kostecki V, Reddick RL, Melby PC, et al. CC chemokine receptor (CCR)2 is required for langerhans cell migration and localization of T helper cell type 1 (Th1)-inducing dendritic cells. Absence of CCR2 shifts the Leishmania major-resistant phenotype to a susceptible state dominated by Th2 cytokines, b cell outgrowth, and sustained neutrophilic inflammation. J Exp Med. (2000) 192:205–18. doi: 10.1084/jem.192.2.205
145. Graham GJ. D6 and the atypical chemokine receptor family: novel regulators of immune and inflammatory processes. Eur J Immunol. (2009) 39:342–51. doi: 10.1002/eji.200838858
146. Lee KM, McKimmie CS, Gilchrist DS, Pallas KJ, Nibbs RJ, Garside P, et al. D6 facilitates cellular migration and fluid flow to lymph nodes by suppressing lymphatic congestion. Blood. (2011) 118:6220–9. doi: 10.1182/blood-2011-03-344044
147. McKimmie CS, Singh MD, Hewit K, Lopez-Franco O, Le Brocq M, Rose-John S, et al. An analysis of the function and expression of D6 on lymphatic endothelial cells. Blood. (2013) 121:3768–77. doi: 10.1182/blood-2012-04-425314
148. Bryce SA, Wilson RA, Tiplady EM, Asquith DL, Bromley SK, Luster AD, et al. ACKR4 on stromal cells scavenges CCL19 to enable CCR7-dependent trafficking of APCs from inflamed skin to lymph nodes. J Immunol. (2016) 196:3341–53. doi: 10.4049/jimmunol.1501542
149. Chiba K, Matsuyuki H, Maeda Y, Sugahara K. Role of sphingosine 1-phosphate receptor type 1 in lymphocyte egress from secondary lymphoid tissues and thymus. Cell Mol Immunol. (2006) 3:11–9.
150. Pham TH, Baluk P, Xu Y, Grigorova I, Bankovich AJ, Pappu R, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. (2010) 207:17–27. doi: 10.1084/jem.20091619
151. Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol. (2008) 9:42–53. doi: 10.1038/ni1534
152. Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D, et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity. (2009) 30:458–69. doi: 10.1016/j.immuni.2008.12.022
153. Brinkman CC, Iwami D, Hritzo MK, Xiong Y, Ahmad S, Simon T, et al. Treg engage lymphotoxin beta receptor for afferent lymphatic transendothelial migration. Nat Commun. (2016) 7:12021. doi: 10.1038/ncomms12021
154. Heng TS, Painter MW, Immunological Genome Project C. The immunological genome project: networks of gene expression in immune cells. Nat Immunol. (2008) 9:1091–4. doi: 10.1038/ni1008-1091
155. Teijeira A, Halin C. Editorial: breaching their way through: neutrophils destroy intercellular junctions to transmigrate rapidly across lymphatic endothelium. J Leukoc Biol. (2015) 98:880–2. doi: 10.1189/jlb.3CE0615-273
156. Kerjaschki D, Bago-Horvath Z, Rudas M, Sexl V, Schneckenleithner C, Wolbank S, et al. Lipoxygenase mediates invasion of intrametastatic lymphatic vessels and propagates lymph node metastasis of human mammary carcinoma xenografts in mouse. J Clin Invest. (2011) 121:2000–12. doi: 10.1172/JCI44751
157. Berk DA, Swartz MA, Leu AJ, Jain RK. Transport in lymphatic capillaries. II. Microscopic velocity measurement with fluorescence photobleaching. Am J Physiol. (1996) 270(1 Pt 2):H330–7.
158. Swartz MA, Berk DA, Jain RK. Transport in lymphatic capillaries. I. Macroscopic measurements using residence time distribution theory. Am J Physiol. (1996) 270(1 Pt 2):H324–9.
159. Russo E, Nitschke M, Halin C. Dendritic cell interactions with lymphatic endothelium. Lymphat Res Biol. (2013) 11:172–82. doi: 10.1089/lrb.2013.0008
160. Russo E, Teijeira A, Vaahtomeri K, Willrodt AH, Bloch JS, Nitschke M, et al. Intralymphatic CCL21 promotes tissue egress of dendritic cells through afferent lymphatic vessels. Cell Rep. (2016) 14:1723–34. doi: 10.1016/j.celrep.2016.01.048
161. Breiteneder-Geleff S, Matsui K, Soleiman A, Meraner P, Poczewski H, Kalt R, et al. Podoplanin, novel 43-kd membrane protein of glomerular epithelial cells, is down-regulated in puromycin nephrosis. Am J Pathol. (1997) 151:1141–52.
162. Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol. (2004) 15:603–12. doi: 10.1097/01.ASN.0000113316.52371.2E
163. Yin X, Johns SC, Kim D, Mikulski Z, Salanga CL, Handel TM, et al. Lymphatic specific disruption in the fine structure of heparan sulfate inhibits dendritic cell traffic and functional T cell responses in the lymph node. J Immunol. (2014) 192:2133–42. doi: 10.4049/jimmunol.1301286
164. Yin X, Johns SC, Lawrence R, Xu D, Reddi K, Bishop JR, et al. Lymphatic endothelial heparan sulfate deficiency results in altered growth responses to vascular endothelial growth factor-C (VEGF-C). J Biol Chem. (2011) 286:14952–62. doi: 10.1074/jbc.M110.206664
165. Yin X, Truty J, Lawrence R, Johns SC, Srinivasan RS, Handel TM, et al. A critical role for lymphatic endothelial heparan sulfate in lymph node metastasis. Mol Cancer. (2010) 9:316. doi: 10.1186/1476-4598-9-316
166. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. (2004) 427:355–60. doi: 10.1038/nature02284
167. Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. (2012) 30:69–94. doi: 10.1146/annurev-immunol-020711-075011
168. Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bolter J, et al. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol. (2011) 12:879–87. doi: 10.1038/ni.2085
169. Ulvmar MH, Werth K, Braun A, Kelay P, Hub E, Eller K, et al. The atypical chemokine receptor CCRL1 shapes functional CCL21 gradients in lymph nodes. Nat Immunol. (2014) 15:623–30. doi: 10.1038/ni.2889
170. Rantakari P, Auvinen K, Jappinen N, Kapraali M, Valtonen J, Karikoski M, et al. The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat Immunol. (2015) 16:386–96. doi: 10.1038/ni.3101
171. Iftakhar EKI, Fair-Makela R, Kukkonen-Macchi A, Elima K, Karikoski M, Rantakari P, et al. Gene-expression profiling of different arms of lymphatic vasculature identifies candidates for manipulation of cell traffic. Proc Natl Acad Sci USA. (2016) 113:10643–8. doi: 10.1073/pnas.1602357113
172. Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, et al. B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity. (2006) 24:203–15. doi: 10.1016/j.immuni.2006.01.003
173. Halin C, Tobler NE, Vigl B, Brown LF, Detmar M. VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood. (2007) 110:3158–67. doi: 10.1182/blood-2007-01-066811
174. Kataru RP, Jung K, Jang C, Yang H, Schwendener RA, Baik JE, et al. Critical role of CD11b+ macrophages and VEGF in inflammatory lymphangiogenesis, antigen clearance, and inflammation resolution. Blood. (2009) 113:5650–9. doi: 10.1182/blood-2008-09-176776
175. Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. (2006) 7:344–53. doi: 10.1038/ni1330
176. Kataru RP, Kim H, Jang C, Choi DK, Koh BI, Kim M, et al. T lymphocytes negatively regulate lymph node lymphatic vessel formation. Immunity. (2011) 34:96–107. doi: 10.1016/j.immuni.2010.12.016
177. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
178. Ivanov S, Scallan JP, Kim KW, Werth K, Johnson MW, Saunders BT, et al. CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. J Clin Invest. (2016) 126:1581–91. doi: 10.1172/JCI84518
179. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. (2010) 121:2437–45. doi: 10.1161/CIRCULATIONAHA.109.916346
180. Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in cardiovascular medicine. Circ Res. (2016) 118:515–30. doi: 10.1161/CIRCRESAHA.115.306544
181. Henri O, Pouehe C, Houssari M, Galas L, Nicol L, Edwards-Levy F, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. (2016) 133:1484–97. Discussion 97. doi: 10.1161/CIRCULATIONAHA.115.020143
Keywords: lymphatic, trafficking, transmigration, dendritic cell, endothelium, chemokine, hyaluronan, LYVE-1
Citation: Jackson DG (2019) Leucocyte Trafficking via the Lymphatic Vasculature— Mechanisms and Consequences. Front. Immunol. 10:471. doi: 10.3389/fimmu.2019.00471
Received: 02 February 2019; Accepted: 21 February 2019;
Published: 14 March 2019.
Edited by:
Amanda W. Lund, Oregon Health & Science University, United StatesReviewed by:
Cornelia Halin, University of Zurich, SwitzerlandAlvaro Teijeira, ETH Zürich, Switzerland
Copyright © 2019 Jackson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David G. Jackson, ZGF2aWQuamFja3NvbkBpbW0ub3guYWMudWs=