 Simone de Leve
Simone de Leve Florian Wirsdörfer
Florian Wirsdörfer Verena Jendrossek
Verena Jendrossek- Institute of Cell Biology (Cancer Research), University Hospital Essen, University of Duisburg-Essen, Essen, Germany
Extracellular adenosine is a potent endogenous immunosuppressive mediator critical to the maintenance of homeostasis in various normal tissues including the lung. Adenosine is either released from stressed or injured cells or generated from extracellular adenine nucleotides by the concerted action of the ectoenzymes ectoapyrase (CD39) and 5′ ectonucleotidase (CD73) that catabolize ATP to adenosine. An acute CD73-dependent increase of adenosine in normal tissues mostly exerts tissue protective functions whereas chronically increased adenosine-levels in tissues exposed to DNA damaging chemotherapy or radiotherapy promote pathologic remodeling processes and fibrosis for example in the skin and the lung. Importantly, cancer cells also express CD73 and high CD73 expression in the tumor tissue has been linked to poor overall survival and recurrence free survival in patients suffering from breast and ovarian cancer. CD73 and adenosine support growth-promoting neovascularization, metastasis, and survival in cancer cells. In addition, adenosine can promote tumor intrinsic or therapy-induced immune escape by various mechanisms that dampen the immune system. Consequently, modulating CD73 or cancer-derived adenosine in the tumor microenvironment emerges as an attractive novel therapeutic strategy to limit tumor progression, improve antitumor immune responses, avoid therapy-induced immune deviation, and potentially limit normal tissue toxicity. However, the role of CD73/adenosine signaling in the tumor and normal tissue responses to radiotherapy and its use as therapeutic target to improve the outcome of radiotherapy approaches is less understood. The present review will highlight the dual role of CD73 and adenosine in tumor and tissue responses to radiotherapy with a special focus to the lung. It will also discuss the potential benefits and risks of pharmacologic modulation of the CD73/adenosine system to increase the therapeutic gain of radiotherapy or combined radioimmunotherapy in cancer treatment.
Introduction
Radiotherapy is a mainstay in the treatment of cancer patients. About 60% of all cancer patients receive radiotherapy during the course of their disease alone or in multimodal combinations of surgery, radiotherapy, and chemotherapy, with beneficial effects of these highly effective treatments on long-term survival and tumor cure (1–5). Moreover, much progress has been made with technical improvements in treatment planning that increase accuracy of dose delivery, as well as by the development of particle therapy approaches (6). Nevertheless, cure rates still need to be improved for prevalent cancer types with high loco-regional failure-rates or a high risk for invasive growth or metastatic spread. For example, patients suffering from locally advanced non-small cell lung cancer (NSCLC) are typically treated with fractionated radiotherapy to the thoracic region, or concurrent platinum-based radiochemotherapy (RCT) yielding local control rates of 40–66% with doses of 60–66 Gray (Gy) (7–9). But loco-regional failures upon definitive RCT or disease progression by distant metastases are common and it is thought that improving local control rates will directly improve survival rates (9, 10).
Herein biological factors such as the high intrinsic tumor cell radio resistance, a pronounced tumor heterogeneity, diversity in radiation responses, and a resistance-promoting microenvironment reduce the efficacy of radiotherapy and thus contribute to failures. Otherwise, the high radio sensitivity of the normal lung tissue limits the application of curative radiation doses to the thoracic region and therapy intensification efforts of RCT (9, 11). Technical advances in image guidance and modern radiation techniques have significantly increased the safety profile of thoracic radiotherapy (12–14); but radiation-induced lung disease (RILD) still represents a serious normal tissue complication associated with radio(chemo)therapy of thoracic neoplasms or total body irradiation in conditioning regimens for hematopoietic stem cell transplantation (15–17). Moreover, toxicity rates can increase or new toxicities can be observed when using molecularly targeted drugs in combination with radiotherapy (18–21). Thus, there is a high need for further innovations in radiotherapy practice that improve the tumor response without increasing toxicity.
The progress in cancer immunotherapy and the discovery that radiotherapy activates T-cell-mediated antitumor immune responses under certain conditions, particularly when combined with established immune checkpoint blockade, expedited interest, and research in exploiting a potential benefit of combining radiotherapy with immunotherapy in pre-clinical and clinical cancer research (22–30). However, there are still major challenges in defining optimal dosing and treatment schedules and understanding the dual face of radiation-induced immune changes with potential impact on immune-related adverse effects. Moreover, only a fraction of patients responds to the treatment with immune checkpoint blockade alone or in combination with radiotherapy as tumors may not be immunogenic, dispose of efficient strategies to escape from tumor immune surveillance, or responses may not be durable (31–34).
In this context, accumulation of extracellular adenosine through activation of 5′ectonucleotidase (CD73) and subsequent signaling through adenosine receptors is a common mechanism how tumors escape from tumor immune surveillance. This makes CD73/adenosine signaling an attractive target in immuno-oncology and the related studies and underlying principles are well covered in various reviews (35–39).
But the role of CD73/adenosine signaling in the response of tumors and normal tissues to radiotherapy and its potential impact on the outcome of radiotherapy and combined radioimmunotherapy are less well described. Herein it is important to consider that the effects of CD73/adenosine activation on the immune system and reconstitution of tissue homeostasis might well differ among tissues of different origins as well as between acute and chronic activation stages. Therefore, we will first introduce the contribution of radiotherapy-induced changes in the innate and adaptive immune cell compartments to acute and chronic tumor and normal tissue responses and point to beneficial and adverse roles to the outcome of radiotherapy. We will then summarize current knowledge about the role of CD73 and adenosine in tumor and normal tissue responses to radiotherapy, and highlight the potential of targeting CD73/adenosine for improving the therapeutic gain of radio (immuno)therapy in thorax-associated tumors with high risk of adverse late effects in the highly radiosensitive normal lung tissue.
Paradigm Change: Radiation Activates Local and Systemic Immune Effects
The broad use of radiotherapy as standard treatment option in the therapy of solid human tumors is based on its ability to damage cellular macromolecules, particularly the DNA, thereby effectively inducing growth arrest and cell death locally in irradiated tumor cells. But bystander effects such as the transmission of lethal signals between cells via gap junctions or the production of diffusible cytotoxic mediators can also contribute to local antineoplastic action of radiotherapy. However, despite reported transient immunosuppressive effects by local induction of immune cell death (40) and or immune impairment (41, 42), multiple reports highlight the ability of radiotherapy to induce systemic effects that involve activation of the innate and adaptive immune systems (22, 23, 43, 44).
In the context of tumor therapy, exposure to ionizing radiation can modulate immunosuppressive barriers in the tumor microenvironment, trigger the recruitment of immune effector cells to the local tumor, render tumors accessible to infiltration of immune effector cells by modulating restrictive tumor vessels, and even elicit tumor-specific immune responses leading to the regression of tumor lesions locally and at tumor sites outside the radiation field (abscopal effects) (22, 45–50). Elegant pre-clinical investigations helped to reveal the importance of T-cell responses to the local and abscopal antitumor effects in response to radio(immuno)therapy and to uncover the underlying mechanisms (47, 51–57).
Since abscopal effects seem to be rare in the clinical situation (49, 58–61), current clinical trials focus on combining radiotherapy with different immunotherapies (30, 47, 48, 62–64). Notably, there is hope from first clinical studies that blockade of the programmed cell death 1/programmed death-ligand 1 (PD-1/PD-L1) immune checkpoint might improve progression-free survival in lung patients with an acceptable safety profile, when given after radiotherapy or platinum-based RCT (65, 66). But further studies are needed to explore the efficacy and the safety profile of combined therapy of cancer patients suffering from thorax-associated neoplasms with radiotherapy and immunotherapies, to define biomarkers for patient selection and potential compensatory immune-tolerance mechanisms in malignant tumors (27, 67), and to define optimal treatment schedules.
It appears that the local induction of damage to highly radiosensitive resident cells in the lung with subsequent activation of non-targeted immune effector mechanisms might also contribute to the adverse effects of ionizing radiation in normal tissues such as the development of pneumonitis and pulmonary fibrosis (68–74). Similar to other models of sterile inflammation radiation-induced damage to resident normal lung tissue cells triggers a multifaceted damage-signaling cascade including a multifactorial secretory program in order to stimulate repair and recovery (74). However, radiation induces chronic changes in irradiated tissues that presumably result from a persistent damage signaling. These chronic environmental changes impact not only the phenotype of resident cells but also the recruitment and polarization of immune cells infiltrating the previously irradiated lung tissue, thereby disturbing the balance between inflammatory and repair processes and promoting chronic fibrosis progression (73).
Dual Face of Radiation-Induced Immune Changes: Balance Between Immunoactivating and Immunosuppressive Effects
As outlined above, exposure to ionizing radiation has the capacity to induce immune responses in normal and tumor tissues. These changes involve a complex interplay between cells of the irradiated malignant or healthy normal tissues and cells of the innate and adaptive immune systems. But, depending on the type (tumor vs. normal) and origin of the irradiated tissue, the temporal appearance (acute vs. chronic), and the basal immune status of the tissue before exposure to ionizing radiation (pro- vs. anti-inflammatory), the response of the immune system can either adopt immunostimulatory or immunosuppressive effects and have either a positive impact (anti-tumor; normal tissue protection) or a negative impact (pro-tumor; normal tissue toxicity) on treatment outcome.
In the following paragraphs we will highlight the dual roles of the immune system in the response of tumor and normal tissues after irradiation that are mostly derived from pre-clinical studies.
Tumor Tissue
Radiation-induced immune changes in the tumor involve the direct activation of innate and adaptive immune responses influencing tumor growth; but radiation-induced immune responses also include indirect responses such as radiation-induced changes in the tumor vasculature or tumor microenvironment that impact the recruitment and activation state of cells from the innate and adaptive immune system [for a review see (64, 75–79)].
Tumor irradiation induces damage and death of cancer cells resulting in the surface exposure of immunogenic molecules as well as the release of damage associated molecular patterns (DAMPs) such as ATP or High-Mobility-Group-Protein B1 (HMGB1), and potentially tumor antigens, to activate innate and adaptive immune responses (80). Nuclear release and cytoplasmic sensing of altered nuclear acids via Toll-like receptor (TLR)9 or cyclic GMP-AMP Synthase/Stimulator of Interferon Genes (cGAS/STING) is intimately connected to the secretion of cytokines that support innate and adaptive antitumor immunity. Priming of tumor-specific T cell responses requires uptake of tumor antigens by antigen presenting cells e.g., dendritic cells. Furthermore, priming of tumor-specific T cells depends on sensing of cancer-cell derived cytoplasmic DNA. e.g., by the cGAS/STING pathway that is connected to the activation of the interferon (IFN) I response to support antitumor immunity. The initiated migration and antigen presentation of dendritic cells then triggers the activation of B and T cells in secondary lymphoid organs. Activated T and B cells subsequently exert anti-tumor effects by several mechanisms like CD8+ T cell mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and antibody-induced complement-mediated lysis. These processes have been excellently described in more detail elsewhere (27, 43, 47, 57, 75, 80–82).
Thus, the direct induction of anti-tumor immunity in response to ionizing radiation requires a complex interplay between the innate and adaptive immune system and the tumor microenvironment. Moreover, the recruitment and activation of dendritic cells in irradiated tumors that are required for the priming of tumor-specific T cell responses largely depends on the dose and fractionation of radiation in a tumor-dependent manner. Finally, tumor cells dispose of multiple mechanisms to evade this response so that the direct induction of anti-tumor immunity by radiotherapy is a rare event (41, 57, 83).
Besides these beneficial radiation-induced anti-tumor immune responses, local irradiation can also induce subacute or chronic immune changes that mostly exert tumor-promoting effects. Pro-inflammatory cytokines released in tissues as a damage response after radiotherapy as well as the humoral immune response from activated B cells can activate cells of the innate immunity, such as granulocytes, macrophages, and mast cells (84, 85). These cells release molecules that modulate gene expression programs in favor of pro-survival signaling and cell cycle progression in neoplastic cells thereby supporting malignant tissue expansion (86, 87). Moreover, cells from the innate immunity have the capacity to induce repair, regeneration, and tissue remodeling. By releasing various mediators these cells influence and initiate fibroblast activation, angiogenesis and matrix metabolism thereby indirectly fostering tumor growth (84, 88–90).
Finally, the tumor itself responds to radiation-induced stress or damage through a panel of phenotypic changes. By releasing several cytokines, chemokines, or growth factors as well as up-regulating specific surface receptors e.g., immunosuppressive PD-L1, cytotoxic T-lymphocyte-associated Protein 4 (CTLA-4), carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1), and others, tumor cells become proficient in dampening immune responses and to escape the immune system (91–96). Detailed reviews from Sharma et al. as well as Wennerberg et al. recently summarized the role of these tumor cell-extrinsic factors for primary and adaptive resistance so that these mechanisms will not be further addressed here (34, 41).
Normal Tissue
Despite technological improvements ionizing radiation still directly hits to some extent tumor-surrounding healthy tissue during treatment, leading to local damage, stress, or cell death in normal resident cells. Moreover, the damage response initiated in malignant tissues and healthy tissues residing in the radiation field not only contributes to the local effects of radiotherapy but can also exert strong systemic effects promoting normal tissue complications (22, 97–99).
Radiation-induced immune changes in normal tissues also include acute and chronic immune effects that will be discussed below. While the effects of radiation-induced normal tissue inflammation are well described (100–106), the contribution of the complex immune mechanisms that support chronic, pathological changes e.g., fibrosis, are less investigated and still not yet completely understood.
Similar to the situation in tumors, radiation-induced acute damage and cell death in normal tissues also results in the release of DAMPs as well as pro-inflammatory cytokines and chemokines which have the capacity to modulate immune responses (105, 107). These “danger signals” trigger the activation and influx of innate and adaptive immune cells at the site of damage resulting in normal tissue inflammation. An excessive inflammation during the acute phase after radiation as a result of an overwhelming secretion of pro-inflammatory cytokines and the release of reactive oxygen species (ROS) supports normal tissue toxicity and severe side effects in treated patients (108).
In addition DAMPs can activate tissue regeneration in normal tissues as well as in tumor tissues. It is known that the extracellular DAMPs HMGB1 and ATP can activate and recruit cells, thus stimulating tissue repair (109). Of these, innate immune cells invade into the damaged tissue to clear dead cells and cellular debris (110). Moreover, stem cells and tissue-associated cells, e.g., fibroblasts, keratinocytes, endothelial cells, and vascular smooth muscle cells, are stimulated by these DAMPs to support angiogenesis and tissue regeneration (111–117). In addition, several DAMPs (e.g., HMGB1, S100A4, uric acid) can also enhance the expression of immunosuppressive mediators like interleukin (IL)-10 and indoleamine 2,3-dioxygenase (IDO) in stem cells, thereby inhibiting lymphocyte responses and contributing to tumor promotion (118). Excellent detailed reviews about the role of DAMPs in mediating regenerative pathways can be found elsewhere (109, 119).
Radiation-induced damage to normal tissues furthermore triggers chronic environmental changes e.g., hypoxia and senescence, that are reminiscent of the changes observed in the tumor microenvironment. These changes support chronic inflammation and repair processes, promote alternative polarization of recruited immune cells, pathologic immune cell interactions and excessive tissue remodeling, and thereby trigger not only the development of tissue scaring and fibrosis but also the development of secondary tumors (120, 121). For more detailed reviews about the acute and chronic events during radiation-induced normal tissue toxicity please refer to Wirsdorfer and Jendrossek (73), McKelvey et al. (79), Schaue et al. (105), Stone et al. (122), Barnett et al. (123), Kim et al. (124), and Ruhle and Huber (125).
The dual face of radiotherapy-induced immune changes in normal tissues can be nicely demonstrated in murine models of RILD. The acute phase of pneumonitis and the chronic event of fibrosis dramatically reveal how complex the radiation-induced alterations of the tissue micromilieu and the immune system impact disease pathogenesis (73).
Own studies in the murine C57BL/6 model of RILD revealed that whole thorax irradiation (WTI) with 15 Gy triggered an acute early immune suppression characterized by a pronounced reduction in the number of lymphocytes and myeloid cells that was followed by an influx of CD3+ T cell lymphocytes into the lung tissue during the pneumonitic phase Interestingly, WTI also enhanced numbers of regulatory T cells (Treg) in the lung tissue of irradiated animals both, during the early inflammatory state as well as at the time of fibrosis development. Of note, radiation-induced pulmonary fibrosis was more severe in recombination-activating gene 2 (RAG2)-deficient mice that lack mature T- and B-lymphocytes suggesting that lymphocytes may have beneficial effects (126). Instead, depletion of CD4+ T cells during the pneumonitic phase decreased radiation-induced lung fibrosis in rats pointing to a contribution of CD4+ T cells to disease pathogenesis (68). These data suggest a causal link between the recruitment or local expansion of specific T-lymphocyte populations and the course of RILD that are also observed in other fibrosis models (127). But further work is required to define the beneficial and adverse effects of recruited and induced T cell subsets during the course of RILD (128).
Thoracic irradiation induces not only changes in the T cell compartment but also in the myeloid compartment and the lung environment. For example, others and we detected a significant reduction in the levels of total pulmonary macrophages and an almost complete eradication of alveolar macrophages early after irradiation as well as time-dependent changes in the macrophage phenotype with increased expression of markers for alternative macrophage activation [e.g., macrophage mannose receptor and Arginase-1] particularly during the fibrotic phase (102, 126, 129–132). Further, macrophages accumulated in organized clusters and expressed pro-fibrotic mediators such as alpha smooth muscle actin (α-SMA) and transforming growth factor beta (TGF-β) at ≥25 weeks post-irradiation (131). Importantly, a recent report confirmed the formation of organized clusters of CD163+ macrophages also in lung tissue of irradiated patients. Intriguingly, pharmacologic inhibition of colony stimulating factor-1 (CSF-1) inhibited macrophage influx and attenuated RT-induced lung fibrosis in mice supporting a pathologic relevance of macrophages in RT-induced lung fibrosis (132). Similarly, pharmacological treatment with as connective tissue growth factor (CTGF) antibody before or after 20 Gy thoracic irradiation reduced acute and chronic radiation toxicity in mice and abrogated M2-like macrophage infiltration (133). The combined inhibition of TGF-β and Platelet-Derived Growth Factor (PDGF) blockade in a pre-clinical murine model attenuated radiation-induced pneumonitis and lung fibrosis and was accompanied by reduced osteopontin expression and leukocyte infiltration (134). Instead strategies using anti- Vascular Endothelial Growth Factor (VEGF) to target the tumor vasculature in combination with radiotherapy turned out to be highly toxic to normal lung tissue in pre-clinical murine models (21).
The Immunomodulatory CD73/Adenosine System as Therapeutic Target for Improving Radiotherapy Outcome
Various observations from pre-clinical and clinical studies summarized in the former paragraphs suggest that targeting tumor-induced or radiation-induced immune deviation may offer novel and attractive opportunities for improving the outcome of radiotherapy by modulating the tumor radiation response, radiation-induced adverse late effects, or both. But the complexity of the tumor-induced and radiation-induced changes in the microenvironment as well as the time- and tissue-dependent “dual face” of radiotherapy-induced immune changes highlight the importance to identify strategies suited to balance adverse pro-inflammatory and immunosuppressive effects of radiotherapy and outweigh the beneficial effects of radioimmunotherapy with optimal tumor control and normal tissue protection. In this context, the purinergic CD73/adenosine system recently moved into the focus of research as it is an important endogenous regulator of the innate and adaptive immune systems with a documented role in tumor immune escape but also in adverse late effects of radiotherapy (36, 38, 131, 135–138).
We therefore hypothesized that the purinergic system might offer novel opportunities to interfere with normal tissue and tumor responses to radiotherapy and radiation-induced immune deviation. Extracellular ATP is a danger signal released by dying and damaged cells, and belongs to the earlier mentioned DAMPs, that function as immunostimulatory pro-inflammatory signals (139). In contrast, extracellular adenosine mostly exerts anti-inflammatory, immunosuppressive or regulatory functions and is a critical mediator for the maintenance of tissue homeostasis in various tissues including the lung and to avoid overwhelming inflammation for example in response to infection (140–143). But balancing pro-inflammatory ATP and anti-inflammatory adenosine might also to be crucial for maintaining or re-establishing immune homeostasis and to orchestrate tissue inflammation and repair under conditions of damage-induced sterile inflammation (73, 144).
CD73 and Adenosine Have Physiological Roles in Maintaining and Restoring Tissue Homeostasis
The purinergic signaling pathway is an evolutionary conserved mechanism that regulates immune homeostasis by the conversion of extracellular ATP to extracellular adenosine by using the sequential degradation via the ectoenzymes ectoapyrase (CD39, ectonucleoside triphosphate diphosphohydrolase 1) and CD73. Adenosine is either released from stressed or injured cells, or generated from extracellular adenine nucleotides by the concerted action of CD39 and CD73. While CD39 catalyzes the breakdown of ATP and ADP to AMP, CD73 converts AMP to adenosine. But the action of CD39 in degrading ATP can alternatively be executed by ectonucleotide pyrophosphatase (ENPP1, phosphodiesterase 1) (145).
CD39 and CD73 are expressed on the surface of specific lymphocyte subpopulations such as Treg and regulatory B cells (Breg) and endothelial cells and are important to their regulatory functions (143, 146–149). But CD73 is also expressed on stromal cells, mesenchymal stem cells (MSCs), and tumor-associated stem cells (150–153). Pre-clinical studies demonstrated that CD73 on stromal cells and tumor cells participates in the suppression of immune-mediated responses (152) as well as in homing and stemness of cancer stem cells (151, 154, 155). Furthermore, CD73 on MSCs promoted their immunosuppressive function and MSC were even able to upregulate CD73 expression on T cells (150). Inhibition of CD73 in a pre-clinical model of pancreatic neuroendocrine tumors led to reduced tumor growth and metastatic potential of cancer stem cells (151). Thus, stem cell-mediated immunosuppressive or regenerative processes might help cancer cells to escape natural anti-tumor immune responses, anti-cancer immunotherapies, or both. Table 1 shows detailed information on the expression of CD73 on multiple cell types in various tissues and their reported prognostic findings. Adenosine suppresses inflammatory functions of cells from innate and adaptive immune system and triggers expansion or differentiation of myeloid-derived suppressor cells (MDSC), M2-like macrophages as well as Treg and Breg and thereby participates in the creation of regulatory environments (144, 146, 149, 180–184). In addition, CD39/CD73-dependent generation of adenosine may also affect other processes in T-cell biology such as naive T-cell homeostasis, memory cell survival, and potentially T cell differentiation (168).
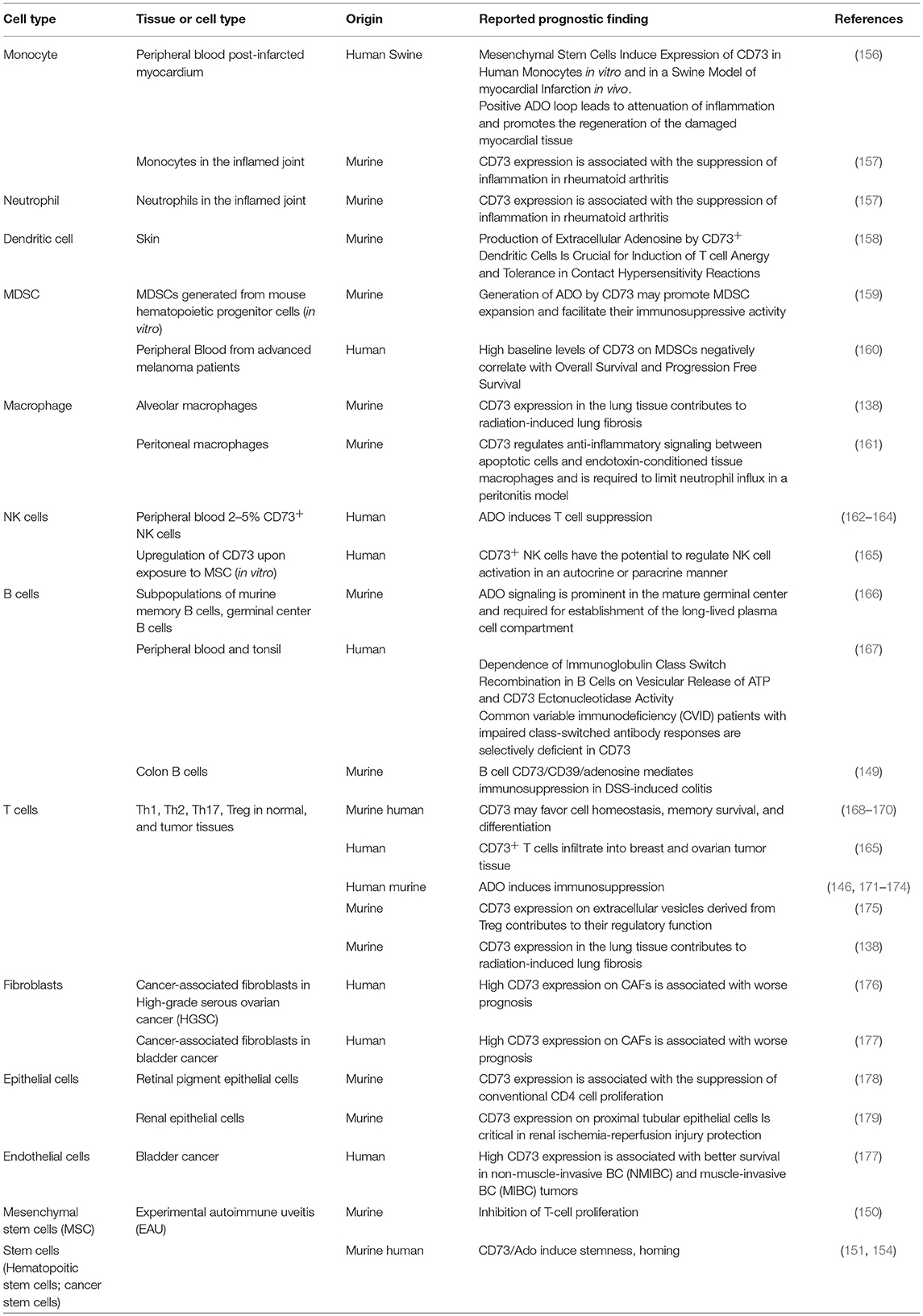
Table 1. CD73 expression on various cell types and tissues and its prognostic finding.
Extracellular adenosine can either be removed by enzymatic inactivation or cellular uptake or exert its actions through receptor binding. Adenosine deaminase (ADA) is responsible for the conversion of adenosine to inosine, a process that can happen either extracellularly or intracellularly (144). Adenosine may also be transported into its target cells via four different adenosine transporters, the so-called equilibrative nucleoside transporters (ENT) 1-4. Instead, adenosine exerts its actions by binding to one of four different G-protein-coupled adenosine receptors (ADORA1, ADORA2A, ADORA2B, and ADORA3) that are widely expressed on immune cells and resident tissue cells (185). ADORA1 and ADORA2A are high-affinity receptors responding to low concentrations of extracellular adenosine, while ADORA2B and ADORA3 are low affinity receptors and are mainly activated if the extracellular adenosine concentration rises above physiological levels (186). The adenosine receptors have various biological functions aimed at maintaining or restoring tissue homeostasis by triggering context-dependent pro- or anti-inflammatory effects (187–189).
Role of CD73 and Adenosine in Radiation-Induced Adverse Late Effects in the Lung
There is evidence from pre-clinical studies in models of injury-induced sterile inflammation that an acute CD73-dependent increase in adenosine mostly exerts tissue protective functions (142, 181, 190, 191). Herein, the role of purinergic signaling to self-terminate TLR-responses in macrophages might contribute to the observed effects (187, 192).
In contrast, chronically increased adenosine-levels induced for example by genetic deficiency of the adenosine-degrading enzyme ADA or chronic treatment with the chemotherapeutic drug Bleomycin (BLM) can promote pathologic remodeling processes in various tissues leading to fibrosis development (136, 193–200). The pathologic effects of BLM-induced chronic adenosine-accumulation in the lung have been attributed to alternatively activated myeloid cells (201, 202).
So far the role of purinergic signaling for radiation-induced adverse late effects in the lung has only been addressed in own investigations (131, 138) while others investigated its role the skin (136). Our work demonstrated a pathologic role of chronically increased CD73/adenosine signaling in irradiated lungs of C57BL/6 mice, presumably by promoting or amplifying profibrotic signaling cascades. Pathologic signaling involved a time-dependent increase in the expression and activity of the CD73 in the lung tissue that could be confined to resident CD45− cells as well as CD45+ immune cells (CD4+ T cells including Treg, alveolar macrophages) and was associated with a progressive increase in adenosine levels in the bronchioalveolar lavage fluid C57BL/6 mice with a knockout of CD73 (CD73−/−) failed to accumulate high levels of adenosine in response to WTI resulting in decreased levels of fibrosis-associated proteins and mediators, reduced recruitment/formation of Treg, and attenuated pulmonary fibrosis with absence of clusters with alternatively activated macrophages. A similar protective effect was obtained by treatment of irradiated C57BL/6 mice either with pegylated ADA (PEG-ADA) to catabolize adenosine, or with the CD73 monoclonal antibody (mAb) TY/23 as of week 16 post-irradiation (131, 138).
Taken together, the progressive up-regulation of CD73/adenosine signaling in the irradiated lung environment promotes the accumulation of immunosuppressive cell types of the innate and adaptive immune system, e.g., Treg and M2-like macrophages and supports a pro-fibrotic cross-talk between damaged resident cells and infiltrating immune cells. Thereby, CD73/adenosine signaling helps to amplify radiation-induced lung fibrosis as a late normal tissue complication (Figure 1). In support of our findings, adenosine also promoted radiation-induced skin fibrosis; but here the pro-fibrotic effects had mainly been attributed to T-cell infiltrates and signaling via ADORA2A, without a role for alternatively activated macrophages (136).
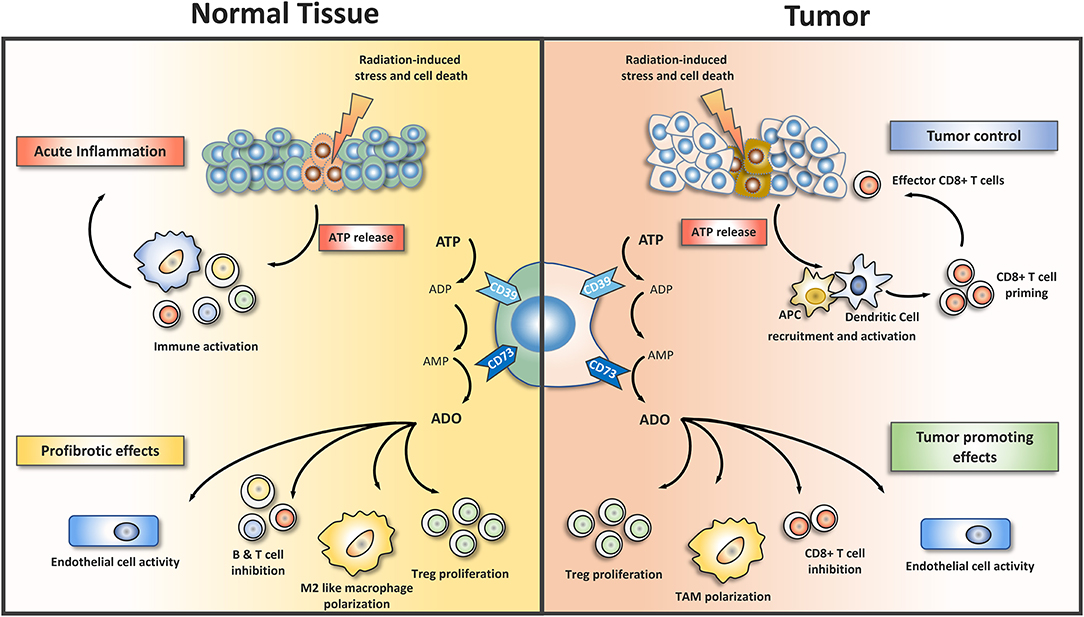
Figure 1. Purinergic signaling shapes the microenvironment in irradiated normal and tumor tissues. Exposure of normal tissues to ionizing radiation induces damage to tissue resident cells, e.g., endothelial cells and epithelial lung cells, as well as in resident immune cells. Equally exposure of tumor tissue results in radiation-induced damage to tumor cells and stromal cells. The resulting cell damage initiates stress responses and/or cell death with subsequent release of damage associated molecular patterns (DAMP). Release of ATP from dying cells is one component of radiation-induced tissue damage. Extracellular ATP acts as a potent inflammatory mediator that promotes inflammation and subsequent further damage to normal tissues. In tumor tissues extracellular ATP is an important mediator of anti-tumor CD8+ T cell responses as it participates in activation of dendritic antigen presenting cells (APC). To avoid excessive inflammation in normal tissues pro-inflammatory ATP is rapidly removed from the extracellular room by a two-step enzymatic conversion into adenosine, involving CD39 (or alternatively ectonucleotide pyrophosphatase) and CD73. Extracellular adenosine is an important endogenous regulator of inflammatory and repair processes as well as vascular functions. Adenosine exerts its pleiotropic actions in a tissue- and context-dependent manner through 4 different adenosine receptors that are expressed on various resident cells and immune cells (not shown). The immunosuppressive actions of adenosine involve the polarization of recruited immune cells toward regulatory or alternatively activated phenotypes, e.g., regulatory T cells (Treg), or M2-like macrophages. Moreover, adenosine mediates the inhibitory action of Treg and other regulatory cell types on proliferation and activation of cytotoxic T cells. By regulating endothelial cell activity CD73 and adenosine impact not only endothelial cell proliferation/angiogenesis but also vascular barrier function and the transmigration of leukocytes into damaged tissues. The expression of CD39 and CD73 thus balances the levels of pro-inflammatory ATP and immunosuppressive adenosine in normal tissues and tumors. The chronic activation of adenosine-driven processes observed in irradiated normal tissues promotes pathologic tissue remodeling and fibrosis development. Tumors coopt the CD73/adenosine system as a mechanism for promoting tumor growth and progression, angiogenesis, and immune escape. ADO, adenosine; CD39, ectonucleoside triphosphate diphosphohydrolase 1; CD73, 5′ ectonucleotidase; TAM, tumor associated macrophages.
Although radiation-induced intestinal injury models exist, the role of CD73/adenosine has only been studied so far in acute inflammatory disease models where CD73/adenosine executes tissue protective functions (203–208). Moreover, while CD73/adenosine had protective effects in acute renal disease models, chronic kidney injury in patients and murine studies was again linked to up-regulation of CD73 and ADORA2B (179, 198, 209, 210).
In summary, these studies point to disease-promoting effects of chronic CD73/adenosine signaling with tissue-specific and damage-specific mediators and immune changes.
Role of CD73 and Adenosine in the Control of Tumor Growth and Response to Therapy
Analyses of patient biopsies have shown that immune cell infiltrates in human tumors exhibit pronounced differences in cell types and numbers, not only intratumorally but also between patients and different tumor entities (211, 212). Interestingly, distribution and type of infiltrating immune cells turned out to have prognostic relevance; for example, the presence of infiltrating T cells was mostly linked to a favorable clinical outcome (213–218). Further pre-clinical and clinical studies showing that tumors can be strongly or poorly immunogenic supported these findings (31, 219). Moreover, the degree of immunogenicity positively correlated with reduced tumor growth and increased survival of tumor-bearing mice in response to immunotherapy indicating that the immune status can be seen as a predictive factor for therapy outcome (220).
High numbers of tumor-infiltrating cytotoxic lymphocytes were also predictive for the response of head and neck cancer patients to treatments involving radiotherapy whereas relapse after chemoradiotherapy and early recurrence correlated to infiltration with myeloid cells (217, 221–224). Local or systemic increases in Treg, high numbers of tumor associated macrophages, or recruitment of CD11b+ myeloid cells have also been associated with poor tumor response to radiotherapy and tumor relapse in pre-clinical models (88, 225). As a proof of concept for the synergistic interaction of radiotherapy and immunotherapy it has been shown that the combination with cancer vaccines, immune checkpoint blockade or inhibition of CD11b+ cell recruitment can improve the outcome of radiotherapy (52, 62, 89, 226–228).
Of note, tumors coopt the activities of the purinergic CD39/CD73/adenosine system to shape the immune landscape in the tumor microenvironment at multiple levels (Figure 1): For example, tumor cells and tumor-associated Treg use CD73-dependent adenosine generation to dampen intratumoral immune responses, particularly in hypoxic tumors (229, 230). The re-direction of the immune response involved suppression of T cell effector functions through CD73-dependent production of extracellular adenosine by CD39+/CD73+ Treg and signaling via stimulation of the ADORA2A on effector T cells (229). Adenosine and ADORA2A thus participate in shaping an immunosuppressive tumor microenvironment by negatively regulating CD8+ T cells (231–233). An adenosine-dependent suppression of immunosurveillance via IFN-γ, NK cells, and CD8+ T cells had also been demonstrated in other pre-clinical models (35, 162). Finally, the creation of an immunosuppressive tumor microenvironment involved the expansion of immunosuppressive myeloid cells, e.g., myeloid-derived suppressor cells, M2-like macrophages, and potentially N2-like neutrophils (234–236). More details about the various effects of CD73 and adenosine on cells from the innate and adaptive immune systems in the tumor microenvironment and the involved ADOR receptors can be found in the following reviews: (137, 143).
In addition, the CD73/adenosine system also supports tumor growth-promoting neovascularization, tumor metastasis, and chemotherapy resistance though part of these actions could also be attributed to the CD73/adenosine-induced modulation of immune cell types in the tumor microenvironment (36, 143, 229, 237–244).
For example, CD73−/− mice were strongly resistant to growth of subcutaneous MC38-ova colon and EG7 lymphoma tumors as well as carcinogen-induced or de novo growth of endogenous prostate tumors in transgenic TRAMP mice (162, 245, 246). These interesting observations pointed to a role of CD73+ host cells in tumor growth. However, CD73−/− mice were less resistant to growth of AT-3 mammary and B16F10 melanoma tumors revealing that the effect of host CD73 on the growth of experimental tumors also depends on the tumor type (245, 246). Of note, treatment with an anti-CD73 mAb reduced the growth of experimental 4T1.2 and E0771 breast tumors in wild-type mice, but not in severe combined immunodeficient (SCID) mice, suggesting a role of the adaptive immune system (245, 246). Anti-CD73 treatment also inhibited growth of carcinogen-induced fibrosarcoma tumors and of transgenic prostate tumors in transgenic TRAMP mice (162). The authors could further attribute the efficient tumor rejection to the action of CD8+ T cells whereas CD4+ T cells and NK cells were not involved (162, 246). These data highlight immunosuppressive CD73+ Treg as an important component of the tumor growth-promoting effects of CD73 and adenosine (162, 246).
Interestingly, CD73−/− mice also developed less lung metastases after intravenous injection of B16F10 or TRAMP-C1 cells (162, 246) suggesting that host CD73 also supports metastasis. In line with these observations treatment with an anti-CD73 mAb (TY/23) strongly reduced the lung metastases after injection of 4T1.2 or TRAMP-C1 tumor cells (162, 245). However, the suppression of metastasis formation was observed in both, immunocompetent and in SCID mice, and turned out to be independent of CD8+ T cells and NK cells (162, 245). Thereby the authors revealed a role of CD73+ non-hematopoietic host cells in metastasis formation, potentially endothelial cells, they could further link the pro-metastatic effect to signaling of tumor-derived extracellular adenosine via ADORA2B activation, at least in the 4T1.2 model (245, 246).
In further studies, tumor-derived adenosine attracted myeloid cells and promoted their differentiation into adenosine-generating tumor-associated macrophages (TAM) to amplify adenosine-dependent tumor-immune escape (247). In support of these findings, in vitro exposure to adenosine promoted alternative activation of macrophages and enhanced the immunosuppressive responses of macrophages to danger signals, particularly if stimulated in the presence of TLR ligands (141, 187). Interestingly, tumor-derived CD73-dependent adenosine promoted growth, neovascularization, and metastasis of subcutaneous B16F10 melanoma tumors and this was linked to infiltration and polarization of macrophages: genetic or pharmacologic inhibition of CD73 on the B16F10 melanoma cells significantly reduced the number of tumor-infiltrating macrophages recruited to subcutaneous B16F10 melanoma tumors on CD73−/− mice when compared to untreated B16F10 wildtype tumors on CD73−/− mice. Cytokine measurements in CD73+ B16F10 wildtype tumor lysates grown on CD73−/− mice revealed a down-regulation of pro-inflammatory cytokines [Granulocyte-macrophage colony-stimulating factor (GM-CSF) and IFN-γ] and enhanced expression of anti-inflammatory/pro-angiogenic cytokines (IL-4, IL-10, IL-13, M-CSF) (248). Although the number of infiltrating macrophages did not change in CD73+ B16F10 WT tumors on CD73−/− mice, less MMR+ macrophages were found inside the tumor. Only a pharmacological CD73 inhibition or knockdown of CD73 in the tumor host reduced the amount of infiltrating macrophages (248, 249). The results indicate a role for CD73 in activation and polarization of macrophages that promote tumor progression. Furthermore, it was shown, that the recruitment and activation of tumor-infiltrating macrophages was dependent on ADORA1, ADORA2A, and ADORA3 (250).
Taken together, CD73-dependent adenosine from host cells and tumor cells participates in the support of tumor growth amongst others by promoting tumor immune escape whereas loss of CD73/adenosine signaling enhances tumor immunity. As nicely summarized in a recent review from Allard et al. CD73/adenosine has become an attractive therapeutic target in (immuno)-oncology (38). Several early-phase clinical trials currently evaluate the therapeutic potential of CD73/adenosine inhibitors to inhibit tumor growth and increase tumor immunity. Besides the direct inhibition of CD73 the identification of the respective ADOR involved in promoting tumor immune escape will offer additional opportunities for therapeutic intervention (38, 251, 252).
Intriguingly, co-inhibition of adenosine signaling via CD73 and ADORA2A achieved better anti-tumor immune responses compared to single treatments, at least in pre-clinical models of breast and colon cancer (253). These effects were associated with improved immune cell infiltration, DC-priming and CD8+ T cell expansion. In line with these findings, Young et al. also observed increased tumor growth delay in CD73/ADORA2A double knockout mice (254). Furthermore, investigations with the human monoclonal anti-CD73 antibody MEDI9447 that is currently in Phase I clinical trials, showed high efficacy in inhibiting CD73 in vitro and potent inhibition of pre-clinical syngeneic tumor models in vivo as well as additive activity in combination with immune checkpoint inhibitors. Interestingly, MEDI9447 efficiently modulated the tumor microenvironment with significant alterations in the number of both, CD8+ effector T cells and activated macrophages (255).
The immunosuppressive actions of CD73 and adenosine in the microenvironment of established tumors also attract major attention as an interesting target for combined treatment approaches, particularly with immunotherapy. In this context, inhibition of CD73 enhanced efficacy of immunotherapy with α-PD-1 or α-CTLA4 in pre-clinical models (251, 256). The synergistic effect of the combined treatment involved improved T cell effector function as well as reduced CD73 expression on tumor-infiltrating lymphocytes and was dependent on interferon gamma (IFN-γ) and perforin (253, 256). Therapeutic inhibition of ADORA2A was also able to modulate expression of T cell co-inhibitory receptors and to improve effector function for enhanced efficacy of immune checkpoint blockade and adoptive cell therapy in murine cancer models (251, 252).
Since the focus of this review is to highlight the therapeutic potential of CD73 and adenosine inhibition to improve the therapeutic gain in radiotherapy we will not discuss such approaches in more detail here. For further information please refer to reviews discussing the therapeutic potential of the purinergic pathway in immunotherapy in more detail (38, 251, 252).
Instead the therapeutic potential of combining radiotherapy or radioimmunotherapy with CD73/adenosine-inhibition in cancer has been highlighted as an attractive approach but sound data are missing so far (41, 137). Pre-clinical studies are now underway to test such approaches including investigations in our own laboratory (257).
Several pre-clinical studies addressing the role of CD39 in cancer revealed that genetic deficiency of CD39 in mice promotes resistance to metastasis of melanoma and colorectal cancer models (258). Similarly, inhibition of angiogenesis in a CD39-deficient background resulted in reduced growth and pulmonary metastasis of LLC and B16F10 tumors (259). Expression of CD39 was important for angiogenesis and the suppression of NK cell-mediated antitumor activity (260, 261). In line with these findings, overexpression of CD39 enhanced the metastatic potential in pre-clinical models whereas the pharmacological inhibition of CD39 reduced metastasis and enhanced antitumor immunity (261). Of note, clinical data also support a correlation of high CD39 expression with poor prognosis indicating that CD39 might be another promising target for cancer therapy (262–264). But CD39 is much less investigated as a cancer target compared to CD73 underlining the need for further pre-clinical studies.
Taken together various pre-clinical studies highlight the potential of CD39/CD73/adenosine-signaling as promising therapeutic target in immuno-oncology. So far, the observed effects have been associated in multiple studies with activation of T-cell dependent tumor immunity. However, it is important to consider further immunoregulatory actions of CD73 and adenosine in the tumor microenvironment, particularly their influence on the biology of myeloid cells and macrophages, respectively.
Targeting CD73 in Lung Cancer
Only limited data are available so far of the role of CD73 and adenosine in lung cancer. Herein, CD73 was found to be expressed in tumor tissue from NSCLC patients on tumor cells, tumor-promoting mesenchymal stromal cells and myeloid-derived suppressor cells, respectively (265–267). Tumor-derived TGF-β stimulated CD39 and CD73 expression in CD11b+CD33+ MDSC in tumor tissues and peripheral blood of NSCLC patients, thereby inhibiting activity of T cells and NK cells and protecting tumor cells from the cytotoxic effect of chemotherapy (267).
Moreover, the prognostic value of high CD73 expression for the survival in lung cancer patients remains controversial: Although one study reported a correlation of high CD73 gene expression and improved overall survival of NSCLC patients (268) another study identified high CD73 protein expression as an independent prognostic marker for poor overall survival and shorter recurrence free survival in NSCLC (269). Interestingly, in the same study, high ADORA2A gene expression was an independent predictor of favorable prognosis for overall survival (269). Own in silico analyses of publicly available datasets for gene expression of CD73 in lung cancer confirmed the positive correlation between high CD73 gene expression and better overall survival of NSCLC patients. Of note, if radiotherapy-treated patients were excluded from the analysis the correlation to an improved overall survival was abrogated. In addition, the in silico analyses revealed poorer overall survival in lung cancer patients with high gene expression of ADORA1, ADORA2A, and ADORA2B (Figure 2). Again, the results about the prognostic value of ADORA2A using immunohistochemical data revealed opposite results (269, 270). The discrepancy in the above findings may be due to the use of gene expression analyses vs. immunohistochemical data as CD73 expression in tumor samples turned out to be highly heterogenous (269). We speculate that the heterogeneity in CD73 protein expression in distinct tumor areas might be linked to heterogeneous tumor oxygenation and make the acquisition of representative gene expression data challenging. So far, CD39 inhibitors are not yet involved in clinical trials for cancer patients but such studies are underway (259).
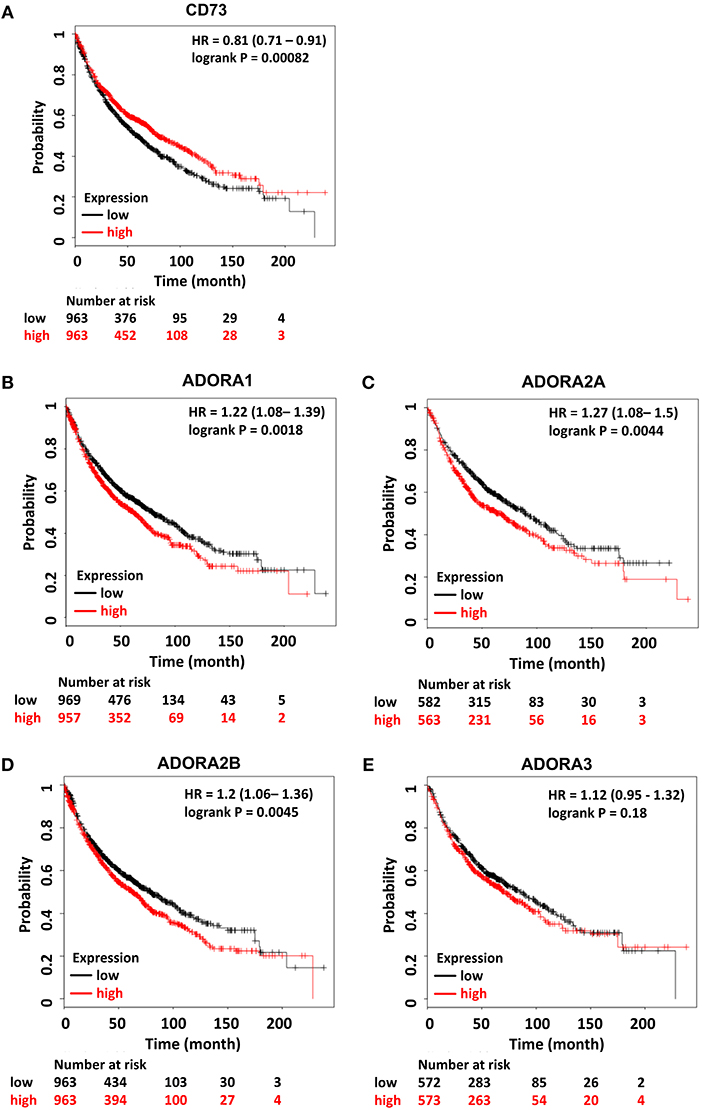
Figure 2. Prognostic relevance of components of the CD73/adenosine signaling system in lung cancer. Kaplan–Meier survival curves relative to (A) NT5E, (B) ADORA1, (C) ADORA2A, (D) ADORA2B, and (E) ADORA3 expression from publically available datasets for lung cancer. Data were analyzed using the KM-plotter tool (270). Red and black lines indicate patients with higher and lower gene expression, respectively. The total number of patients in the two categories are shown below the graph. Hazard ratios (HR) and p-values (log rank p) are shown inside the graph. Patient data is not restricted and includes all datasets.
Taken together, adenosine released in an inflammatory milieu or generated by the CD39/CD73 axis will impact the immune landscape of lung tumors presumably by limiting T cell immunity and promoting immunosuppressive and tumor-promoting lymphoid and myeloid immune cell phenotypes (Figure 1). We thus speculate that modulating CD73/adenosine signaling in the lung tumor microenvironment is an attractive strategy to limit tumor progression, improve antitumor immune responses, and avoid escape from therapy in combination with radiotherapy and potentially radioimmunotherapy. On the other hand, the pathologic role of the radiation-induced increase in CD73/adenosine signaling in promoting chronic inflammation and fibrosis in the normal lung tissue strongly suggest that pharmacologic inhibition of CD73/adenosine offers the opportunity for widening the therapeutic window by reducing radiation-induced lung toxicity, particularly in CD73-rich thoracic tumors with a high risk for CD73-dependent normal tissue toxicity.
Targeting the CD73/adenosine pathway or the involved receptors may thus provide a clear therapeutic gain in the treatment of lung cancer and other CD73/adenosine-rich thorax-associated neoplasms: we expect that inhibition of CD73/adenosine signaling will limit lung toxicity during thoracic irradiation without protecting the tumor or even reinstall anti-tumor immunity when applied during therapeutic irradiation of adenosine-rich tumors with high radioresistance such as NSCLC (138). However, a tight regulation of pro- and anti-inflammatory actions of resident and immune cells is necessary to protect the lung from inflammation-induced loss in its vital function (271–273). For example, immunosuppressive Treg are also to be part of a protective response limiting inflammation-induced collateral normal tissue damage after radiotherapy (44, 274). Therefore, pharmacologic strategies targeting the CD73/adenosine pathway in combination with radiotherapy or combined radioimmunotherapy will require careful validation of potential normal tissue complications. Such complications might include excessive inflammation or autoimmunity by abrogating protective signals mediated by various ADORAs, particularly during acute disease stages. Moreover, the dual effects of acute and chronic CD73 activation as well as spatiotemporal heterogeneity of CD73 and ADOR expression in normal and tumor tissues need to be considered when designing combination treatments for therapeutic intervention.
Current Research and Future Perspectives
So far work from our group identified CD73/adenosine signaling as a novel mechanism promoting RILD through local and systemic actions. Consequences of pathologic CD73/adenosine signaling involved amongst others the accumulation and/or alternative activation of macrophages in organized clusters, their expression of pro-fibrotic mediators, or both. We speculate that the radiation-induced increase in CD73/adenosine is necessary to amplify pro-fibrotic signaling in the irradiated lung environment by fueling the multifaceted cross-talk between damaged resident cells, local and infiltrating immune cells, immunosuppressive Treg and other pro-fibrotic mediators such as hyaluronic acid and TGF-β.
Though immunomodulatory effects of adenosine had been linked to CD73/adenosine-induced adverse effects in other injury models (136, 202) the tissue specific effector and target cells of CD73/adenosine-signaling in response to genotoxic treatment (BLM, radiotherapy) are still controversial and need to be further investigated (73, 200). In this context radiation-induced normal tissue toxicity had also been linked to endothelial cell damage and dysfunction as well as endothelial cell loss as long-term complication (275, 276). As a direct consequence of impaired vascular function, WTI increased numbers of total CD45+ leukocytes, particularly profibrotic CD11b+ myeloid cells and Ly6C+ inflammatory monocytes, in lungs of irradiated mice. However, on the long term, persistence of an activated pro-coagulant endothelial cell type, thickening of the basement membrane, endothelial loss, and collapse of microvessels will contribute to the creation of a hypoxic, pro-inflammatory disease-promoting environment. We assume that the pathologic environment involves a hypoxia-induced up-regulation of CD73 and pathology-associated ADOR on resident cells and immune cells. It is tempting to speculate that therapeutic inhibition of CD73 might also impact adverse late effects in the lung by reducing radiation-induced vascular impairment, but this remains to be determined. Interestingly, further work demonstrates that locally irradiated MSC play a role in the pathogenesis of radiotherapy-induced pulmonary fibrosis by acquiring a pro-fibrotic myofibroblast-like phenotype that promotes extracellular matrix deposition, tissue remodeling, and the development of pulmonary fibrosis upon WTI (276). Since CD73 is expressed on endothelial cells and on MSC of healthy lungs (153) future studies should explore whether the expression of CD73 on the surface of endothelial cells or resident MSC impacts the development of RILD. The same holds true for the expression of CD39 and CD73 on cancer exosomes, which have also been shown to suppress T cells through adenosine production (239).
Adenosine released in an inflammatory milieu or generated by the CD39/CD73 axis impacts the tumor microenvironment and limits tumor immunity at multiple levels. Thus, modulating cancer-derived adenosine in the tumor microenvironment emerges as an attractive strategy to limit tumor progression and improve antitumor immune responses and our own studies suggest that this might be possible without excessively increasing late normal tissue complications (36, 187, 242, 243, 277). Fortunately, multiple approaches for pharmacologic modulation of adenosine levels exist or are being developed and multiple clinical studies have been initiated to evaluate the use of novel inhibitors of CD73 or ADORA2A signaling in cancer therapy alone and in combination with immune checkpoint blockade (38, 39, 143, 188). These studies will give insight into efficacy, compatibility, and potential side effects.
Herein, major attention in oncology has so far been attributed to adenosine signaling via ADORA2A as it is known to effectively dampen immune responses in tumors and normal tissues. However, it has to be taken into account that depending on the tissue of origin and the molecular and immune signature of the tumor, other ADOR may be more important. Moreover, the role of purinergic signaling in the radiation response of malignant tumors and the potential of CD73 or ADOR inhibitors to enhance the efficacy of RT alone and in combination with immunotherapy is still largely unknown. Finally, no reliable biomarkers for the prediction or diagnosis for the individual risk of RILD upon treatment are available to date. Thus, further studies are needed that correlate the gene and protein expression of CD73 and the ADORAs to the outcome after radio(chemo)therapy or immunotherapy. Moreover, as mentioned before, the receptors differ in their affinity for adenosine and extracellular adenosine levels will vary depending on the tissue, the treatment modality and intensity in a spatiotemporal manner. It would therefore be highly beneficial to perform an immunoscore of tissues from pre-clinical studies and test association of high or low expression of CD73 and the ADORAs with the presence of immunosuppressive lymphoid and myeloid cell subsets, and potentially tissue hypoxia. Such knowledge could later be translated into patient samples. Here, it was an intriguing observation that a high expression of CD73 in normal tissues was indicative for a poor infiltration of prostate tumors with CD8+ T cells whereas high CD73 expression in the tumor stroma was indicative for a longer recurrence-free survival (278). This highlights that CD73 expression in both, normal and tumor tissue should be evaluated.
Final Remarks
Nowadays it is increasingly recognized that strategies for a biology-based optimization and individualization of radiotherapy should include not only the available knowledge about tumor promoting mutations, tumor heterogeneity, tumor cell plasticity, and unfavorable gene expression profiles indicative of the individual radiosensitivity of tumor and normal tissues, but also consider knowledge about the modulation of the radiation response by the immune system and vice-versa. Such a comprehensive view shall allow to harness the combined potential of high precision local radiotherapy, cytotoxic chemotherapy, molecularly targeted small molecule signal transduction inhibitors, and immunotherapy approaches for biologically optimized therapeutic strategies with acceptable safety profile and durable responses in the future (22, 30, 41, 279–284).
The observation that radiotherapy can help to reactivate anti-tumor immunity in immunogenic tumors or increase the potential of immunotherapy has attracted major attention to the use of radiotherapy in combination with various immunotherapies, particularly immune checkpoint blockade immunostimulatory antibodies, and cancer vaccines (24–26, 28–30, 62, 67). However, tumors have evolved effective strategies to escape from immune surveillance and therapy-induced enhancement of tumor immunity is balanced by feed-back inhibition of immune activation in residual tumors, the mobilization of tissue regeneration mechanisms with tumor promoting actions, or both (41, 88, 89, 285–287).
We believe that the identification of mediators driving both, adverse immune changes in irradiated normal tissues and tumor immune escape, will allow us to uncover attractive new therapeutic targets for improving the outcome of radiotherapy. The CD73/adenosine pathway is such a signaling system that regulates adverse immune responses in tumors and normal tissues to microenvironmental stress (e.g., tumor hypoxia) and radiotherapy. So far, CD73/adenosine is mostly considered as a metabolic immune checkpoint that supports immunosuppressive signaling of Treg via ADORA2A. However, there is evidence that CD39, CD73 and adenosine are involved in further immunosuppressive and tumor-promoting signals in the tumor microenvironment beyond modulating Treg function. Intriguingly, radiochemotherapy was also shown to trigger up-regulation of CD73 and CD39 in circulating immune cells of cancer patients (288). This suggests that a radiotherapy-induced systemic upregulation of CD73/adenosine signaling may additionally dampen systemic anti-tumor immune responses during standard fractionated radiotherapy.
Thus, pharmacologic inhibition of CD73/adenosine signaling is an attractive approach to increase the therapeutic ratio in the RT of thoracic tumors with high risk of adverse late effects in the highly radiosensitive normal lung tissue by (i) dampening growth and metastasis of lung tumors, (ii) enhancing the radiation-induced activation of the antitumor immune response, (iii) by restricting the immunosuppressive action of CD39/CD73 on circulating immune cells, and (iv) attenuating adverse late effects in the lung. Moreover, pharmacologic modulation of CD73, adenosine or the four adenosine receptors might offer opportunities to enhance the potential of combined radioimmunotherapy to mount efficient and durable responses with acceptable safety profile.
But the complexity of the tumor-induced and radiation-induced changes in the microenvironment and the multifaceted interactions between damaged resident cells and recruited immune cells outlined above underline the necessity of further work suited to identify strategies that achieve the required balance between pro-immunogenic and immunosuppressive effects of radiotherapy and outweigh the beneficial effects of radioimmunotherapy with optimal tumor control and normal tissue protection. Moreover, further work is required to gain a better mechanistic understanding of the tissue-, injury-, and disease stage-dependent beneficial or adverse effects of CD73/adenosine as well as the identification of involved ADORAs and effector cells for a successful restriction of lung damage during therapeutic lung irradiation by targeting CD73, adenosine or specific ADORAs (73).
Finally, it remains to be determined which approach for targeting the CD73/adenosine axis might be best suited to be used in combination with RT. Above all, the immune effects of RT also depend on physical parameters such as total dose, fractionation schemes (43, 57, 225, 289) and potentially the quality of radiation (290). Thus, attention has also to be given to the best sequence of application, as well as appropriate radiation doses and fractionation-schemes as they may largely impact the effects of radiotherapy on microvessels, immunogenic cell death, immune cell infiltration, the production of immune modulatory mediators, and the activation of CD73/adenosine signaling in both, normal and tumor tissues. Here, the major challenge will be to therapeutically redirect the immune response toward anti-tumor action and avoid tumor recurrence without enhancing collateral normal tissue damage.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The work was supported by grants of the DFG (GRK1739/2; JE275/1) and the BMBF (ZISStrans 02NUK047D). We acknowledge support by the Open Access Publication Fund of the University of Duisburg-Essen.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADA, adenosine deaminase; ADOR, adenosine receptor; α-SMA, alpha smooth muscle actin; BLM, Bleomycin; Breg, regulatory B cells; CD39, ectoapyrase, ectonucleoside triphosphate diphosphohydrolase 1; CD73, 5′ ectonucleotidase; CEACAM1, carcinoembryonic antigen-related cell adhesion molecule 1; cGAS/STING, cyclic GMP-AMP Synthase/Stimulator of Interferon Genes; CSF-1, colony-stimulating factor 1; CTGF, connective tissue growth factor; CTLA-4, cytotoxic T-lymphocyte-associated Protein 4; DAMPs, damage associated molecular patterns; ENPP1, ectonucleotidepyrophosphatase (phosphodiesterase 1); ENT, equilibrative nucleoside transporter; Gy, Gray; HMGB1, high-mobility-group-protein B1; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon gamma; IL, Interleukin; mAb, monoclonal antibody; MDSC, myeloid-derived suppressor cells; MMR, macrophage mannose receptor; MSC, mesenchymal stem cell; NSCLC, non-small cell lung cancer; PD-1, programmed cell death 1; PD-L1, programmed death-ligand 1; PEG-ADA, pegylated ADA; PDGF, platelet-Derived Growth Factor; RILD, radiation-induced lung disease; ROS, reactive oxygen species; RT, Radiotherapy; SCID, severe combined immunodeficiency; TAM, tumor-associated macrophages; TGF-β, transforming growth factor beta; TLR, toll-like receptor; Treg, regulatory T cells; VEGF, vascular Endothelial Growth Factor; WTI, whole thorax irradiation.
References
1. Bartelink H, Roelofsen F, Eschwege F, Rougier P, Bosset JF, Gonzalez DG, et al. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastrointestinal Cooperative Groups. J Clin Oncol. (1997) 15:2040–9. doi: 10.1200/JCO.1997.15.5.2040
2. Green JA, Kirwan JM, Tierney JF, Symonds P, Fresco L, Collingwood M, et al. Survival and recurrence after concomitant chemotherapy and radiotherapy for cancer of the uterine cervix: a systematic review and meta-analysis. Lancet. (2001) 358:781–6. doi: 10.1016/S0140-6736(01)05965-7
3. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
4. Bosset JF, Collette L, Calais G, Mineur L, Maingon P, Radosevic-Jelic L, et al. Chemotherapy with preoperative radiotherapy in rectal cancer. N Engl J Med. (2006) 355:1114–23. doi: 10.1056/NEJMoa060829
5. Blanchard P, Baujat B, Holostenco V, Bourredjem A, Baey C, Bourhis J, et al. Meta-analysis of chemotherapy in head and neck cancer (MACH-NC): a comprehensive analysis by tumour site. Radiother Oncol. (2011) 100:33–40. doi: 10.1016/j.radonc.2011.05.036
6. Orth M, Lauber K, Niyazi M, Friedl AA, Li M, Maihofer C, et al. Current concepts in clinical radiation oncology. Radiat Environ Biophys. (2014) 53:1–29. doi: 10.1007/s00411-013-0497-2
7. Curran WJ Jr., Paulus R, Langer CJ, Komaki R, Lee JS, Hauser S, et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst. (2011) 103:1452–60. doi: 10.1093/jnci/djr325
8. Bradley JD, Paulus R, Komaki R, Masters G, Blumenschein G, Schild S, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. (2015) 16:187–99. doi: 10.1016/S1470-2045(14)71207-0
9. Ohri N. Radiotherapy dosing for locally advanced non-small cell lung carcinoma: “MTD” or “ALARA”? Front Oncol. (2017) 7:205. doi: 10.3389/fonc.2017.00205
10. Auperin A, Le Pechoux C, Rolland E, Curran WJ, Furuse K, Fournel P, et al. Meta-analysis of concomitant versus sequential radiochemotherapy in locally advanced non-small-cell lung cancer. J Clin Oncol. (2010) 28:2181–90. doi: 10.1200/JCO.2009.26.2543
11. Pfeifer GP. How the environment shapes cancer genomes. Curr Opin Oncol. (2015) 27:71–7. doi: 10.1097/CCO.0000000000000152
12. Shirvani SM, Jiang J, Gomez DR, Chang JY, Buchholz TA, Smith BD. Intensity modulated radiotherapy for stage III non-small cell lung cancer in the United States: predictors of use and association with toxicities. Lung Cancer. (2013) 82:252–9. doi: 10.1016/j.lungcan.2013.08.015
13. Chun SG, Hu C, Choy H, Komaki RU, Timmerman RD, Schild SE, et al. Impact of intensity-modulated radiation therapy technique for locally advanced non-small-cell lung cancer: a secondary analysis of the NRG oncology RTOG 0617 randomized clinical trial. J Clin Oncol. (2017) 35:56–62. doi: 10.1200/JCO.2016.69.1378
14. Higgins KA, O'Connell K, Liu Y, Gillespie TW, McDonald MW, Pillai RN, et al. National cancer database analysis of proton versus photon radiation therapy in non-small cell lung cancer. Int J Radiat Oncol Biol Phys. (2017) 97:128–37. doi: 10.1016/j.ijrobp.2016.10.001
15. Kelsey CR, Horwitz ME, Chino JP, Craciunescu O, Steffey B, Folz RJ, et al. Severe pulmonary toxicity after myeloablative conditioning using total body irradiation: an assessment of risk factors. Int J Radiat Oncol Biol Phys. (2011) 81:812–8. doi: 10.1016/j.ijrobp.2010.06.058
16. Farr KP, Khalil AA, Knap MM, Moller DS, Grau C. Development of radiation pneumopathy and generalised radiological changes after radiotherapy are independent negative prognostic factors for survival in non-small cell lung cancer patients. Radiother Oncol. (2013) 107:382–8. doi: 10.1016/j.radonc.2013.04.024
17. Jackson IL, Baye F, Goswami CP, Katz BP, Zodda A, Pavlovic R, et al. Gene expression profiles among murine strains segregate with distinct differences in the progression of radiation-induced lung disease. Dis Model Mech. (2017) 10:425–37. doi: 10.1242/dmm.028217
18. Seiwert TY, Haraf DJ, Cohen EE, Stenson K, Witt ME, Dekker A, et al. Phase I study of bevacizumab added to fluorouracil- and hydroxyurea-based concomitant chemoradiotherapy for poor-prognosis head and neck cancer. J Clin Oncol. (2008) 26:1732–41. doi: 10.1200/JCO.2007.13.1706
19. Spigel DR, Hainsworth JD, Yardley DA, Raefsky E, Patton J, Peacock N, et al. Tracheoesophageal fistula formation in patients with lung cancer treated with chemoradiation and bevacizumab. J Clin Oncol. (2010) 28:43–8. doi: 10.1200/JCO.2009.24.7353
20. Niyazi M, Maihoefer C, Krause M, Rodel C, Budach W, Belka C. Radiotherapy and “new” drugs-new side effects? Radiat Oncol. (2011) 6:177. doi: 10.1186/1748-717X-6-177
21. Mangoni M, Vozenin MC, Biti G, Deutsch E. Normal tissues toxicities triggered by combined anti-angiogenic and radiation therapies: hurdles might be ahead. Br J Cancer. (2012) 107:308–14. doi: 10.1038/bjc.2012.236
22. Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. (2009) 10:718–26. doi: 10.1016/S1470-2045(09)70082-8
23. Liang H, Deng L, Chmura S, Burnette B, Liadis N, Darga T, et al. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J Immunol. (2013) 190:5874–81. doi: 10.4049/jimmunol.1202612
24. Crittenden M, Kohrt H, Levy R, Jones J, Camphausen K, Dicker A, et al. Current clinical trials testing combinations of immunotherapy and radiation. Semin Radiat Oncol. (2015) 25:54–64. doi: 10.1016/j.semradonc.2014.07.003
25. Sharabi AB, Lim M, DeWeese TL, Drake CG. Radiation and checkpoint blockade immunotherapy: radiosensitisation and potential mechanisms of synergy. Lancet Oncol. (2015) 16:e498–509. doi: 10.1016/S1470-2045(15)00007-8
26. Vacchelli E, Bloy N, Aranda F, Buque A, Cremer I, Demaria S, et al. Trial watch: immunotherapy plus radiation therapy for oncological indications. Oncoimmunology. (2016) 5:e1214790. doi: 10.1080/2162402X.2016.1214790
27. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin. (2017) 67:65–85. doi: 10.3322/caac.21358
28. Cadena A, Cushman TR, Anderson C, Barsoumian HB, Welsh JW, Cortez MA. Radiation and anti-cancer vaccines: a winning combination. Vaccines. (2018) 6:E9. doi: 10.3390/vaccines6010009
29. Rodriguez-Ruiz ME, Vanpouille-Box C, Melero I, Formenti SC, Demaria S. Immunological mechanisms responsible for radiation-induced abscopal effect. Trends Immunol. (2018) 39:644–55. doi: 10.1016/j.it.2018.06.001
30. Vanpouille-Box C, Formenti SC, Demaria S. Toward precision radiotherapy for use with immune checkpoint blockers. Clin Cancer Res. (2018) 24:259–65. doi: 10.1158/1078-0432.CCR-16-0037
31. Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. (2015) 42:663–71. doi: 10.1053/j.seminoncol.2015.05.011
32. Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. (2016) 2:286–94. doi: 10.1016/j.trecan.2016.05.002
33. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
34. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
35. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. (2010) 70:2245–55. doi: 10.1158/0008-5472.CAN-09-3109
36. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
37. Antonioli L, Blandizzi C, Malavasi F, Ferrari D, Hasko G. Anti-CD73 immunotherapy: a viable way to reprogram the tumor microenvironment. Oncoimmunology. (2016) 5:e1216292. doi: 10.1080/2162402X.2016.1216292
38. Allard D, Chrobak P, Allard B, Messaoudi N, Stagg J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol Lett. (2018) 205:31–9. doi: 10.1016/j.imlet.2018.05.001
39. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
40. Belka C, Ottinger H, Kreuzfelder E, Weinmann M, Lindemann M, Lepple-Wienhues A, et al. Impact of localized radiotherapy on blood immune cells counts and function in humans. Radiother Oncol. (1999) 50:199–204. doi: 10.1016/S0167-8140(98)00130-3
41. Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones KA, Garcia-Martinez E, Rudqvist NP, et al. Barriers to radiation-induced in situ tumor vaccination. Front Immunol. (2017) 8:229. doi: 10.3389/fimmu.2017.00229
42. Mouw KW, Konstantinopoulos PA. From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer. (2018) 118:933–5. doi: 10.1038/s41416-018-0017-x
43. Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, et al. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. (2012) 189:558–66. doi: 10.4049/jimmunol.1200563
44. Wirsdorfer F, Cappuccini F, Niazman M, de Leve S, Westendorf AM, Ludemann L, et al. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat Oncol. (2014) 9:98. doi: 10.1186/1748-717X-9-98
45. Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. (1953) 26:234–41. doi: 10.1259/0007-1285-26-305-234
46. Vacchelli E, Vitale I, Tartour E, Eggermont A, Sautes-Fridman C, Galon J, et al. Trial watch: anticancer radioimmunotherapy. Oncoimmunology. (2013) 2:e25595. doi: 10.4161/onci.25595
47. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. (2014) 41:843–52. doi: 10.1016/j.immuni.2014.10.019
48. Derer A, Frey B, Fietkau R, Gaipl US. Immune-modulating properties of ionizing radiation: rationale for the treatment of cancer by combination radiotherapy and immune checkpoint inhibitors. Cancer Immunol Immunother. (2016) 65:779–86. doi: 10.1007/s00262-015-1771-8
49. Brix N, Tiefenthaller A, Anders H, Belka C, Lauber K. Abscopal, immunological effects of radiotherapy: narrowing the gap between clinical and preclinical experiences. Immunol Rev. (2017) 280:249–79. doi: 10.1111/imr.12573
50. Schaue D. A century of radiation therapy and adaptive immunity. Front Immunol. (2017) 8:431. doi: 10.3389/fimmu.2017.00431
51. Ganss R, Ryschich E, Klar E, Arnold B, Hammerling GJ. Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. (2002) 62:1462–70.
52. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. (2005) 11:728–34.
53. Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. (2009) 114:589–95. doi: 10.1182/blood-2009-02-206870
54. Filatenkov A, Baker J, Mueller AM, Kenkel J, Ahn GO, Dutt S, et al. Ablative tumor radiation can change the tumor immune cell microenvironment to induce durable complete remissions. Clin Cancer Res. (2015) 21:3727–39. doi: 10.1158/1078-0432.CCR-14-2824
55. Galluzzi L, Zitvogel L, Kroemer G. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol Res. (2016) 4:895–902. doi: 10.1158/2326-6066.CIR-16-0197
56. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. (2016) 16:759–73. doi: 10.1038/nrc.2016.91
57. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. (2017) 8:15618. doi: 10.1038/ncomms15618
58. Kingsley DP. An interesting case of possible abscopal effect in malignant melanoma. Br J Radiol. (1975) 48:863–6. doi: 10.1259/0007-1285-48-574-863
59. Ohba K, Omagari K, Nakamura T, Ikuno N, Saeki S, Matsuo I, et al. Abscopal regression of hepatocellular carcinoma after radiotherapy for bone metastasis. Gut. (1998) 43:575–7. doi: 10.1136/gut.43.4.575
60. Wersall PJ, Blomgren H, Pisa P, Lax I, Kalkner KM, Svedman C. Regression of non-irradiated metastases after extracranial stereotactic radiotherapy in metastatic renal cell carcinoma. Acta Oncol. (2006) 45:493–7. doi: 10.1080/02841860600604611
61. Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. (2016) 40:25–37. doi: 10.1016/j.currproblcancer.2015.10.001
62. Frey B, Rubner Y, Kulzer L, Werthmoller N, Weiss EM, Fietkau R, et al. Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol Immunother. (2014) 63:29–36. doi: 10.1007/s00262-013-1474-y
63. Reynders K, Illidge T, Siva S, Chang JY, De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. (2015) 41:503–10. doi: 10.1016/j.ctrv.2015.03.011
64. Frey B, Ruckert M, Deloch L, Ruhle PF, Derer A, Fietkau R, et al. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol Rev. (2017) 280:231–48. doi: 10.1111/imr.12572
65. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. (2017) 377:1919–29. doi: 10.1056/NEJMoa1709937
66. Shaverdian N, Lisberg AE, Bornazyan K, Veruttipong D, Goldman JW, Formenti SC, et al. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: a secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. (2017) 18:895–903. doi: 10.1016/S1470-2045(17)30380-7
67. Ngwa W, Irabor OC, Schoenfeld JD, Hesser J, Demaria S, Formenti SC. Using immunotherapy to boost the abscopal effect. Nat Rev Cancer. (2018) 18:313–22. doi: 10.1038/nrc.2018.6
68. Westermann W, Schobl R, Rieber EP, Frank KH. Th2 cells as effectors in postirradiation pulmonary damage preceding fibrosis in the rat. Int J Radiat Biol. (1999) 75:629–38. doi: 10.1080/095530099140276
69. Paun A, Fox J, Balloy V, Chignard M, Qureshi ST, Haston CK. Combined Tlr2 and Tlr4 deficiency increases radiation-induced pulmonary fibrosis in mice. Int J Radiat Oncol Biol Phys. (2010) 77:1198–205. doi: 10.1016/j.ijrobp.2009.12.065
70. Brickey WJ, Neuringer IP, Walton W, Hua X, Wang EY, Jha S, et al. MyD88 provides a protective role in long-term radiation-induced lung injury. Int J Radiat Biol. (2012) 88:335–47. doi: 10.3109/09553002.2012.652723
71. Schaue D, McBride WH. T lymphocytes and normal tissue responses to radiation. Front Oncol. (2012) 2:119. doi: 10.3389/fonc.2012.00119
72. Jackson IL, Zhang Y, Bentzen SM, Hu J, Zhang A, Vujaskovic Z. Pathophysiological mechanisms underlying phenotypic differences in pulmonary radioresponse. Sci Rep. (2016) 6:36579. doi: 10.1038/srep36579
73. Wirsdorfer F, Jendrossek V. Modeling DNA damage-induced pneumopathy in mice: insight from danger signaling cascades. Radiat Oncol. (2017) 12:142. doi: 10.1186/s13014-017-0865-1
74. Wiesemann A, Ketteler J, Slama A, Wirsdorfer F, Hager T, Roeck K, et al. Inhibition of radiation-induced Ccl2 signaling protects lungs from vascular dysfunction and endothelial cell loss. Antioxid Redox Signal. (2018) 30:213–31. doi: 10.1089/ars.2017.7458
75. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. (2006) 6:24–37. doi: 10.1038/nrc1782
76. Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. (2015) 12:527–40. doi: 10.1038/nrclinonc.2015.120
77. Jeong H, Bok S, Hong BJ, Choi HS, Ahn GO. Radiation-induced immune responses: mechanisms and therapeutic perspectives. Blood Res. (2016) 51:157–63. doi: 10.5045/br.2016.51.3.157
78. Ma Y, Yang H, Pitt JM, Kroemer G, Zitvogel L. Therapy-induced microenvironmental changes in cancer. J Mol Med. (2016) 94:497–508. doi: 10.1007/s00109-016-1401-8
79. McKelvey KJ, Hudson AL, Back M, Eade T, Diakos CI. Radiation, inflammation and the immune response in cancer. Mamm Genome. (2018) 29:843–65. doi: 10.1007/s00335-018-9777-0
80. Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol. (2012) 2:88. doi: 10.3389/fonc.2012.00088
81. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
82. Tsou P, Katayama H, Ostrin EJ, Hanash SM. The emerging role of B cells in tumor immunity. Cancer Res. (2016) 76:5597–601. doi: 10.1158/0008-5472.CAN-16-0431
83. Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, et al. TGFbeta is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. (2015) 75:2232–42. doi: 10.1158/0008-5472.CAN-14-3511
84. de Visser KE, Coussens LM. The interplay between innate and adaptive immunity regulates cancer development. Cancer Immunol Immunother. (2005) 54:1143–52. doi: 10.1007/s00262-005-0702-5
85. de Visser KE, Coussens LM. The inflammatory tumor microenvironment and its impact on cancer development. Contrib Microbiol. (2006) 13:118–37. doi: 10.1159/000092969
86. Barbera-Guillem E, May KF Jr., Nyhus JK, Nelson MB. Promotion of tumor invasion by cooperation of granulocytes and macrophages activated by anti-tumor antibodies. Neoplasia. (1999) 1:453–60. doi: 10.1038/sj.neo.7900054
87. Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer. (2016) 2:747–57. doi: 10.1016/j.trecan.2016.10.010
88. Ahn GO, Brown JM. Matrix metalloproteinase-9 is required for tumor vasculogenesis but not for angiogenesis: role of bone marrow-derived myelomonocytic cells. Cancer Cell. (2008) 13:193–205. doi: 10.1016/j.ccr.2007.11.032
89. Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci USA. (2010) 107:8363–8. doi: 10.1073/pnas.0911378107
90. Hagerling C, Casbon AJ, Werb Z. Balancing the innate immune system in tumor development. Trends Cell Biol. (2015) 25:214–20. doi: 10.1016/j.tcb.2014.11.001
91. Nouvion AL, Beauchemin N. [CEACAM1 as a central modulator of metabolism, tumor progression, angiogenesis and immunity]. Med Sci. (2009) 25:247–52. doi: 10.1051/medsci/2009253247
92. Gao ZW, Dong K, Zhang HZ. The roles of CD73 in cancer. Biomed Res Int. (2014) 2014:460654. doi: 10.1155/2014/460654
93. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) (Suppl. 35):S185–98. doi: 10.1016/j.semcancer.2015.03.004
94. Qin A, Coffey DG, Warren EH, Ramnath N. Mechanisms of immune evasion and current status of checkpoint inhibitors in non-small cell lung cancer. Cancer Med. (2016) 5:2567–78. doi: 10.1002/cam4.819
95. Lhuillier C, Vanpouille-Box C, Galluzzi L, Formenti SC, Demaria S. Emerging biomarkers for the combination of radiotherapy and immune checkpoint blockers. Semin Cancer Biol. (2018) 52 (Pt 2):125–34. doi: 10.1016/j.semcancer.2017.12.007
96. Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res. (2018) 6:139–50. doi: 10.1158/2326-6066.CIR-17-0134
97. Lumniczky K, Safrany G. The impact of radiation therapy on the antitumor immunity: local effects and systemic consequences. Cancer Lett. (2015) 356:114–25. doi: 10.1016/j.canlet.2013.08.024
98. Mavragani IV, Laskaratou DA, Frey B, Candeias SM, Gaipl US, Lumniczky K, et al. Key mechanisms involved in ionizing radiation-induced systemic effects. A current review Toxicol Res. (2016) 5:12–33. doi: 10.1039/c5tx00222b
99. Jelonek K, Pietrowska M, Widlak P. Systemic effects of ionizing radiation at the proteome and metabolome levels in the blood of cancer patients treated with radiotherapy: the influence of inflammation and radiation toxicity. Int J Radiat Biol. (2017) 93:683–96. doi: 10.1080/09553002.2017.1304590
100. Down JD, Steel GG. The expression of early and late damage after thoracic irradiation: a comparison between CBA and C57B1 mice. Radiat Res. (1983) 96:603–10. doi: 10.2307/3576125
101. Johnston CJ, Williams JP, Elder A, Hernady E, Finkelstein JN. Inflammatory cell recruitment following thoracic irradiation. Exp Lung Res. (2004) 30:369–82. doi: 10.1080/01902140490438915
102. Chiang CS, Liu WC, Jung SM, Chen FH, Wu CR, McBride WH, et al. Compartmental responses after thoracic irradiation of mice: strain differences. Int J Radiat Oncol Biol Phys. (2005) 62:862–71. doi: 10.1016/j.ijrobp.2005.02.037
103. O'Brien TJ, Letuve S, Haston CK. Radiation-induced strain differences in mouse alveolar inflammatory cell apoptosis. Can J Physiol Pharmacol. (2005) 83:117–22. doi: 10.1139/y05-005
104. Paun A, Kunwar A, Haston CK. Acute adaptive immune response correlates with late radiation-induced pulmonary fibrosis in mice. Radiat Oncol. (2015) 10:45. doi: 10.1186/s13014-015-0359-y
105. Schaue D, Micewicz ED, Ratikan JA, Xie MW, Cheng G, McBride WH. Radiation and inflammation. Semin Radiat Oncol. (2015) 25:4–10. doi: 10.1016/j.semradonc.2014.07.007
106. Kainthola A, Haritwal T, Tiwari M, Gupta N, Parvez S, Tiwari M, et al. Immunological aspect of radiation-induced pneumonitis, current treatment strategies, and future prospects. Front Immunol. (2017) 8:506. doi: 10.3389/fimmu.2017.00506
107. Ratikan JA, Micewicz ED, Xie MW, Schaue D. Radiation takes its Toll. Cancer Lett. (2015) 368:238–45. doi: 10.1016/j.canlet.2015.03.031
108. Hekim N, Cetin Z, Nikitaki Z, Cort A, Saygili EI. Radiation triggering immune response and inflammation. Cancer Lett. (2015) 368:156–63. doi: 10.1016/j.canlet.2015.04.016
109. Venereau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. (2015) 6:422. doi: 10.3389/fimmu.2015.00422
110. Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. (2010) 207:1807–17. doi: 10.1084/jem.20101157
111. Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, et al. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ Res. (2007) 100:204–12. doi: 10.1161/01.RES.0000257774.55970.f4
112. Ranzato E, Patrone M, Pedrazzi M, Burlando B. HMGb1 promotes scratch wound closure of HaCaT keratinocytes via ERK1/2 activation. Mol Cell Biochem. (2009) 332:199–205. doi: 10.1007/s11010-009-0192-4
113. Hill LM, Gavala ML, Lenertz LY, Bertics PJ. Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J Immunol. (2010) 185:3028–34. doi: 10.4049/jimmunol.1001298
114. Lin Q, Yang XP, Fang D, Ren X, Zhou H, Fang J, et al. High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol. (2011) 31:1024–32. doi: 10.1161/ATVBAHA.111.224048
115. van Beijnum JR, Nowak-Sliwinska P, van den Boezem E, Hautvast P, Buurman WA, Griffioen AW. Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene. (2013) 32:363–74. doi: 10.1038/onc.2012.49
116. Jin H, Seo J, Eun SY, Joo YN, Park SW, Lee JH, et al. P2Y2 R activation by nucleotides promotes skin wound-healing process. Exp Dermatol. (2014) 23:480–5. doi: 10.1111/exd.12440
117. Nakagawa S, Omura T, Yonezawa A, Yano I, Nakagawa T, Matsubara K. Extracellular nucleotides from dying cells act as molecular signals to promote wound repair in renal tubular injury. Am J Physiol Renal Physiol. (2014) 307:F1404–11. doi: 10.1152/ajprenal.00196.2014
118. Eisenbacher JL, Schrezenmeier H, Jahrsdorfer B, Kaltenmeier C, Rojewski MT, Yildiz T, et al. S100A4 and uric acid promote mesenchymal stromal cell induction of IL-10+/IDO+ lymphocytes. J Immunol. (2014) 192:6102–10. doi: 10.4049/jimmunol.1303144
119. Pandolfi F, Altamura S, Frosali S, Conti P. Key role of DAMP in inflammation, cancer, and tissue repair. Clin Ther. (2016) 38:1017–28. doi: 10.1016/j.clinthera.2016.02.028
120. Citrin D, Cotrim AP, Hyodo F, Baum BJ, Krishna MC, Mitchell JB. Radioprotectors and mitigators of radiation-induced normal tissue injury. Oncologist. (2010) 15:360–71. doi: 10.1634/theoncologist.2009-S104
121. Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol. (2010) 97:149–61. doi: 10.1016/j.radonc.2010.09.002
122. Stone HB, Coleman CN, Anscher MS, McBride WH. Effects of radiation on normal tissue: consequences and mechanisms. Lancet Oncol. (2003) 4:529–36. doi: 10.1016/S1470-2045(03)01191-4
123. Barnett GC, West CM, Dunning AM, Elliott RM, Coles CE, Pharoah PD, et al. Normal tissue reactions to radiotherapy: towards tailoring treatment dose by genotype. Nat Rev Cancer. (2009) 9:134–42. doi: 10.1038/nrc2587
124. Kim JH, Jenrow KA, Brown SL. Mechanisms of radiation-induced normal tissue toxicity and implications for future clinical trials. Radiat Oncol J. (2014) 32:103–15. doi: 10.3857/roj.2014.32.3.103
125. Ruhle A, Huber PE. [Normal tissue: radiosensitivity, toxicity, consequences for planning]. Radiologe. (2018) 58:746–53. doi: 10.1007/s00117-018-0430-4
126. Cappuccini F, Eldh T, Bruder D, Gereke M, Jastrow H, Schulze-Osthoff K, et al. New insights into the molecular pathology of radiation-induced pneumopathy. Radiother Oncol. (2011) 101:86–92. doi: 10.1016/j.radonc.2011.05.064
127. Lo Re S, Lison D, Huaux F. CD4+ T lymphocytes in lung fibrosis: diverse subsets, diverse functions. J Leukoc Biol. (2013) 93:499–510. doi: 10.1189/jlb.0512261
128. Wirsdorfer F, Jendrossek V. The role of lymphocytes in radiotherapy-induced adverse late effects in the lung. Front Immunol. (2016) 7:591. doi: 10.3389/fimmu.2016.00591
129. Zhang H, Han G, Liu H, Chen J, Ji X, Zhou F, et al. The development of classically and alternatively activated macrophages has different effects on the varied stages of radiation-induced pulmonary injury in mice. J Radiat Res. (2011) 52:717–26. doi: 10.1269/jrr.11054
130. Groves AM, Johnston CJ, Misra RS, Williams JP, Finkelstein JN. Whole-lung irradiation results in pulmonary macrophage alterations that are subpopulation and strain specific. Radiat Res. (2015) 184:639–49. doi: 10.1667/RR14178.1
131. de Leve S, Wirsdorfer F, Cappuccini F, Schutze A, Meyer AV, Rock K, et al. Loss of CD73 prevents accumulation of alternatively activated macrophages and the formation of prefibrotic macrophage clusters in irradiated lungs. FASEB J. (2017) 31:2869–80. doi: 10.1096/fj.201601228R
132. Meziani L, Mondini M, Petit B, Boissonnas A, Thomas de Montpreville V, Mercier O, et al. CSF1R inhibition prevents radiation pulmonary fibrosis by depletion of interstitial macrophages. Eur Respir J. (2018) 51:1702120. doi: 10.1183/13993003.02120-2017
133. Bickelhaupt S, Erbel C, Timke C, Wirkner U, Dadrich M, Flechsig P, et al. Effects of CTGF blockade on attenuation and reversal of radiation-induced pulmonary fibrosis. J Natl Cancer Inst. (2017) 109:djw339. doi: 10.1093/jnci/djw339
134. Dadrich M, Nicolay NH, Flechsig P, Bickelhaupt S, Hoeltgen L, Roeder F, et al. Combined inhibition of TGFbeta and PDGF signaling attenuates radiation-induced pulmonary fibrosis. Oncoimmunology. (2016) 5:e1123366. doi: 10.1080/2162402X.2015.1123366
135. Allard B, Turcotte M, Stagg J. CD73-generated adenosine: orchestrating the tumor-stroma interplay to promote cancer growth. J Biomed Biotechnol. (2012) 2012:485156. doi: 10.1155/2012/485156
136. Perez-Aso M, Mediero A, Low YC, Levine J, Cronstein BN. Adenosine A2A receptor plays an important role in radiation-induced dermal injury. FASEB J. (2016) 30:457–65. doi: 10.1096/fj.15-280388
137. Vaupel P, Multhoff G. Adenosine can thwart antitumor immune responses elicited by radiotherapy: therapeutic strategies alleviating protumor ADO activities. Strahlenther Onkol. (2016) 192:279–87. doi: 10.1007/s00066-016-0948-1
138. Wirsdorfer F, de Leve S, Cappuccini F, Eldh T, Meyer AV, Gau E, et al. Extracellular adenosine production by ecto-5′-nucleotidase (cd73) enhances radiation-induced lung fibrosis. Cancer Res. (2016) 76:3045–56. doi: 10.1158/0008-5472.CAN-15-2310
139. Kaufmann A, Musset B, Limberg SH, Renigunta V, Sus R, Dalpke AH, et al. “Host tissue damage” signal ATP promotes non-directional migration and negatively regulates toll-like receptor signaling in human monocytes. J Biol Chem. (2005) 280:32459–67. doi: 10.1074/jbc.M505301200
140. Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. (2001) 414:916–20. doi: 10.1038/414916a
141. Hasko G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. (2004) 25:33–9. doi: 10.1016/j.it.2003.11.003
142. Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. (2006) 2:351–60. doi: 10.1007/s11302-005-5302-5
143. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
144. Della Latta V, Cabiati M, Rocchiccioli S, Del Ry S, Morales MA. The role of the adenosinergic system in lung fibrosis. Pharmacol Res. (2013) 76:182–9. doi: 10.1016/j.phrs.2013.08.004
145. Lim HM, Heo W, Han JW, Lee MG, Kim JY. NPP1 is responsible for potent extracellular ATP hydrolysis as NTPDase1 in primary cultured murine microglia. Purinergic Signal. (2018) 14:157–66. doi: 10.1007/s11302-018-9601-z
146. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
147. Thompson LF, Takedachi M, Ebisuno Y, Tanaka T, Miyasaka M, Mills JH, et al. Regulation of leukocyte migration across endothelial barriers by ECTO-5′-nucleotidase-generated adenosine. Nucleosides Nucleotides Nucleic Acids. (2008) 27:755–60. doi: 10.1080/15257770802145678
148. Hasko G, Csoka B, Nemeth ZH, Vizi ES, Pacher P. A(2B) adenosine receptors in immunity and inflammation. Trends Immunol. (2009) 30:263–70. doi: 10.1016/j.it.2009.04.001
149. Kaku H, Cheng KF, Al-Abed Y, Rothstein TL. A novel mechanism of B cell-mediated immune suppression through CD73 expression and adenosine production. J Immunol. (2014) 193:5904–13. doi: 10.4049/jimmunol.1400336
150. Chen X, Shao H, Zhi Y, Xiao Q, Su C, Dong L, et al. CD73 Pathway Contributes to the immunosuppressive ability of mesenchymal stem cells in intraocular autoimmune responses. Stem Cells Dev. (2016) 25:337–46. doi: 10.1089/scd.2015.0227
151. Katsuta E, Tanaka S, Mogushi K, Shimada S, Akiyama Y, Aihara A, et al. CD73 as a therapeutic target for pancreatic neuroendocrine tumor stem cells. Int J Oncol. (2016) 48:657–69. doi: 10.3892/ijo.2015.3299
152. Turcotte M, Allard D, Mittal D, Bareche Y, Buisseret L, Jose V, et al. CD73 Promotes resistance to HER2/ErbB2 antibody therapy. Cancer Res. (2017) 77:5652–63. doi: 10.1158/0008-5472.CAN-17-0707
153. Breitbach M, Kimura K, Luis TC, Fuegemann CJ, Woll PS, Hesse M, et al. In vivo labeling by CD73 marks multipotent stromal cells and highlights endothelial heterogeneity in the bone marrow niche. Cell Stem Cell. (2018) 22:262–76.e267. doi: 10.1016/j.stem.2018.01.008
154. Aponte PM, Caicedo A. Stemness in cancer: stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. (2017) 2017:5619472. doi: 10.1155/2017/5619472
155. Lupia M, Angiolini F, Bertalot G, Freddi S, Sachsenmeier KF, Chisci E, et al. CD73 Regulates stemness and epithelial-mesenchymal transition in ovarian cancer-initiating cells. Stem Cell Rep. (2018) 10:1412–25. doi: 10.1016/j.stemcr.2018.02.009
156. Monguio-Tortajada M, Roura S, Galvez-Monton C, Franquesa M, Bayes-Genis A, Borras FE. Mesenchymal stem cells induce expression of CD73 in human monocytes in vitro and in a swine model of myocardial infarction In vivo. Front Immunol. (2017) 8:1577. doi: 10.3389/fimmu.2017.01577
157. Flogel U, Burghoff S, van Lent PL, Temme S, Galbarz L, Ding Z, et al. Selective activation of adenosine A2A receptors on immune cells by a CD73-dependent prodrug suppresses joint inflammation in experimental rheumatoid arthritis. Sci Transl Med. (2012) 4:146ra108. doi: 10.1126/scitranslmed.3003717
158. Silva-Vilches C, Ring S, Schrader J, Clausen BE, Probst HC, Melchior F, et al. Production of extracellular adenosine by CD73+ dendritic cells is crucial for induction of tolerance in contact hypersensitivity reactions. J Invest Dermatol. (2018) 139:541–51. doi: 10.1016/j.jid.2018.10.016
159. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
160. Fratangelo F, Morello S, Madonna G, Capone M, Mallardo D, Falcone R, et al. Expression of CD73 on MDSCs and CD8+/PD-1+ cells as new possible biomarkers for advanced melanoma patients treated with nivolumab. Eur J Cancer. (2018) 92:S24–5. doi: 10.1016/j.ejca.2018.01.060
161. Murphy PS, Wang J, Bhagwat SP, Munger JC, Janssen WJ, Wright TW, et al. CD73 regulates anti-inflammatory signaling between apoptotic cells and endotoxin-conditioned tissue macrophages. Cell Death Differ. (2017) 24:559–70. doi: 10.1038/cdd.2016.159
162. Stagg J, Beavis PA, Divisekera U, Liu MC, Moller A, Darcy PK, et al. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. (2012) 72:2190–6. doi: 10.1158/0008-5472.CAN-12-0420
163. Morandi F, Horenstein AL, Chillemi A, Quarona V, Chiesa S, Imperatori A, et al. CD56brightCD16- NK cells produce adenosine through a CD38-mediated pathway and act as regulatory cells inhibiting autologous CD4+ T cell proliferation. J Immunol. (2015) 195:965–72. doi: 10.4049/jimmunol.1500591
164. Resta R, Jiang H, Hooker SW, Laurent AB, Thompson LF. Insights into adenosine deaminase deficiency provided by murine fetal thymic organ culture with 2'-deoxycoformycin. Adv Exp Med Biol. (1998) 431:451–4. doi: 10.1007/978-1-4615-5381-6_89
165. Chatterjee D, Marquardt N, Tufa DM, Beauclair G, Low HZ, Hatlapatka T, et al. Role of gamma-secretase in human umbilical-cord derived mesenchymal stem cell mediated suppression of NK cell cytotoxicity. Cell Commun Signal. (2014) 12:63. doi: 10.1186/s12964-014-0063-9
166. Conter LJ, Song E, Shlomchik MJ, Tomayko MM. CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS ONE. (2014) 9:e92009. doi: 10.1371/journal.pone.0092009
167. Schena F, Volpi S, Faliti CE, Penco F, Santi S, Proietti M, et al. Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity. Cell Rep. (2013) 3:1824–31. doi: 10.1016/j.celrep.2013.05.022
168. Bono MR, Fernandez D, Flores-Santibanez F, Rosemblatt M, Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. (2015) 589:3454–60. doi: 10.1016/j.febslet.2015.07.027
169. Yang L, Kobie JJ, Mosmann TR. CD73 and Ly-6A/E distinguish In vivo primed but uncommitted mouse CD4 T cells from type 1 or type 2 effector cells. J Immunol. (2005) 175:6458–64. doi: 10.4049/jimmunol.175.10.6458
170. Flores-Santibanez F, Fernandez D, Meza D, Tejon G, Vargas L, Varela-Nallar L, et al. CD73-mediated adenosine production promotes stem cell-like properties in mouse Tc17 cells. Immunology. (2015) 146:582–94. doi: 10.1111/imm.12529
171. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol. (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
172. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. (2010) 285:7176–86. doi: 10.1074/jbc.M109.047423
173. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Vegran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. (2012) 36:362–73. doi: 10.1016/j.immuni.2011.12.019
174. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73+CD4+ effector T cells devoid of immune checkpoints. Cancer Res. (2018) 78:3604–18. doi: 10.1158/0008-5472.CAN-17-2405
175. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. (2013) 43:2430–40. doi: 10.1002/eji.201242909
176. Gaudreau PO, Allard B, Turcotte M, Stagg J. CD73-adenosine reduces immune responses and survival in ovarian cancer patients. Oncoimmunology. (2016) 5:e1127496. doi: 10.1080/2162402X.2015.1127496
177. Koivisto M, Tervahartiala M, Kenessey I, Jalkanen S, Bostrom PJ, Salmi M. Cell-type specific CD73 expression is an independent prognostic factor in bladder cancer. Carcinogenesis. (2018) 40:84–92. doi: 10.1093/carcin/bgy154
178. Chen S, Zhou S, Zang K, Kong F, Liang D, Yan H. CD73 expression in RPE cells is associated with the suppression of conventional CD4 cell proliferation. Exp Eye Res. (2014) 127:26–36. doi: 10.1016/j.exer.2014.05.008
179. Sung SJ, Li L, Huang L, Lawler J, Ye H, Rosin DL, et al. Proximal tubule CD73 is critical in renal ischemia-reperfusion injury protection. J Am Soc Nephrol. (2017) 28:888–902. doi: 10.1681/ASN.2016020229
180. Cronstein BN, Kramer SB, Weissmann G, Hirschhorn R. Adenosine: a physiological modulator of superoxide anion generation by human neutrophils. J Exp Med. (1983) 158:1160–77. doi: 10.1084/jem.158.4.1160
181. Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. (2006) 176:4449–58. doi: 10.4049/jimmunol.176.7.4449
182. Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signal. (2013) 9:145–65. doi: 10.1007/s11302-012-9349-9
183. Regateiro FS, Cobbold SP, Waldmann H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin Exp Immunol. (2013) 171:1–7. doi: 10.1111/j.1365-2249.2012.04623.x
184. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
185. Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal. (2014) 10:529–64. doi: 10.1007/s11302-014-9427-2
186. Fredholm BB, AP IJ, Jacobson KA, Linden J, Muller CE. International union of basic and clinical pharmacology. LXXXI Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev. (2011) 63:1–34. doi: 10.1124/pr.110.003285
187. Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. (2008) 7:759–70. doi: 10.1038/nrd2638
188. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
189. Ferrari D, Gambari R, Idzko M, Muller T, Albanesi C, Pastore S, et al. Purinergic signaling in scarring. FASEB J. (2016) 30:3–12. doi: 10.1096/fj.15-274563
190. Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. (2004) 200:1395–405. doi: 10.1084/jem.20040915
191. Hoegl S, Brodsky KS, Blackburn MR, Karmouty-Quintana H, Zwissler B, Eltzschig HK. Alveolar epithelial A2B adenosine receptors in pulmonary protection during acute lung injury. J Immunol. (2015) 195:1815–24. doi: 10.4049/jimmunol.1401957
192. Hamidzadeh K, Mosser DM. Purinergic signaling to terminate TLR responses in macrophages. Front Immunol. (2016) 7:74. doi: 10.3389/fimmu.2016.00074
193. Blackburn MR, Kellems RE. Adenosine deaminase deficiency: metabolic basis of immune deficiency and pulmonary inflammation. Adv Immunol. (2005) 86:1–41. doi: 10.1016/S0065-2776(04)86001-2
194. Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol. (2005) 175:1937–46. doi: 10.4049/jimmunol.175.3.1937
195. Sun CX, Zhong H, Mohsenin A, Morschl E, Chunn JL, Molina JG, et al. Role of A2B adenosine receptor signaling in adenosine-dependent pulmonary inflammation and injury. J Clin Invest. (2006) 116:2173–82. doi: 10.1172/JCI27303
196. Cronstein BN. Adenosine receptors and fibrosis: a translational review. F1000 Biol Rep. (2011) 3:21. doi: 10.3410/B3-21
197. Zhou Y, Schneider DJ, Morschl E, Song L, Pedroza M, Karmouty-Quintana H, et al. Distinct roles for the A2B adenosine receptor in acute and chronic stages of bleomycin-induced lung injury. J Immunol. (2011) 186:1097–106. doi: 10.4049/jimmunol.1002907
198. Zhang W, Zhang Y, Wang W, Dai Y, Ning C, Luo R, et al. Elevated ecto-5′-nucleotidase-mediated increased renal adenosine signaling via A2B adenosine receptor contributes to chronic hypertension. Circ Res. (2013) 112:1466–78. doi: 10.1161/CIRCRESAHA.111.300166
199. Luo F, Le NB, Mills T, Chen NY, Karmouty-Quintana H, Molina JG, et al. Extracellular adenosine levels are associated with the progression and exacerbation of pulmonary fibrosis. FASEB J. (2016) 30:874–83. doi: 10.1096/fj.15-274845
200. Shaikh G, Cronstein B. Signaling pathways involving adenosine A2A and A2B receptors in wound healing and fibrosis. Purinergic Signal. (2016) 12:191–7. doi: 10.1007/s11302-016-9498-3
201. Karmouty-Quintana H, Zhong H, Acero L, Weng T, Melicoff E, West JD, et al. The A2B adenosine receptor modulates pulmonary hypertension associated with interstitial lung disease. FASEB J. (2012) 26:2546–57. doi: 10.1096/fj.11-200907
202. Karmouty-Quintana H, Philip K, Acero LF, Chen NY, Weng T, Molina JG, et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. FASEB J. (2015) 29:50–60. doi: 10.1096/fj.14-260182
203. Hauer-Jensen M, Poulakos L, Osborne JW. Effects of accelerated fractionation on radiation injury of the small intestine: a new rat model. Int J Radiat Oncol Biol Phys. (1988) 14:1205–12. doi: 10.1016/0360-3016(88)90399-9
204. Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. (2011) 186:4367–74. doi: 10.4049/jimmunol.0903617
205. Colgan SP, Fennimore B, Ehrentraut SF. Adenosine and gastrointestinal inflammation. J Mol Med. (2013) 91:157–64. doi: 10.1007/s00109-012-0990-0
206. Francois V, Shehade H, Acolty V, Preyat N, Delree P, Moser M, et al. Intestinal immunopathology is associated with decreased CD73-generated adenosine during lethal infection. Mucosal Immunol. (2015) 8:773–84. doi: 10.1038/mi.2014.108
207. Loinard C, Vilar J, Milliat F, Levy B, Benderitter M. Monocytes/macrophages mobilization orchestrate neovascularization after localized colorectal irradiation. Radiat Res. (2017) 187:549–61. doi: 10.1667/RR14398.1
208. Verginadis II, Kanade R, Bell B, Koduri S, Ben-Josef E, Koumenis C. A novel mouse model to study image-guided, radiation-induced intestinal injury and preclinical screening of radioprotectors. Cancer Res. (2017) 77:908–17. doi: 10.1158/0008-5472.CAN-16-2724
209. Rajakumar SV, Lu B, Crikis S, Robson SC, d'Apice AJ, Cowan PJ, et al. Deficiency or inhibition of CD73 protects in mild kidney ischemia-reperfusion injury. Transplantation. (2010) 90:1260–4. doi: 10.1097/TP.0b013e3182003d9b
210. Jian R, Sun Y, Wang Y, Yu J, Zhong L, Zhou P. CD73 protects kidney from ischemia-reperfusion injury through reduction of free radicals. APMIS. (2012) 120:130–8. doi: 10.1111/j.1600-0463.2011.02827.x
211. Galon J, Fridman WH, Pages F. The adaptive immunologic microenvironment in colorectal cancer: a novel perspective. Cancer Res. (2007) 67:1883–6. doi: 10.1158/0008-5472.CAN-06-4806
212. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. (2012) 12:298–306. doi: 10.1038/nrc3245
213. Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. (2010) 29:1093–102. doi: 10.1038/onc.2009.416
214. Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. (2011) 29:610–8. doi: 10.1200/JCO.2010.30.5425
215. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. (2011) 331:1565–70. doi: 10.1126/science.1203486
216. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. (2013) 34:67–73. doi: 10.1016/j.it.2012.10.004
217. Balermpas P, Rodel F, Liberz R, Oppermann J, Wagenblast J, Ghanaati S, et al. Head and neck cancer relapse after chemoradiotherapy correlates with CD163+ macrophages in primary tumour and CD11b+ myeloid cells in recurrences. Br J Cancer. (2014b) 111:1509–18. doi: 10.1038/bjc.2014.446
218. Martin D, Rodel F, Winkelmann R, Balermpas P, Rodel C, Fokas E. Peripheral leukocytosis is inversely correlated with intratumoral CD8+ T-cell infiltration and associated with worse outcome after chemoradiotherapy in anal cancer. Front Immunol. (2017) 8:1225. doi: 10.3389/fimmu.2017.01225
219. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. (2015) 348:56–61. doi: 10.1126/science.aaa8172
220. Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, et al. Immunogenicity of murine solid tumor models as a defining feature of In vivo behavior and response to immunotherapy. J Immunother. (2013) 36:477–89. doi: 10.1097/01.cji.0000436722.46675.4a
221. Milas L, Wike J, Hunter N, Volpe J, Basic I. Macrophage content of murine sarcomas and carcinomas: associations with tumor growth parameters and tumor radiocurability. Cancer Res. (1987) 47:1069–75.
222. Trellakis S, Bruderek K, Dumitru CA, Gholaman H, Gu X, Bankfalvi A, et al. Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int J Cancer. (2011) 129:2183–93. doi: 10.1002/ijc.25892
223. Balermpas P, Michel Y, Wagenblast J, Seitz O, Weiss C, Rodel F, et al. Tumour-infiltrating lymphocytes predict response to definitive chemoradiotherapy in head and neck cancer. Br J Cancer. (2014a) 110:501–9. doi: 10.1038/bjc.2013.640
224. Okubo M, Kioi M, Nakashima H, Sugiura K, Mitsudo K, Aoki I, et al. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci Rep. (2016) 6:27548. doi: 10.1038/srep27548
225. Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing tumor immunity with fractionated radiation. Int J Radiat Oncol Biol Phys. (2012) 83:1306–10. doi: 10.1016/j.ijrobp.2011.09.049
226. Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. (2010) 120:694–705. doi: 10.1172/JCI40283
227. Wada S, Harris TJ, Tryggestad E, Yoshimura K, Zeng J, Yen HR, et al. Combined treatment effects of radiation and immunotherapy: studies in an autochthonous prostate cancer model. Int J Radiat Oncol Biol Phys. (2013) 87:769–76. doi: 10.1016/j.ijrobp.2013.07.015
228. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. (2013) 86:343–9. doi: 10.1016/j.ijrobp.2012.12.025
229. Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. (2009) 30:102–8. doi: 10.1016/j.it.2008.12.002
230. Sitkovsky MV, Hatfield S, Abbott R, Belikoff B, Lukashev D, Ohta A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol Res. (2014) 2:598–605. doi: 10.1158/2326-6066.CIR-14-0075
231. Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3′,5′-monophosphate and phosphatases. J Immunol. (2004) 173:932–44. doi: 10.4049/jimmunol.173.2.932
232. Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, et al. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. (2009) 183:5487–93. doi: 10.4049/jimmunol.0901247
233. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. (2014) 74:7239–49. doi: 10.1158/0008-5472.CAN-13-3581
234. Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. (2015) 25:18–27. doi: 10.1016/j.semradonc.2014.07.008
235. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. (2016) 37:208–20. doi: 10.1016/j.it.2016.01.004
236. Takeshima T, Pop LM, Laine A, Iyengar P, Vitetta ES, Hannan R. Key role for neutrophils in radiation-induced antitumor immune responses: potentiation with G-CSF. Proc Natl Acad Sci USA. (2016) 113:11300–5. doi: 10.1073/pnas.1613187113
237. Merighi S, Mirandola P, Varani K, Gessi S, Leung E, Baraldi PG, et al. A glance at adenosine receptors: novel target for antitumor therapy. Pharmacol Ther. (2003) 100:31–48. doi: 10.1016/S0163-7258(03)00084-6
238. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. (2010) 29:5346–58. doi: 10.1038/onc.2010.292
239. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. (2011) 187:676–83. doi: 10.4049/jimmunol.1003884
240. Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochim Biophys Acta. (2011) 1808:1400–12. doi: 10.1016/j.bbamem.2010.09.020
241. Zhang B. CD73 promotes tumor growth and metastasis. Oncoimmunology. (2012) 1:67–70. doi: 10.4161/onci.1.1.18068
242. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov. (2014) 4:879–88. doi: 10.1158/2159-8290.CD-14-0341
243. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
244. Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. (2017) 77:4684–96. doi: 10.1158/0008-5472.CAN-17-0393
245. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
246. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.CAN-10-4246
247. Montalban Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wockel A, et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages - a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J Immunother Cancer. (2016) 4:49. doi: 10.1186/s40425-016-0154-9
248. Koszalka P, Golunska M, Stanislawowski M, Urban A, Stasilojc G, Majewski M, et al. CD73 on B16F10 melanoma cells in CD73-deficient mice promotes tumor growth, angiogenesis, neovascularization, macrophage infiltration and metastasis. Int J Biochem Cell Biol. (2015) 69:1–10. doi: 10.1016/j.biocel.2015.10.003
249. Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemela J, Laurila JP, Elima K, et al. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol. (2011) 41:1231–41. doi: 10.1002/eji.201041292
250. Koszalka P, Golunska M, Urban A, Stasilojc G, Stanislawowski M, Majewski M, et al. Specific activation of A3, A2A and A1 adenosine receptors in CD73-knockout mice affects B16F10 melanoma growth, neovascularization, angiogenesis and macrophage infiltration. PLoS ONE. (2016) 11:e0151420. doi: 10.1371/journal.pone.0151420
251. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. (2014) 4:172–81.
252. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother. (2018) 67:1271–84. doi: 10.1007/s00262-018-2186-0
253. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res. (2015) 3:506–17. doi: 10.1158/2326-6066.CIR-14-0211
254. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
255. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
256. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
257. Wennerberg E, Kawashima N, Demaria S. Adenosine regulates radiation therapy-induced anti-tumor immunity. J Immunother Cancer. (2015) 3 (Suppl. 2):p378. doi: 10.1186/2051-1426-3-s2-p378
258. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
259. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
260. Jackson SW, Hoshi T, Wu Y, Sun X, Enjyoji K, Cszimadia E, et al. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1-null mice. Am J Pathol. (2007) 171:1395–404. doi: 10.2353/ajpath.2007.070190
261. Sun X, Han L, Seth P, Bian S, Li L, Csizmadia E, et al. Disordered purinergic signaling and abnormal cellular metabolism are associated with development of liver cancer in Cd39/ENTPD1 null mice. Hepatology. (2013) 57:205–16. doi: 10.1002/hep.25989
262. Perry C, Hazan-Halevy I, Kay S, Cipok M, Grisaru D, Deutsch V, et al. Increased CD39 expression on CD4+ T lymphocytes has clinical and prognostic significance in chronic lymphocytic leukemia. Ann Hematol. (2012) 91:1271–9. doi: 10.1007/s00277-012-1425-2
263. Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. (2013) 32:1743–51. doi: 10.1038/onc.2012.269
264. Cai XY, Wang XF, Li J, Dong JN, Liu JQ, Li NP, et al. High expression of CD39 in gastric cancer reduces patient outcome following radical resection. Oncol Lett. (2016) 12:4080–6. doi: 10.3892/ol.2016.5189
265. Ampollini L, Madeddu D, Falco A, Frati C, Lorusso B, Graiani G, et al. Lung mesenchymal cells function as an inductive microenvironment for human lung cancer propagating cellsdagger. Eur J Cardiothorac Surg. (2014) 46:e103–12. doi: 10.1093/ejcts/ezu359
266. Bichsel CA, Wang L, Froment L, Berezowska S, Muller S, Dorn P, et al. Increased PD-L1 expression and IL-6 secretion characterize human lung tumor-derived perivascular-like cells that promote vascular leakage in a perfusable microvasculature model. Sci Rep. (2017) 7:10636. doi: 10.1038/s41598-017-09928-1
267. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
268. Jiang T, Xu X, Qiao M, Li X, Zhao C, Zhou F, et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer. (2018) 18:267. doi: 10.1186/s12885-018-4073-7
269. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget. (2017) 8:8738–51. doi: 10.18632/oncotarget.14434
270. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. (2010) 123:725–31. doi: 10.1007/s10549-009-0674-9
271. Sun CX, Young HW, Molina JG, Volmer JB, Schnermann J, Blackburn MR. A protective role for the A1 adenosine receptor in adenosine-dependent pulmonary injury. J Clin Invest. (2005) 115:35–43. doi: 10.1172/JCI22656
272. Morschl E, Molina JG, Volmer JB, Mohsenin A, Pero RS, Hong JS, et al. A3 adenosine receptor signaling influences pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol. (2008) 39:697–705. doi: 10.1165/rcmb.2007-0419OC
273. Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, et al. CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB J. (2013) 27:2207–19. doi: 10.1096/fj.12-225201
274. Kachikwu EL, Iwamoto KS, Liao YP, DeMarco JJ, Agazaryan N, Economou JS, et al. Radiation enhances regulatory T cell representation. Int J Radiat Oncol Biol Phys. (2011) 81:1128–35. doi: 10.1016/j.ijrobp.2010.09.034
275. Klein D, Schmetter A, Imsak R, Wirsdorfer F, Unger K, Jastrow H, et al. Therapy with multipotent mesenchymal stromal cells protects lungs from radiation-induced injury and reduces the risk of lung metastasis. Antioxid Redox Signal. (2016) 24:53–69. doi: 10.1089/ars.2014.6183
276. Klein D, Steens J, Wiesemann A, Schulz F, Kaschani F, Rock K, et al. Mesenchymal stem cell therapy protects lungs from radiation-induced endothelial cell loss by restoring superoxide dismutase 1 expression. Antioxid Redox Signal. (2017) 26:563–82. doi: 10.1089/ars.2016.6748
277. Kepp O, Loos F, Liu P, Kroemer G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol Rev. (2017) 280:83–92. doi: 10.1111/imr.12571
278. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 expression is an independent prognostic factor in prostate cancer. Clin Cancer Res. (2016) 22:158–66. doi: 10.1158/1078-0432.CCR-15-1181
279. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. (2008) 8:545–54. doi: 10.1038/nrc2419
280. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
281. Holzel M, Bovier A, Tuting T. Plasticity of tumour and immune cells: a source of heterogeneity and a cause for therapy resistance? Nat Rev Cancer. (2013) 13:365–76. doi: 10.1038/nrc3498
282. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. (2013) 23:811–25. doi: 10.1016/j.ccr.2013.05.003
283. Leder K, Pitter K, Laplant Q, Hambardzumyan D, Ross BD, Chan TA, et al. Mathematical modeling of PDGF-driven glioblastoma reveals optimized radiation dosing schedules. Cell. (2014) 156:603–16. doi: 10.1016/j.cell.2013.12.029
284. Bristow RG, Alexander B, Baumann M, Bratman SV, Brown JM, Camphausen K, et al. Combining precision radiotherapy with molecular targeting and immunomodulatory agents: a guideline by the American Society for Radiation Oncology. Lancet Oncol. (2018) 19:e240–51. doi: 10.1016/S1470-2045(18)30096-2
285. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. (2015) 520:373–7. doi: 10.1038/nature14292
286. Bhatia S, Sharma J, Bukkapatnam S, Oweida A, Lennon S, Phan A, et al. Inhibition of EphB4-ephrin-B2 signaling enhances response to cetuximab-radiation therapy in head and neck cancers. Clin Cancer Res. (2018) 24:4539–50. doi: 10.1158/1078-0432.CCR-18-0327
287. Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, et al. Resistance to radiotherapy and PD-L1 blockade is mediated by TIM-3 upregulation and regulatory T-cell infiltration. Clin Cancer Res. (2018) 78:2766. doi: 10.1158/1078-0432.CCR-18-1038
288. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Lenzner DE, Jackson EK, et al. Increased ectonucleotidase expression and activity in regulatory T cells of patients with head and neck cancer. Clin Cancer Res. (2009) 15:6348–57. doi: 10.1158/1078-0432.CCR-09-1143
289. Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. (2009) 15:5379–88. doi: 10.1158/1078-0432.CCR-09-0265
Keywords: CD73, adenosine, radiotherapy, therapeutic window, normal tissue toxicity, Treg, macrophages, tumor microenvironment
Citation: de Leve S, Wirsdörfer F and Jendrossek V (2019) Targeting the Immunomodulatory CD73/Adenosine System to Improve the Therapeutic Gain of Radiotherapy. Front. Immunol. 10:698. doi: 10.3389/fimmu.2019.00698
Received: 31 October 2018; Accepted: 14 March 2019;
Published: 05 April 2019.
Edited by:
Catherine Sautes-Fridman, INSERM U1138 Centre de Recherche des Cordeliers, FranceReviewed by:
Frederique Vegran, INSERM U1231 Lipides, Nutrition, Cancer (LNC), FranceZoltan Vereb, University of Szeged, Hungary
Copyright © 2019 de Leve, Wirsdörfer and Jendrossek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Verena Jendrossek, dmVyZW5hLmplbmRyb3NzZWtAdW5pLWR1ZS5kZQ==