Abstract
The identification of Receptor activator of nuclear factor kappa B ligand (RANKL) and its cognate receptor Receptor activator of nuclear factor kappa B (RANK) during a search for novel tumor necrosis factor receptor (TNFR) superfamily members has dramatically changed the scenario of bone biology by providing the functional and biochemical proof that RANKL signaling via RANK is the master factor for osteoclastogenesis. In parallel, two independent studies reported the identification of mouse RANKL on activated T cells and of a ligand for osteoprotegerin on a murine bone marrow-derived stromal cell line. After these seminal findings, accumulating data indicated RANKL and RANK not only as essential players for the development and activation of osteoclasts, but also for the correct differentiation of medullary thymic epithelial cells (mTECs) that act as mediators of the central tolerance process by which self-reactive T cells are eliminated while regulatory T cells are generated. In light of the RANKL-RANK multi-task function, an antibody targeting this pathway, denosumab, is now commonly used in the therapy of bone loss diseases including chronic inflammatory bone disorders and osteolytic bone metastases; furthermore, preclinical data support the therapeutic application of denosumab in the framework of a broader spectrum of tumors. Here, we discuss advances in cellular and molecular mechanisms elicited by RANKL-RANK pathway in the bone and thymus, and the extent to which its inhibition or augmentation can be translated in the clinical arena.
Introduction
Receptor activator of nuclear factor kappa B (RANK) and its ligand (RANKL), encoded, respectively, by the Tumor necrosis factor receptor superfamily member 11A (Tnfrsf11a) and the Tumor necrosis factor ligand superfamily member 11 (Tnfsf11) genes, constitute a receptor-ligand pair initiating a signaling pathway of paramount relevance in many pathophysiological contexts (1). They have been described in the context of T cell-dendritic cell interactions (2), in bone and in the immune system (3, 4), thus triggering the start of the osteoimmunology era. This axis has revealed an unexpected role in the thermoregulation by the central nervous system (5) and in mammary epithelium development during pregnancy and progesterone-driven breast cancer (3, 6). The RANKL-RANK axis has also been involved in diverse immune-mediated diseases affecting the bone (7–9) as well as other tissues (10), and in cancer settings (11). Overall, this pathway has emerged as a potential target of therapy in a wide range of conditions; which at the same time implies monitoring many different physiological functions when interfering with this axis.
As schematically depicted in Figure 1, here we focus on advances in cellular and molecular mechanisms elicited by RANKL-RANK signaling in two functionally related compartments: the bone and the thymus. Moreover, we review novel perspectives to translate inhibition or enhancement of this pathway in the clinic.
Figure 1
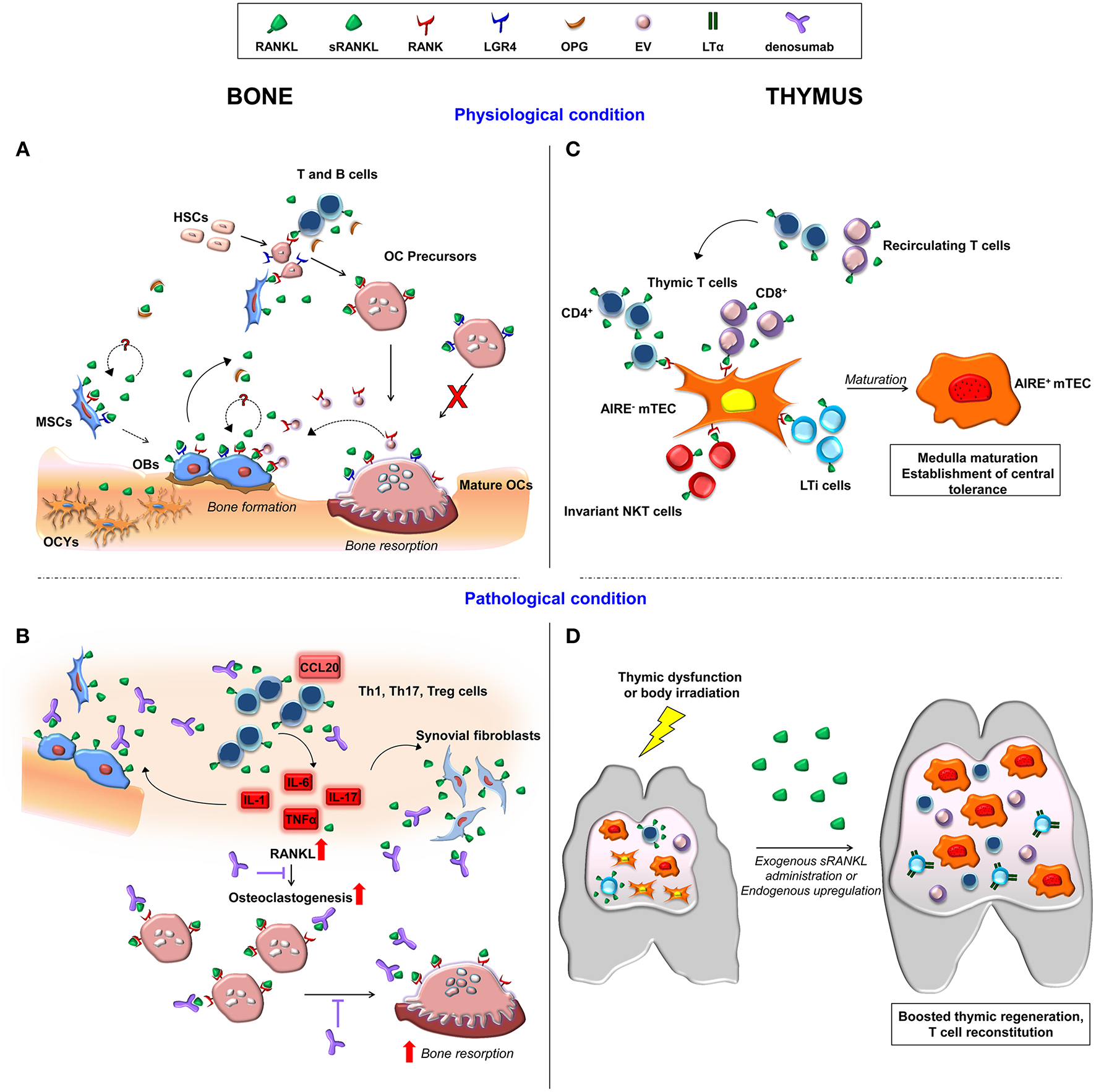
Schematic representation of cellular and molecular players involved in RANKL-RANK signaling axis in the bone and thymus in physiological and in pathological conditions. (A) Membrane-bound and soluble RANKL produced by cells of the osteoblast lineage and by immune cells induce osteoclastogenesis upon its binding to RANK on osteoclast precursors. OPG is the soluble decoy receptor for RANKL. Moreover, RANKL binding to LGR4 on osteoclasts hinders their maturation. RANK expression by MSC and osteoblasts points to a potential RANKL autoregulatory mechanism affecting bone formation. In addition, osteoclast-derived RANK-expressing extracellular vesicles (EV) trigger a reverse signaling on osteoblast. (B) The inflammatory bone environment in pathological condition, such as osteoporosis and rheumatoid arthritis, results in increased production of RANKL by immune cells, osteoblastic cells and synovial fibroblasts. This exacerbates osteoclast generation and bone loss, which are target of denosumab treatment. (C) In the thymus, RANKL produced by resident and recirculating T cells, invariant NKT and LTi cells fosters mTEC AIRE expression and maturation via RANK receptor, allowing correct establishment of central tolerance. (D) In the presence of thymic dysfunction, pharmacological sRANKL administration boosts thymic regeneration, and T cell reconstitution. Similarly, in the early phases of thymic regeneration after body irradiation, CD4+ and LTi cells upregulate RANKL. This results in increased expression of LTα in LTi cells. OBs, osteoblasts; OCs, osteoclasts; OCYs, osteocytes.
RANKL-RANK Axis in the Bone
The identification of RANKL-RANK signaling in bone represents a milestone in bone biology (12, 13). Its indispensable role in osteoclast formation is clearly demonstrated by the complete absence of osteoclasts in the Rankl−/− and Rank−/− murine models (3, 4, 14, 15), as well as in their human counterpart, i.e., patients affected by RANKL-deficient and RANK-deficient osteoclast-poor Autosomal Recessive Osteopetrosis (16, 17). Nonetheless, the possibility of RANKL-independent osteoclastogenesis, particularly in pathologic conditions, has been a matter of a long-lasting debate (18–22) and a general consensus in the field has not been reached, yet.
RANKL is mainly produced by stromal cells in bone, in normal conditions, and primarily by osteocytes (23–25). RANKL is mostly membrane-bound and can be shed to form a soluble protein; the former is sufficient for most functions, while the latter contributes to physiological bone remodeling, as recently demonstrated in mice expressing a sheddase-resistant form of RANKL (26).
The membrane functional receptor RANK is mainly expressed by cells of hematopoietic origin, including also osteoclasts and their precursors, and has been recently detected also in Mesenchymal Stem Cells (MSCs) (27, 28), raising the intriguing hypothesis of an autocrine/paracrine loop in these cells (Figure 1A).
The RANKL-RANK signaling pathway in the osteoclast lineage comprises a plethora of molecules (29). Essentially, upon engagement by its ligand, RANK recruits a number of adaptors (most importantly, TNF Receptor-Associated Factor 6, TRAF6) (30), which converge on kinases activation, including Phosphoinositide-3-Kinase (PI3K) and Mitogen Activated Protein (MAP) kinases. This promotes nuclear translocation and activation of transcription factors, Nuclear Factor of Activated T cell 1 (NFATc1) (31), c-fos (32), and Nuclear Factor kappa B (NF-κB) (33), comprising the master regulator of the osteoclast-specific transcriptional program. The RANKL-RANK pathway interacts with costimulatory signals from immunoreceptor tyrosine based activation (ITAM)-motif containing proteins, further regulating NFATc1 activation (34, 35).
RANKL signaling during osteoclastogenesis results in the generation of reactive oxygen species (ROS), which further stimulate osteoclast formation and bone resorption (36). On the other hand, a variety of antioxidant mechanisms monitors ROS levels and the reciprocal control between these opposite functions (i.e., ROS production and scavenging) importantly impacts on bone homeostasis (37–39).
The RANKL-RANK axis is counterbalanced by the soluble decoy receptor osteoprotegerin (OPG) (40), which is itself controlled by many ligands, including the TNF-Related Apoptosis Inducing Ligand (TRAIL), von Willebrand factor (vWF), and glycosaminoglycans (GAGs) (41). Moreover, the Leucine-rich repeat-containing G protein-coupled receptor 4 (LGR4) is an additional membrane receptor for RANKL, competing with RANK for ligand binding and negatively regulating osteoclastogenesis through the inhibition of NFATc1 activation (42). LGR4 acts also as an R-spondin receptor in bone marrow MSCs and has been recently demonstrated as a key molecule in mesoderm-derived tissue development and MSC differentiation (43), whether RANKL might be involved in this specific context has to be investigated (Figure 1A).
The recognition of the crucial role of RANKL-RANK signaling in osteoclast biology led to the development of the anti-RANKL antibody denosumab, a fully human Immunoglobulin (Ig) G2 monoclonal antibody with high affinity and specificity for human soluble and membrane-bound RANKL (44). Specifically, denosumab binds to the DE loop region of the ligand, which is one of the surface loop structures interacting with the functional receptor on responding cells (44). Denosumab is used as an antiresorptive drug for diverse indications, such as osteoporosis (45), primary bone tumors (46), and osteolytic bone metastases (47). Its use is under evaluation also in other fields, such as solid tumors (11) and Rheumatoid Arthritis (48), and has been very recently proposed in the prevention of BRCA1-associated breast cancer (49). Finally, denosumab administration has been considered in the field of rare diseases too, for example for the treatment of persistent severe hypercalcemia after hematopoietic stem cell transplantation in patients affected by Autosomal Recessive Osteopetrosis (50), in patients affected by Fibrous Dysplasia (51), or by Osteogenesis Imperfecta, even though some variability in the clinical outcome has been reported (52) (Figure 1B).
Clinical case series and a recent analysis of the FREEDOM and FREEDOM Extension Trials about osteoporosis treatment with the anti-RANKL antibody have pointed to an increased risk of multiple vertebral fractures after denosumab discontinuation due to a rebound in bone resorption (53, 54), thus raising a note of caution. In an attempt to identify potential alternative antiresorptive therapies, scientific interest about natural compounds possibly interfering with the RANKL-RANK axis (e.g., flavonoids, alkaloid compounds, triterpenoids, polysaccharides as well as monomeric sugars) has been growing exponentially, as demonstrated by the number of publications evaluating this kind of approach (55–58).
In parallel, recent papers pointed to an unexpected osteogenic function of RANKL through (at least) two different, not mutually exclusive mechanisms: an autocrine-paracrine loop activated by RANKL binding to its receptor(s) on MSCs (27); and a reverse signaling elicited by osteoclast-derived RANK-expressing extracellular vesicles, which might induce membrane-RANKL clustering on osteoblasts (59, 60). This might represent an additional means for osteoblast-osteoclast crosstalk. As a perspective, it might be exploited by means of a new drug with two simultaneous activities: dampening of bone resorption by preventing RANKL binding to RANK receptors on the osteoclasts, and stimulating osteogenesis by triggering RANKL signaling in the osteoblasts (Figure 1A).
Actually, the biological relevance of these new findings in the framework of the overall bone homeostasis has to be clearly defined; for the sake of completeness, opposite results have been reported by others (28). Nevertheless, the possibility of an osteogenic function of RANKL is worth further investigations since it could pave the way to the development of new therapeutic strategies, thus fulfilling a medical need.
RANKL-RANK Axis in the Thymus
The thymus is a primary lymphoid organ responsible for the development of T lymphocytes expressing a T cell repertoire capable of responding to a diverse array of foreign antigens but tolerant to self-antigens (61, 62). Migrant lymphoid progenitors, arising in the liver during embryonic life and in the bone marrow in postnatal life, enter the thymus where they undergo different phases of differentiation throughout a complex journey from the cortical region to the medullary compartment (63). The early phases of thymocyte differentiation strictly depend on stromal derived signals mediated by the interaction of CD4+CD8+ double positive (DP) T cell precursors with cortical thymic epithelial cells (cTECs) and indirectly by the production of soluble factors (Figure 1C). cTECs foster lineage commitment during the early stages of T cell differentiation (double negative, DN, stage) through the expression of Notch ligand Delta-like 4 (64, 65) and mediate positive selection of DP T cells by presenting a broad array of self-peptides via major histocompatibility complex (MHC) class I and II molecules. This process results in the survival of thymocytes, which migrate into the thymic medulla where T cells are negatively selected to single positive (SP) CD4+CD8− and CD8+CD4− T cells (66). Mature medullary thymic epithelial cells (mTECs) mediate central tolerance process by expressing the transcriptional coactivator AutoImmune Regulator (AIRE), which drives the expression of self-antigens, including tissue restricted antigens (TRAs) leading to the clonal deletion of autoreactive T cells, while inducing the generation of regulatory T cells (67, 68), and the intra-thymic positioning of X-C Motif Chemokine Ligand 1 (XCL1)+ dendritic cells (69).
Various factors modulate the development and maturation of the thymic epithelial compartment, including several signal transducers regulating NF-κB pathway and the NF-κB family member RelB (70–76). Signaling mediated by four receptors of the tumor necrosis factor family [RANK, OPG, CD40, and lymphotoxin (LT) β receptor] acts as important modulator of thymic microenvironment along with the cross talk between thymocytes and TECs (77–79). In addition, the Ets transcription factor family member Spi-B, which was found to be associated with autoimmune phenomena (80), mediates OPG expression via a negative feedback regulatory loop thus limiting the development of mature TECs (81). RANKL is mainly produced by CD4+ cells, a small subset of CD8+ cells, invariant (Natural Killer T) NKT cells and CD4+CD3− lymphoid tissue inducer (LTi) cells (82, 83). Of note, during embryonic life at the initial stages of thymus development, invariant Vγ5+ dendritic epidermal T cells (DETCs) and Vγ5+ γδ T cells T cells contribute to central tolerance establishment by promoting CD80−Aire− mTECs to become CD80+Aire+ mTECs (84–86) thus supporting a critical role for RANK signaling in the interaction between fetal γδ T cell progenitors and mTECs (87, 88). Of note, these immune cell subsets provide different physiological levels of RANKL and CD40 Ligand (CD40L) during ontogeny. During fetal life, mTEC development is controlled by the expression of RANKL by LTi and invariant Vγ5+ DETC progenitors, while after birth is controlled by RANKL and CD40L produced by αβ T Cell Receptor (TCR)high CD4+ thymocytes (89).
Transgenic mice expressing Venus, a fluorescent protein to track RANK expression, showed that this receptor is mainly expressed by mTECs at different stages of differentiation (90). Moreover, activated T cells recirculating to the thymus further contribute to the production of RANKL (91). Thus, it is tempting to speculate that the increased production of RANKL may support the skewing toward mTEC lineage, with consequent maturation of T cells leading to the exhaustion of the progenitor pool. These observations might explain the age-related changes observed in thymic epithelium during aging or thymic dysmorphology found in some pathological conditions (92, 93).
Extensive in vitro and in vivo studies have further confirmed the relevant role of the RANKL-RANK axis in the establishment and maintenance of the central tolerance process. In vitro stimulation of fetal thymic organ culture (FTOC) with recombinant RANKL or agonistic anti-RANK antibody results in the upregulation of CD80 and Aire expression by mTECs (87, 94). In parallel, mice deficient in TCRα or murine models with a reduced number of CD4+ T cells for instance lacking molecules of the MHC II complex have a dramatic reduction in Aire+ cells and decreased mTEC compartment (95, 96). Other molecular players contribute to TEC differentiation and among them a peculiar role is played by the interferon regulatory factor 7/interferon β/ interferon-α/β receptor/signal transducer and activator of transcription 1 (IRF7/IFNβ/IFNAR/STAT1) pathway (97). During embryonic life, the absence of RANK or RANKL severely affects mTEC maturation resulting in the complete loss of Aire+ mTECs (87, 94, 98). However, after birth other factors compensate the absence of RANK signaling allowing the maturation of few Aire+ mTECs (94). Furthermore, OPG is expressed by mTECs and genetically deletion in mice causes enlargement of the medulla area (82, 90). Overall, these data indicate that the RANKL-RANK axis is essential for the correct differentiation and development of mTECs and for the formation of the thymic medulla and consequent establishment of self-tolerance (Figure 1C). Consistently with the role of RANKL as a potent mTEC inducer and indirectly as a key player in the control of central tolerance, systemic administration of soluble RANKL (sRANKL) can be considered to treat primary or secondary thymic dysfunction (99). Transgenic mice constitutively overexpressing human sRANKL displayed thymic medulla enlargement (100) and increased number of Aire+ mTECs (101). Interestingly, during in vivo administration of recombinant soluble RANKL (sRANKL) to cure the bone defect in Rankl−/− mice, we observed a dramatic effect of the cytokine on thymic architecture (102) further confirming data reported in literature. Pharmacological sRANKL treatment induced expansion of the medulla in Rankl−/− mice and increase of Aire+ mTECs. Improvement of thymic epithelium resulted in higher frequency of CD4+ and CD8+ SP and reduction of double positive thymocytes (102). These data suggest that the exogenous administration of RANKL may be a new therapeutic strategy to boost thymic regeneration. In line with this, compelling evidence indicate that upon body irradiation CD4+ cells and LTi cells up-regulate RANKL in the early phase of thymic regeneration. Upon tissue damage, RANKL mediates the increased expression of LTα by LTi cells and reduces the expression of pro-apoptotic genes while increases the expression of the B-cell lymphoma-extra large (Bcl-xl) anti-apoptotic gene (103). The administration of RANKL to wild-type animals confirmed its crucial role in thymic recovery by enhancing TECs, thymocyte numbers, and in parallel increasing vasculature. Improved T cell reconstitution is also mediated by the increased expression of adhesion molecules and chemokines, which foster thymus homing of lymphoid progenitors. Remarkably, since RANKL is the master gene of osteoclastogenesis, it is tempting to speculate that the increased osteoclast activity may also boost hematopoiesis and consequent migration of thymic progenitors. Overall, these in vivo findings confirm the therapeutic effect of RANKL suggesting its putative use to boost immune reconstitution in transplanted elderly patients or in patients affected by primary thymic epithelial defects (104–106) (Figure 1D). Conversely, transient inhibition of RANKL in murine models indicate its effect on thymic negative selection of self-reactive T cells specific for tumor antigens, and resulting in an improvement of antitumor immune response (107, 108). However, in vivo inhibition of RANKL during prenatal life in rats and mice or long-life inhibition after birth did not show gross effects on innate or humoral immune response (109), thus supporting a possible repurposing of denosumab as anti-tumoral agent in combinatorial treatments and extending its use in the clinical arena.
T Cells and RANKL-RANK Signaling in Bone Pathology
The overall picture described highlighted the importance of the RANKL-RANK axis in the bone and thymus compartments: in the former, RANKL-RANK signaling influences the bone remodeling process regulating bone cells activities; in the latter, it is pivotal in thymic cell development and T cell maturation and functioning.
After maturation, T cells exert their function centrally and in all the other peripheral organs, going back also to the bone. Although T cell levels represent about 3–8% of total nucleated bone marrow cells in homeostatic conditions (110), in pathological settings T cell recruitment from the periphery may occur and induce molecular and metabolic changes in bone cells, contributing to the bone loss phenotype associated with various conditions such as post-menopausal osteoporosis and Rheumatoid Arthritis (RA) (Figure 1B).
In post-menopausal osteoporotic patients an increase in RANKL production by activated T cells (and B cells, too), alone or in combination with TNFα, has been reported (111, 112). A similar finding has been shown in surgically ovariectomized (OVX) pre-menopausal women (113), further confirming the causative link between estrogen deprivation, T cell activation and RANKL-mediated bone loss previously observed in the murine model (114). Accordingly, 17β-estradiol inhibits thymic expansion after OVX in mice and T cell development, and protects against bone loss, while selective estrogen receptor modulators exhibit agonistic activity on bone but do not affect T lymphopoiesis (115). Of note, a study in thymectomized pre-menopausal women showed a drop in T cell counts after surgery, as expected, with enhanced activation and production of osteoclastogenic factors by the remaining T cells (116). On the other hand, the authors of the study hypothesized that the establishment of not clarified compensatory mechanisms could be responsible for maintaining bone density at levels similar to euthymic age-matched controls.
Another example of bone-thymus interplay is RA, a chronic inflammatory autoimmune disease characterized by joint inflammation, involving mainly synovial membranes, and bone and cartilage destruction (117, 118). In this condition, the synovium and articular tissues are highly enriched in inflammatory leukocytes, likely due to cell recruitment in the inflamed tissue (119), sustained by resident stromal cells of mesenchymal origin (120). The inflammatory process in the joints is suggested to enhance bone loss in patients with RA, in particular when Anti-Citrullinated Protein Antibodies (ACPA), Rheumatoid Factor (RF) and anti-Carbamylated Protein Antibodies (anti-CarP) are present (121, 122). Most of the T cells recruited from the circulation are T helper 1 (Th1), Th17, and Treg cells (123), which express C-X-C Motif Chemokine Receptor 3 (CXCR3), CXCR4, C-C chemokine receptor type 5 (CCR5), and CCR6 (mainly on Th17 cells) receptors that permit their entry into the inflammatory site upon attraction by the high levels of chemokines (e.g., CCL20) found in arthritic joints (124–126). The relevant presence of these cells exacerbates bone erosion by osteoclasts located at the interface between the synovial membrane and bone (48). The pathological bone loss is not compensated by osteoblast-repairing activity since this process is inhibited by synovial inflammation (127). Pro-inflammatory cytokines, such as IL-1, IL-6, and more importantly TNFα and IL-17 are produced in the inflamed synovium and strongly induce RANKL production through the activation of NF-κB pathway in synovial cells and T cells, which in turn massively activate osteoclasts (22, 48, 128). In patients with early RA, RANKL plasma levels have been associated with bone destruction and with radiological progression of the disease after 24 months of follow-up (129). Moreover, the combined presence of increased RANKL levels and the positivity for anti-Cyclic Citrullinated Peptide 2 (anti-CCP2) antibodies correlated with a more destructive process. These data were confirmed in a case-control study conducted in RA pre-symptomatic patients, where RANKL plasma levels were higher in pre-symptomatic individuals as compared to control subjects, increased over time until the onset of RA symptoms and were associated with levels of inflammatory cytokines. However, the positivity for ACPA/RF/anti-CarP preceded the rise of RANKL plasma levels (129).
Based on this, preventing bone erosion by targeting RANKL-RANK axis could be an effective strategy for intervention (130). In fact, taking into account that RANKL-RANK axis is a pivotal immune modulator in DC development and function, in memory B cells, Th17, and Treg cells (131), RANKL blockade might modulate the immune response thus contributing to limit pathological bone erosion and joint damage occurring in RA.
In a phase II trial (Denosumab in patients with RheumatoId arthritis on methotrexate to Validate inhibitory effect on bone Erosion -DRIVE- study) on Japanese RA patients treated with methotrexate, denosumab significantly inhibited the progression of bone erosion at 12 months, and preserved the bone mineral density (132). In addition, in a retrospective cohort trial, the decrease of bone erosion in patients treated with denosumab in combination with biological disease-modifying anti-rheumatic drugs (bDMARDs), at 12 months was significantly higher as compared to denosumab alone, with no adverse effects. Therefore, blocking RANKL-RANK signaling in RA patients by the addition of denosumab to conventional treatment agents may represent a potential new therapeutic option for patients to limits RA pathological outcome (Figure 1B).
Importantly, RA is primarily an autoimmune disease, in which defects in central and peripheral T cell tolerance are involved. Altered intra-thymic selection for the removal of autoreactive T cells may have a great impact on the onset of T cell mediated autoimmune disease (133). In the SKG strain murine model of autoimmune arthritis, bearing a spontaneous point mutation in Zeta Chain of T Cell Receptor Associated Protein Kinase 70 (ZAP-70), alterations in αβ TCR signaling in the thymus have been linked to the escape of autoreactive T cells from negative selection, playing an essential role in immune response in the periphery (134). In turn, the onset of RA may be due to impaired peripheral tolerance mechanisms, mainly elicited by Treg cells, in controlling autoreactive T cells (135, 136). In addition, recirculation of peripheral T cells back to the thymus has been described, and the re-entering cells (mainly Treg cells) might alter central tolerance and induce the deletion of thymic antigen presenting cell populations. This could be considered a mechanism for silencing autoreactive T cells in an RA setting where impaired thymic functions are present (133). Whether alteration of this process may be linked to T cell mediated autoimmunity is still not clear and how T cell production in the thymus and their effector functions in the periphery regulate tolerance maintenance needs further investigation from a therapeutic point of view.
Overall, although targeting RANKL-RANK axis in RA with a RANKL antagonist can improve bone and joints pathological features, it remains to be defined whether an effect on central tolerance and autoimmune reactions is achieved too, because of RANKL requirement for the correct thymic development and production of functional T cells.
Finally, interest has recently grown in another field, i.e., regarding the possibility to exploit immune-related mechanisms based on RANKL-RANK signaling in cancer settings for therapeutic purposes (11). In malignancies with enhanced RANKL expression, such as Multiple Myeloma, denosumab alone is well-known to be effective in terms of overall survival and skeletal-related events (137). In different tumor types that usually have low expression of RANKL, denosumab treatment combined with immune check-point inhibitors might lead to a cross-modulation of antitumor immunity (138, 139). The mechanisms proposed are various: denosumab might act on RANKL-expressing tumor infiltrating lymphocytes and relieve their anticancer activity that is otherwise blocked by engagement of the ligand with RANK receptor on cells of the tumor microenvironment (138, 139). Moreover, RANKL antagonists might put a break on central tolerance by transiently inhibiting negative selection in the thymus, resulting in the release of self-specific T cells in the periphery (108). Finally, the activation of reverse-signaling pathways might be proposed (140, 141), in line with mechanisms described in bone (142). At present, all these possibilities require further investigations; their elucidation might shed light on novel therapeutic perspectives.
Conclusions
The RANKL-RANK axis exerts pleiotropic effects and consistently involves an ever-increasing number of molecular and cellular players. In the bone and thymus compartments, where the crucial role of RANKL signaling was recognized first, novel functions have recently been discovered. This extends our understanding of the basic biology of these tissues and has translational implications in terms of current therapies monitoring. In particular, opposite effects are expected in the case of blocking or activating the RANKL-RANK pathway on bone and immune tolerance: while used as an antiresorptive drug, the anti-RANKL antibody denosumab might have adverse effects on the establishment of central tolerance, which would deserve attention. On the other hand, recent advances might support efforts toward drug repurposing strategies and development of new medicines, based on limitations of those currently available.
Statements
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The original work was supported by the Italian Telethon Grant C5, the European Community's Seventh Framework Program (FP7/2007-2013, SYBIL Project), PRIN Project (2015F3JHMB_004), and by Programma Nazionale per la Ricerca-Consiglio Nazionale delle Ricerche Aging Project to AV. CM is recipient of an ECTS Basic Research Fellowship.
Acknowledgments
We acknowledge the many authors whose original contribution in the field has not been cited in this mini review for the sake of brevity.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- ACPA
Anti-Citrullinated Protein Antibodies
- AIRE
Auto Immune Regulator
- anti-CarP
anti-Carbamylated Protein
- anti-CCP2
anti-Cyclic Citrullinated Peptide 2
- Bcl-xl
B-cell lymphoma-extra-large
- CCL20
C-C Motif Chemokine Ligand 20
- CCR5
C-C chemokine receptor type 5
- CCR6
C-C chemokine receptor type 6
- cTEC
cortical Thymic Epithelial Cell
- CXCR3
C-X-C Motif Chemokine Receptor 3
- CXCR4
C-X-C Motif Chemokine Receptor 4
- bDMARDs
biological Disease-Modifying Anti-Rheumatic Drugs
- DETC
dendritic epidermal T cell
- DP
Double Positive
- FTOC
fetal thymic organ culture
- GAGs
glycosaminoglycans
- IL-1
Interleukin 1
- IL-6
Interleukin 6
- IL-17
Interleukin 17
- IFNβ
Interferon beta
- IFNAR
Interferon-α/β receptor
- IRF7
Interferon Regulatory Factor 7
- ITAM
Immunoreceptor Tyrosine-based Activation Motif
- LGR4
Leucine Rich Repeat Containing G Protein-Coupled Receptor 4
- LTα
Lymphotoxin α
- LTi
Lymphoid Tissue inducer cells
- MAP kinase
Mitogen Activated Protein kinase
- MHC
Major Histocompatibility Complex
- MSCs
Mesenchymal Stem Cells
- mTEC
medullary Thymic Epithelial Cell
- NFATc1
Nuclear Factor Of Activated T Cells 1
- NF-κB
Nuclear Factor kappa B
- OPG
Osteoprotegerin
- OVX
ovariectomized
- PI3K
Phosphoinositide 3-kinase
- RA
Rheumatoid Arthritis
- RANK
Receptor Activator of Nuclear Factor kappa B
- RANKL
Receptor Activator of Nuclear Factor kappa B Ligand
- RF
Rheumatoid Factor
- ROS
Reactive Oxygen Species
- SP
Single Positive
- STAT1
Signal Transducer and Activator of Transcription 1
- TCR
T Cell Receptor
- Th1
T helper 1 cells
- Th17
T helper 17 cells
- TNFα
Tumor Necrosis Factor α
- TNFR
Tumor Necrosis Factor Receptor
- TNFRSF11A
Tumor Necrosis Factor Receptor Superfamily Member 11A
- TNFSF11
Tumor Necrosis Factor Ligand Superfamily Member 11
- TRAF6
TNF Receptor-Associated Factor 6
- TRAIL
TNF-Related Apoptosis Inducing Ligand
- TRAs
Tissue Restricted Antigens
- Treg
T regulatory cells
- vWF
von Willebrand factor
- XCL-1
X-C Motif Chemokine Ligand 1
- ZAP-70
Zeta Chain Of T Cell Receptor Associated Protein Kinase 70.
Abbreviations
References
1.
Rao S Cronin SJF Sigl V Penninger JM . RANKL and RANK: from mammalian physiology to cancer treatment. Trends Cell Biol. (2018) 28:213–23. 10.1016/j.tcb.2017.11.001
2.
Anderson DM Maraskovsky E Billingsley WL Dougall WC Tometsko ME Roux ER et al . A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. (1997) 390:175–9. 10.1038/36593
3.
Kong YY Yoshida H Sarosi I Tan HL Timms E Capparelli C et al . OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. (1999) 397:315–23. 10.1038/16852
4.
Kim N Odgren PR Kim DK Marks SC Jr Choi Y . Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci USA. (2000) 97:10905–10. 10.1073/pnas.200294797
5.
Hanada R Leibbrandt A Hanada T Kitaoka S Furuyashiki T Fujihara H et al . Central control of fever and female body temperature by RANKL/RANK. Nature. (2009) 462:505–9. 10.1038/nature08596
6.
Schramek D Leibbrandt A Sigl V Kenner L Pospisilik JA Lee HJ et al . Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature. (2010) 468:98–102. 10.1038/nature09387
7.
Hofbauer LC Schoppet M . Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA. (2004) 292:490–5. 10.1001/jama.292.4.490
8.
Han YK Jin Y Miao YB Shi T Lin XP . Improved RANKL production by memory B cells: a way for B cells promote alveolar bone destruction during periodontitis. Int Immunopharmacol. (2018) 64:232–7. 10.1016/j.intimp.2018.08.033
9.
Tanaka S Tanaka Y Ishiguro N Yamanaka H Takeuchi T . RANKL: a therapeutic target for bone destruction in rheumatoid arthritis. Mod Rheumatol. (2018) 28:9–16. 10.1080/14397595.2017.1369491
10.
Guerrini MM Okamoto K Komatsu N Sawa S Danks L Penninger JM et al . Inhibition of the TNF family cytokine RANKL prevents autoimmune inflammation in the central nervous system. Immunity. (2015) 43:1174–85. 10.1016/j.immuni.2015.10.017
11.
Ahern E Smyth MJ Dougall WC Teng MWL . Roles of the RANKL-RANK axis in antitumour immunity - implications for therapy. Nat Rev Clin Oncol. (2018) 15:676–93. 10.1038/s41571-018-0095-y
12.
Nakagawa N Kinosaki M Yamaguchi K Shima N Yasuda H Yano K et al . RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun. (1998) 253:395–400. 10.1006/bbrc.1998.9788
13.
Yasuda H Shima N Nakagawa N Yamaguchi K Kinosaki M Mochizuki S et al . Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. (1998) 95:3597–602. 10.1073/pnas.95.7.3597
14.
Dougall WC Glaccum M Charrier K Rohrbach K Brasel K De Smedt T et al . RANK is essential for osteoclast and lymph node development. Genes Dev. (1999) 13:2412–24. 10.1101/gad.13.18.2412
15.
Li J Sarosi I Yan XQ Morony S Capparelli C Tan HL et al . RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA. (2000) 97:1566–71. 10.1073/pnas.97.4.1566
16.
Sobacchi C Frattini A Guerrini MM Abinun M Pangrazio A Susani L et al . Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat Genet. (2007) 39:960–2. 10.1038/ng2076
17.
Guerrini MM Sobacchi C Cassani B Abinun M Kilic SS Pangrazio A et al . Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet. (2008) 83:64–76. 10.1016/j.ajhg.2008.06.015
18.
Kobayashi K Takahashi N Jimi E Udagawa N Takami M Kotake S et al . Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. (2000) 191:275–86. 10.1084/jem.191.2.275
19.
Kim N Kadono Y Takami M Lee J Lee SH Okada F et al . Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. (2005) 202:589–95. 10.1084/jem.20050978
20.
O'Brien W Fissel BM Maeda Y Yan J Ge X Gravallese EM et al . RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. (2016) 68:2889–900. 10.1002/art.39837
21.
Tsukasaki M Hamada K Okamoto K Nagashima K Terashima A Komatsu N et al . LOX fails to substitute for RANKL in osteoclastogenesis. J Bone Miner Res. (2017) 32:434–9. 10.1002/jbmr.2990
22.
Mbalaviele G Novack DV Schett G Teitelbaum SL . Inflammatory osteolysis: a conspiracy against bone. J Clin Invest. (2017) 127:2030–9. 10.1172/JCI93356
23.
Nakashima T Hayashi M Fukunaga T Kurata K Oh-Hora M Feng JQ et al . Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. (2011) 17:1231–4. 10.1038/nm.2452
24.
Xiong J Onal M Jilka RL Weinstein RS Manolagas SC O'Brien CA . Matrix-embedded cells control osteoclast formation. Nat Med. (2011) 17:1235–41. 10.1038/nm.2448
25.
Fujiwara Y Piemontese M Liu Y Thostenson JD Xiong J O'Brien CA . RANKL (receptor activator of NFkB ligand) produced by osteocytes is required for the increase in B cells and bone loss caused by estrogen deficiency in mice. J Biol Chem. (2016) 291:24838–50. 10.1074/jbc.M116.742452
26.
Xiong J Cawley K Piemontese M Fujiwara Y Zhao H Goellner JJ et al . Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun. (2018) 9:2909. 10.1038/s41467-018-05244-y
27.
Schena F Menale C Caci E Diomede L Palagano E Recordati C et al . Murine Rankl(-/-) mesenchymal stromal cells display an osteogenic differentiation defect improved by a RANKL-expressing lentiviral vector. Stem Cells. (2017) 35:1365–77. 10.1002/stem.2574
28.
Chen X Zhi X Wang J Su J . RANKL signaling in bone marrow mesenchymal stem cells negatively regulates osteoblastic bone formation. Bone Res. (2018) 6:34. 10.1038/s41413-018-0035-6
29.
Okamoto K Nakashima T Shinohara M Negishi-KOGA T Komatsu N Terashima A et al . Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev. (2017) 97:1295–349. 10.1152/physrev.00036.2016
30.
Kobayashi N Kadono Y Naito A Matsumoto K Yamamoto T Tanaka S et al . Segregation of TRAF6-mediated signaling pathways clarifies its role in osteoclastogenesis. EMBO J. (2001) 20:1271–80. 10.1093/emboj/20.6.1271
31.
Kim K Lee SH Ha Kim J Choi Y Kim N . NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol Endocrinol. (2008) 22:176–85. 10.1210/me.2007-0237
32.
Matsuo K Owens JM Tonko M Elliott C Chambers TJ Wagner EF . Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nat Genet. (2000) 24:184–7. 10.1038/72855
33.
Abu-Amer Y . NF-kappaB signaling and bone resorption. Osteoporos Int. (2013) 24:2377–86. 10.1007/s00198-013-2313-x
34.
Kim HS Kim DK Kim AR Mun SH Lee SK Kim JH et al . Fyn positively regulates the activation of DAP12 and FcRgamma-mediated costimulatory signals by RANKL during osteoclastogenesis. Cell Signal. (2012) 24:1306–14. 10.1016/j.cellsig.2012.02.014
35.
Li S Miller CH Giannopoulou E Hu X Ivashkiv LB Zhao B . RBP-J imposes a requirement for ITAM-mediated costimulation of osteoclastogenesis. J Clin Invest. (2014) 124:5057–73. 10.1172/JCI71882
36.
Lee NK Choi YG Baik JY Han SY Jeong DW Bae YS et al . A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. (2005) 106:852–9. 10.1182/blood-2004-09-3662
37.
Gambari L Lisignoli G Cattini L Manferdini C Facchini A Grassi F . Sodium hydrosulfide inhibits the differentiation of osteoclast progenitor cells via NRF2-dependent mechanism. Pharmacol Res. (2014) 87:99–112. 10.1016/j.phrs.2014.06.014
38.
Tan P Guan H Xie L Mi B Fang Z Li J et al . FOXO1 inhibits osteoclastogenesis partially by antagnozing MYC. Sci Rep. (2015) 5:16835. 10.1038/srep16835
39.
Kim HS Nam ST Mun SH Lee SK Kim HW Park YH et al . DJ-1 controls bone homeostasis through the regulation of osteoclast differentiation. Nat Commun. (2017) 8:1519. 10.1038/s41467-017-01527-y
40.
Simonet WS Lacey DL Dunstan CR Kelley M Chang MS Luthy R et al . Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. (1997) 89:309–19. 10.1016/S0092-8674(00)80209-3
41.
Baud'huin M Duplomb L Teletchea S Lamoureux F Ruiz-Velasco C Maillasson M et al . Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev. (2013) 24:401–9. 10.1016/j.cytogfr.2013.06.001
42.
Luo J Yang Z Ma Y Yue Z Lin H Qu G et al . LGR4 is a receptor for RANKL and negatively regulates osteoclast differentiation and bone resorption. Nat Med. (2016) 22:539–46. 10.1038/nm.4076
43.
Sun P Jia K Zheng C Zhu X Li J He L et al . Loss of Lgr4 inhibits differentiation, migration and apoptosis, and promotes proliferation in bone mesenchymal stem cells. J Cell Physiol. (2018). 10.1002/jcp.27927 [Epub ahead of print].
44.
Lacey DL Boyle WJ Simonet WS Kostenuik PJ Dougall WC Sullivan JK et al . Bench to bedside: elucidation of the OPG-RANK-RANKL pathway and the development of denosumab. Nat Rev Drug Discov. (2012) 11:401–19. 10.1038/nrd3705
45.
Lewiecki EM . New and emerging concepts in the use of denosumab for the treatment of osteoporosis. Ther Adv Musculoskelet Dis. (2018) 10:209–23. 10.1177/1759720X18805759
46.
Savvidou OD Bolia IK Chloros GD Papanastasiou J Koutsouradis P Papagelopoulos PJ . Denosumab: current use in the treatment of primary bone tumors. Orthopedics. (2017) 40:204–10. 10.3928/01477447-20170627-04
47.
Zheng H Li W Kang Y . Tumor-stroma interactions in bone metastasis: molecular mechanisms and therapeutic implications. Cold Spring Harb Symp Quant Biol. (2016) 81:151–61. 10.1101/sqb.2016.81.030775
48.
Tanaka Y Ohira T . Mechanisms and therapeutic targets for bone damage in rheumatoid arthritis, in particular the RANK-RANKL system. Curr Opin Pharmacol. (2018) 40:110–9. 10.1016/j.coph.2018.03.006
49.
Sigl V Jones LP Penninger JM . RANKL/RANK: from bone loss to the prevention of breast cancer. Open Biol. (2016) 6:160230. 10.1098/rsob.160230
50.
Shroff R Beringer O Rao K Hofbauer LC Schulz A . Denosumab for post-transplantation hypercalcemia in osteopetrosis. N Engl J Med. (2012) 367:1766–7. 10.1056/NEJMc1206193
51.
Rotman M Hamdy NAT Appelman-Dijkstra NM . Clinical and translational pharmacological aspects of the management of fibrous dysplasia of bone. Br J Clin Pharmacol. (2018). 10.1111/bcp.13820 [Epub ahead of print].
52.
Li G Jin Y Levine MAH Hoyer-Kuhn H Ward L Adachi JD . Systematic review of the effect of denosumab on children with osteogenesis imperfecta showed inconsistent findings. Acta Paediatr. (2018) 107:534–7. 10.1111/apa.14154
53.
Tsourdi E Langdahl B Cohen-Solal M Aubry-Rozier B Eriksen EF Guanabens N et al . Discontinuation of Denosumab therapy for osteoporosis: a systematic review and position statement by ECTS. Bone. (2017) 105:11–7. 10.1016/j.bone.2017.08.003
54.
Roux S Massicotte MH Huot Daneault A Brazeau-Lamontagne L Dufresne J . Acute hypercalcemia and excessive bone resorption following anti-RANKL withdrawal: case report and brief literature review. Bone. (2019) 120:482–6. 10.1016/j.bone.2018.12.012
55.
An J Hao D Zhang Q Chen B Zhang R Wang Y et al . Natural products for treatment of bone erosive diseases: the effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int Immunopharmacol. (2016) 36:118–31. 10.1016/j.intimp.2016.04.024
56.
Hong G Zhou L Shi X He W Wang H Wei Q et al . Bajijiasu abrogates osteoclast differentiation via the suppression of RANKL signaling pathways through NF-kappaB and NFAT. Int J Mol Sci. (2018) 18:E203. 10.3390/ijms18010203
57.
Wang Q Yao L Xu K Jin H Chen K Wang Z et al . Madecassoside inhibits estrogen deficiency-induced osteoporosis by suppressing RANKL-induced osteoclastogenesis. J Cell Mol Med. (2019) 23:380–94. 10.1111/jcmm.13942
58.
Xu Q Chen G Liu X Dai M Zhang B . Icariin inhibits RANKL-induced osteoclastogenesis via modulation of the NF-kappaB and MAPK signaling pathways. Biochem Biophys Res Commun. (2019) 508:902–6. 10.1016/j.bbrc.2018.11.201
59.
Ikebuchi Y Aoki S Honma M Hayashi M Sugamori Y Khan M et al . Coupling of bone resorption and formation by RANKL reverse signalling. Nature. (2018) 561:195–200. 10.1038/s41586-018-0482-7
60.
Sone E Noshiro D Ikebuchi Y Nakagawa M Khan M Tamura Y et al . The induction of RANKL molecule clustering could stimulate early osteoblast differentiation. Biochem Biophys Res Commun. (2018) 509:435–40. 10.1016/j.bbrc.2018.12.093
61.
Koch U Radtke F . Mechanisms of T cell development and transformation. Annu Rev Cell Dev Biol. (2011) 27:539–62. 10.1146/annurev-cellbio-092910-154008
62.
Shah DK Zuniga-Pflucker JC . An overview of the intrathymic intricacies of T cell development. J Immunol. (2014) 192:4017–23. 10.4049/jimmunol.1302259
63.
Petrie HT Zuniga-Pflucker JC . Zoned out: functional mapping of stromal signaling microenvironments in the thymus. Annu Rev Immunol. (2007) 25:649–79. 10.1146/annurev.immunol.23.021704.115715
64.
Koch U Fiorini E Benedito R Besseyrias V Schuster-Gossler K Pierres M et al . Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med. (2008) 205:2515–23. 10.1084/jem.20080829
65.
Hirano K Negishi N Yazawa M Yagita H Habu S Hozumi K . Delta-like 4-mediated Notch signaling is required for early T-cell development in a three-dimensional thymic structure. Eur J Immunol. (2015) 45:2252–62. 10.1002/eji.201445123
66.
Rodrigues PM Peterson P Alves NL . Setting up the perimeter of tolerance: insights into mTEC physiology. Trends Immunol. (2018) 39:2–5. 10.1016/j.it.2017.11.001
67.
Aschenbrenner K D'Cruz LM Vollmann EH Hinterberger M Emmerich J Swee LK et al . Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol. (2007) 8:351–8. 10.1038/ni1444
68.
Anderson G Takahama Y . Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol. (2012) 33:256–63. 10.1016/j.it.2012.03.005
69.
Lei Y Ripen AM Ishimaru N Ohigashi I Nagasawa T Jeker LT et al . Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med. (2011) 208:383–94. 10.1084/jem.20102327
70.
Burkly L Hession C Ogata L Reilly C Marconi LA Olson D et al . Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. (1995) 373:531–6. 10.1038/373531a0
71.
Weih F Carrasco D Durham SK Barton DS Rizzo CA Ryseck RP et al . Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. (1995) 80:331–40. 10.1016/0092-8674(95)90416-6
72.
Kajiura F Sun S Nomura T Izumi K Ueno T Bando Y et al . NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol. (2004) 172:2067–75. 10.4049/jimmunol.172.4.2067
73.
Akiyama T Maeda S Yamane S Ogino K Kasai M Kajiura F et al . Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science. (2005) 308:248–51. 10.1126/science.1105677
74.
Nitta T Ohigashi I Nakagawa Y Takahama Y . Cytokine crosstalk for thymic medulla formation. Curr Opin Immunol. (2011) 23:190–7. 10.1016/j.coi.2010.12.002
75.
Sun L Luo H Li H Zhao Y . Thymic epithelial cell development and differentiation: cellular and molecular regulation. Protein Cell. (2013) 4:342–55. 10.1007/s13238-013-3014-0
76.
Bichele R Kisand K Peterson P Laan M . TNF superfamily members play distinct roles in shaping the thymic stromal microenvironment. Mol Immunol. (2016) 72:92–102. 10.1016/j.molimm.2016.02.015
77.
Lkhagvasuren E Sakata M Ohigashi I Takahama Y . Lymphotoxin beta receptor regulates the development of CCL21-expressing subset of postnatal medullary thymic epithelial cells. J Immunol. (2013) 190:5110–7. 10.4049/jimmunol.1203203
78.
Danzl NM Jeong S Choi Y Alexandropoulos K . Identification of novel thymic epithelial cell subsets whose differentiation is regulated by RANKL and Traf6. PLoS ONE. (2014) 9:e86129. 10.1371/journal.pone.0086129
79.
Fujihara C Williams JA Watanabe M Jeon H Sharrow SO Hodes RJ . T cell-B cell thymic cross-talk: maintenance and function of thymic B cells requires cognate CD40-CD40 ligand interaction. J Immunol. (2014) 193:5534–44. 10.4049/jimmunol.1401655
80.
Liu X Invernizzi P Lu Y Kosoy R Lu Y Bianchi I et al . Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet. (2010) 42:658–60. 10.1038/ng.627
81.
Akiyama N Shinzawa M Miyauchi M Yanai H Tateishi R Shimo Y et al . Limitation of immune tolerance-inducing thymic epithelial cell development by Spi-B-mediated negative feedback regulation. J Exp Med. (2014) 211:2425–38. 10.1084/jem.20141207
82.
Hikosaka Y Nitta T Ohigashi I Yano K Ishimaru N Hayashi Y et al . The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity. (2008) 29:438–50. 10.1016/j.immuni.2008.06.018
83.
White AJ Lucas B Jenkinson WE Anderson G . Invariant NKT cells and control of the thymus medulla. J Immunol. (2018) 200:3333–9. 10.4049/jimmunol.1800120
84.
Zuklys S Balciunaite G Agarwal A Fasler-Kan E Palmer E Hollander GA . Normal thymic architecture and negative selection are associated with Aire expression, the gene defective in the autoimmune-polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). J Immunol. (2000) 165:1976–83. 10.4049/jimmunol.165.4.1976
85.
White AJ Withers DR Parnell SM Scott HS Finke D Lane PJ et al . Sequential phases in the development of Aire-expressing medullary thymic epithelial cells involve distinct cellular input. Eur J Immunol. (2008) 38:942–7. 10.1002/eji.200738052
86.
Montero-Herradon S Garcia-Ceca J Zapata AG . Altered maturation of medullary TEC in EphB-deficient Thymi is recovered by RANK signaling stimulation. Front Immunol. (2018) 9:1020. 10.3389/fimmu.2018.01020
87.
Rossi SW Kim MY Leibbrandt A Parnell SM Jenkinson WE Glanville SH et al . RANK signals from CD4(+)3(−) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med. (2007) 204:1267–72. 10.1084/jem.20062497
88.
Roberts NA White AJ Jenkinson WE Turchinovich G Nakamura K Withers DR et al . Rank signaling links the development of invariant gammadelta T cell progenitors and Aire(+) medullary epithelium. Immunity. (2012) 36:427–37. 10.1016/j.immuni.2012.01.016
89.
Desanti GE Cowan JE Baik S Parnell SM White AJ Penninger JM et al . Developmentally regulated availability of RANKL and CD40 ligand reveals distinct mechanisms of fetal and adult cross-talk in the thymus medulla. J Immunol. (2012) 189:5519–26. 10.4049/jimmunol.1201815
90.
Mccarthy NI Cowan JE Nakamura K Bacon A Baik S White AJ et al . Osteoprotegerin-mediated homeostasis of rank+ Thymic epithelial cells does not limit Foxp3+ regulatory T cell development. J Immunol. (2015) 195:2675–82. 10.4049/jimmunol.1501226
91.
Yin C Pei XY Shen H Gao YN Sun XY Wang W et al . Thymic homing of activated CD4(+) T cells induces degeneration of the thymic epithelium through excessive RANK signaling. Sci Rep. (2017) 7:2421. 10.1038/s41598-017-02653-9
92.
Shanley DP Aw D Manley NR Palmer DB . An evolutionary perspective on the mechanisms of immunosenescence. Trends Immunol. (2009) 30:374–81. 10.1016/j.it.2009.05.001
93.
Sato K Kato A Sekai M Hamazaki Y Minato N . Physiologic thymic involution underlies age-dependent accumulation of senescence-associated CD4(+) T cells. J Immunol. (2017) 199:138–48. 10.4049/jimmunol.1602005
94.
Akiyama T Shimo Y Yanai H Qin J Ohshima D Maruyama Y et al . The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity. (2008) 29:423–37. 10.1016/j.immuni.2008.06.015
95.
Irla M Hugues S Gill J Nitta T Hikosaka Y Williams IR et al . Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity. Immunity. (2008) 29:451–63. 10.1016/j.immuni.2008.08.007
96.
Irla M Guerri L Guenot J Serge A Lantz O Liston A et al . Antigen recognition by autoreactive CD4(+) thymocytes drives homeostasis of the thymic medulla. PLoS ONE. (2012) 7:e52591. 10.1371/journal.pone.0052591
97.
Otero DC Baker DP David M . IRF7-dependent IFN-beta production in response to RANKL promotes medullary thymic epithelial cell development. J Immunol. (2013) 190:3289–98. 10.4049/jimmunol.1203086
98.
Mouri Y Yano M Shinzawa M Shimo Y Hirota F Nishikawa Y et al . Lymphotoxin signal promotes thymic organogenesis by eliciting RANK expression in the embryonic thymic stroma. J Immunol. (2011) 186:5047–57. 10.4049/jimmunol.1003533
99.
Lin J Yang L Silva HM Trzeciak A Choi Y Schwab SR et al . Increased generation of Foxp3(+) regulatory T cells by manipulating antigen presentation in the thymus. Nat Commun. (2016) 7:10562. 10.1038/ncomms10562
100.
Mizuno A Kanno T Hoshi M Shibata O Yano K Fujise N et al . Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J Bone Miner Metab. (2002) 20:337–44. 10.1007/s007740200049
101.
Ohigashi I Nitta T Lkhagvasuren E Yasuda H Takahama Y . Effects of RANKL on the thymic medulla. Eur J Immunol. (2011) 41:1822–7. 10.1002/eji.201141480
102.
Lo Iacono N Blair HC Poliani PL Marrella V Ficara F Cassani B et al . Osteopetrosis rescue upon RANKL administration to Rankl(-/-) mice: a new therapy for human RANKL-dependent ARO. J Bone Min Res. (2012) 27:2501–10. 10.1002/jbmr.1712
103.
Lopes N Vachon H Marie J Irla M . Administration of RANKL boosts thymic regeneration upon bone marrow transplantation. Embo Mol Med. (2017) 9:835–51. 10.15252/emmm.201607176
104.
Poliani PL Vermi W Facchetti F . Thymus microenvironment in human primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. (2009) 9:489–95. 10.1097/ACI.0b013e3283327e5c
105.
Toubert A Glauzy S Douay C Clave E . Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: never say never again. Tissue Antigens. (2012) 79:83–9. 10.1111/j.1399-0039.2011.01820.x
106.
Da Rocha LKA De Barros SF Bandeira F Bollini A Testa LHD Simione AJ et al . Thymopoiesis in pre- and post-hematopoietic stem cell transplantation. Front Immunol. (2018) 9:1889. 10.3389/fimmu.2018.01889
107.
Khan IS Mouchess ML Zhu ML Conley B Fasano KJ Hou Y et al . Enhancement of an anti-tumor immune response by transient blockade of central T cell tolerance. J Exp Med. (2014) 211:761–8. 10.1084/jem.20131889
108.
Bakhru P Zhu ML Wang HH Hong LK Khan I Mouchess M et al . Combination central tolerance and peripheral checkpoint blockade unleashes antimelanoma immunity. JCI Insight. (2017) 2:93265. 10.1172/jci.insight.93265
109.
Stolina M Dwyer D Ominsky MS Corbin T Van G Bolon B et al . Continuous RANKL inhibition in osteoprotegerin transgenic mice and rats suppresses bone resorption without impairing lymphorganogenesis or functional immune responses. J Immunol. (2007) 179:7497–505. 10.4049/jimmunol.179.11.7497
110.
Di Rosa F Gebhardt T . Bone marrow T cells and the integrated functions of recirculating and tissue-resident memory T cells. Front Immunol. (2016) 7:51. 10.3389/fimmu.2016.00051
111.
Eghbali-Fatourechi G Khosla S Sanyal A Boyle WJ Lacey DL Riggs BL . Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. (2003) 111:1221–30. 10.1172/JCI200317215
112.
D'Amelio P Grimaldi A Di Bella S Brianza SZM Cristofaro MA Tamone C et al . Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone. (2008) 43:92–100. 10.1016/j.bone.2008.02.017
113.
Adeel S Singh K Vydareny KH Kumari M Shah E Weitzmann MN et al . Bone loss in surgically ovariectomized premenopausal women is associated with T lymphocyte activation and thymic hypertrophy. J Investig Med. (2013) 61:1178–83. 10.2310/JIM.0000000000000016
114.
Toraldo G Roggia C Qian WP Pacifici R Weitzmann MN . IL-7 induces bone loss in vivo by induction of receptor activator of nuclear factor kappa B ligand and tumor necrosis factor alpha from T cells. Proc Natl Acad Sci USA. (2003) 100:125–30. 10.1073/pnas.0136772100
115.
Bernardi AI Andersson A Stubelius A Grahnemo L Carlsten H Islander U . Selective estrogen receptor modulators in T cell development and T cell dependent inflammation. Immunobiology. (2015) 220:1122–8. 10.1016/j.imbio.2015.05.009
116.
D'Amelio P Grimaldi A Bernabei P Pescarmona GP Isaia G . Immune system and bone metabolism: does thymectomy influence postmenopausal bone loss in humans?Bone. (2006) 39:658–65. 10.1016/j.bone.2006.03.009
117.
Takayanagi H . New developments in osteoimmunology. Nat Rev Rheumatol. (2012) 8:684–9. 10.1038/nrrheum.2012.167
118.
Smolen JS Aletaha D Barton A Burmester GR Emery P Firestein GS et al . Rheumatoid arthritis. Nat Rev Dis Primers. (2018) 4:18001. 10.1038/nrdp.2018.1
119.
Mcinnes IB Schett G . The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. 10.1056/NEJMra1004965
120.
Dakin SG Coles M Sherlock JP Powrie F Carr AJ Buckley CD . Pathogenic stromal cells as therapeutic targets in joint inflammation. Nat Rev Rheumatol. (2018) 14:714–26. 10.1038/s41584-018-0112-7
121.
Harre U Georgess D Bang H Bozec A Axmann R Ossipova E et al . Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest. (2012) 122:1791–802. 10.1172/JCI60975
122.
Bugatti S Bogliolo L Vitolo B Manzo A Montecucco C Caporali R . Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res Ther. (2016) 18:226. 10.1186/s13075-016-1116-9
123.
Komatsu N Takayanagi H . Immune-bone interplay in the structural damage in rheumatoid arthritis. Clin Exp Immunol. (2018) 194:1–8. 10.1111/cei.13188
124.
Hirota K Yoshitomi H Hashimoto M Maeda S Teradaira S Sugimoto N et al . Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. (2007) 204:2803–12. 10.1084/jem.20071397
125.
Komatsu N Okamoto K Sawa S Nakashima T Oh-Hora M Kodama T et al . Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. 10.1038/nm.3432
126.
Nevius E Gomes AC Pereira JP . Inflammatory cell migration in rheumatoid arthritis: a comprehensive review. Clin Rev Allergy Immunol. (2016) 51:59–78. 10.1007/s12016-015-8520-9
127.
Baum R Gravallese EM . Bone as a target organ in rheumatic disease: impact on osteoclasts and osteoblasts. Clin Rev Allergy Immunol. (2016) 51:1–15. 10.1007/s12016-015-8515-6
128.
Hayer S Bauer G Willburger M Sinn K Alasti F Plasenzotti R et al . Cartilage damage and bone erosion are more prominent determinants of functional impairment in longstanding experimental arthritis than synovial inflammation. Dis Model Mech. (2016) 9:1329–38. 10.1242/dmm.025460
129.
Boman A Kokkonen H Arlestig L Berglin E Rantapaa-Dahlqvist S . Receptor activator of nuclear factor kappa-B ligand (RANKL) but not sclerostin or gene polymorphisms is related to joint destruction in early rheumatoid arthritis. Clin Rheumatol. (2017) 36:1005–12. 10.1007/s10067-017-3570-4
130.
Chiu YG Ritchlin CT . Denosumab: targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin Biol Ther. (2017) 17:119–28. 10.1080/14712598.2017.1263614
131.
Walsh MC Choi Y . Biology of the RANKL-RANK-OPG System in Immunity, Bone, and Beyond. Front Immunol. (2014) 5:511. 10.3389/fimmu.2014.00511
132.
Takeuchi T Tanaka Y Ishiguro N Yamanaka H Yoneda T Ohira T et al . Effect of denosumab on Japanese patients with rheumatoid arthritis: a dose-response study of AMG 162 (Denosumab) in patients with RheumatoId arthritis on methotrexate to Validate inhibitory effect on bone Erosion (DRIVE)-a 12-month, multicentre, randomised, double-blind, placebo-controlled, phase II clinical trial. Ann Rheum Dis. (2016) 75:983–90. 10.1136/annrheumdis-2015-208052
133.
Cosway E Anderson G Garside P Prendergast C . The thymus and rheumatology: should we care?Curr Opin Rheumatol. (2016) 28:189–95. 10.1097/BOR.0000000000000251
134.
Tanaka S Maeda S Hashimoto M Fujimori C Ito Y Teradaira S et al . Graded attenuation of TCR signaling elicits distinct autoimmune diseases by altering thymic T cell selection and regulatory T cell function. J Immunol. (2010) 185:2295–305. 10.4049/jimmunol.1000848
135.
Flores-Borja F Jury EC Mauri C Ehrenstein MR . Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci USA. (2008) 105:19396–401. 10.1073/pnas.0806855105
136.
Cribbs AP Kennedy A Penn H Read JE Amjadi P Green P et al . Treg cell function in rheumatoid arthritis is compromised by ctla-4 promoter methylation resulting in a failure to activate the indoleamine 2,3-dioxygenase pathway. Arthritis Rheumatol. (2014) 66:2344–54. 10.1002/art.38715
137.
Raje N Terpos E Willenbacher W Shimizu K Garcia-Sanz R Durie B et al . Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: an international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. (2018) 19:370–81. 10.1016/S1470-2045(18)30072-X
138.
Ahern E Harjunpaa H Barkauskas D Allen S Takeda K Yagita H et al . Co-administration of RANKL and CTLA4 antibodies enhances lymphocyte-mediated antitumor immunity in mice. Clin Cancer Res. (2017) 23:5789–801. 10.1158/1078-0432.CCR-17-0606
139.
Ahern E Harjunpaa H O'Donnell JS Allen S Dougall WC Teng MWL et al . RANKL blockade improves efficacy of PD1-PD-L1 blockade or dual PD1-PD-L1 and CTLA4 blockade in mouse models of cancer. Oncoimmunology. (2018) 7:e1431088. 10.1080/2162402X.2018.1431088
140.
Chen NJ Huang MW Hsieh SL . Enhanced secretion of IFN-gamma by activated Th1 cells occurs via reverse signaling through TNF-related activation-induced cytokine. J Immunol. (2001) 166:270–6. 10.4049/jimmunol.166.1.270
141.
Secchiero P Corallini F Barbarotto E Melloni E Di Iasio MG Tiribelli M et al . Role of the RANKL/RANK system in the induction of interleukin-8 (IL-8) in B chronic lymphocytic leukemia (B-CLL) cells. J Cell Physiol. (2006) 207:158–64. 10.1002/jcp.20547
142.
Zhang S Wang X Li G Chong Y Zhang J Guo X et al . Osteoclast regulation of osteoblasts via RANKRANKL reverse signal transduction in vitro. Mol Med Rep. (2017) 16:3994–4000. 10.3892/mmr.2017.7039
Summary
Keywords
osteoclasts, denosumab, thymus, central tolerance, rheumatoid arthritis, osteoporosis, tumor
Citation
Sobacchi C, Menale C and Villa A (2019) The RANKL-RANK Axis: A Bone to Thymus Round Trip. Front. Immunol. 10:629. doi: 10.3389/fimmu.2019.00629
Received
11 January 2019
Accepted
08 March 2019
Published
29 March 2019
Volume
10 - 2019
Edited by
Claudine Blin-Wakkach, UMR7370 Laboratoire de Physio Médecine Moléculaire (LP2M), France
Reviewed by
Frederic Lezot, INSERM UMR957, France; Magali Irla, Institut National de la Santé et de la Recherche Médicale (INSERM), France; Eleni Douni, Agricultural University of Athens, Greece
Updates
Copyright
© 2019 Sobacchi, Menale and Villa.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Villa villa.anna@hsr.it
This article was submitted to Inflammation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.