 Agata Gabryelska
Agata Gabryelska Piotr Kuna2
Piotr Kuna2 Michał Panek
Michał Panek- 1Department of Sleep Medicine and Metabolic Disorders, Medical University of Lodz, Łódz, Poland
- 2Department of Internal Medicine, Asthma and Allergy, Medical University of Lodz, Łódz, Poland
- 3Department of General and Oncological Pulmonology, Medical University of Lodz, Łódz, Poland
Interleukin 33 (IL-33) is an alarmin cytokine from the IL-1 family. IL-33 is localized in the nucleus and acts there as a gene regulator. Following injury, stress or cell death, it is released from the nucleus, and exerts its pro-inflammatory biological functions via the transmembrane form of the ST2 receptor, which is present mainly as attached to immune cells. In recent years, IL-33 became a focus of many studies due to its possible role in inflammatory disorders. Among respiratory disorders, the contribution of IL-33 to the development of asthma, in particular, has been most identified. Increased level of IL-33 in lung epithelial cells and blood serum has been observed in asthma patients. The IL-33/ST2 interaction activated the Th2 mediated immune response and further production of many pro-inflammatory cytokines. Single nucleotide polymorphisms in the IL-33 gene cause a predisposition to the development of asthma. Similarly, in chronic pulmonary obstructive disease (COPD), both increased expression of IL-33 and the ST2 receptor has been observed. Interestingly, cigarette smoke, a key inducer of COPD, not only activates IL-33 production by epithelial and endothelial cells, but also induces the expression of IL-33 in peripheral blood mononuclear cells. Knowledge regarding its contribution in other respiratory disorders, such as obstructive sleep apnea, remains greatly limited. Recently it was shown that IL-33 is one of the inflammatory mediators by which levels in blood serum are increased in OSA patients, compared to healthy control patients. This mini review summarizes current knowledge on IL-33 involvement in chosen chronic respiratory disorders and proposes this interleukin as a possible link in the pathogenesis of these diseases.
Introduction
The inteleukin-1 (IL-1) superfamily of cytokines plays a pivotal role in both innate and adaptive immunity by regulating host defense, inflammation and injury (1, 2). Similarly to the transforming growth factor, the Smad (TGF-Smad) superfamily, members of the IL-1 superfamily are associated with chronic pulmonary disorders and fibrosis (3).The IL-1 superfamily includes 11 members: IL-1α, IL-1β, IL-1 receptor antagonist (IL-1Ra), IL-18, IL-33, IL-36α, IL-36β, IL-36γ, IL-36Ra, IL-37, and IL-38 (4, 5). All family members are characterized by a similar gene structure, in some cases having identical intron positioning and amino acids sequences, enabling folding of the protein into the three dimensional structure of a 12-stranded-β-barrel (6). Furthermore, all genes encoding members of IL-1 other than IL-18 and IL-33 occupy approximately a 400 kB interval on chromosome 2 (7). All the biologically active cytokines of the family are extracellular molecules, while their precursors are primarily intracellular with an exception of IL-1Ra, which encodes classical signal peptide. IL-1β and IL-18 achieve biological activity only after their precursors are cleavaged from the pro-domain by inflammasome. In the case of IL-36 α, β, γ, IL-36Ra, and IL-33, the full-length molecules already possess biological activity, however, following the cleavage of the N-terminus they obtain full biological potency (8, 9). Origin and function is not the only characteristic feature uniting the cytokines within IL-1 super family, as they also relate to each other by transduction pathways and receptor structure, all consisting of three Ig-like domains and Toll/IL-1R domain (1).
IL-33
IL-33, one of the most recently discovered members of IL-1 superfamily, is an alarmin cytokine promoting inflammatory responses (5). Originally it was known as a nuclear factor from high endothelium venules (HF-HEV) (10), and only later has its cytokine activity been defined (11). It mainly expresses itself through epithelial and endothelial cells, fibroblasts-like cell and myofibroblasts (10–13).
Differently to most of the members of IL-1 superfamily, the gene encoding IL-33 is localized on the short arm of chromosome 9, at 9p41.1 (14). The gene is comprised of 8 exons, poses interferon stimulated response element (ISRE), and several gamma interferon activation sites (GAS) in the promotor area (15). It is constitutively expressed in its pro form of 270 amino acids (full length IL-33; f-IL 33), with its molecular mass of 30 kD being stored in the nucleus of cells where it acts as a nuclear regulator (10, 11, 16). This function is facilitated by its N-terminal nuclear domain, which contains a chromatin binding motif that binds to histones (17, 18). Two other domains building up IL-33 are the IL-1-like cytokine domain, and the central domain (11, 19). IL-33 has been identified as a dual function cytokine, also possessing a full bioactivity as an alarmin upon its release from a cell (20). Furthermore, f-IL-33 serves as a substrate for serine proteases of neutrophils and mast cells. It results in shorter forms of a peptide comprised of IL-1 like domain and part of central domain, forming a mature form of IL-33 (m-IL-33). It is estimated that bioactivity of m-IL-33 increases ~10–30 times compared to f-IL-33 (8, 19). During apoptosis caspase-3 and caspase-7 cleave f-IL-33, resulting in peptide, which lacks biological activity (9). It has been shown that the expression of IL-33 can be increased following the exposure to cigarette smoke (21). Other factors that can induce the same effect include viral infection and exposure to allergens such as pollen, chitin, fungi, or Alternaria spp. (22–28). The main mechanism in which the activity of IL-33 is limited is sequestration by soluble isoform of its suppression of tumorigenicity 2 (sST2) receptor that acts as a decoy, inhibiting the expansion of IL-33 mediated inflammation (29–31). Regulation of IL-33 activity also includes oxidation of cysteine residues following extracellular release and formation of disulphide bridges, resulting in the inactivation of IL-33 (24).
IL-33 signaling pathway begins with binding of the cytokine to the ST2 receptor. This transmembrane form of the receptor is generated by the same mRNA as the sST2 and the distinction is made through a different promoter and alternative splicing of the transcript (32, 33). Following the attachment of the protein to ST2, the corrector IL-1 receptor accessory protein IL-1RAcP is recruited and further heterodimeric signaling complex is formed, which involves myeloid differentiation primary response protein 88 (MYD88), IL-1R-assosiated kinase 1, and 4 (IRAK1, IRAK4), as well as tumor necrosis factor (TNF) receptor-assosiatedreceptor-6 (TRAF6) (11, 34, 35). This results in nuclear factor κB (NFκB) transcription and activation of mitogen-activated protein kinases (MAPK), including c-Jun N-terminal kinases (JNK) and p38 (11), which then in a signaling pathway drive processes such as proliferation, cell survival, cytokine secretion (IL-4, IL-5, and IL-13) and amphiregulin (AREG) expression (36–38). Only ST2+ cells are affected by IL-33 stimulation. The type of response differs depending on the type of stimulated cell, as T helper 2 (Th2) cells respond by the secretion of Il-5 and IL-13 (11, 39), while mast cells release a different profile of cytokines, including IL-4, IL-5, and IL-6 (40). On the other hand, neutrophils following IL-33 stimulation migrate through chemotaxis (41). In the case of eosinophils, their response consists of degranulation and reactive oxygen species (ROS) production (36, 42, 43), while macrophages in response to IL-33 develop an alternatively activated phenotype (44, 45). IL-33 has also been shown to activate NFκB and p38 in regulatory lymphocytes T (Tregs), which further leads to the expansion of ST2+ Tregs (46). Additionally, IL-33 stimulates TGF-β-mediated Treg differentiation (47). Activation of Tregs through MYD88 has been observed only in the IL-33/ST2 independent manner (48). It remains unknown if Tregs function can be affected in IL-33/ST2- MYD88 dependent pathways (49). Main directions of IL-33 molecular function in induction and maintenance of chronic respiratory disorders: asthma, COPD, and OSA are shown in Figure 1.
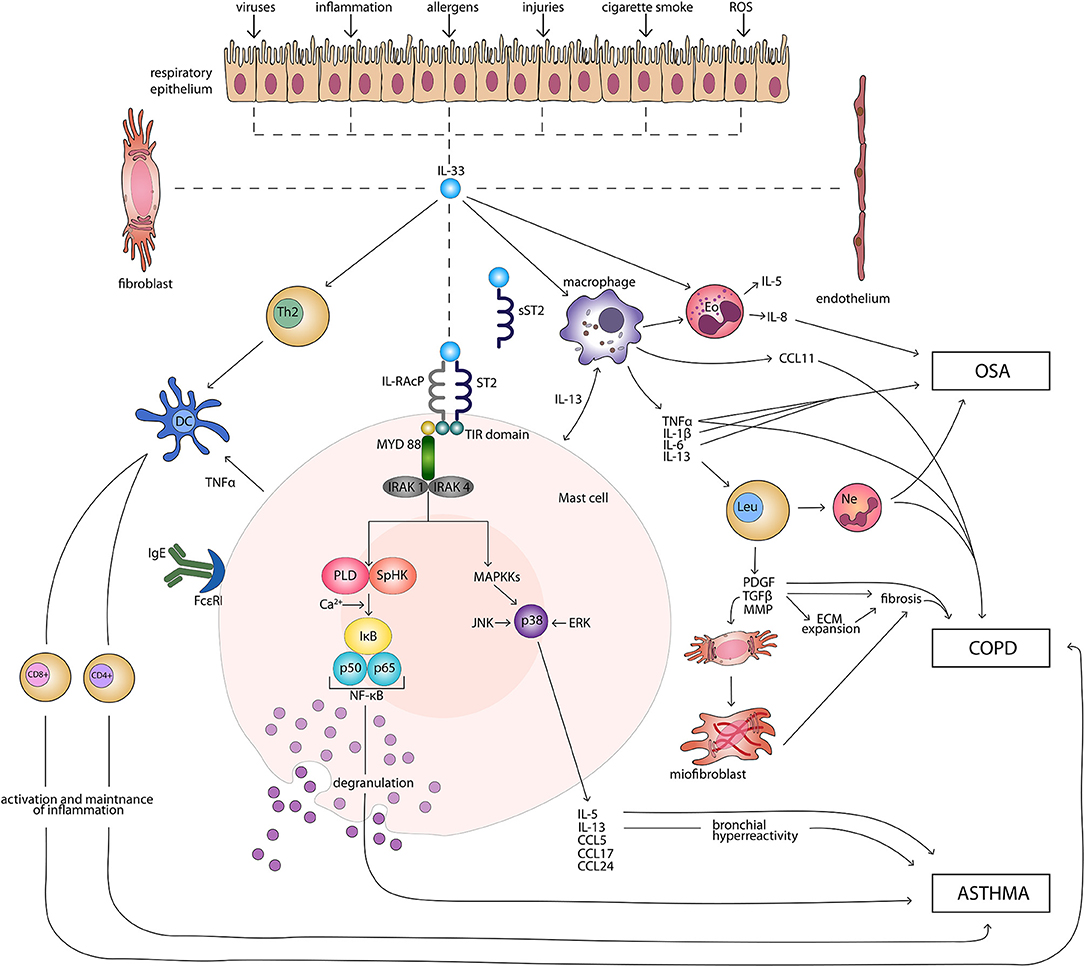
Figure 1. Main directions of IL-33 molecular function in induction and maintenance of chronic respiratory disorders: asthma, COPD, and OSA. CD4+, CD4+ T cell; CD8+, CD8+ T cell; CCL, CC-chemokine ligand; COPD, chronic obstructive pulmonary disease; DC, dendritic cell; ECM, extracellular matrix; Eo, eosinophil; ERK, extracellular signal-regulated kinase; FcεRI, high-affinity receptor for the Fc region of immunoglobulin E; IgE, immunoglobulin E; IκB, inhibitor of κB; IL, interleukin; IL-RAcP, interleukin 1 receptor accessory protein; IRAK, interleukin-1 receptor associated kinase; JNK, c-Jun N-terminal kinases; Leu, leucocyte; MAPKKs, mitogen-activated protein kinase kinases; MMP, matrix metalloproteinases; MYD 88, myeloid differentiation primary response protein 88; Ne, neutrophil; NF-κB, nuclear factor κB; OSA, obstructive sleep apnea; PDGF, platelet-derived growth factor; PLD, phospholipase D; ROS, reactive oxygen species; SpHK, sphingosine kinase; ST2, suppression of tumorigenicity 2 transmembrane receptor; sST2, suppression of tumorigenicity 2 soluble receptor; Th2, T helper 2 cell; TIR, Toll-like/interleukin 1receptor; TGF-β, transforming growth factor β; TNFα, tumor necrosis factor α.
Asthma
Among chronic respiratory disorders, the IL-33 contribution to disease development has been furthest identified in bronchial asthma. Increased levels of IL-33 in lung epithelial cells and blood serum has been observed in asthma patients. In several genome wide association (GWA) studies it has been established that both genes encoding IL-33 and its receptor IL1RL1 are susceptibility loci for asthma (50–55). The GWA studies have identified single nucleotide polymorphisms (SNPs) associated with asthma in general, as well as in different clinical phenotypes (56–59). A significant connection was identified, for example, between SNP of IL-33—rs3939286—and atopic asthma, as well as eosinophil blood count (60). Similar observations were made for multiple SNPs in the gene encoding ST2 receptor. One example of the SNP of IL1RL1 that predisposes to both atopic and non-atopic asthma and is connected with eosinophilia, causing an increased level of serum IgE and airway hyperresponsiveness, is rs950880 for non-Islandic population (in Islandic population it is rs1420101) (60, 61). The SNP (rs10204137) in the Toll-like/IL-1 (TIR) domain of ST2 can enhance the IL-33/ST2L pathway through further cascade (62). What is very interesting, and highlights the role of IL-33 in the development of asthma, is a rare loss of function mutation (rs146587587-C), in which premature STOP codon causes truncation of the last 66 amino acids of the cytokine. This results in a reduced eosinophil blood count and protection against asthma (63).
Several mechanisms in which IL-33 participates in the development of asthma have been described.
It has been shown that IL-33 promotes lung fibrosis and bronchial remodeling, causing further advancement of asthma (64, 65). IL-33 participates in the process of fibrosis by directly activating cells to produce the profibrotic factors as well as by inducing inflammation. Following damage, cells secrete IL-33, which activates a response from cells that possess IL1RL1 receptor. In a Th2 mediated immune response, innate lymphoid cells (ILC) and Th2 cells produce pro-inflammatory cytokines (IL-4, IL-5, and IL-13) (66), which have been identified as important mediators of fibrosis (67, 68). IL-4 and IL-13 are responsible for recruitment of basophils and eosinophils that cause differentiation of fibroblasts to myofibroblasts and collagen deposition (69). Same cytokines induce macrophage polarization to an alternatively activated profibrotic phenotype (44). IL-5 secreted by Th2 cells also takes part in the recruitment and activation of eosinophils, which are the source of profibrotic factors such as TGF-β, PDGF, and IL-13 (70). Interestingly, it has been shown that IL-33 can induce IL-5-producing T cells and promote airway inflammation independent of IL-4, which was shown in IL-4 deficient mice (45).The increased tissue fibrosis can be enhanced by interaction between major histocompatibility complex II (MHCII) of ILCs and Th2 cell receptor (TCR), which activates Th2 cells. Additionally, IL-33 itself recruits basophil, eosinophils, and mast cells to the local inflammatory site and activities them into the production of profibrotic factors (71). Moreover, it was observed that IL-33 directly promotes fibroblasts to produce fibronectin 1 and collagen type 1, factors involved in airway remodeling (65). Interestingly, it has been shown that IL-33, similarly to TGF-β, not only activates the fibroblasts, but also promotes epithelial-to-mesenchymal transition (EMT), which is associated with overexpression of profibrotic markers (72, 73). IL-33 can also contribute to the development of allergen driven airway inflammation by dysregulating Treg and impairing immunologic tolerance to inhaled antigens. Treg cells in lungs exposed to IL-33 not only upregulate their expression of Th2 transcription factor GATA binding protein 3 (GATA3) and ST2, but also enhance production of type 2 cytokines. In effect, Tregs lose their ability to supress effector T cells resulting in development of airway inflammation (74).
Bronchial hyperreactivity is typical in the course of asthma and IL-33 has been identified as inducing this process. It has been shown that through the activation of IL-5 and IL-13 secretion, IL-33 promotes airway hyperresponsiveness (75). It has been suggested that this is dependent on smooth muscle cells (76, 77). Other possible mechanisms involve irritation caused by secretion of serotonin by mast cells following the IL-33 activation (78). Feedback circuit involving both IL-33 and ILCs responsible for bronchial hyperactivity and persistence of asthma has been suggested based on the animal model of the disease (79).
Chronic Obstructive Pulmonary Disease
Chronic pulmonary obstructive disease (COPD), is a progressive inflammatory condition, in which an increased expression of IL-33 and the ST2 receptor, similarly to asthma, has been observed (27, 80). Cigarette smoke has been identified as the key inducer of COPD. In the mouse model of cigarette smoke-induced COPD, both IL-33 and the ST2 receptor expression was increased in lungs (27, 81, 82). Inhibition of cigarette-induced pathogenic changes in lungs have been observed following introduction of anti-IL-33 antibody (81), which suggests an important role of IL-33 in the pathogenesis of COPD. Furthermore, mice treated with IL-33 developed histopathological changes in the lungs such as lining hypertrophy, goblet cell hypertrophy, and mucus hypersecretion, which are typical for COPD patients (11). Interestingly, cigarette smoke not only activates IL-33 production by epithelial and endothelial cells, but also induces the expression of IL-33 in peripheral blood mononuclear cells (PBMC) (82) and peripheral blood lymphocytes (PBL) (80). This systemic response trough activation of the immune system might enhance processes involved in the development of COPD. Additionally, cigarette smoke alters the distribution of the expression of IL-33 receptor ST2 (21). Decreased expression of the receptor was observed on ILC2s, while macrophages and natural killer cells presented elevated expression of this receptor. This change significantly amplified type I proinflammatory response within the lungs, exaggerating exacerbations of COPD in the course of infections (21). Infections play an important role in progression of COPD and greatly increase the risk of death. Lipopolysaccharide (LPS) has been shown to enhance IL-33 expression and release not only in epithelial cells, but also in PBMC and PBL (80). This suggests that IL-33 is an important mediator of immunological response during inflammations and exacerbations in the course of COPD. It has been widely established that COPD patients suffer from chronic inflammation, which leads to alveolar disruption. It has been shown that IL-33 induces IL-6 and IL-8 production and release in lung epithelial and endothelial cells (82, 83). The same two interleukins have been observed to be increased in bronchoalveolar lavage (BAL) and the lungs of COPD patients, compared to healthy controls (84–86). This causes an influx of neutrophils to the lungs which, through the secretion of elastases and proteases, cause lung tissue damage, further resulting in lung fibrosis and decreased lung function (87). Additionally, it has been shown that IL-33 is involved in the development of eosinophilic airway inflammation in non-atopic COPD patients (88). Moreover, IL-33 plays a part in mucus production, advancing the inflammatory process and decreasing respiratory capacity of COPD patients (27). What is more, IL-33 increases vascular endothelial permeability, which further intensifies inflammatory effect (89). It has been shown that IL-33 can directly contribute to lung tissue damage trough triggering cortactin degradation mediated apoptosis in alveolar epithelial cells (90).
Obstructive Sleep Apnea
Obstructive sleep apnea (OSA) is a chronic condition characterized by recurrent pauses in breathing during sleep caused by collapse of the upper airways. The disorder is highly associated with chronic low-grade systemic inflammation. The most prominent inflammatory mediators present in OSA patients include IL-1, IL-6, and CRP (91). Literature concerning IL-33 in OSA patients is greatly limited. In recent studies, it has been observed that IL-33 is one of the inflammatory mediators causing levels in blood serum to be increased, in comparison to healthy controls (92). Interestingly, increased IL-33 levels have also been shown in the saliva of OSA patients (93). This emphasizes the contribution of IL-33 to systemic inflammation in this group of patients. Substantial obesity is a well-known risk factor for OSA (94). Excessive amounts of central adipose tissue also contributes to levels of systemic inflammation among OSA (95). Adipocytes have been identified to possess ability to produce IL-33 (96), therefore adipose tissue in OSA patients offers an extensive source of IL-33. Additionally, intermittent hypoxia contributes to systemic inflammation (97). It can possibly affect the IL-33 production too, however the mechanisms of this relationship are not known yet. As described before, IL-33 highly contributes to the development of chronic inflammatory diseases such as asthma and COPD. Several studies have shown increased prevalence of these diseases among OSA patients compared to the general population (98–100). This suggests that OSA patients are vulnerable to the development of comorbidities of inflammatory etiology which are not only limited to respiratory disorders but also others, such as diabetes or psoriasis (97, 101–103). It has been shown that OSA with severe co-morbid asthma intensifies airway remodeling and is associated with more frequent exacerbations (104, 105). Even though IL-33 has not been investigated as a mediator in a group suffering from these two co-morbid diseases, it can be hypothesized that this alarmin enhances airway remodeling and fibrosis occurring in the course of asthma due to its increased level caused by co-morbid OSA. Similar observations have been made regarding co-morbid OSA and COPD, as the BAL fluid of patients with the overlapping syndromes showed a significantly increased proportion of neutrophils, higher TNFα concentrations, and IL-8 levels than that of COPD. Co-morbid OSA exacerbated the course of COPD (106). Yet again IL-33 was not investigated in the study. However, it may again be assumed that systemic inflammation present in OSA with increased IL-33 levels intensifies processes involved in the development and progression of COPD.
IL-33 as a Potential Therapeutic Target
The murine models of both asthma and COPD suggest that IL-33 and the ST2 receptor might be prominent new therapeutic targets for these chronic inflammatory respiratory diseases. In a murine model of asthma, the use of anti-IL-33 antibody reduced Th2 cytokines production by ILC2 (107), while the antibody blocking ST2 receptor caused a decrease of interleukin 4 expression in the lungs of mice undergoing ovalbumin challenge and extenuated airway hyperresponsiveness (108). In another study a decrease in eosinophil count in BAL fluid and reduced airway hyperresponsiveness to methacholine was observed following anti-IL-33 antibody and sST2 receptors treatment (109). It has been shown, in vitro, that pre-treatment with anti-ST2 antibody supressed the production of fibronectin 1 and type I collagen by human lung fibroblasts (65). Anti-IL-33 antibody has also been shown to significantly inhibit cigarette smoke induced lung inflammation as, following the treatment, neutrophil and macrophage infiltration along with cytokines (IL-1β, TNF, and IL-17) expression decreased (81).
Several clinical trials targeting either IL-33 or its receptor in asthma and COPD are ongoing at the moment. In COPD two trials concerning IL-33/ST2 have reached phase 2. One investigates the effects of anti-IL-33 monoclonal antibody compared with placebo, on the annualized rate of moderate-to-severe acute exacerbations of COPD over up to 52 weeks of treatment (ClinicalTrials.gov: NCT03546907). The second evaluates the efficacy of anti-ST2 antibody vs. placebo on the frequency of moderate-to-severe exacerbations of COPD (ClinicalTrials.gov: NCT03615040). At phase 2 of the asthma clinical trial, the anti-IL-33 receptor monoclonal antibody is investigated in subjects with moderately severe asthma, and is compared to the placebo, fluticasone propionate/salmeterol combination and fluticasone propionate (ClinicalTrials.gov: NCT03207243). Another clinical trial concerning anti-IL-33 antibody in asthma patients is in phase 1 and compares it to the placebo Dupilumab and fluticasone propionate (ClinicalTrials.gov: NCT03112577) (Table 1).
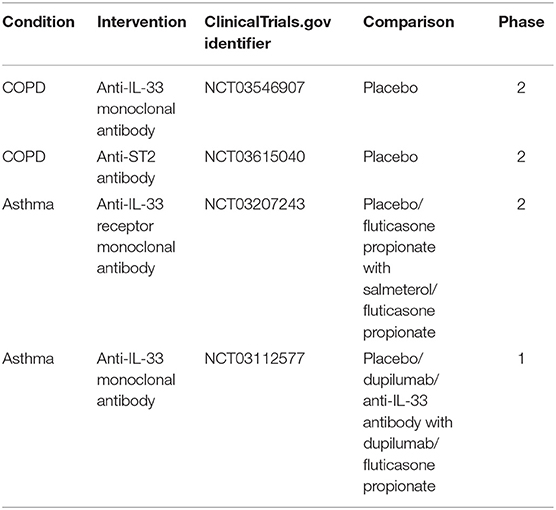
Table 1. Summary of ongoing clinical trials targeting IL-33 pathway in COPD and asthma.
Conclusion
Analysis of the pleiotropic effects of IL-33 on multiple immunological cells (macrophages, mastocytes), as well as neurological cells of medulla oblongata, dorsal root ganglion, antigen-induced arthritis system, carrageen, and formalin, shows that this alarmin plays curtail, yet not fully known role in mediating inflammation, especially in chronic inflammatory pulmonary diseases such as asthma, COPD, and OSA. Taken into consideration the engagement in this process, in particular of mastocytes and their secretion of CXCL2, 4, 8, and other cytokines, there is no doubt regarding the etiopathogenic role of IL-33 in the development of asthma in response to various stimuli damaging bronchial epithelial cells. Additionally, IL-33 intensifies recruitment of eosinophils, macrophages, and Th2 lymphocytes, which again confirms its inflammatory role. Particularly important in the context of aggravation of chronic inflammation and progression of respiratory disease is IL-33 mediated influx of neutrophils and macrophages. As these cells secrete IL-1β, TNF-α, and release proteases (elastases, metalloproteinases, cathepsins, and proteinases), it is only a logical consequence that IL-33 is involved in development of lung emphysema and chronic bronchial inflammation. It is worth mentioning that IL-33 expression is enhanced following exposure to cigarette smoke, which correlates with increased number of lymphocytes CD8+, resulting in the release of perforin and granzymes. However, surprisingly, this model of COPD development was only confirmed during experiments on mice and was observed following the introduction of anti-IL-33 antibody. Among discussed respiratory disorders, the role of IL-33 in OSA is the least known. The alarmin indirectly increases the expression of IL-1, IL-6, CRP, and enhances systemic inflammation present in OSA. Importantly, adipocytes have been identified to possess the ability produce IL-33, which in case of OSA patients seems crucial as the vast majority of them are obese, making adipose tissue an considerable source of the interleukin. However, the effect it exerts is not yet fully understood, and further research is needed, especially focusing on the mRNA expression of chosen genetic markers and animal models. Nevertheless, IL-33 through the intensification of TGF-β expression is involved in the stimulation of fibrosis and bronchial remodeling, including EMT, which might further contribute to the development and comorbidity of asthma and OSA.
The mechanisms of IL-33 activity described in this short review highlight its important role in the development of multidirectional inflammation. Therefore, direct or indirect blockade of this cytokine might greatly increase conservative therapy quality among patients with chronic respiratory diseases. At the moment, there are clinical trials involving anti-IL-33 antibodies which are very promising. Nonetheless, authors of this review indicate a necessity to undertake new research focusing on the development of peptide inhibitors for the ST2 receptors. The establishment of several interface regions between IL-33 and ST2 receptor, based on the available complex crystallographic structure, enables the creation of peptide library in silico through bioinformatics methods. Creation of peptide domain for the interface of IL-33 may be a universal method of safe, non-toxic and well-tolerated treatment for patients suffering from chronic inflammatory diseases of the respiratory system. At present, there is no investigation carried out in the direction suggested by the authors.
Author Contributions
AG and MP created the concept of the paper. AG conducted the literature research and wrote the manuscript. PK, AA, PB, and MP revised the paper.
Funding
AG is supported by grant no. 0067/DIA/2018/47 from the Polish Ministry of Science and Higher Education.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AHI, apnea-hypopnea index; AREG, amphiregulin; BAL, bronchoalveolar lavage; COPD, chronic obstructive pulmonary sleep apnea; CRP, C-reactive protein; CXCL, chemokine (C-X-C motif) ligand; EMT, epithelial-to-mesenchymal transition; f-IL-33, full length interleukin-33; GAS, gamma interferon activation sites; GWS, genome-wide studies; IAK, interleukin-1 receptor associated kinase; IL, interleukin; IL-1Ra, interleukin 1 receptor antagonist; IL-1RacP, interleukin 1 receptor accessory protein; IL-36Ra, interleukin 36 receptor antagonist; ILC, innate lymphoid cell; ISRE, interferon stimulated response element; JNK, c-Jun N-terminal kinases; HF-HEV, nuclear factor from high endothelial venules; MAPK, mitogen-activated protein kinases; m-IL-33, mature interleukin 33; MHCII, major histocompatibility complex II; MYD88, myeloid differentiation primary response protein 88; NFκB, nuclear factor κB; OSA, obstructive sleep apnea; PDGF, platelet-derived growth factor; ROS, reactive oxygen species; SNP, single nucleotide polymorphism; sST2, suppression of tumorigenicity 2 soluble receptor; ST2, suppression of tumorigenicity 2 transmembrane receptor; TCR, Th2 cell receptor; TGF-β, transforming growth factor β; Th2, T helper 2 cell; TIR, Toll-like/interleukin 1receptor; TNF, tumor necrosis factor; TRAF6, tumor necrosis factor receptor-assosiatedreceptor-6.
References
1. O'Neill LAJ. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. (2008) 226:10–8. doi: 10.1111/j.1600-065X.2008.00701.x
2. Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. (2010) 10:89–102. doi: 10.1038/nri2691
3. Hams E, Bermingham R, Fallon PG. Macrophage and innate lymphoid cell interplay in the genesis of fibrosis. Front Immunol. (2015) 6:597. doi: 10.3389/fimmu.2015.00597
4. Dinarello C, Arend W, Sims J, Smith D, Blumberg H, O'Neill L, et al. IL-1 family nomenclature. Nat Immunol. (2010) 11:973. doi: 10.1038/ni1110-973
5. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
6. Taylor SL, Renshaw BR, Garka KE, Smith DE, Sims JE. Genomic organization of the interleukin-1 locus. Genomics. (2002) 79:726–33. doi: 10.1006/geno.2002.6752
7. Nicklin MJH, Barton JL, Nguyen M, FitzGerald MG, Duff GW, Kornman K. A sequence-based map of the nine genes of the human interleukin-1 cluster. Genomics. (2002) 79:718–25. doi: 10.1006/geno.2002.6751
8. Lefrançais E, Roga S, Gautier V, Gonzalez-de-Peredo A, Monsarrat B, Girard J-P, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci USA. (2012) 109:1673–8. doi: 10.1073/pnas.1115884109
9. Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. (2009) 284:19420–6. doi: 10.1074/jbc.M901744200
10. Baekkevold ES, Roussigné M, Yamanaka T, Johansen F-E, Jahnsen FL, Amalric F, et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol. (2003) 163:69–79. doi: 10.1016/S0002-9440(10)63631-0
11. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
12. Yagami A, Orihara K, Morita H, Futamura K, Hashimoto N, Matsumoto K, et al. IL-33 mediates inflammatory responses in human lung tissue cells. J Immunol. (2010) 185:5743–50. doi: 10.4049/jimmunol.0903818
13. Moussion C, Ortega N, Girard J-P. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel 'alarmin'? PLoS ONE. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
14. Liew FY, Girard J-P, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. (2016) 16:676–89. doi: 10.1038/nri.2016.95
15. Tsuda H, Komine M, Tominaga S-I, Ohtsuki M. Identification of the promoter region of human IL-33 responsive to induction by IFNγ. J Dermatol Sci. (2017) 85:137–40. doi: 10.1016/j.jdermsci.2016.11.002
16. Hong J, Bae S, Jhun H, Lee S, Choi J, Kang T, et al. Identification of constitutively active interleukin 33 (IL-33) splice variant. J Biol Chem. (2011) 286:20078–86. doi: 10.1074/jbc.M111.219089
17. Carriere V, Roussel L, Ortega N, Lacorre D-A, Americh L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. (2007) 104:282–7. doi: 10.1073/pnas.0606854104
18. Roussel L, Erard M, Cayrol C, Girard J-P. Molecular mimicry between IL-33 and KSHV for attachment to chromatin through the H2A-H2B acidic pocket. EMBO Rep. (2008) 9:1006–12. doi: 10.1038/embor.2008.145
19. Lefrançais E, Duval A, Mirey E, Roga S, Espinosa E, Cayrol C, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci USA. (2014) 111:15502–7. doi: 10.1073/pnas.1410700111
20. Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, et al. The dual function cytokine IL-33 interacts with the transcription factor NF-κB to dampen NF-κB-stimulated gene transcription. J Immunol. (2011) 187:1609–16. doi: 10.4049/jimmunol.1003080
21. Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. (2015) 42:566–79. doi: 10.1016/j.immuni.2015.02.011
22. Mohapatra A, Van Dyken SJ, Schneider C, Nussbaum JC, Liang H-E, Locksley RM. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. (2016) 9:275–86. doi: 10.1038/mi.2015.59
23. Hardman CS, Panova V, McKenzie ANJ. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. (2013) 43:488–98. doi: 10.1002/eji.201242863
24. Cohen ES, Scott IC, Majithiya JB, Rapley L, Kemp BP, England E, et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat Commun. (2015) 6:8327. doi: 10.1038/ncomms9327
25. Yasuda K, Muto T, Kawagoe T, Matsumoto M, Sasaki Y, Matsushita K, et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci USA. (2012) 109:3451–56. doi: 10.1073/pnas.1201042109
26. Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. (2013) 132:933–41. doi: 10.1016/j.jaci.2013.05.012
27. Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. (2013) 123:3967–82. doi: 10.1172/JCI65570
28. Uchida M, Anderson EL, Squillace DL, Patil N, Maniak PJ, Iijima K, et al. Oxidative stress serves as a key checkpoint for IL-33 release by airway epithelium. Allergy. (2017) 72:1521–31. doi: 10.1111/all.13158
29. Hayakawa H, Hayakawa M, Kume A, Tominaga S-I. Soluble ST2 blocks interleukin-33 signaling in allergic airway inflammation. J Biol Chem. (2007) 282:26369–80. doi: 10.1074/jbc.M704916200
30. Bandara G, Beaven MA, Olivera A, Gilfillan AM, Metcalfe DD. Activated mast cells synthesize and release soluble ST2-a decoy receptor for IL-33. Eur J Immunol. (2015) 45:3034–44. doi: 10.1002/eji.201545501
31. Lécart S, Lecointe N, Subramaniam A, Alkan S, Ni D, Chen R, et al. Activated, but not resting human Th2 cells, in contrast to Th1 and T regulatory cells, produce soluble ST2 and express low levels of ST2L at the cell surface. Eur J Immunol. (2002) 32:2979–87. doi: 10.1002/1521-4141(2002010)32:10<2979::AID-IMMU2979>3.0.CO;2-5
32. Bergers G, Reikerstorfer A, Braselmann S, Graninger P, Busslinger M. Alternative promoter usage of the Fos-responsive gene Fit-1 generates mRNA isoforms coding for either secreted or membrane-bound proteins related to the IL-1 receptor. EMBO J. (1994) 13:1176–88. doi: 10.1002/j.1460-2075.1994.tb06367.x
33. Lott JM, Sumpter TL, Turnquist HR. New dog and new tricks: evolving roles for IL-33 in type 2 immunity. J Leukoc Biol. (2015) 97:1037–48. doi: 10.1189/jlb.3RI1214-595R
34. Yanagisawa K, Takagi T, Tsukamoto T, Tetsuka T, Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett. (1993) 318:83–7. doi: 10.1016/0014-5793(93)81333-U
35. Liu X, Hammel M, He Y, Tainer JA, Jeng U-S, Zhang L, et al. Structural insights into the interaction of IL-33 with its receptors. Proc Natl Acad Sci USA. (2013) 110:14918–23. doi: 10.1073/pnas.1308651110
36. Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. (2015) 42:1005–19. doi: 10.1016/j.immuni.2015.06.006
37. Zaiss DMW, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. (2015) 42:216–26. doi: 10.1016/j.immuni.2015.01.020
38. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
39. Xu D, Chan WL, Leung BP, Huang FP, Wheeler R, Piedrafita D, et al. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. (1998) 187:787–94. doi: 10.1084/jem.187.5.787
40. Hsu C-L, Neilsen CV, Bryce PJ. IL-33 is produced by mast cells and regulates IgE-dependent inflammation. PLoS ONE. (2010) 5:e11944. doi: 10.1371/journal.pone.0011944
41. Alves-Filho JC, Sônego F, Souto FO, Freitas A, Verri WA, Auxiliadora-Martins M, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. (2010) 16:708–12. doi: 10.1038/nm.2156
42. Cherry WB, Yoon J, Bartemes KR, Iijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. (2008) 121:1484–90. doi: 10.1016/j.jaci.2008.04.005
43. Borish L, Steinke JW. Interleukin-33 in asthma: how big of a role does it play? Curr Allergy Asthma Rep. (2011) 11:7–11. doi: 10.1007/s11882-010-0153-8
44. Li D, Guabiraba R, Besnard A-G, Komai-Koma M, Jabir MS, Zhang L, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. (2014) 134:1422–32.e11. doi: 10.1016/j.jaci.2014.05.011
45. Kurowska-Stolarska M, Stolarski B, Kewin P, Murphy G, Corrigan CJ, Ying S, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. (2009) 183:6469–77. doi: 10.4049/jimmunol.0901575
46. Matta BM, Reichenbach DK, Zhang X, Mathews L, Koehn BH, Dwyer GK, et al. Peri-alloHCT IL-33 administration expands recipient T-regulatory cells that protect mice against acute GVHD. Blood. (2016) 128:427–39. doi: 10.1182/blood-2015-12-684142
47. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. (2014) 513:564–8. doi: 10.1038/nature13577
48. Negishi H, Fujita Y, Yanai H, Sakaguchi S, Ouyang X, Shinohara M, et al. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc Natl Acad Sci USA. (2006) 103:15136–41. doi: 10.1073/pnas.0607181103
49. Griesenauer B, Paczesny S. The ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. (2017) 8:475. doi: 10.3389/fimmu.2017.00475
50. Grotenboer NS, Ketelaar ME, Koppelman GH, Nawijn MC. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J Allergy Clin Immunol. (2013) 131:856–65. doi: 10.1016/j.jaci.2012.11.028
51. Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. (2011) 43:887–92. doi: 10.1038/ng.888
52. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
53. Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet. (2009) 41:342–7. doi: 10.1038/ng.323
54. Bønnelykke K, Sleiman P, Nielsen K, Kreiner-Møller E, Mercader JM, Belgrave D, et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat Genet. (2013) 46:51–5. doi: 10.1038/ng.2830
55. Ramasamy A, Kuokkanen M, Vedantam S, Gajdos ZK, Couto Alves A, Lyon HN, et al. Genome-wide association studies of asthma in population-based cohorts confirm known and suggested loci and identify an additional association near HLA. PLoS ONE. (2012) 7:e44008. doi: 10.1371/journal.pone.0044008
56. Jonakowski M, Zioło J, Koćwin M, Przemecka M, Mokros Ł, Panek M, et al. Role of IL-15 in the modulation of TGF-β1-mediated inflammation in asthma. Exp Ther Med. (2017) 14:4533–40. doi: 10.3892/etm.2017.5108
57. Panek M, Jonakowski M, Zioło J, Wieteska Ł, Małachowska B, Pietras T, et al. A novel approach to understanding the role of polymorphic forms of the NR3C1 and TGF-β1 genes in the modulation of the expression of IL-5 and IL-15 mRNA in asthmatic inflammation. Mol Med Rep. (2016) 13:4879–87. doi: 10.3892/mmr.2016.5104
58. Panek M, Pietras T, Fabijan A, Zioło J, Wieteska L, Małachowska B, et al. Identification and association of the single nucleotide polymorphisms, C-509T, C+466T and T+869C, of the TGF-β1 gene in patients with asthma and their influence on the mRNA expression level of TGF-β1. Int J Mol Med. (2014) 34:975–86. doi: 10.3892/ijmm.2014.1894
59. Panek M, Pietras T, Szemraj J, Fabijan A, Kuna P. Identification and association of TGFβ-1 expression in patients with asthma in a Polish population - Lodz metropolitan area study. Int J Biochem Mol Biol. (2013) 4:67–74.
60. Savenije OEM, Kerkhof M, Reijmerink NE, Brunekreef B, de Jongste JC, Smit HA, et al. Interleukin-1 receptor-like 1 polymorphisms are associated with serum IL1RL1-a, eosinophils, and asthma in childhood. J Allergy Clin Immunol. (2011) 127:750–6.e1–5. doi: 10.1016/j.jaci.2010.12.014
61. Reijmerink NE, Postma DS, Bruinenberg M, Nolte IM, Meyers DA, Bleecker ER, et al. Association of IL1RL1, IL18R1, and IL18RAP gene cluster polymorphisms with asthma and atopy. J Allergy Clin Immunol. (2008) 122:651–4.e8. doi: 10.1016/j.jaci.2008.06.030
62. Ho JE, Chen W-Y, Chen M-H, Larson MG, McCabe EL, Cheng S, et al. Common genetic variation at the IL1RL1 locus regulates IL-33/ST2 signaling. J Clin Invest. (2013) 123:4208–18. doi: 10.1172/JCI67119
63. Smith D, Helgason H, Sulem P, Bjornsdottir US, Lim AC, Sveinbjornsson G, et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLoS Genet. (2017) 13:e1006659. doi: 10.1371/journal.pgen.1006659
64. Hamzaoui A, Berraies A, Kaabachi W, Haifa M, Ammar J, Kamel H. Induced sputum levels of IL-33 and soluble ST2 in young asthmatic children. J Asthma. (2013) 50:803–9. doi: 10.3109/02770903.2013.816317
65. Guo Z, Wu J, Zhao J, Liu F, Chen Y, Bi L, et al. IL-33 promotes airway remodeling and is a marker of asthma disease severity. J Asthma. (2014) 51:863–9. doi: 10.3109/02770903.2014.921196
66. Sjöberg LC, Nilsson AZ, Lei Y, Gregory JA, Adner M, Nilsson GP. Interleukin 33 exacerbates antigen driven airway hyperresponsiveness, inflammation and remodeling in a mouse model of asthma. Sci Rep. (2017) 7:716. doi: 10.1038/s41598-017-03674-0
67. Kumar RK, Herbert C, Yang M, Koskinen AML, McKenzie ANJ, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy. (2002) 32:1104–11. doi: 10.1046/j.1365-2222.2002.01420.x
68. Fallon PG, Richardson EJ, McKenzie GJ, McKenzie ANJ. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. (2000) 164:2585–91. doi: 10.4049/jimmunol.164.5.2585
69. Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol. (2003) 132:168–76. doi: 10.1159/000073718
70. Gharaee-Kermani M, Phan SH. Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. (1997) 16:438–47. doi: 10.1165/ajrcmb.16.4.9115755
71. Rothenberg ME, Saito H, Peebles RS. Advances in mechanisms of allergic disease in 2016. J Allergy Clin Immunol. (2017) 140:1622–31. doi: 10.1016/j.jaci.2017.08.029
72. Chen S-F, Nieh S, Jao S-W, Wu M-Z, Liu C-L, Chang Y-C, et al. The paracrine effect of cancer-associated fibroblast-induced interleukin-33 regulates the invasiveness of head and neck squamous cell carcinoma. J Pathol. (2013) 231:180–9. doi: 10.1002/path.4226
73. Camelo A, Dunmore R, Sleeman MA, Clarke DL. The epithelium in idiopathic pulmonary fibrosis: breaking the barrier. Front Pharmacol. (2014) 4:173. doi: 10.3389/fphar.2013.00173
74. Chen CC, Kobayashi T, Iijima K, Hsu FC, Kita H. IL-33 dysregulates regulatory T cells and impairs established immunologic tolerance in the lungs. J Allergy Clin Immunol. (2017) 140:1351–63.e7. doi: 10.1016/j.jaci.2017.01.015
75. Kim HY, Chang Y-J, Subramanian S, Lee H-H, Albacker LA, Matangkasombut P, et al. Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. (2012) 129:216–27.e6. doi: 10.1016/j.jaci.2011.10.036
76. Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13–producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. (2012) 129:191–8.e4. doi: 10.1016/j.jaci.2011.09.041
77. Prefontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. (2009) 183:5094–103. doi: 10.4049/jimmunol.0802387
78. Zoltowska AM, Lei Y, Fuchs B, Rask C, Adner M, Nilsson GP. The interleukin-33 receptor ST2 is important for the development of peripheral airway hyperresponsiveness and inflammation in a house dust mite mouse model of asthma. Clin Exp Allergy. (2016) 46:479–90. doi: 10.1111/cea.12683
79. Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT Jr, Rollins DR, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. (2015) 136:59–68.e14. doi: 10.1016/j.jaci.2014.11.037
80. Xia J, Zhao J, Shang J, Li M, Zeng Z, Zhao J, et al. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L619–27. doi: 10.1152/ajplung.00305.2014
81. Qiu C, Li Y, Li M, Li M, Liu X, McSharry C, et al. Anti-interleukin-33 inhibits cigarette smoke-induced lung inflammation in mice. Immunology. (2012) 138:76–82. doi: 10.1111/imm.12020
82. Wu H, Yang S, Wu X, Zhao J, Zhao J, Ning Q, et al. Interleukin-33/ST2 signaling promotes production of interleukin-6 and interleukin-8 in systemic inflammation in cigarette smoke-induced chronic obstructive pulmonary disease mice. Biochem Biophys Res Commun. (2014) 450:110–6. doi: 10.1016/j.bbrc.2014.05.073
83. Shang J, Zhao J, Wu X, Xu Y, Xie J, Zhao J. Interleukin-33 promotes inflammatory cytokine production in chronic airway inflammation. Biochem Cell Biol. (2015) 93:359–66. doi: 10.1139/bcb-2014-0163
84. Bhowmik A. Relation of sputum inflammatory markers to symptoms and lung function changes in COPD exacerbations. Thorax. (2000) 55:114–20. doi: 10.1136/thorax.55.2.114
85. Bucchioni E, Kharitonov SA, Allegra L, Barnes PJ. High levels of interleukin-6 in the exhaled breath condensate of patients with COPD. Respir Med. (2003) 97:1299–302. doi: 10.1016/j.rmed.2003.07.008
86. Hacievliyagil SS, Gunen H, Mutlu LC, Karabulut AB, Temel I. Association between cytokines in induced sputum and severity of chronic obstructive pulmonary disease. Respir Med. (2006) 100:846–54. doi: 10.1016/j.rmed.2005.08.022
87. Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. (2013) 48:531–9. doi: 10.1165/rcmb.2012-0492TR
88. Tworek D, Majewski S, Szewczyk K, Kiszałkiewicz J, Kurmanowska Z, Górski P, et al. The association between airway eosinophilic inflammation and IL-33 in stable non-atopic COPD. Respir Res. (2018) 19:108. doi: 10.1186/s12931-018-0807-y
89. Choi YS, Choi HJ, Min JK, Pyun BJ, Maeng YS, Park H, et al. Interleukin-33 induces angiogenesis and vascular permeability through ST2/TRAF6-mediated endothelial nitric oxide production. Blood. (2009) 114:3117–26. doi: 10.1182/blood-2009-02-203372
90. Zhao J, Wei J, Mialki RK, Mallampalli DF, Chen BB, Coon T, et al. F-box protein FBXL19–mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat Immunol. (2012) 13:651–8. doi: 10.1038/ni.2341
91. Nadeem R, Molnar J, Madbouly EM, Nida M, Aggarwal S, Sajid H, et al. Serum inflammatory markers in obstructive sleep apnea: a meta-analysis. J Clin Sleep Med. (2013) 9:1003–12. doi: 10.5664/jcsm.3070
92. Sozer V, Kutnu M, Atahan E, Caliskaner Ozturk B, Hysi E, Cabuk C, et al. Changes in inflammatory mediators as a result of intermittent hypoxia in obstructive sleep apnea syndrome. Clin Respir J. (2018) 12:1615–22. doi: 10.1111/crj.12718
93. Nizam N, Basoglu OK, Tasbakan MS, Nalbantsoy A, Buduneli N. Salivary cytokines and the association between obstructive sleep apnea syndrome and periodontal disease. J Periodontol. (2014) 85:e251–8. doi: 10.1902/jop.2014.130579
94. Mokros Ł, Kuczynski W, Gabryelska A, Franczak Ł, Spałka J, Białasiewicz P. High negative predictive value of normal body mass index for obstructive sleep apnea in the lateral sleeping position. J Clin Sleep Med. (2018) 14:985–90. doi: 10.5664/jcsm.7166
95. Unnikrishnan D, Jun J, Polotsky V. Inflammation in sleep apnea: an update. Rev Endocr Metab Disord. (2015) 16:25–34. doi: 10.1007/s11154-014-9304-x
96. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab. (2015) 21:543–57. doi: 10.1016/j.cmet.2015.03.005
97. Gabryelska A, Łukasik ZM, Makowska JS, Białasiewicz P. Obstructive sleep apnea: from intermittent hypoxia to cardiovascular complications via blood platelets. Front Neurol. (2018) 9:635. doi: 10.3389/fneur.2018.00635
98. Owens RL, Macrea MM, Teodorescu M. The overlaps of asthma or COPD with OSA: a focused review. Respirology. (2017) 22:1073–83. doi: 10.1111/resp.13107
99. Prasad B, Nyenhuis SM, Weaver TE. Obstructive sleep apnea and asthma: associations and treatment implications. Sleep Med Rev. (2014) 18:165–71. doi: 10.1016/j.smrv.2013.04.004
100. Soler X, Gaio E, Powell FL, Ramsdell JW, Loredo JS, Malhotra A, et al. High prevalence of obstructive sleep apnea in patients with moderate to severe chronic obstructive pulmonary disease. Annals ATS. (2015) 12:1219–25. doi: 10.1513/AnnalsATS.201506-379LE
101. Seetho IW, Wilding JPH. Sleep-disordered breathing, type 2 diabetes and the metabolic syndrome. Chron Respir Dis. (2014) 11:257–75. doi: 10.1177/1479972314552806
102. Gabryelska A, Sochal M, Wasik B, Białasiewicz P. Patients with obstructive sleep apnea are over four times more likely to suffer from psoriasis than the general population. J Clin Sleep Med. (2018) 14:153. doi: 10.5664/jcsm.6908
103. Gabryelska A, Białasiewicz P. Is autonomic activation a middleman between obstructive sleep apnea syndrome and psoriasis? J Clin Sleep Med. (2018) 14:1087–8. doi: 10.5664/jcsm.7196
104. Taillé C, Rouvel-Tallec A, Stoica M, Danel C, Dehoux M, Marin-Esteban V, et al. Obstructive Sleep Apnoea Modulates Airway Inflammation and Remodelling in Severe Asthma. PLoS ONE. (2016) 11:e0150042. doi: 10.1371/journal.pone.0150042
105. Wang Y, Liu K, Hu K, Yang J, Li Z, Nie M, et al. Impact of obstructive sleep apnea on severe asthma exacerbations. Sleep Med. (2016) 26:1–5. doi: 10.1016/j.sleep.2016.06.013
106. Wang Y, Hu K, Liu K, Li Z, Yang J, Dong Y, et al. Obstructive sleep apnea exacerbates airway inflammation in patients with chronic obstructive pulmonary disease. Sleep Med. (2015) 16:1123–30. doi: 10.1016/j.sleep.2015.04.019
107. Halim TYF, Krauß RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. (2012) 36:451–63. doi: 10.1016/j.immuni.2011.12.020
108. Kearley J, Buckland KF, Mathie SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST2–IL-33 pathway. Am J Respir Crit Care Med. (2009) 179:772–81. doi: 10.1164/rccm.200805-666OC
Keywords: asthma, COPD—Chronic obstructive pulmonary disease, OSA (Obstructive sleep apnea), IL-33, inflammation
Citation: Gabryelska A, Kuna P, Antczak A, Białasiewicz P and Panek M (2019) IL-33 Mediated Inflammation in Chronic Respiratory Diseases—Understanding the Role of the Member of IL-1 Superfamily. Front. Immunol. 10:692. doi: 10.3389/fimmu.2019.00692
Received: 01 December 2018; Accepted: 13 March 2019;
Published: 16 April 2019.
Edited by:
Elizabeth Brint, University College Cork, IrelandReviewed by:
Remo Castro Russo, Federal University of Minas Gerais, BrazilJarek T. Baran, Jagiellonian University Medical College, Poland
Copyright © 2019 Gabryelska, Kuna, Antczak, Białasiewicz and Panek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agata Gabryelska, YWdhdGEuZ2FicnllbHNrYUBnbWFpbC5jb20=