Abstract
Chronic HIV-infection modulates the expression of Fc gamma receptors (FcγRs) on immune cells and their antibody-dependent effector function capability. Given the increasingly recognized importance of antibody-dependent cellular cytotoxicity (ADCC) in HIV-specific immunity, we investigated the cellular distribution of FcγRIIIa on cytotoxic lymphocytes—natural killer cells and CD8+ T cells—and the effect of the FcγRIIIa-F158V variant on ADCC capacity in HIV-infected individuals (n = 23) and healthy controls (n = 23). Study participants were matched for F158V genotypes, carried two copies of the FCGR3A gene and were negative for FcγRIIb expression on NK cells. The distribution of CD56dimFcγRIIIabright and CD56negFcγRIIIabright NK cell subsets, but not FcγRIIIa surface expression, differed significantly between HIV-1 negative and HIV-1 positive donors. NK cell-mediated ADCC responses negatively correlated with the proportion of the immunoregulatory CD56brightFcγRIIIadim/neg cells and were lower in the HIV-1 positive group. Intriguingly, the FcγRIIIa-F158V variant differentially affected the NK-mediated ADCC responses for HIV-1 negative and HIV-1 positive donors. Healthy donors bearing at least one 158V allele had higher ADCC responses compared to those homozygous for the 158F allele (48.1 vs. 34.1%), whereas the opposite was observed for the HIV-infected group (26.4 vs. 34.6%), although not statistically significantly different. Furthermore, FcγRIIIa+CD8bright and FcγRIIIa+CD8dim T cell subsets were observed in both HIV-1 negative and HIV-1 positive donors, with median proportions that were significantly higher in HIV-1 positive donors compared to healthy controls (15.7 vs. 8.3%; P = 0.016 and 18.2 vs. 14.1%; P = 0.038, respectively). Using an HIV-1-specific GranToxiLux assay, we demonstrate that CD8+ T cells mediate ADCC through the delivery of granzyme B, which was overall lower compared to that of autologous NK cells. In conclusion, our findings demonstrate that in the presence of an HIV-1 infection, the cellular distribution of FcγRIIIa is altered and that the functional consequence of FcγRIIIa variant is affected. Importantly, it underscores the need to characterize FcγR expression, cellular distribution and functional consequences of FcγR genetic variants within a specific environment or disease state.
Introduction
Receptors for the Fc domain of immunoglobulin G (IgG), so called Fc gamma receptors (FcγRs), link the specificity of IgG with potent effector functions of the innate immune system. FcγRs comprise a family of activating (FcγRI, FcγRIIa, and FcγRIIIa) and inhibiting (FcγRIIb) receptors that are differentially expressed on innate immune cells such as natural killer (NK) cells, monocytes, dendritic cells, neutrophils, and granulocytes (1–3). During an infection, these receptors play an important role in activating IgG-induced protective inflammatory processes and regulating immune responses (4–7).
Increasing evidence support an important role for FcγR-mediated effector functions, in particular antibody-dependent cellular cytotoxicity (ADCC), in HIV-1-specific immunity (6, 8). In adults, ADCC has been associated with a reduced risk of HIV-1 acquisition in the RV144 vaccine trial, whereas in infants born to HIV-1 infected mothers, passively acquired ADCC activity associated with reduced mortality (9). Similarly, ADCC responses associate with slower disease progression in HIV-1 infected adults (10–14).
In the periphery, CD56dimFcγRIIIabright NK cells are the primary effectors of ADCC responses (15). During a chronic HIV-1 infection, however, shedding of FcγRIIIa from the surface of cytotoxic CD56dimFcγRIIIabright NK cells together with an increase in the CD56negFcγRIIIabright NK cell subset leads to a significant reduction in NK cell function (16, 17). It is unknown whether this loss in NK cell-mediated ADCC capacity is compensated for by other cytotoxic cells, where FcγR expression is induced upon immune cell activation. For chronic hepatitis C virus or Epstein Barr virus infections, for instance, it has been shown that FcγRIIIa expression is induced on an effector memory CD8+ T cell subset (18, 19). This FcγRIIIa+CD8+ T cell subset acquires NK cell-like functional properties, including ADCC activity accompanied by the release of pore forming perforin and serine protease granzyme B (18, 19). The presence and role of this CD8+ T cell subset in HIV-1 infection is currently undefined.
In addition to the effect of an actively replicating virus on FcγRIIIa expression, host genetics also contribute to variability of FcγRIIIa expression and/or ADCC responses. FCGR3A copy number variation directly correlates with the surface density of FcγRIIIa, with individuals bearing a single FCGR3A copy and correspondingly lower FcγRIIIa surface densities, having reduced ADCC responses compared to individuals with two or more gene copies (20). In addition, a phenylalanine (F) to valine (V) substitution at amino acid 158 in the proximal Ig-like domain of FcγRIIIa confers increased binding for IgG1, IgG3, and IgG4, which has been associated with higher NK cell activation and ADCC responses (21–23). Unlike FCGR3A copy number and the FcγRIIIa-F158V variant, a deletion of a copy number variable region (CNR) encompassing FCGR3B and FCGR2C—known as CNR1—does not affect FcγRIIIa directly (24). However, it juxtaposes the 5′-regulatory sequences of FCGR2C with the open reading frame of FCGR2B, creating a chimeric FCGR2B' gene (25). This results in the expression of the inhibitory FcγRIIb on NK cells where it regulates FcγRIIIa-mediated ADCC responses (25, 26).
FCGR variants are rarely adjusted for in studies that compare NK cell-mediated ADCC capacity between HIV-positive and HIV-negative individuals. Moreover, it is unclear if the altered immune milieu accompanying an HIV-1 infection modulates the functional consequences of the aforementioned variants. In this study, we sought to characterize FcγRIIIa expression on cytotoxic lymphocytes—NK cells and CD8+ T cells—and associated ADCC responses in healthy donors and viraemic HIV-1 individuals matched for FCGR genetic variants.
Materials and Methods
Cohort
All study participants were black South Africans recruited from the city of Johannesburg, Gauteng province, South Africa (Table 1). Self-reported HIV-1 uninfected individuals who did not have an acute or chronic illness at the time of sample collection were prospectively recruited from the National Institute for Communicable Diseases as healthy controls. Viraemic, treatment naïve HIV-1 infected individuals were identified from an existing cohort recruited from hospitals in Johannesburg and Soweto. This study was carried out in accordance with the recommendations of the National Health Research Ethics Council (NHREC) of the South African Department of Health. The protocol was approved by the University of the Witwatersrand Ethics Committee (Ethics clearance certificate no. M1511102). All participants provided written informed consent in accordance with the Declaration of Helsinki.
Table 1
| HIV-1 negative | HIV-1 positive | P-value | |
|---|---|---|---|
| N | 23 | 23 | |
| Age [mean (SD)] | 36.3 [8.5] | 40.2 (8.8) | 0.143* |
| Gender [% Females] | 69.6 | 82.6 | 0.491* |
| HIV-1 VIRAL LOAD [MEDIAN (IQR)] | |||
| 158FF | – | 6,931 (3,598–15,090) |
0.563† |
| 158FV/VV | – | 4,732 (1,134–11,430) |
|
| CD4 T CELL COUNT [MEAN ( SD )] | |||
| 158FF | – | 508 (267) | 0.629† |
| 158FV/VV | – | 562 (261) | |
| OTHER DISEASES OR INFECTIONS | |||
| Mycobacterium tuberculosis | 0 (0%) | 2 (8.7%) | |
Clinical and demographic characteristics of study cohort.
Comparison between HIV-1 negative and HIV-1 positive groups.
Comparison between genotypes.
FCGR Variant Genotyping
Genomic DNA was isolated from ethylenediaminetetraacetic acid (EDTA)-anticoagulated whole blood. Study participants were genotyped for FCGR variants using the FCGR-specific multiplex ligation-dependent probe amplification (MLPA) assay (MRC Holland, Amsterdam, The Netherlands) as previously described (27, 28). This assay detects the genomic copy number of FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B and functional allelic variants that include FcγRIIa-H131R (alias H166R, c.497A>G, rs1801274); FcγRIIb-I232T (c.695T>C, rs1050501), FcγRIIIa-F158V (alias F176V, c.634T>G, rs396991), FcγRIIIb-HNA1a/b/c, FCGR2C gene expression variants c.169T>C (X57Q) and c.798+1A>G (rs76277413), and the FCGR2B/C promoter variants c.-386G>C (rs3219018) and c.-120T>A (rs34701572) in two multiplex reactions. Capillary electrophoresis was used to separate the MLPA assay amplicons on an ABI Genetic Analyzer 3500. Data were analyzed with Coffalyser.NET software created by the MLPA assay manufacturer, MRC Holland.
Monoclonal Antibodies and Flow Cytometric Analysis
The following monoclonal antibodies were used: CD3-PerCP (SK7), CD56-AF647 (B159), CD8-APC-H7 (SK1), CD16-PE (3G8), CD16-FITC (NKP15), CD32-FITC (2B6), and CD32-PE (FLI8.26). All antibodies were obtained from BD Biosciences (San Jose, CA). Dead cells were labeled with the BD Horizon™ Fixable Viability Stain 510. Samples were acquired on a BD Fortessa X20 (BD biosciences) and analyzed on FlowJo version 9.8.1 software (Tree Star, San Carlos, CA). Fluorescence-minus-one (FMO) controls were used to set the appropriate gates for analyses.
Isolation of Specific Cell Populations
Peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-anticoagulated whole blood using Ficoll-Paque™ PLUS density gradient centrifugation (GE Healthcare) and stored at −80°C. NK cells and CD8+ T cells were positively selected from overnight rested PBMCs using MACS® magnetic cell separation technology (Miltenyl Biotec). To limit the presence of NK cells in the enriched CD8+ T cell preparation, NK cells were first isolated from PBMCs using CD56+ beads prior to isolation of CD8+ T cells with CD8+ beads. Viability and number of each participant's NK and CD8+ T cells were determined by trypan blue exclusion through direct cell counting on a haemocytometer. The purity of enriched cell fractions was determined by flow cytometric phenotyping assays.
Antibodies
ADCC capacity was evaluated using pooled IgG isolated from the HIV-1 infected individuals included in the study. IgG was isolated from plasma using the Melon Gel IgG Purification kit according to the manufacturer's instructions (Thermo Scientific) and quantitated using the Bicinchoninic acid (BCA) assay. HIV immune globulin (HIVIG; NIH AIDS reagent program) was used as a positive control.
ADCC
NK cells and CD8+ T cells both utilize the pore forming perforin and serine protease granzyme B during cytotoxic responses. It is therefore possible to evaluate ADCC responses of both NK cells and CD8+ T cells with the HIV-specific GranToxiLux assay (15). In brief, CEM.NKR.CCR5 cells were coated with recombinant HIV-1 ConC gp120, followed by opsonisation with HIV-1-specific antibodies and incubation for 45 min in the presence of NK cells or CD8+ T cells at an effector-to-target (E:T) ratio of 10:1. The optimal concentration of isolated IgG (30 μg/ml) was determined empirically. Samples were acquired on BD Fortessa X20 flow cytometer and data analyzed on FlowJo version 9.8.1 software (Tree Star, San Carlos, CA). Granzyme B activity in the absence of any antibody (background killing) was determined for each subject—NK cells and CD8+ T cells—and subtracted from granzyme B activity in the presence of antibody. The inter assay coefficient of variation, as calculated from a HIVIG-specific ADCC response measured for a single donor included in every run, was <10%.
To assess CD8+ T cell mediated granzyme B responses, HIV-1 positive donors with <5% NK cell contamination in their enriched CD8+ T cell fractions were identified and selected for further analysis. This proportion of NK cells equates to a <0.5:1 NK-to-target ratio in a 10:1 CD8-to-target preparation. In our experience, the NK cell-mediated granzyme B activity at 0.5:1 is on average 14.1% (standard deviation [SD]: 2.8%) of that observed for NK cells at a ratio of 10:1 (Figure 1). Using these data, a positivity threshold for CD8+ T cell-mediated granzyme B responses was calculated relative to autologous NK cell-mediated granzyme B responses whereby the mean granzyme B activity for autologous NK cells (E:T = 10:1) was multiplied with 22.4% (14.1% + 3 × SD:2.8%).
Figure 1
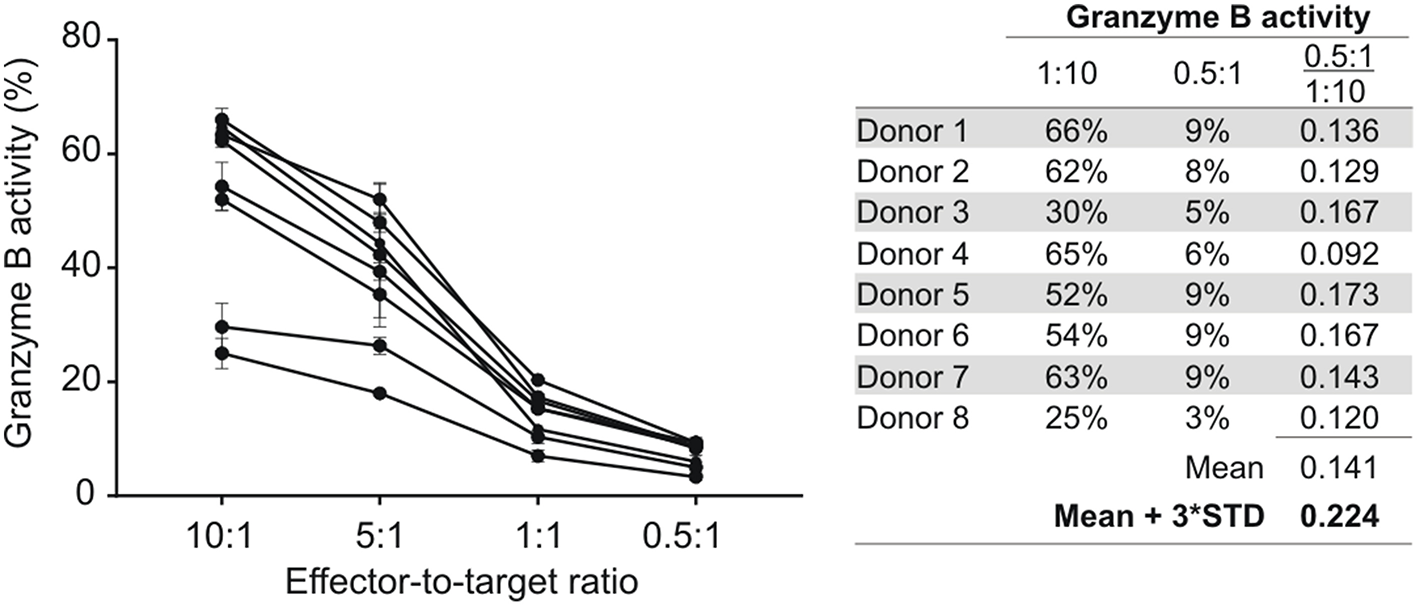
NK cell-mediated ADCC responses at different effector-to-target (E:T) cell ratios. NK cells isolated from eight HIV-1 negative donors were tested at different E:T ratios in an HIV-specific GranToxiLux assay using an ADCC-mediating monoclonal antibody (A32) at 2.5 μg/ml. The mean fraction of Granzyme B activity observed at an E:T of 0.5:1 relative to 10:1 was used to calculate a 99% confidence level cut-off value for NK cell-mediated ADCC responses at an E:T of 0.5:1. Data points represent the mean of triplicate measures and the standard deviation indicated by error bars.
Statistics
All statistical analyses were performed using GraphPad Prism 7 software version 7.04 (GraphPad Software). P-values from 2-tailed tests <0.05 were considered statistically significant. The significance of differences between unpaired data sets were analyzed with the Mann-Whitney U tests and paired data sets with the Wilcoxon matched-pairs signed rank test. The significance of differences between more than two data sets were analyzed using Kruskal-Wallis tests. Correlation analyses of data between two groups were assessed using the non-parametric Spearman rank correlation coefficient. To determine the role of FcγRIIIa-F158V alleles in ADCC responses, the V allele was studied under a dominant model due to the low prevalence of VV homozygotes (29).
Results
Study Population
Thirty-seven chronic HIV-1 infected, viraemic, and antiretroviral naïve individuals with sufficient sample available were identified from an existing cohort of HIV-1 positive individuals (Figure 2). Following FCGR genotyping, seven individuals were excluded due to the possession of an FCGR3A gene duplication (n = 1), FCGR3A gene deletion (n = 2), or CNR1 deletion that results in the expression of the inhibitory FcγRIIb on NK cells (n = 4). To ensure that individuals carrying an undetectable CNR1 deletion—possess a duplication of this region on a single chromosome and a deletion on the other—were also excluded, expression of FcγRIIb on NK cells was monitored using flow cytometry. During the eligibility screening of individuals, such an individual was indeed identified and excluded from the study; thus, five HIV-1 positive individuals in total expressed FcγRIIb on their NK cells and were excluded.
Figure 2
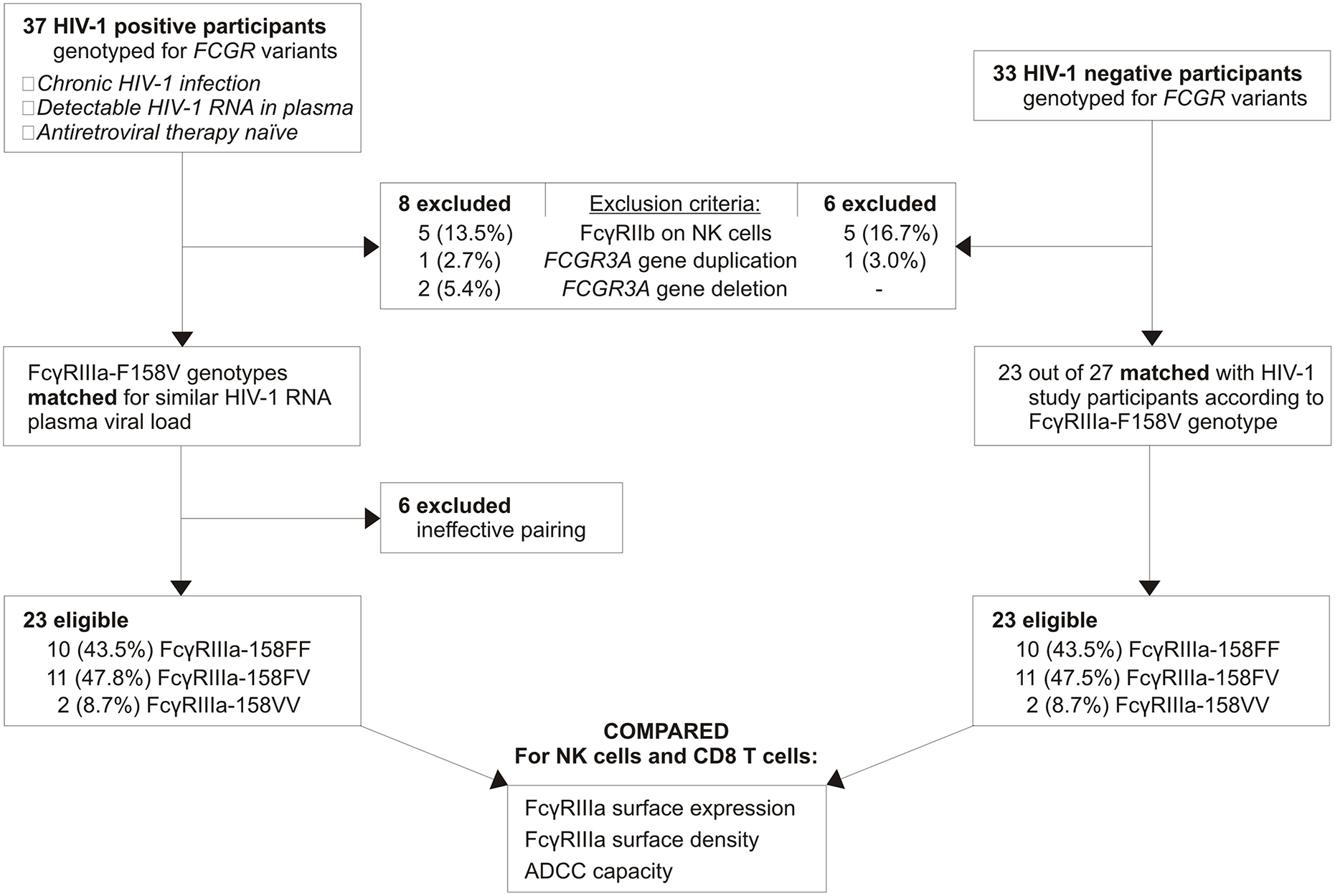
Study participant selection.
Within the HIV-1 positive group, FcγRIIIa-158FF donors were paired with FcγRIIIa-158FV donors according to their HIV-1 RNA plasma viral load. Twenty-three HIV-1 positive individuals were eligible for further analysis, with the FcγRIIIa-F158V genotype distribution closely resembling that observed in the general black population from the same region in South Africa (29). As a comparative group, thirty-three HIV-1 negative individuals were genotyped for FCGR variants. Six individuals were excluded due to the possession of an FCGR3A duplication (n = 1) or CNR1 deletion (n = 5). Twenty-three eligible HIV-1 negative individuals were subsequently paired with HIV-1 positive individuals based on the FcγRIIIa-F158V genotype. None of the study participants expressed the activating FcγRIIc on their NK cells, as determined by the FCGR2C c.798+1A>G splice-site variant, or carried the FCGR3A intragenic haplotype previously associated with increased FcγRIIIa surface density (26, 30).
Age and gender did not differ significantly between HIV-1 negative and positive donors (Table 1). Two HIV-1 positive individuals had tuberculosis, of which one was an FcγRIIIa-158FF donor and the other an FcγRIIIa-158FV donor. No other infections were noted for these patients. Human cytomegalovirus (HCMV) infection status was not determined for study participants. However, the prevalence is likely 100% for the HIV-1 positive individuals and >85% for HIV-1 negative individuals as observed in other cohorts in rural and urban South Africa [(31) and Tiemessen, unpublished data]. Other sexually transmitted infections were not tested for in the study cohort.
FcγRIIIa Expression on NK Cell Subsets
NK cell subsets were defined based on the relative surface expression of CD56 and FcγRIIIa. Four subsets were identified that include CD56brightFcγRIIIadim/neg, CD56dimFcγRIIIabright, CD56negFcγRIIIabright, and CD56dimFcγRIIIadim/neg (Figure 3A). The distribution of these subsets within the NK cell population differed significantly between HIV-1 positive and HIV-1 negative donors. Compared to the HIV-1 negative group, the HIV-1 positive group had a smaller median proportion of CD56dimFcγRIIIabright cells (60.2 vs. 77.0%, P = 0.0002, Figure 3B) that was offset by a larger median proportion of CD56negFcγRIIIabright cells (14.9 vs. 2.1%, P < 0.0001, Figure 3B). The inverse relationship between the NK cell subsets in HIV-1 positive individuals, but not HIV-1 negative individuals, was further observed in a correlation analysis (R = −0.633, P = 0.001, Figure 3F). The relative proportions of CD56brightFcγRIIIadim/neg and CD56dimFcγRIIIadim/neg did not differ significantly between HIV-1 positive and HIV-1 negative donors. In contrast to the observed differences in NK cell subsets, FcγRIIIa surface density on neither cytotoxic CD56dimFcγRIIIabright nor CD56negFcγRIIIabright NK cells differed between the two groups (P = 0.948 and P = 0.486, respectively; Figure 3C).
Figure 3
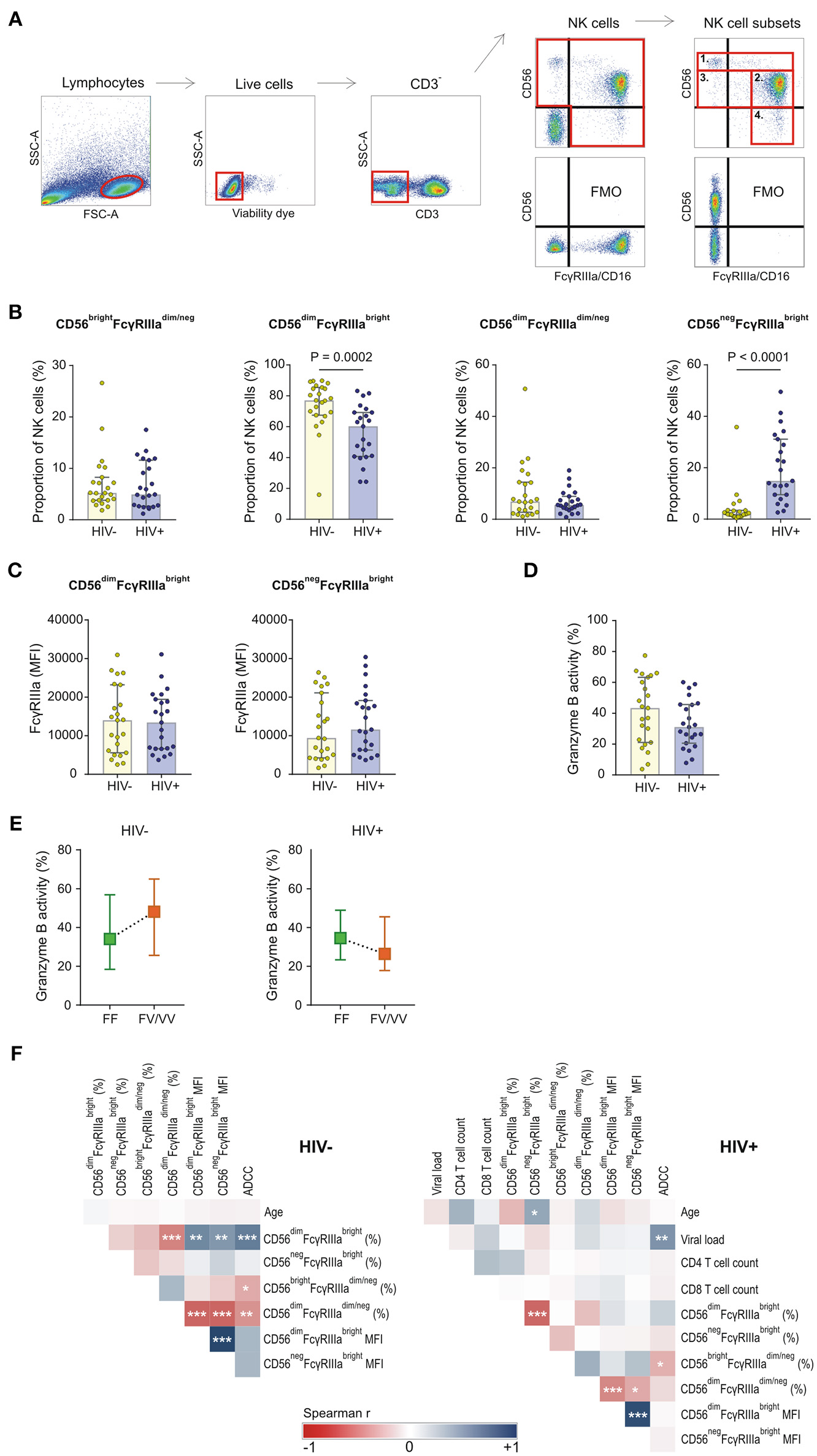
FcγRIIIa expression on NK cell subsets in HIV-1 uninfected and HIV-1 infected individuals matched for FCGR genetic variants. (A) Gating strategy for defining four NK cell subsets: 1. CD56brightFcγRIIIadim/neg, 2. CD56dimFcγRIIIabright, 3. CD56dimFcγRIIIadim/neg, and 4. CD56negFcγRIIIabright; (B) Comparison of NK cell subsets between HIV-1 uninfected and infected individuals; (C) FcγRIIIa surface density on FcγRIIIabright NK cell subsets; (D) ADCC activity of NK cells at a target-to-effector cell ratio of 10:1 with isolated IgG pooled from HIV-1 study participants; (E) Median ADCC responses for individuals homozygous for the FcγRIIIa-158F allele and individuals bearing at least one FcγRIIIa-158V allele; (F) Correlation analysis between demographic, clinical, phenotypic and functional variables in HIV-1 uninfected and HIV-1 infected individuals (***P < 0.001; **P < 0.01; *P < 0.05).
NK Cell-Mediated ADCC Responses
The capacity of CD56+ NK cells to mediate ADCC was tested in an HIV-1-specific granzyme B assay in the presence of pooled IgG isolated from HIV-1-infected South Africans. ADCC responses, measured as granzyme B activity in target cells, were reduced in the HIV-1 positive group compared to the HIV-1 negative group, although not statistically significantly different (31.0 vs. 43.3%, P = 0.184, Figure 3D). In both groups, the ADCC responses were affected by the FcγRIIIa-F158V variant (Figure 3E). Healthy donors bearing at least one V allele had a higher median NK cell-mediated granzyme B activity compared to those homozygous for the F allele (48.1 vs. 34.1%, P = 0.284). This trend was, however, not observed in the HIV-1 positive group. In contrast, HIV-1 positive donors bearing at least one V allele had reduced granzyme B activity compared to those homozygous for the F allele (26.4 vs. 34.6%, P = 0.522).
ADCC responses of both HIV-1 negative and HIV-1 positive donors negatively correlated with the proportion of immunoregulatory CD56brightFcγRIIIadim/neg cells (R = −0.486, P = 0.019; and R = −0.454, P = 0.030, respectively; Figure 3F). Furthermore, ADCC responses of HIV-1 negative donors, but not HIV-1 positive donors, positively correlated with the proportion of CD56dimFcγRIIIabright cells (R = 0.665, P = 0.0005; and R = 0.233, P = 0.284, respectively; Figure 3F). FcγRIIIa expression levels on the cytotoxic CD56dimFcγRIIIabright cell subset did not correlate with ADCC responses in either HIV-1 negative or HIV-1 positive group.
Expression of FcγRIIIa on CD8+ T Cells
During the course of characterizing FcγRIIIa expression on peripheral leukocytes in whole blood obtained from a preliminary cohort of HIV-1 negative and HIV-1 positive donors, we observed a CD8bright T cell subset expressing FcγRIIIa in both groups (Figure 4). The proportion of FcγRIIIa+CD8bright T cells within the CD8bright T cell population varied extensively, ranging from 4.4 to 45.9%, with the median proportion significantly higher in the HIV-1 infected group compared to the healthy control group (17.8 vs. 9.8%, P = 0.002; Figure 4).
Figure 4
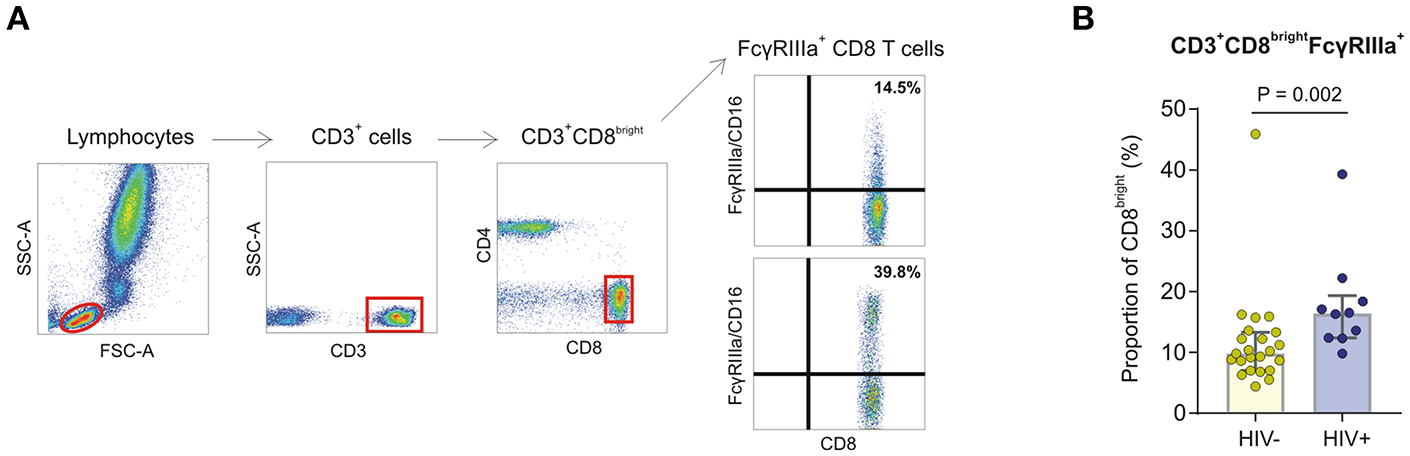
FcγRIIIa expression on CD8 T cells in whole blood obtained from HIV-1 negative and HIV-1 positive donors. (A) Gating strategy showing two representative individuals with low and high proportions of FcγRIIIa+CD8bright T cell subsets. (B) Frequencies of FcγRIIIa+CD8bright T cells in whole blood isolated from HIV-negative healthy controls (n = 23) and HIV-infected individuals (n = 10).
In the present validation study, two CD8+ T cell subsets are identified, designated CD8bright and CD8dim (also referred to as CD8high and CD8low), according to the relative expression of CD8 on CD3+ T cells (Figure 5). The CD8dim subset accounted for 13.9 and 13.4% of the total CD8+ T cell population in HIV-1 negative and HIV-1 positive donors, respectively (P = 0.978). In agreement with our observations in whole blood, 2–44.8% of CD8bright cells and 2–68.4% of CD8dim cells expressed FcγRIIIa. Compared to its FcγRIIIa+CD8bright counterpart, the FcγRIIIa+CD8dim subset expressed significantly higher levels of FcγRIIIa in both the HIV-1 positive and negative group (471 vs. 1610, P < 0.001; and 377 vs. 740, P < 0.001, respectively; data not shown).
Figure 5
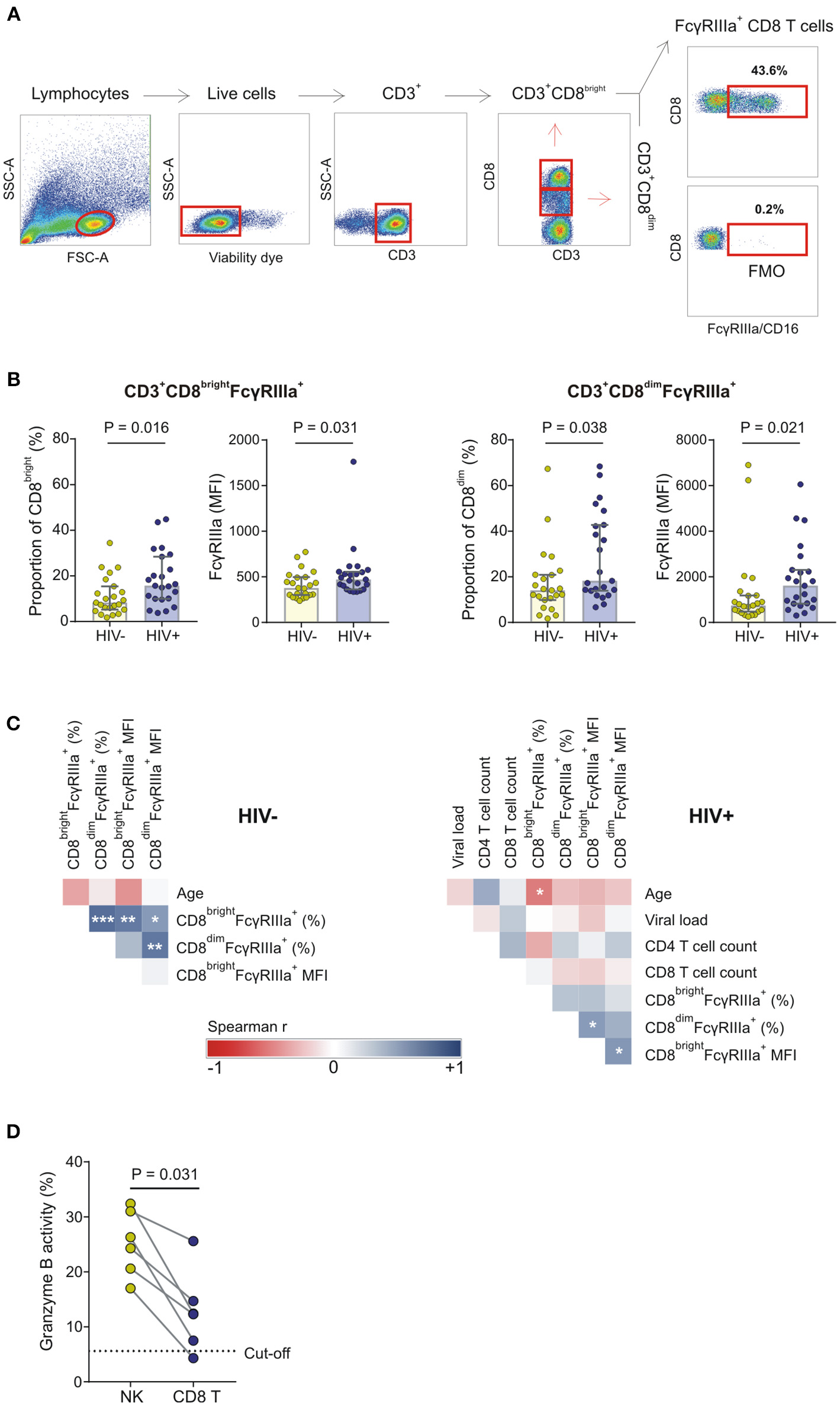
FcγRIIIa expression on CD8 T cells in HIV-1 uninfected and HIV-1 infected individuals matched for FCGR genetic variants. (A) Gating strategy; (B) The proportion of FcγRIIIa+CD8bright and FcγRIIIa+CD8dim T cells and corresponding median fluorescence intensity (MFI) of FcγRIIIa on these FcγRIIIa+CD8+ T cell subsets in a cohort of HIV-1 uninfected and infected individuals; (C) Correlation analysis between demographic, clinical, phenotypic, and functional variables in HIV-1 uninfected and HIV-1 infected individuals; (D) CD8+ T cell-mediated ADCC responses of HIV-1 positive donors relative to autologous NK cell-mediated ADCC responses at the same effector-to-target cell ratio (***P < 0.001; **P < 0.01; *P < 0.05).
Compared to the HIV-1 negative group, the HIV-1 positive group had a significantly higher median proportion of FcγRIIIa+CD8bright and FcγRIIIa+CD8dim T cell subsets (15.7 vs. 8.3%; P = 0.016 and 18.2 vs. 14.1%; P = 0.038, respectively) and correspondingly higher median FcγRIIIa surface densities on both FcγRIIIa+CD8+ T cell subsets (471 vs. 377, P = 0.031; and 1,610 vs. 740, P = 0.021, respectively) (Figure 5B). The proportion of FcγRIIIa+CD8bright and FcγRIIIa+CD8dim cells positively correlated in the HIV-1 negative group (R = 0.671, P = 0.001; Figure 5C). However, this relationship was not observed for the HIV-1 positive group. In the latter group, neither the presence nor levels of FcγRIIIa correlated with CD4+ T cell count or HIV-1 plasma viral load (Figure 5C). However, the proportion of FcγRIIIa on CD8bright T cells negatively correlated with age in both groups, although this correlation was only statistically significant for the HIV-1 positive group (R = −0.510, P = 0.013; Figure 5C).
CD8+ T Cells Mediate ADCC
To assess CD8+ T cell-mediated ADCC responses, CD8+ T cells were positively selected from PBMCs after depletion of NK cells. The proportion of potential contaminating NK cells was determined using flow cytometry and six HIV-1 positive donors were identified with <5% NK cells in their enriched CD8+ T cell fractions (mean: 1.3%; range: 0.3–2.9%). A threshold for positive CD8+ T cell-mediated granzyme B responses (5.6%) was calculated based on the corresponding NK cell granzyme B responses at an NK-to-target ratio of 10:1 (described in section Materials and Methods). Five out of six donors had positive CD8+ T cell-mediated granzyme B responses that ranged from 7.5 to 25.6% (mean: 14.5%; Figure 5E). Overall, the CD8+ T cell-mediated granzyme B activity was comparatively lower than that observed for their NK cell counterparts (P = 0.031).
Discussion
This study set out to comprehensively characterize FcγRIIIa variability and its effect on ADCC responses in HIV-1 positive and HIV-1 negative South African individuals, the population that bears the largest HIV-1 epidemic. Accurate assessment of FcγRIIIa variability requires careful selection of study participants to exclude confounding genetic variables that modify FcγRIIIa expression and FcγRIIIa-mediated cell activation. Study participants within the comparative groups were further matched for the FcγRIIIa-F158V variant that not only alters ADCC responses, but also affects measurements of FcγRIIIa surface expression with the most commonly used anti-FcγRIIIa antibody, clone 3G8 (32).
In comparing a cohort of Black South African HIV-1 negative and HIV-1 positive donors matched for genotypic variants, we confirmed previously described significant differences in the distribution of NK cell subsets, while differences in FcγRIIIa surface density on cytotoxic NK cells were not observed. NK cell-mediated ADCC responses of HIV-1 positive donors were both reduced and differentially affected by the FcγRIIIa-F158V variant compared to HIV-1 negative donors. In addition, FcγRIIIa expression was identified on a subset of cytotoxic CD8 T cells where it potentially contributes to HIV-1-specific ADCC responses in a granzyme B-dependent manner.
In healthy individuals, NK cells typically comprise a dominant CD56dimFcγRIIIabright population and minor populations that include CD56brightFcγRIIIadim/neg, CD56dimFcγRIIIdim/neg and CD56negFcγRIIIabright. Perturbation of NK cell subsets in the presence of an HIV-1 infection has been extensively described (16, 17, 33). In HIV-1 infected individuals, the cytotoxic CD56dimFcγRIIIabright subset contracts with an associated expansion of the CD56negFcγRIIIabright subset. The latter is characterized by higher levels of inhibitory NK cell receptors, lower levels of natural cytotoxicity receptors, and reduced secretion of cytokines compared to the CD56dimFcγRIIIabright subset (16, 17). This hyporesponsive NK cell subset was similarly increased in the South African HIV-1 positive cohort; however, it did not associate with HIV-1 viral load as observed by Mavilio et al. (16, 34). The lack of an association with HIV-1 viral load may be explained by an ~5- to 6-fold lower median HIV-1 plasma viral load of the South African cohort compared to the other cohorts. Moreover, in the present study, NK cell subsets were studied for overnight rested PBMCs as opposed to freshly isolated negatively-selected NK cells.
The dysregulation of NK cell subsets is typically associated with reduced ADCC responses in HIV-1 infected individuals (16, 17, 35, 36). In the present study, lower ADCC responses were similarly observed for HIV-1 infected individuals. Since ADCC capacity was studied for CD56+ NK cells it precluded an analysis of the association between the CD56negFcγRIIIabright subset and ADCC responses, but not for the CD56dimFcγRIIIabright and CD56brightFcγRIIIadim/neg subsets. A negative correlation observed between the CD56brightFcγRIIIadim/neg subset and ADCC responses for both HIV-1 negative and positive donors would suggest that ADCC capacity is similarly affected by the immunoregulatory CD56brightFcγRIIIadim/neg subset in both groups. However, as demonstrated by a positive correlation between the cytotoxic CD56dimFcγRIIIabright subset and ADCC responses for HIV-1 negative donors, but not HIV-1 positive donors, not all factors modulating ADCC capacity may be shared between healthy individuals and HIV-1 infected individuals.
Given the association of ADCC-mediating antibody responses with HIV-1 protective immunity, it could be hypothesized that genetic determinants of NK cell-mediated ADCC capacity, in particular the FcγRIIIa-F158V variant, may associate with HIV-1 acquisition risk or disease progression. The FcγRIIIa-158V isoform has greater avidity for complexes comprising IgG1, IgG3, and IgG4 than the FcγRIIIa-158F isoform and confers increased NK cell activation and ADCC responses in healthy individuals (21–23). Despite its potential contribution to ADCC responses, the FcγRIIIa-158V isoform is yet to be positively associated with HIV-1 acquisition and disease progression. On the contrary, the FcγRIIIa-158V isoform has been associated with an increased risk of HIV-1 infection (37), disease progression (37), and HIV-1-associated Kaposi's sarcoma (KS) and Cryptococcal disease (38, 39). Furthermore, homozygosity for the FcγRIIIa-158V allele associated with a higher rate of HIV-1 infection among vaccinated men in the VAX004 trial (40), while homozygosity for the FcγRIIIa-158F allele associated with greater protection from HIV-1 disease progression in male participants in the RV144 vaccine trial (41). Taken together, these findings are more indicative of FcγRIIIa-158V-mediated antibody-dependent enhancement of infection rather than improved ADCC responses to the benefit of the individual. Alternatively, it is possible that the functional consequence of the FcγRIIIa-F158V isoforms may be different in the presence of an HIV-1 infection and that a different mechanism(s) may underlie the aforementioned associations.
FcγRIIa/FcγRIIIb polymorphic variants, for example, show distinct differences in oxidative burst responses of resting neutrophils; however, once neutrophils were pre-activated with IFNγ and G-CSF these differences were no longer observed (42). Other non-FcγR genetic variants have also been shown to differentially affect gene expression or cytokine production of activated and resting immune cells (43). In the present study, we show a similar trend for the FcγRIIIa-F158V variant, higher ADCC responses for HIV-1 negative donors bearing the FcγRIIIa-158V allele compared to those homozygous for the FcγRIIIa-158F allele, whereas in HIV-1 positive donors this trend was lost or even slightly reversed. The effect of the variant on ADCC responses was, however, not significant in either group. It is possible that the independent effect size of the FcγRIIIa-F158V variant is too small to detect with the current sample size. Larger cohort studies that adjust for other NK cell activation and inhibitory receptors are required to further define the role of this variant in HIV-1 infection. Nonetheless, these findings may partially explain the inconclusive role of FcγRIIIa genetic variants in HIV-1-specific immunity [reviewed by Cocklin and Schmitz (44)].
In addition to modulating the function of an allelic variant, infection can lead to the induction of FcγRIIIa on other cytotoxic cells, including CD8+ T cells. An FcγRIIIa+CD8+ T cell population was first described in the 1980's and has since been characterized in the context of hepatitis C virus and Epstein Barr virus infections (18, 19, 45). These terminally differentiated CD8+ T cells belong to the T effector memory CD45RA+ lymphocyte subset, are perforin positive, directly mediate ADCC ex vivo, and increase in vivo during hyperlymphocytosis (18, 19). In addition to FcγRIIIa, this cell subset also has increased expression of other NK-like receptors including NKG2A, NKG2D, KIR2DL2/L3 and KIR2DL1/S1 when compared to FcγRIIIa−CD8+ T cells (46). The present study validates the expression of FcγRIIIa on cytotoxic CD8+ T lymphocytes and is in agreement with other studies that have consistently showed an increase in the proportion of FcγRIIIa+CD8+ T cells in the presence of a virus infection (18, 19, 46). Compared to CD8bright cells, the surface density of FcγRIIIa was significantly higher on CD8dim cells, a subset characterized by higher activation levels, increased cytotoxicity and increased cytokine production (47–49). The higher proportions of FcγRIIIa+CD8+ T cells in HIV-1-infected individuals suggests that HIV-1 in its own right is a driver of these cell expansions, whereas increasing age associated with reduced proportions of FcγRIIIa-expressing CD8+ T cells. The increase in FcγRIIIa expression on CD8+ T cells in HIV infection contrasts with decreased proportions of FcγRIIIa expressing NK cells. This suggests the development of an ADCC capacity by CD8+ T cells that could compensate to some extent for the reduction in NK cell ADCC function.
Attributing the otherwise innate cell function of ADCC to CD8+ T cells—mediated through expression of FcγRIIIa and engagement of HIV-1-specific antibodies—is reminiscent of other known examples of innate-like unconventional T cell populations. Among these are invariant NKT cells, mucosal-associated invariant T cells, and γδT cells, that recognize foreign/self-lipid presented by non-classical MHC molecules (50, 51). Another interesting unconventional CD8+ T cell subset is one with a prominent innate/memory phenotype identified by co-expression of eomesodermin (Eomes) and KIR/NKG2A (52). The current study highlights the addition of another unconventional CD8+ T cell population, capable of ADCC function, that warrants further investigation.
In conclusion, our findings underscore the importance of expanding studies of HIV-specific antibodies to include the influence of different host cell types that share expression of FcγRs (constitutive or induced), the respective functional cellular capabilities, as well as host genotypes, in the context of the presiding immune milieu which is altered as a consequence of chronic HIV infection. Continuing investigations are warranted to further define effector functions, cytokine production and activation status of these different FcγRIIIa+ NK and CD8 T cell subsets in similarly selected individuals.
Statements
Author contributions
NP recruited HIV-1 negative donors, performed the majority of the experiments, analyzed the data and wrote the manuscript. RL designed the study, contributed to the data analysis, and writing of the manuscript. BD contributed to the flow cytometry experiments. ZW and NM recruited HIV-1 positive donors, CT in her capacity as head of the laboratory, allocated funds toward the study, supervised the research, and provided the necessary infrastructure to perform the work.
Funding
This work is based on the research supported by the Poliomyelitis Research Foundation and the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa, and the Strategic Health Innovation Partnerships (SHIP) Unit of the South African Medical Research Council (a grantee of the Bill & Melinda Gates Foundation). NP is the recipient of bursaries from the South African National Research Foundation, the Poliomyelitis Research Foundation and a University of the Witwatersrand postgraduate merit award.
Acknowledgments
The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Catalog #3957, HIV-IG from NABI and NHLBI. The anti-FcγRIIb/c clone 2B6 was a gift from MacroGenics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- NK
natural killer cell
- IgG
immunoglobulin G
- HIV-1
human immunodeficiency virus 1
- ADCC
antibody-dependent cellular cytotoxicity
- FCGR
Fc gamma receptor gene
- FcγR
Fc gamma receptor protein
- CNR1
gene copy number variable region 1
- EDTA
ethylenediaminetetraacetic acid
- HNA
Human neutrophil antigen
- MLPA
multiplex ligation-dependent probe amplification
- FMO
flurescence minus one
- PBMCs
peripheral blood mononuclear cells
- KIR2DL2/L3
killer immunoglobulin-like receptor 2 DL2/L3
- NKG2A/D
natural killer group 2A or−2D.
Abbreviations
References
1.
Nimmerjahn F Ravetch JV . Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. 10.1038/nri2206
2.
Bournazos S Ravetch JV . Fcγ receptor function and the design of vaccination strategies. Immunity. (2017) 47:224–33. 10.1016/j.immuni.2017.07.009
3.
Rosales C . Fcγ receptor heterogeneity in leukocyte functional responses. Front Immunol. (2017) 8:280. 10.3389/fimmu.2017.00280
4.
Ravetch JV Bolland S . IgG Fc receptors. Annu Rev Immunol. (2001) 19:275–90. 10.1146/annurev.immunol.19.1.275
5.
Nimmerjahn F Ravetch JV . Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. (2005) 310:1510–2. 10.1126/science.1118948
6.
Ackerman ME Dugast A-S Alter G . Emerging concepts on the role of innate immunity in the prevention and control of HIV infection. Annu Rev Med. (2012) 63:113–30. 10.1146/annurev-med-050310-085221
7.
Wren LH Chung AW Isitman G Kelleher AD Parsons MS Amin J et al . Specific antibody-dependent cellular cytotoxicity responses associated with slow progression of HIV infection. Immunology. (2013) 138:116–23. 10.1111/imm.12016
8.
Lewis GK . Role of Fc-mediated antibody function in protective immunity against HIV-1. Immunology. (2014) 142:46–57. 10.1111/imm.12232
9.
Milligan C Richardson BA John-Stewart G Nduati R Overbaugh J . Passively acquired antibody-dependent cellular cytotoxicity (ADCC) activity in HIV-infected infants is associated with reduced mortality. Cell Host Microbe. (2015) 17:500–6. 10.1016/j.chom.2015.03.002
10.
Ahmad R Sindhu ST Toma E Morisset R Vincelette J Menezes J et al . Evidence for a correlation between antibody-dependent cellular cytotoxicity-mediating anti-HIV-1 antibodies and prognostic predictors of HIV infection. J Clin Immunol. (2001) 21:227–33. 10.1023/A:1011087132180
11.
Banks ND Kinsey N Clements J Hildreth JE . Sustained antibody-dependent cell-mediated cytotoxicity (ADCC) in SIV-infected macaques correlates with delayed progression to AIDS. AIDS Res Hum Retrovirus. (2002) 18:1197–205. 10.1089/08892220260387940
12.
Gomez-Roman VR Patterson LJ Venzon D Liewehr D Aldrich K Florese R et al . Vaccine-elicited antibodies mediate antibody-dependent cellular cytotoxicity correlated with significantly reduced acute viremia in rhesus macaques challenged with SIVmac251. J Immunol. (2005) 174:2185–9. 10.4049/jimmunol.174.4.2185
13.
Hessell AJ Hangartner L Hunter M Havenith CE Beurskens FJ Bakker JM et al . Fc receptor but not complement binding is important in antibody protection against HIV. Nature. (2007) 449:101–4. 10.1038/nature06106
14.
Lambotte O Ferrari G Moog C Yates NL Liao HX Parks RJ et al . Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS. (2009) 23:897–906. 10.1097/QAD.0b013e328329f97d
15.
Pollara J Hart L Brewer F Pickeral J Packard BZ Hoxie JA et al . High-throughput quantitative analysis of HIV-1 and SIV-specific ADCC-mediating antibody responses. Cytometry A. (2011) 79:603–12. 10.1002/cyto.a.21084
16.
Mavilio D Lombardo G Benjamin J Kim D Follman D Marcenaro E et al . Characterization of CD56-/CD16+ natural killer (NK) cells: a highly dysfunctional NK subset expanded in HIV-infected viremic individuals. Proc Natl Acad Sci USA. (2005) 102:2886–91. 10.1073/pnas.0409872102
17.
Brunetta E Hudspeth KL Mavilio D . Pathologic natural killer cell subset redistribution in HIV-1 infection: new insights in pathophysiology and clinical outcomes. J Leukoc Biol. (2010) 88:1119–30. 10.1189/jlb.0410225
18.
Bjorkstrom NK Gonzalez VD Malmberg KJ Falconer K Alaeus A Nowak G et al . Elevated numbers of Fc gamma RIIIA+ (CD16+) effector CD8 T cells with NK cell-like function in chronic hepatitis C virus infection. J Immunol. (2008) 181:4219–28. 10.4049/jimmunol.181.6.4219
19.
Clemenceau B Vivien R Berthome M Robillard N Garand R Gallot G et al . Effector memory alphabeta T lymphocytes can express FcgammaRIIIa and mediate antibody-dependent cellular cytotoxicity. J Immunol. (2008) 180:5327–34. 10.4049/jimmunol.180.8.5327
20.
Breunis WB van Mirre E Geissler J Laddach N Wolbink G van der Schoot E et al . Copy number variation at the FCGR locus includes FCGR3A, FCGR2C and FCGR3B but not FCGR2A and FCGR2B. Hum Mutat. (2009) 30:E640–650. 10.1002/humu.20997
21.
Koene HR Kleijer M Algra J Roos D von dem Borne AE de Haas M . Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood. (1997) 90:1109–14.
22.
Wu J Edberg JC Redecha PB Bansal V Guyre PM Coleman K et al . A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. (1997) 100:1059–70. 10.1172/JCI119616
23.
Bruhns P Iannascoli B England P Mancardi DA Fernandez N Jorieux S et al . Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. (2009) 113:3716–25. 10.1182/blood-2008-09-179754
24.
Niederer HA Willcocks LC Rayner TF Yang W Lau YL Williams TN et al . Copy number, linkage disequilibrium and disease association in the FCGR locus. Hum Mol Genet. (2010) 19:3282–94. 10.1093/hmg/ddq216
25.
Mueller M Barros P Witherden AS Roberts AL Zhang Z Schaschl H et al . Genomic pathology of SLE-associated copy-number variation at the FCGR2C/FCGR3B/FCGR2B locus. Am J Hum Genet. (2013) 92:28–40. 10.1016/j.ajhg.2012.11.013
26.
van der Heijden J Breunis WB Geissler J de Boer M van den Berg TK Kuijpers TW . Phenotypic variation in IgG receptors by nonclassical FCGR2C alleles. J Immunol. (2012) 188:1318–24. 10.4049/jimmunol.1003945
27.
Schouten JP McElgunn CJ Waaijer R Zwijnenburg D Diepvens F Pals G . Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. (2002) 30:e57. 10.1093/nar/gnf056
28.
Breunis WB van Mirre E Bruin M Geissler J de Boer M Peters M et al . Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008) 111:1029–38. 10.1182/blood-2007-03-079913
29.
Lassaunière R Tiemessen CT . Variability at the FCGR locus: characterization in Black South Africans and evidence for ethnic variation in and out of Africa. Genes Immun. (2015) 17:93. 10.1038/gene.2015.60
30.
Lassauniere R Shalekoff S Tiemessen CT . A novel FCGR3A intragenic haplotype is associated with increased FcgammaRIIIa/CD16a cell surface density and population differences. Hum Immunol. (2013) 74:627–34. 10.1016/j.humimm.2013.01.020
31.
Schaftenaar E Verjans GM Getu S McIntyre JA Struthers HE Osterhaus AD et al . High seroprevalence of human herpesviruses in HIV-infected individuals attending primary healthcare facilities in rural South Africa. PLoS ONE. (2014) 9:e99243. 10.1371/journal.pone.0099243
32.
Congy-Jolivet N Bolzec A Ternant D Ohresser M Watier H Thibault G . Fc gamma RIIIa expression is not increased on natural killer cells expressing the Fc gamma RIIIa-158V allotype. Cancer Res. (2008) 68:976–80. 10.1158/0008-5472.CAN-07-6523
33.
Fauci AS Mavilio D Kottilil S . NK cells in HIV infection: paradigm for protection or targets for ambush. Nat Rev Immunol. (2005) 5:835–43. 10.1038/nri1711
34.
Mavilio D Benjamin J Daucher M Lombardo G Kottilil S Planta MA et al . Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. Proc Natl Acad Sci USA. (2003) 100:15011–6. 10.1073/pnas.2336091100
35.
Ahmad A Menezes J . Defective killing activity against gp120/41-expressing human erythroleukaemic K562 cell line by monocytes and natural killer cells from HIV-infected individuals. AIDS. (1996) 10:143–9. 10.1097/00002030-199602000-00003
36.
Scott-Algara D Paul P . NK cells and HIV infection: lessons from other viruses. Curr Mol Med. (2002) 2:757–68. 10.2174/1566524023361781
37.
Poonia B Kijak GH Pauza CD . High affinity allele for the gene of FCGR3A is risk factor for HIV infection and progression. PLoS ONE. (2010) 5:e15562. 10.1371/journal.pone.0015562
38.
Rohatgi S Gohil S Kuniholm MH Schultz H Dufaud C Armour KL et al . Fc Gamma receptor 3A polymorphism and risk for HIV-associated cryptococcal disease. mBio. (2013) 4:e00573–13. 10.1128/mBio.00573-13
39.
Foster CB Zhu S Venzon D Steinberg SM Wyvill K Metcalf JA et al . Variant genotypes of FcγRIIIA influence the development of Kaposi's sarcoma in HIV-infected men. Blood. (2000) 95:2386–90.
40.
Forthal DN Gabriel EE Wang A Landucci G Phan TB . Association of Fcgamma receptor IIIa genotype with the rate of HIV infection after gp120 vaccination. Blood. (2012) 120:2836–42. 10.1182/blood-2012-05-431361
41.
Kijak G . (2012). “Modulation of Vaccine Effect by FcGamma Receptor 3a Genetic Polymorphism in RV144”, in 19th Conference on Retroviruses and Opportunistic Infections (CROI). (Seattle, WA).
42.
van der Heijden J Nagelkerke S Zhao X Geissler J Rispens T van den Berg TK et al . Haplotypes of FcgammaRIIa and FcgammaRIIIb polymorphic variants influence IgG-mediated responses in neutrophils. J Immunol. (2014) 192:2715–21. 10.4049/jimmunol.1203570
43.
Jonkers IH Wijmenga C . Context-specific effects of genetic variants associated with autoimmune disease. Hum Mol Genet. (2017) 26:R185–92. 10.1093/hmg/ddx254
44.
Cocklin SL Schmitz JE . The role of Fc receptors in HIV infection and vaccine efficacy. Curr Opin HIV AIDS. (2014) 9:257–62. 10.1097/COH.0000000000000051
45.
Lanier LL Kipps TJ Phillips JH . Functional properties of a unique subset of cytotoxic CD3+ T lymphocytes that express Fc receptors for IgG (CD16/Leu-11 antigen). J Exp Med. (1985) 162:2089–106. 10.1084/jem.162.6.2089
46.
Clemenceau B Vivien R Debeaupuis E Esbelin J Biron C Levy Y et al . FcγRIIIa (CD16) induction on human T lymphocytes and CD16pos T-lymphocyte amplification. J Immunother. (2011) 34:542–9. 10.1097/CJI.0b013e31822801d4
47.
Trautmann A Rückert B Schmid-Grendelmeier P Niederer E Bröcker E-B Blaser K et al . Human CD8 T cells of the peripheral blood contain a low CD8 expressing cytotoxic/effector subpopulation. Immunology. (2003) 108:305–12. 10.1046/j.1365-2567.2003.01590.x
48.
Kienzle N Baz A Kelso A . Profiling the CD8low phenotype, an alternative career choice for CD8 T cells during primary differentiation. Immunol Cell Biol. (2004) 82:75–83. 10.1111/j.1440-1711.2004.01210.x
49.
Falanga YT Frascoli M Kaymaz Y Forconi C Ong'echa JM Bailey JA et al . High pathogen burden in childhood promotes the development of unconventional innate-like CD8+ T cells. JCI Insight. (2017) 2:93814. 10.1172/jci.insight.93814
50.
Lanier LL . Shades of grey–the blurring view of innate and adaptive immunity. Nat Rev Immunol. (2013) 13:73–4. 10.1038/nri3389
51.
Godfrey DI Uldrich AP McCluskey J Rossjohn J Moody DB . The burgeoning family of unconventional T cells. Nat Immunol. (2015) 16:1114–23. 10.1038/ni.3298
52.
Jacomet F Cayssials E Barbarin A Desmier D Basbous S Lefevre L et al . The hypothesis of the human iNKT/Innate CD8(+) T-cell axis applied to cancer: evidence for a deficiency in chronic myeloid leukemia. Front Immunol. (2016) 7:688. 10.3389/fimmu.2016.00688
Summary
Keywords
NK cells, CD8 T cells, antibody-dependent cellular cytotoxicity, Fc gamma receptor, polymorphism, HIV, infection
Citation
Phaahla NG, Lassaunière R, Da Costa Dias B, Waja Z, Martinson NA and Tiemessen CT (2019) Chronic HIV-1 Infection Alters the Cellular Distribution of FcγRIIIa and the Functional Consequence of the FcγRIIIa-F158V Variant. Front. Immunol. 10:735. doi: 10.3389/fimmu.2019.00735
Received
17 December 2018
Accepted
19 March 2019
Published
10 April 2019
Volume
10 - 2019
Edited by
Guido Ferrari, Duke University, United States
Reviewed by
Francisco Borrego, BioCruces Health Research Institute, Spain; Bin Su, Capital Medical University, China
Updates
Copyright
© 2019 Phaahla, Lassaunière, Da Costa Dias, Waja, Martinson and Tiemessen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline T. Tiemessen carolinet@nicd.ac.za
This article was submitted to NK and Innate Lymphoid Cell Biology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.