 Irbaz Bin Riaz
Irbaz Bin Riaz Warda Faridi
Warda Faridi Mrinal M. Patnaik
Mrinal M. Patnaik Roshini S. Abraham
Roshini S. Abraham- 1Division of Hematology, Department of Medicine, Mayo Clinic, Rochester, MN, United States
- 2Department of Hematology, University of Arizona, Tucson, AZ, United States
- 3Department of Pathology and Laboratory Medicine, Nationwide Children's Hospital, Columbus, OH, United States
Primary immunodeficiencies and immune dysregulatory disorders (PIDDs; now referred to as inborn errors in immunity) are rare disorders with a prevalence of 41. 4 or 50.5 per 100,000 persons (1). The incidence of malignancy in PIDD patents is the second-highest cause of death in children as well as adults, after infection, and is higher in certain PIDDs compared to others. We performed a systematic review of the literature to identify reports of B cell and T cell neoplasias in PIDDs and clustered them based on their classification in the IUIS schema. As would be expected, higher susceptibility to malignancies are typically reported in patients with Common Variable Immunodeficiency (CVID), combined immunodeficiencies affecting cellular immunity, in particular, DNA repair defects, or in the context of impaired immune regulatory control. There is not much evidence of increased risk for cancer in patients with innate immune defects, indicating that not all types of infection or genetic susceptibility predispose equally to cancer risk. Viral infections, in particular EBV, HHV and HPV, have been shown to increase susceptibility to developing cancer, but also patients with defects in immune regulation, such as Autoimmune Lymphoproliferative Syndrome (ALPS), activated p110delta syndrome (APDS type 1) and IL-10 receptor deficiency among others have a higher incidence of neoplastic disease, particularly lymphomas. In fact, lymphomas account for two-thirds of all malignancies reported in PIDD patients (2), with either a combined immunodeficiency or DNA repair defect predominating as the underlying immune defect in one registry, or antibody deficiencies in another (3). The vast majority of lymphomas reported in the context of PIDDs are B cell lymphomas, though T cell lymphomas have been reported in a few studies, and tend to largely be associated with chromosomal breakage disorders (4) or Cartilage Hair Hypoplasia (5). There appears to be a much higher prevalence of T cell lymphomas in patients with secondary immunodeficiencies (6), though this could reflect treatment bias. We reviewed the literature and summarized the reports of B and T cell lymphoma in PIDD patients to survey the current state of knowledge in this area.
Introduction
Monogenic and other genetic defects of the immune system, now collectively grouped as primary immunodeficiencies and immune dysregulatory disorders (PIDDs) affect various components of the immune system with susceptibility to infections, but also to autoimmunity, malignancies, and other manifestations of immune dysregulation (7–9). The number of genetically defined PIDDs is increasing with the current tally at well over 300 genes (10), and several of these are associated with an increased predisposition to developing neoplastic disease (11). Early studies have suggested a variable prevalence of malignancies in PIDDs with approximately 25% affected with cancer at the higher end of the spectrum (2). A recent large study spanning 12 years for patients enrolled in a national registry (USIDNET) revealed an age-adjusted cancer risk, as well as a gender-associated cancer risk with male patients predominating in this cohort (12). The largest proportion of risk was conferred by susceptibility to hematopoietic malignancies rather than solid tumors, in particular lymphoma. Herein, we describe a targeted literature review on the associations of B and T cell lymphomas with PIDDs, particularly with regard to specific immune defects. We will also reflect on current state of knowledge as regards to pathogenesis and management of lymphoid neoplasias in PIDDs.
Methods and Results
In these sections, we describe the strategy to identify relevant citations gathered for this review and a summary of the results. A comprehensive search of the MEDLINE database was initially conducted from its inception to October 17th, 2018. The search was updated on February 1st, 2019. Controlled vocabulary supplemented with keywords (which included lymphoma) was used to search for individual primary immunodeficiencies as per the IUIS 2017 classification. Search results and study selection are outlined in Figure 1. Number of cases and type (B, T or unspecified) are shown in Figure 2. The cases of B cell lymphomas, T cell lymphomas and unspecified lymphomas in PIDDs are summarized in Tables 1–3 respectively.
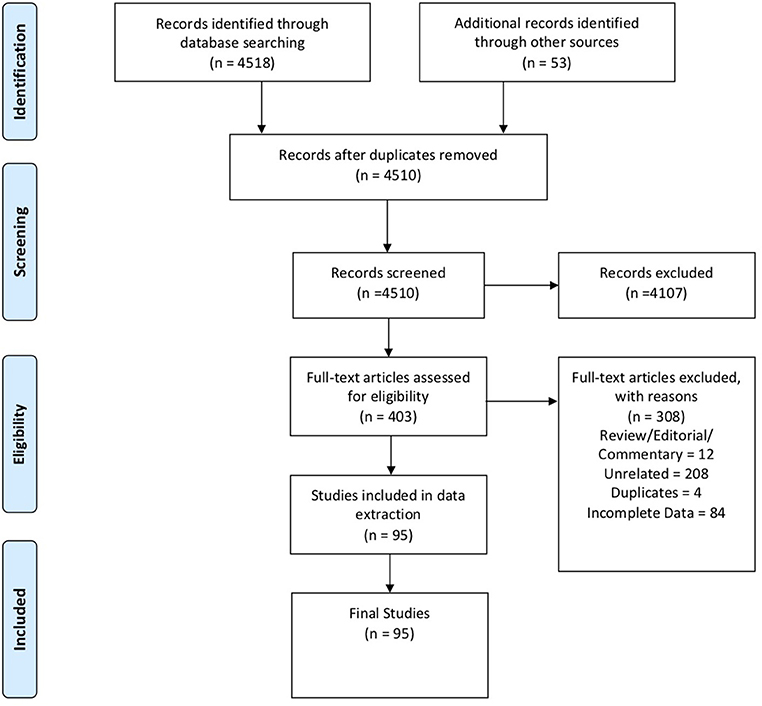
Figure 1. PRISMA flow diagram: The PRIMSA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) diagram details our search and selection process applied during the overview.
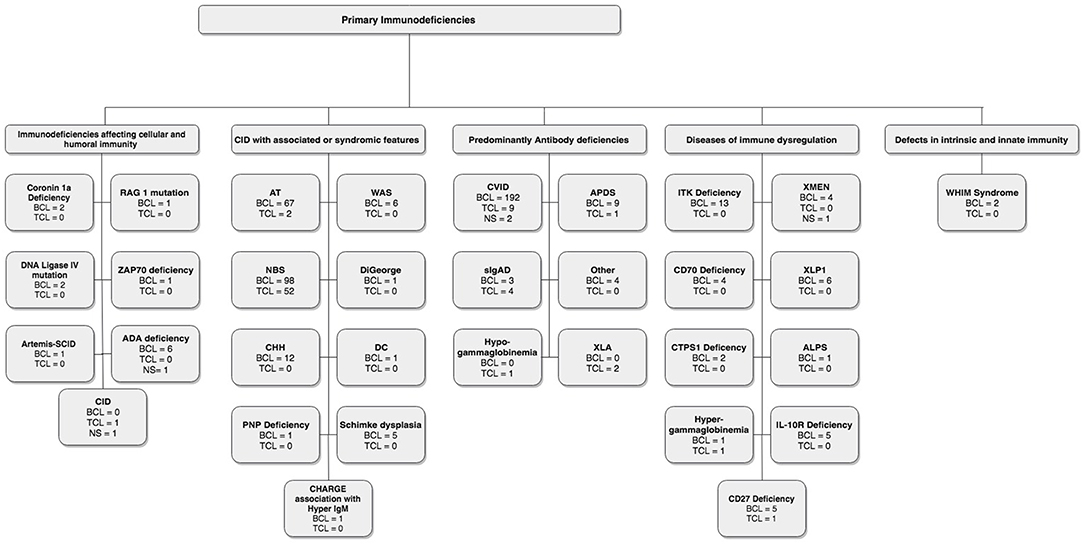
Figure 2. Lymphoma distribution according to IUIS classification.
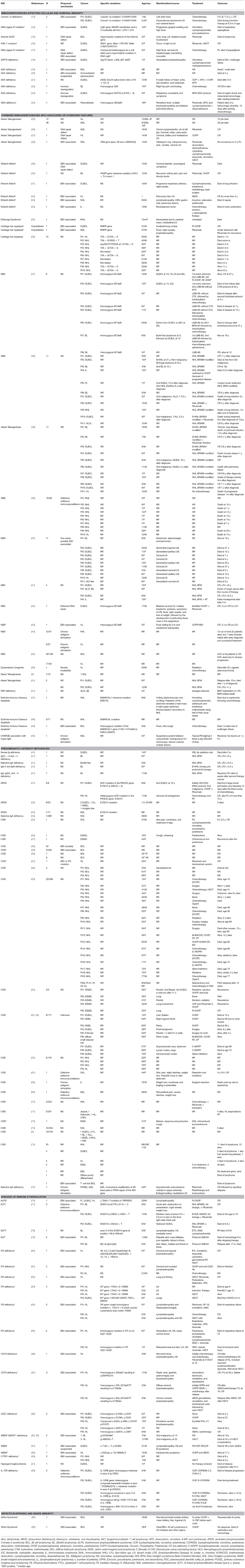
Table 1. Summary of B cell lymphomas in PIDDs.
B Cell Lymphomas in PIDDs
A thorough search of the literature using Medline database (via PubMed; Figure 1) identified 86 studies reporting B cell lymphoma in PIDD patients. Table 1 gives details of the 456 patients identified from literature plus two unpublished cases from our center (Mayo Clinic, Rochester). The types of B cell lymphoma were unspecified non-Hodgkin Lymphoma (NHL) (37%, n = 171), diffuse large B cell lymphoma (DLBCL) (15%, n = 68), Hodgkin lymphoma (HL) (13%, n = 59), marginal zone lymphoma (MZL) including extranodal and intranodal MZL (5%, n = 23), Burkitt lymphoma (BL) (4%, n = 17) and diffuse histiocytic lymphoma (DHL) (0.4%, n = 2). Unlike T cell lymphomas where most of cases were reported in males, gender distribution was similar in males (29%, n = 130) and females (34%, n = 157), it was not specified (NS) in 37% (n = 169). The age of onset/diagnosis of lymphoma ranged from 7 months to 76 years (median age: 12 years). EBV association was seen in 25% (n = 113) of the patients. The majority of patients received combination chemotherapy as a standard treatment. While allogeneic hematopoietic cell transplantation (HCT) was not performed in many of these cases, it appeared to be successful in achieving a complete response (CR) in some cases. Serious infectious complications and death were frequently associated with chemotherapy treatment. Individual groups of PIDDs are examined in further detail based on the IUIS classification.
IUIS: Immunodeficiencies Affecting Cellular and Humoral Immunity
There were 12 studies which included 13 patients with B cell lymphoma. Adenosine deaminase 1 (ADA1) deficiency was most common immunodeficiency disease in this category (19–24). The other 7 cases include patients with an underlying diagnosis of Coronin 1A and DNA Ligase IV deficiencies, Artemis-SCID, RAG1, and ZAP70 defects (13–18, 24, 107). Of these 13 cases, 69% (n = 9) patients were females, 15% (n = 2) were males and 15% (n = 2) were NS. DLBCL (62%, n = 8) was the most common type of B cell neoplasm identified followed by unspecified B cell lymphoma (15%, n = 2). The median age at presentation was 1.5 years (range 0.9–14 years) and all patients were EBV-positive. The most common clinical presentation was lymphadenopathy and high fevers. The underlying etiology for the development of lymphoma appeared to be DNA repair defects and EBV association. All patients were treated with some form of combination chemotherapy, most commonly, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone (R-CHOP). Of these 7 cases, only 2 patients survived and were in complete remission at follow-up. One patient who developed DLBCL in the setting of RAG1 deficiency had a partial response with rituximab, which was consolidated by HCT from an HLA-matched unrelated donor. Though there is no long-term follow-up data in this report, the patient had no evidence of disease at 3 years follow-up (16).
Adenosine Deaminase 1 (ADA1) Deficiency
B cell lymphoma with ADA1 deficiency was seen in six patients, three of whom had EBV association (19–24). Only one patient was reported to be in complete remission 20 months after diagnosis, others died despite treatment.
IUIS: Combined Immunodeficiency Disorders With Associated or Syndromic Features
In this category, 27 studies plus two unpublished cases reported B cell lymphomas in 191 patients with combined immunodeficiency disorders with associated or syndromic features. Of these, 41% (n = 78) cases were associated with EBV infection, and the lymphoma was diagnosed at median age of 10 years. In this cohort there appeared to be a preponderance of males at 38% (n = 73) with females accounting for 32% (n = 62) and 30% (n = 56). Some of the more common genetic disorders are discussed further.
Ataxia Telangiectasia (AT)
Over one-third (35%, n = 67) of the reported cases of lymphoma in this IUIS category were in the setting of ataxia telangiectasia, which is an autosomal recessive disorder with immunodeficiency, DNA repair defects and neurological complications(3, 25–28, 38, 41). Lymphomas were diagnosed across all ages of patients typically manifesting before 10 years of age with the oldest cases being reported at 16 years of age. Original reports reported the histology as DLBCL or unspecified NHL in 75% (n = 50), HL in 21% (n = 14), BL in 3% (n = 2), and MZL in 1% (n = 1) of cases. The most common presenting symptoms included extensive lymphadenopathy. A French study reported a crude incidence of cancer of 24.7% in AT with B cell lymphoid malignancies representing the majority of cases. The pathological diagnosis of lymphoma in these patients can be challenging, and it is relevant to note that three of the four T-cell NHLs, based on pathology, were reclassified as B-cell NHLs after centralized pathology review. The median overall survival (OS) decreased from 24 to 15 years in AT patients with malignant disease. In fact, the common causes of mortality in this group were either cancer (47%) or infectious complications (34%). There was a trend toward increased survival if there was an excellent response to chemotherapy. Though the overall prognosis in AT remains relatively poor, a subset of these of patients might benefit from treatment of the malignancy with an improved survival (25).
Wiskott -Aldrich Syndrome (WAS)
There were 6 studies identified, which reported B cell lymphoma in 6 patients (median age 14 years) with WAS, which is an X-linked PIDD (29–34). DLBCL (33%, n = 2) was the most common type of B cell lymphoma in this group, followed by BL, HL, unspecified NHL and immunoblastic B cell lymphoma. As with many of the other PIDDs, EBV association was present in all these types. It is relevant to note that only 33% patients (n = 2, HL and Burkitt lymphoma) had complete remission on follow-up, while the others died of disease progression or its complications. In a multi-center cohort of X-linked thrombocytopenia (XLT) patients, certain WAS genetic variants associated with XLT, a milder form of WAS, predisposed to a higher incidence of developing lymphoma suggesting that these patients would benefit from treatment with HCT (108).
DiGeorge Syndrome (DGS)
In this literature review, DGS did not appear to be associated with a high incidence of lymphomas, with only one reported case in a 10-month female who developed immunoblastic B cell NHL(35). Similar to other PIDDs this case was also associated with EBV infection. The patient manifested multi-system complications with hemiparesis due to a cerebral mass, mediastinal lymphadenopathy, liver and kidney involvement, and succumbed to the disease prior to initiation of therapy.
Cartilage Hair Hypoplasia (CHH)
CHH is a syndromic PIDD due to genetic defects in the RMRP gene and manifests with variable degree of immunodeficiency. While many patients who manifest with severe cellular immunodeficiency early in life may receive an HCT, patients with initially milder immunodeficiency may develop complications later in life, including malignancy. We identified 10 reported cases, with a median age of 29 years, equally divided between males and females. Majority of these cases were NHL (90%, n = 9) while the remaining (10%, n = 1) were HL. Interestingly, none of these were associated with EBV (36). This is in contrast to the 2 cases of adult CHH (male patients) in our center (unpublished, Mayo Clinic, Rochester), who were both EBV-positive and diagnosed in their 3rd to 4th decade of life. One of these patients was diagnosed with DLBCL after an incidental work-up of a lung nodule, while the other patient had MZL with recurrent fevers and night sweats. The patient with DLBCL was treated with R-CHOP and had CR, while the patients with MZL had recurrent disease, and was treated with rituximab. Therefore, like other PIDDs, CHH may also present with EBV-associated lymphomas, and it remains unclear if the EBV diagnosis could have been missed in other cases reported in the literature.
Nijmegen Breakage Syndrome (NBS)
In this review strategy, 9 studies with 97 cases of lymphomas in patients with NBS were identified (37–45). NBS is a syndromic DNA repair defect, and in these published reports, 76% (n = 74) of patients presented with unclassified NHL and DLBCL. Most cases (82%, n = 80) were EBV-negative. The underlying DNA repair defect puts these patients at high risk, unless they receive HCT early after diagnosis.
Dyskeratosis Congenita (DC)
Among the short telomere syndromes, while one would postulate high-risk of malignancy due to premature cellular senescence, this literature search only yielded a single case of an adult DC patient with HL not associated with EBV (46). The treatment strategy included combination chemotherapy and radiation, which likely was not appropriate in the context of the patient's underlying genetic defect, and presumed radiosensitivity, as the patient subsequently developed a gastric carcinoma and died of the disease. Short telomere syndromes are known to be associated with radiosensitivity, and therefore, radiation therapy is probably not a recommended treatment modality in this group, as well as in the patients with DNA repair defects.
Schimke Immuno-Osseous dysplasia
We identified 3 studies, with 5 cases of B cell lymphoma (47–49). EBV association was seen in a single case (20%, n = 1). Outcome data was given for two patients, both of whom died with complications soon after starting chemotherapy.
CHARGE Association With Hyper IgM Syndrome
We identified one report of a 5-year-old girl, with nonclassical CHARGE association and elevated IgM levels who developed bilateral extranodal (ocular) MZL. She was treated with topical interferon alpha with subsequent complete resolution of disease (50).
IUIS: Predominantly Antibody Deficiencies
In this literature review, we identified 27 studies, which reported B cell lymphomas in 208 patients with predominantly antibody deficiencies. The median age at presentation was 46 years (range: 2–76 years) with a gender distribution of 34% (n = 71) female, 13% (n = 27) male, and 53% (n = 110) NS.
Common Variable Immune Deficiency (CVID)
Twenty-two studies of 192 CVID patients with B-cell lymphoma are presented in Table 1 (3, 54–74). Common lymphomas included unclassified NHL (32%, n = 62), MALT lymphoma (EMZL) (7%, n = 14), DLBCL (5%, n = 9), and HL (4%, n = 8). Of these, 31% (n = 60) cases appeared to be associated with EBV infection. Possible mechanisms of lymphomagenesis in these different cohorts included chronic antigenic stimulation and defective immune surveillance. The treatments of choice in these patients included surgical resection and/or radiotherapy with chemotherapy. Common Variable Immunodeficiency (CVID) is the most common adult humoral immunodeficiency, both in US and European studies. There are several reports documenting an increased risk of lymphoma in these patients. In a large cohort of 176 patients with CVID, an increased incidence of lymphoma (obs = 4; SIR = 12.1; 95% CI = 3.3–31.0) was noted (54). In an ESID (European Society for Immunodeficiencies) registry study of 2,212 CVID patients, a subset (n = 902) analysis identified 3% of patients with lymphoma (106). However, in another study from a single center, which followed 473 CVID patients over four decades, the incidence of lymphoma was higher at 8.2% (74).
Selective IgA Deficiency
Three studies reported three cases of selective IgA deficiency with B cell lymphoma; an adult patient with apparent selective IgA deficiency who presented with a primary cutaneous MZL, a 38-year-old male with unclassified NHL and a 2-year-old boy with Burkitt-like lymphoma (41, 54, 75). It is relevant to note that a combined Danish and Swedish study of 386 patients with IgA deficiency did not show an increased risk for cancer (standardized incidence ratio = 1.0) (54).
Activated Phosphoinositide 3-Kinase D Syndrome (APDS)
Nine patients with gain of function (GOF) mutation in PIK3CD gene (APDS) developed B cell lymphoma in the three studies identified in this review (51–53). DLBCL was the most common type (33%, n = 3). An EBV association was not seen in these particular cases.
Other Ig Deficiencies
Seidemann et al. reported four cases of B cell lymphoma in different Ig subclass deficiencies (excluding selective IgA deficiency) (41). Seventy-Five Percent (n = 3) had Burkitt lymphoma (BL). Two died during treatment with chemotherapy, two were alive and in complete remission at last follow up.
IUS: Diseases of Immune Dysregulation
In this category, 42 cases from 20 studies were identified, and of these, 76% (n = 32) had an association with EBV (41, 76–94). The median age of the reported cases was 5 years (range: 1–33 years), 62% (n = 26) were male, 31% (n = 13) female and 7% (n = 3) were NS. HL (48%, n = 20) was the most common subtype in these disorders of immune dysregulation, followed by DLBCL (29%, n = 12), BL (12%, n = 5), unspecified NHL (7%, n = 3) and composite lymphoma—FL, DLBCL, HL (2%). Specific examples are discussed further.
X-linked Lymphoproliferative Syndrome Type 1 (XLP-1)
In patients with X-linked lymphoproliferative syndrome type 1 due to genetic defects in SH2D1A, there were 3 studies with six patients who developed B cell lymphoma [4 BL, 2 DLBCL (77–79)]. The median age at diagnosis of lymphoma was 5 years (range: 1–14 years), and of these cases, 50% (n = 3) were EBV-associated and 50% (n = 3) had no apparent EBV association. Since XLP1 is associated with a defective immune response to EBV related to impaired cellular cytotoxicity it is not unexpected that there would be impaired immune surveillance in these patients (109). The two patients who had a DLBCL not related to EBV presented with a testicular mass and right chest mass, respectively. In the cases with the BL, clinical presentation involved lymphadenopathy and palpable abdominal mass/obstruction or cerebellar tumor. With the exception of a single case, all patients died within 5 years of diagnosis regardless of treatment modality used.
Interleukin-2-Inducible T-Cell Kinase (ITK) deficiency
Patients with ITK deficiency have an intrinsic susceptibility to EBV. In six studies, 13 patients with ITK deficiency (Table 1) who developed B cell lymphoma were identified, and all were associated with EBV (80–85). The most common was HL (84%, n = 11) followed by NHL (7%, n = 1) and DLBCL (7%, n = 1). The median age at presentation was 5 years (range: 1–6) with 54% (n = 7) female and 46% (n = 6) male, presenting clinically with lymphadenopathy and hepatomegaly. The treatment of choice was chemotherapy and HCT.
Autoimmune Lymphoproliferative Syndrome (ALPS)
While there are several genetic defects associated with ALPS or ALPS-like disease, the most frequent genetic defect associated with a classic ALPS-like phenotype is heterozygous germline pathogenic variants in the FAS gene. In Table 1, there was a single report an EBV-associated composite lymphoma (FL, DLBCL, HL) in an adult male with ALPS, which was treated with R-CHOP resulting in CR (76).
IL10-R Deficiency
Monogenic inflammatory bowel disease (IBD) can be associated with complex presentations, and patients with genetic defects in the IL-10 receptor (IL10RA and IL10RB) have particularly severe disease with additional complications. A single case report (110) and a case series has reported DLBCL in 5 patients with IL10R deficiency (94). There was no EBV association noted in any of these cases, and the median age of developing lymphoma was 5 years (range: 5–6 years). Interestingly, all patients in these series were male. Chemotherapy with COP (cyclophosphamide, vincristine, prednisone), COPADM (cyclophosphamide, vincristine, prednisone, doxorubicin, methotrexate), CYM (cytarabine, methotrexate), and ICE (ifosfamide, carboplatin, etoposide) was used with variable success. Among the five patients, two died due to disease progression during treatment, while three who received HCT remained alive and in remission on follow-up. It is evident that in this context, HCT is not only curative but may very well prevent the occurrence or recurrence of lymphoma (111).
CD70 Deficiency
B cell lymphoma was reported in four male patients under the age of 20 with CD70 deficiency across two studies (86, 87). All patients presented with HL, associated with EBV infection and were managed with chemotherapy and radiotherapy. Two patients underwent HCT and complete remission was achieved in all cases.
CD27 Deficiency
We identified one study reporting five cases (mostly females) with CD27 deficiency who developed B cell lymphoma (88). Three patients were diagnosed with HL, and two with DLBCL. All were related to EBV infection. Only two patients with HL treated with the EuroNet-PHL-C1 (EuroNet-Pediatric Hodgkin's Lymphoma Group-C1) protocol and ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) plus radiotherapy respectively were alive and in complete remission.
XMEN (MAGT1) Deficiency
Four studies with four patients were reported to develop B cell lymphoma in the context of MAGT1 deficiency (89–92). All were males with EBV-associated lymphomas. The ages ranged from 7 to 57 years, and the distribution of B cell lymphomas included 2 HL, 1 BL, and 1 DLBCL.
CTP Synthase 1 (CTPS1) deficiency
One study reported two cases with EBV- associated unspecified NHL in patients with CTPS1 deficiency (93).
IUIS: Defects in Intrinsic and Innate Immunity
WHIM (Warts, Hypogammaglobulinemia, Immunodeficiency, and Myelokathexis) Syndrome
WHIM syndrome is caused by gain-of-function defects in the CXCR4 gene frequently associated with severe neutropenia and variable degree of immunodeficiency. There are two reports (Table 1) of B cell lymphoma (type not specified) in two adult patients with WHIM syndrome (95, 96). Both were associated with EBV infection leading to lymphoproliferation and ultimately lymphoma. Treatment in both cases was with CHOP chemotherapy, and one patient had a CR, while the other patient who presented with intestinal lymphoma had no response and died of intestinal perforation.
T Cell Lymphomas in PIDs
The incidence of T cell lymphomas is infrequent in PIDDs (Table 2). In this particular literature review, 74 patients were identified through 20 different reports. The types of T cell lymphoma identified included T cell non-Hodgkin lymphoma (TNHL) 36%, T cell lymphoblastic lymphoma (T-LL) 32%, peripheral T cell lymphomas (PTCL) 9%, granulomatous cutaneous T-cell lymphoma (G-CTCL) 4%, anaplastic large cell lymphoma (ALCL) 4%, and T cell-diffuse lymphocytic lymphoma (T-DLL) 1%. Of these cases, 41% (n = 30) were male, 10% (n = 10) were females and 46% (n = 34) did not report gender. The median age at the diagnosis of lymphoma was 13 years (range 0.5–68 years). It is interesting and relevant to note that the cases of T cell lymphomas were mostly observed in patients with predominantly antibody deficiencies or combined immunodeficiencies with associated or syndromic features. Only a single report is available of a patient with a combined immunodeficiency (CID) (Table 2), who developed T-LL and was treated with full dose NHL-BFM95 (Non-Hodgkin Lymphoma-Berlin-Frankfurt-Münster 95) on a phase1 clinical trial (4), and was in CR but subsequently died of sepsis.
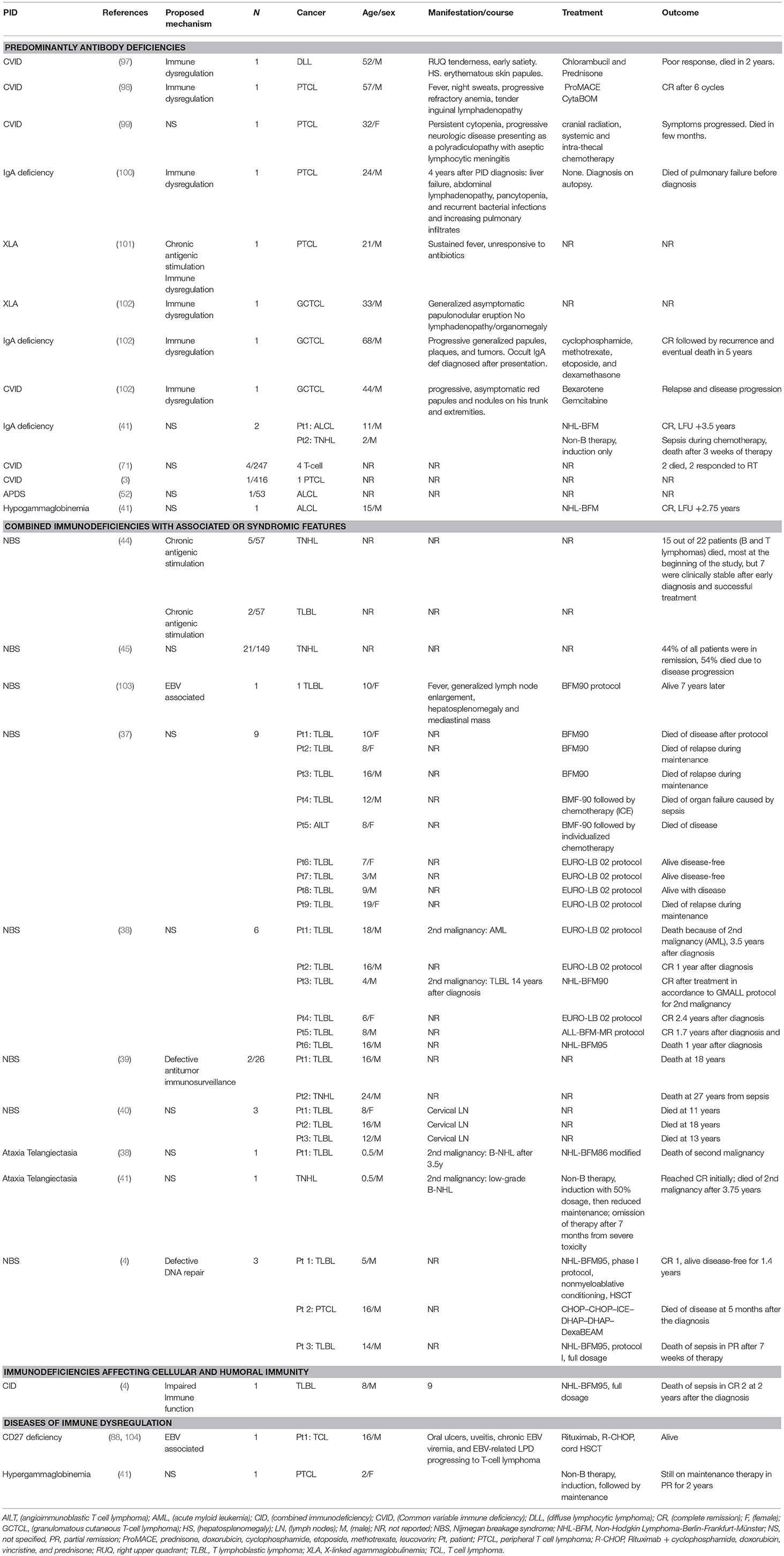
Table 2. Summary of T cell lymphomas in PIDDs.
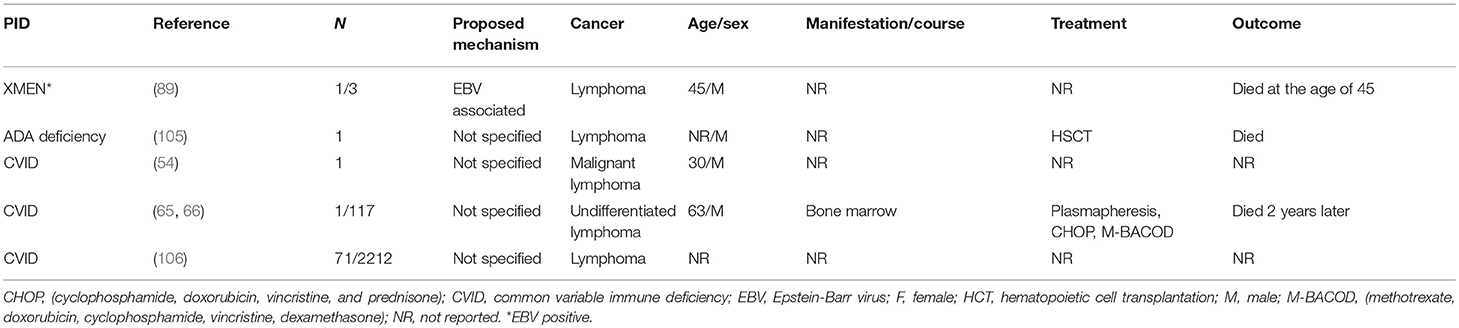
Table 3. Summary of unspecified lymphoma in PIDDs.
IUIS: Diseases of Immune Dysregulation
CD27 Deficiency
A 16 year old male presented with oral ulcers, uveitis, chronic EBV viremia, and EBV-related lymphoproliferative disorder (LPD) progressing to T-cell lymphoma was treated with rituximab, R-CHOP, and subsequently a cord blood HCT, and the patient was alive at last follow-up (104).
IUIS: Immunodeficiencies Affecting Cellular and Humoral Immunity
CID
Only a single report of a patient with a combined immunodeficiency (CID) is reported (Table 2) who developed T-LL and was treated with full dose NHL-BFM95 on a phase1 clinical trial, and was in CR but subsequently died of sepsis (4).
IUIS: Predominantly Antibody Deficiencies
In patients with humoral immune defects, there were 10 studies with 17 patients reported (Table 2). The types of T cell lymphomas observed were PTCL (29% n = 5), G-CTCL (18%, n = 3), ALCL (18%, n = 3), T-DLL (5% n = 1), and TNHL (5%, n = 1). The median age at diagnosis was 32 years (range: 2–68 years) with a distribution of males (59%, n = 10), females (6%, n = 1) and NS (35%, n = 6).
Common Variable Immunodeficiency (CVID)
Of the patients with humoral defects, 53% (n = 9) had CVID across six studies, and the main types of T cell lymphomas included were PTCL, T-DLL, G-CTCL, and 4 unspecified T-cell lymphomas (3, 71, 97–99, 102). The treatment modalities used include Prom ACE (prednisone, doxorubicin, cyclophosphamide, etoposide, methotrexate, leucovorin), CytaBOM (cytarabine bleomycin, vincristine, methotrexate), radiation, systemic and intra-thecal chemotherapy. A single CVID patient presented with G-CTCL, which relapsed and progressed after treatment with bexarotene and gemcitabine. Another CVID patient developed T-DLL and had a poor response to chlorambucil and prednisone and succumbed to disease within 2 years.
Selective IgA Deficiency (sIgAD)
As with B cell lymphoma, there were three reports of four patients with sIgAD who developed T cell lymphoma (G-CTCL, PTCL, ALCL, TNHL) (41, 100, 102). All were males, and the diagnosis was made by autopsy in one patient, while the diagnosis of occult IgA deficiency was made in the second patient after the development of malignant disease. Though this patient was treated with chemotherapy with apparent CR, there was subsequent relapse of disease and mortality within 5 years. In patients classified as having sIgAD and malignant disease, it remains an open question as to whether there was a more severe underlying immunological defect that was not identified at the time.
Hypogammaglobinemia
Seidemann et al. reported a case of 15-year-old male with hypogammaglobulinemia who developed ALCL (41). Complete remission was achieved on treatment with chemotherapy. Last follow up was almost three years after diagnosis. A molecular etiology for the hypogammaglobulinemia was not available in this patient.
XLA
In the 2 patients reported with XLA, the types of T cell lymphoma observed were G-CTCL and PTCL (101, 102).
IUIS: Combined Immunodeficiencies With Associated or Syndromic Features
In this category, we identified nine studies with 54 patients. Median age was 10 years (0.5–24). Similar to other categories with T cell lymphomas, males had a higher percentage (33%, n = 18) compared to females (15%, n = 8) and 52% (n = 28) were NS. TNHL (52%, n = 28) was the most common type of TCL followed by TLBL (44%, n = 24).
Nijmegen Breakage Syndrome (NBS)
Similar to B cell lymphomas (BCL) in NBS, there are reports of T cell lymphoma (TCL) in this group of patients. In Table 2, there are eight studies with 52 patients, all under the age of 20 (4, 37–40, 44, 45, 103). The underlying mechanism for development of malignant disease is the same as for BCL and related to defects in DNA repair. The commonly used chemotherapy protocols in these patients included NHL-BFM-90, NHL-BFM-95, and EURO-LB 02.
AT
We identified two studies reporting two cases of TCL in AT patients (38, 41). A 6-month-old boy was diagnosed with TLBL and treated with a modified NHL-BFM 86 regimen. He developed B-NHL at the age of four leading to death. The second patient was also a 6-month-old boy who developed T-NHL and achieved complete remission initially, but like the first patient died at the age of four due to a second malignancy (B-NHL).
Discussion
In this section, we discuss the mechanisms responsible for lymphomagenesis in the various inborn errors of immunity and provide an overview of the treatment.
Defects in Immune Responses That Predispose to Lymphomagenesis in PIDDs
The complex immune mechanisms and their interplay that predisposes to neoplastic transformation of B or T cells and development of lymphomas in PIDD patients has not been fully elucidated. However, it is expected that the etiology in most cases is multifactorial and related to a dynamic regulation of immune response and environmental triggers (Figure 3). An underlying intrinsic susceptibility to DNA damage in some of these PIDDs, may provide a substrate for uncontrolled cell growth, impaired apoptosis of damaged cells, premature cellular senescence, all of which may be compounded by increased antigenic stimulation of adaptive lymphocytes by viruses, such as EBV (112). This uncontrolled stimulation in the setting of altered immune checkpoints and immune dysregulation characterized by T cell exhaustion, defective anti-tumor immune surveillance and overproduction of inflammatory cytokines provides a fertile setting for neoplastic transformation and lymphoproliferation (Figure 3).
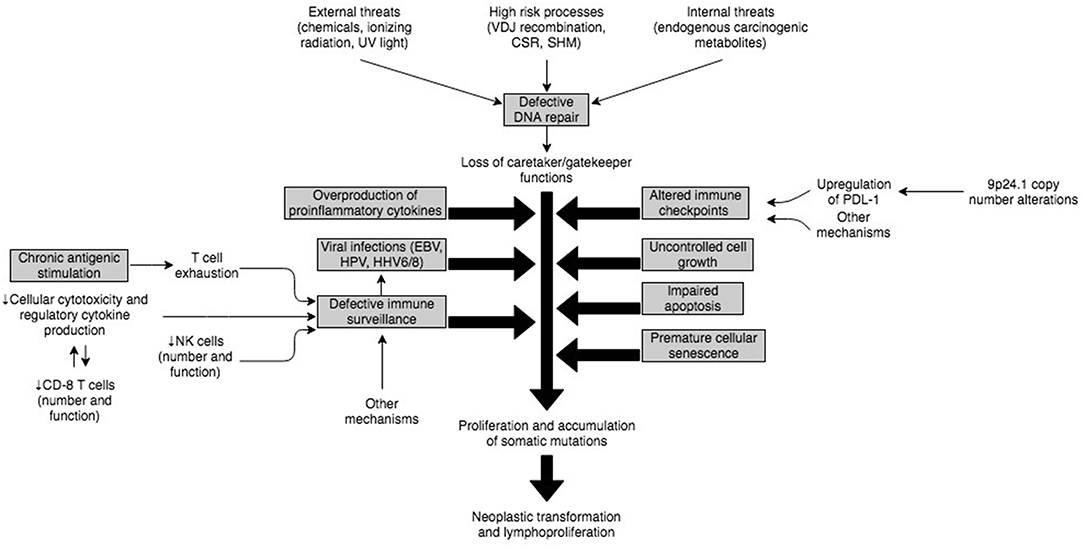
Figure 3. Illustration of potential interplay of mechanisms implicated in pathogenesis of malignancies in PIDs.
Defective DNA Repair
DNA integrity is constantly challenged at various levels as part of physiological processes. In lymphocytes, there is particular susceptibility specifically related to intrinsic mechanisms that are part of the immune diversity generation apparatus, such as V(D)J recombination, class switch recombination (CSR), and somatic hypermutation (SMH) (113). These intrinsic “stress points” coupled with other factors, including exposure to internal mutagens (e.g., endogenous metabolites) and external factors such as ultraviolet rays, ionizing radiation and chemicals (107) sets the stage for development of dysregulated cellular proliferation in B or T cells. Therefore, it is not surprising that normally there is a highly conserved DNA repair system orchestrated by a network of enzymes, which constantly assesses and detects DNA damaging lesions, modifies or removes damaged DNA, and reconstitutes the integrity of DNA through nucleotide resynthesis and ligation (114). When these checks and balances fail in the context of monogenic defects, it promotes the development of lymphoid tumors (113). Patients with premature cellular senescence related to shortened telomeres not only have an intrinsic susceptibility to DNA damage-inducing stimuli but also may have impaired apoptosis of damaged cells, which in turn may promote lymphoproliferation.
Role of Viral Infections and Defective Immune Surveillance, and Immune Dysregulation
Several of the documented PIDDs increase susceptibility to viral infections such as EBV, human papilloma virus (HPV), human herpes virus-6 (HHV-6), HHV-8, human T-cell lymphotropic virus (HTLV), Kaposi sarcoma–associated virus (KSV), and other viruses due to defective immune surveillance and immune dysregulation (115). Among these viruses, EBV represents the biggest threat because of its high prevalence (95%) and ability to transform epithelial cells, B cells, T cells and NK cells (116).
In the majority of cases, EBV virions entering through the oro-pharynx infect epithelial cells and B cells via the CD21 receptor. From a host immune system standpoint, NK cells and antigen-specific CD8+ T-cells are the main defense against primary EBV infection. Therefore, if EBV-infected B cells escape the cellular immune response, it can provoke an inflammatory outburst, resulting in cellular hyperproliferation and abnormal cell survival eventually leading to EBV-associated lymphoproliferative disorders (EBV-LPDs). EBV-LPDs consist of virus-associated hemophagocytic syndrome, non-malignant and malignant B-cell LPDs including non-Hodgkin and Hodgkin's types of B lymphomas and rarely EBV-positive T/NK cell lymphoma (117). The critical role of NK and T cells is demonstrated by the fact that combined B and T cell deficiency PIDDs account for approximately 2/3rd of EBV-associated LPDs in PIDDs, whereas defects in innate immunity do not significantly increase the risk of EBV-associated LPDs (116). Thus, underlying genetic defects in SH2D1A (SAP–XLP-1), ITK, MAGT1, CD27, CD70, CTPS1, RASGRP1, CORO1A and others account for the majority of EBV-LPDs.
The lack of adequate immune surveillance in the context of immune dysregulation is not uncommon in PIDDs, and though these cannot be exhaustively discussed here, failure of both innate (NK cells) and adaptive (T cells) immune cellular mechanisms facilitate viral escape and subsequent viral transformation of lymphocytes with poor cytotoxic clearance of virally-infected cells (89, 117). On the other hand, in certain other PIDDs, for example, the IL-10 receptor defects, there is production of pro-inflammatory cytokines due to the lack of IL-10-based regulation, and NFκB activation, with defective intra-tumoral CD8+ T cell immune surveillance and impaired cytotoxicity. This results in B cell proliferation and accumulation of somatic mutations (94, 111). Therefore, this is associated with a non-EBV-associated lymphoproliferation.
Chronic Antigenic Stimulation
Patients with PIDDs are more likely to develop persistence of antigens, and therefore ongoing stimulation of effector immune cells. T and B cells respond to external stimuli (antigens) and subsequently eliminate non-self-antigens (pathogen-infected and/or transformed cells). Sustained antigenic stimulation, in the context of cellular and systemic immune compromise, is likely to increase T cell exhaustion with progressive loss of cytokine secretion (IL-2, TNF-<), impairment of IFN-γ production, and in extreme cases premature apoptosis of CD8+ T cells (118). This quantitative and qualitative loss of the CD8+ T cell immune response against tumor cells eventually results in the development of lymphomas, and another plausible mechanism for tumor development in PIDDs.
Altered Immune Checkpoints
One of the primary mechanisms by which tumors escape the immune system is by engaging immune checkpoints (119), which is why immune checkpoint inhibitors (ICIs) were approved for the treatment of cancers and are now used in 14 different types of cancer (120). This issue is relevant to the lymphomas, which develop in PIDDs as it has been shown that PDL-1 protein is overexpressed in most EBV-associated tumors (121). Further, copy number variations (CNVs) at 9p24.1 have also been reported, and this is a locus, which harbors PD-1 (122). However, only one of two studies found amplifications of chromosome 9p via array CGH (comparative genomic hybridization) in EBV+ DLBCL (123). These findings raise the possibility that EBV+ DLBCL evades immune surveillance by selectively targeting the PD1/PDL pathway. Therefore, there may be some value to using immune checkpoint inhibitors in PIDDs for the treatment of lymphoma, and this raises the need for clinical trials in this area.
Treatment of B and T Cell Lymphomas in PIDDs
Clinical Presentation, Diagnosis and Staging
PIDD patients often present with advanced disease, and extranodal sites of disease, including bone marrow, liver, and spleen, and the presence of B-symptoms is quite common. There is no difference in how the diagnosis of lymphoma is established, whether in in PIDDs or non-intrinsic lymphoid malignancies, and it usually involves tissue biopsy, most frequently of lymph nodes.
Clinical evaluation should include standard workflow of history and physical, complete blood count with differential, liver and kidney function and baseline pulmonary function test for adult patients, if they will undergo bleomycin-containing regimens in the context of HL. Additional work-up to assess the degree of immune impairment, based on underlying genetic defect is essential for this patient population, and should include evaluation of radiosensitivity (especially if the genetic defect is known to predispose to this) so that appropriate treatment regimens can be formulated. Principles of diagnostic imaging may be extrapolated from imaging recommendations for Hodgkin's lymphoma in HIV patients, which suggests 18FDG-PET with integrated or concurrent CT (computerized tomography) of the neck, chest, abdomen, and pelvis. Since 18FDG-PET imaging has not been validated in this setting, it is unclear whether a bone marrow biopsy can be omitted. The interpretation of 18FDG-PET scan be challenging in presence of concurrent infections, which is not uncommon in these patients (124).
Treatment Options
While there are no specific treatment options for lymphoma in PIDD patients, radiation-based therapies should be avoided in patients with known genetic defects that predispose to radiosensitivity. Most frequently standard treatment options are based on type of lymphoma, such as B or T cell, indolent, aggressive or very aggressive and patient-related characteristics such as age, comorbidities, immune status, and degree of immune compromise. While there are no large or randomized studies to confirm this, small series and case reports show that response to the treatment and prognosis is inferior in PIDD patients when compared to non-PIDD patients, largely related to their inability to tolerate standard chemotherapy, and susceptibility to life-threatening infections (25). HCT remains the only viable curative option for many of these diseases, though its efficacy of HCT is variable across different PIDDs. For example, the role of HCT in severe combined immune deficiencies (SCID) is well established and it has been consistently replicated (125). HCT is not a standard treatment of choice in CVID, especially those without a molecular defect, though it may be warranted in certain cases depending on the presentation and associated co-morbidities. In circumstances, of a specific molecular diagnosis replacing or clarifying the underling CVID, HCT may be the most optimal long-term treatment of choice, even though more targeted therapies may be available. HCT for CVID was reported in a multicenter experience for 25 patients transplanted at 14 centers in Europe, the US, and Japan, and though it may be considered for lymphoma in this context, the risk of transplant-related mortality (TRM) should be clearly weighed against the potential benefits (126). Several retrospective studies have reported outcomes of HCT as across a range of PIDDs, including SCID (127), and Wiskott Aldrich Syndrome (128). Only case reports or small case series are available for successful treatment in patients with other PIDDs such as GATA2 haploinsufficiency, IL-10 receptor or XIAP deficiencies. Despite the limited experience, data strongly favors the early use of HCT in the setting of IL-10 receptor deficiency (111). Recently, a retrospective case series of 29 adult patients with a variety of PIDDs (mean age: 24 years, range: 17–50 years) treated with reduced intensity conditioning (RIC)-HCT was published. This study reported an overall survival of 85.2% at 3 years (129). These data provide support for moving forward with HCT in adult PIDD patients. As such, in addition to the treatment of lymphomas, HCT can be lifesaving in patients with PIDDs who develop very severe complications, including susceptibility to life-threatening infections, bone marrow failure, autoimmune and autoinflammatory diseases, other malignancies, and hemophagocytic syndrome. Despite all these potential benefits, it remains to be ascertained as to whether early HCT can reduce the risk of developing lymphoma in these PIDD patients in the long term.
There is no consistent evidence to support the use of newer immunotherapies for treatment of lymphoma in PIDDs but these therapies hold significant mechanistic promise for personalized, chemotherapy-free treatments. Potential immunotherapy options include monoclonal antibody-based immunotherapy (e.g., Rituximab, Obinutuzumab, Epratuzumab), conjugated antibodies (Brentuximab Vedotin), Bi-Specific T cell engaging (BiTE) antibodies (Blinatumumab), Anti-PD1(Pembrolizumab, Nivolumab), Anti PDL-1 (Atezolizumab and Darvalumab), and anti-CTLA4 checkpoint inhibitors (e.g., Ipilimumab). The role of adoptive T cell therapies such as chimeric antigen receptor (CAR) T-cells remains uncertain at this point (130). While these targeted therapies may have value, they may also pose an increased risk as a result of immune manipulation in the context of an underlying immune deficiency or immune dysregulation.
While there are no reports for successful use of immune checkpoint inhibitors or BiTE antibodies, complete remission with Rituximab and Brentuximab Vedotin was reported in an adult female patient with CVID -associated classic HL, while two other cases of pediatric CVID-associated HL succumbed to severe infection related to chemotherapy (61).
Conclusions
Though this is not a comprehensive summary of malignancies in PIDDs, or even lymphoproliferative disease in this area, this review summarizes the Medline-indexed published reports of B and T lymphomas in patients with PIDDs. This report highlights the diversity of malignant lymphoproliferative disorders in setting of PIDDs, and its associated challenges of diagnosis and treatment. The pathological classification and nomenclature for the lymphoid malignancies with variably reported and postulated underlying mechanisms were inconsistent and inadequate for many of these published reports. A wide range of treatment options were utilized, and response rate was highly variable suggesting an empirical approach rather than a systematic and tailored treatment regimen, based on underlying genetic defect, and degree of immunological impairment. HCT and gene therapy (where available) remains the best treatment option for many, but not all of these patients, and should be promptly initiated after diagnosis, particularly in some conditions, such as SCID, WAS, IL10 receptor deficiency among others. HCT, as a therapeutic option, remains significantly under-utilized in adult patients, likely related to inadequate awareness among adult hematologists, and these patients may benefit from increased utilization of HCT in appropriate settings. We highlight the significant need of unifying nomenclature, pathological analysis, and assessment of mechanisms of lymphomagenesis in these patients to develop better and more personalized treatment regimens. Also, there is a considerable urgency to conduct clinical trials to develop evidence-based treatment plans that considers the underlying immunodeficiency rather than using approaches extrapolated from non-PIDD settings (131). It is recognized that there has been a recent effort to standardize classification and nomenclature in immunodeficiency-associated malignant LPDs, but this is largely focused on secondary immunodeficiency disorders, in contrast to primary, though presumably some of the same standards could be applied (132), but this should be undertaken specifically for PIDDs, to fully understand the similarities and differences in pathology.
Author Contributions
All authors designed the study. IR and WF performed the literature search, conducted data extraction of relevant studies, and wrote the first draft of the manuscript. MP and RA critically reviewed the literature search and made revisions in the manuscript. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kobrynski L, Powell RW, Bowen S. Prevalence and morbidity of primary immunodeficiency diseases, United States 2001-2007. J Clin Immunol. (2014) 34:954–61. doi: 10.1007/s10875-014-0102-8
2. Filipovich AH, Mathur A, Kamat D, Shapiro RS. Primary immunodeficiencies: genetic risk factors for lymphoma. Cancer Res. (1992) 52(19 Suppl.):5465s–7s.
3. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood. (2010) 116:1228–34. doi: 10.1182/blood-2010-03-272351
4. Fedorova A, Sharapova S, Mikhalevskaya T, Aleshkevich S, Proleskovskaya I, Stsegantseva M, et al. Non-Hodgkin lymphoma in children with primary immunodeficiencies: clinical manifestations, diagnosis, and management, belarusian experience. Lymphoma. (2015) 2015:10. doi: 10.1155/2015/123548
5. Taskinen M, Jeskanen L, Karjalainen-Lindsberg ML, Makitie A, Makitie O, Ranki A. Combating cancer predisposition in association with idiopathic immune deficiency: a recurrent nodal and cutaneous T-cell lymphoproliferative disease in a patient with cartilage-hair hypoplasia. Clin Lymph Myeloma Leuk. (2013) 13:73–6. doi: 10.1016/j.clml.2012.06.005
6. Nijland ML, Koens L, Pals ST, Berge I, Bemelman FJ, Kersten MJ. Clinicopathological characteristics of T-cell non-Hodgkin lymphoma arising in patients with immunodeficiencies: a single-center case series of 25 patients and a review of the literature. Haematologica. (2018) 103:486–96. doi: 10.3324/haematol.2017.169987
7. Schmidt RE, Grimbacher B, Witte T. Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol. (2017) 14:7–18. doi: 10.1038/nrrheum.2017.198
8. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers related to immunodeficiencies: update and perspectives. Front Immunol. (2016) 7:365. doi: 10.3389/fimmu.2016.00365
9. Shapiro RS, Robbins N, Cowen LE. Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev. (2011) 75:213–67. doi: 10.1128/MMBR.00045-10
10. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
11. Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res. (2008) 28:1263–9. Retrieved from: http://ar.iiarjournals.org/content/28/2B/1263.long
12. Mayor PC, Eng KH, Singel KL, Abrams SI, Odunsi K, Moysich KB, et al. Cancer in primary immunodeficiency diseases: cancer incidence in the United States immune deficiency network registry. J Allergy Clin Immunol. (2018) 141:1028–35. doi: 10.1016/j.jaci.2017.05.024
13. Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, et al. Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J Allergy Clin Immunol. (2013) 131:1594–603. doi: 10.1016/j.jaci.2013.01.042
14. Toita N, Hatano N, Ono S, Yamada M, Kobayashi R, Kobayashi I, et al. Epstein–Barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. Am J Med Genet A. (2007) 143A:742–5. doi: 10.1002/ajmg.a.31644
15. Moshous D, Pannetier C, Chasseval Rd R, Deist Fl F, Cavazzana-Calvo M, Romana S, et al. Partial T and B lymphocyte immunodeficiency and predisposition to lymphoma in patients with hypomorphic mutations in Artemis. J Clin Investig. (2003) 111:381–7. doi: 10.1172/JCI16774
16. Schuetz C, Huck K, Gudowius S, Megahed M, Feyen O, Hubner B, et al. An immunodeficiency disease with RAG mutations and granulomas. New Engl J Med. (2008) 358:2030–8. doi: 10.1056/NEJMoa073966
17. Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. (2006) 176:5060–8.
18. Newell A, Dadi H, Goldberg R, Ngan B-Y, Grunebaum E, Roifman CM. Diffuse large B-cell lymphoma as presenting feature of Zap-70 deficiency. J Allergy Clin Immunol. (2011) 127:517–20. doi: 10.1016/j.jaci.2010.09.016
19. Kapoor N, Jung LKL, Engelhard D, Filler J, Shalit I, Landreth KS, et al. Lymphoma in a patient with severe combined immunodeficiency with adenosine deaminase deficiency, following unsustained engraftment of histoincompatible T cell-depleted bone marrow. J Pediatr. (1986) 108:435–8. doi: 10.1016/S0022-3476(86)80892-7
20. Ratech H, Hirschhorn R, Greco MA. Pathologic findings in adenosine deaminase deficient-severe combined immunodeficiency. II. Thymus, spleen, lymph node, and gastrointestinal tract lymphoid tissue alterations. Am J Pathol. (1989) 135:1145–56.
21. Kaufman DA, Moissidis IJ, Bocchini JA Jr, Hershfield MS, Bahna SL. Case report: cerebral lymphoma in an ADA-deficient SCID patient receiving PEG-ADA. J Allergy Clin Immunol. (2005) 115:S154. doi: 10.1016/j.jaci.2004.12.626
22. Husain M, Grunebaum E, Naqvi A, Atkinson A, Ngan BY, Aiuti A, et al. Burkitt's lymphoma in a patient with adenosine deaminase deficiency-severe combined immunodeficiency treated with polyethylene glycol-adenosine deaminase. J Pediatr. (2007) 151:93–5. doi: 10.1016/j.jpeds.2007.03.059
23. Genel F, Ozbek E, Ozek G, Vergin C, Ortac R, Santisteban I, et al. Adenosine deaminase-deficient severe combined immunodeficiency and diffuse large B-cell lymphoma. Pediatr AllergyImmunol Pulmonol. (2015) 28:138–42. doi: 10.1089/ped.2014.0478
24. Migliavacca M, Assanelli A, Ponzoni M, Pajno R, Barzaghi F, Giglio F, et al. First occurrence of plasmablastic lymphoma in adenosine deaminase-deficient severe combined immunodeficiency disease patient and review of the literature. Front Immunol. (2018) 9:113. doi: 10.3389/fimmu.2018.00113
25. Suarez F, Mahlaoui N, Canioni D, Andriamanga C, Dubois d'Enghien C, Brousse N, et al. Incidence, presentation, and prognosis of malignancies in ataxia-telangiectasia: a report from the French national registry of primary immune deficiencies. J Clin Oncol. (2015) 33:202–8. doi: 10.1200/JCO.2014.56.5101
26. Bennett JA, Bayerl MG. Epstein-barr virus-associated extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT Lymphoma) arising in the parotid gland of a child with ataxia telangiectasia. J Pediatr Hematol Oncol. (2015) 37:e114–7. doi: 10.1097/MPH.0b013e31829f3496
27. Meister MT, Voss S, Schwabe D. Treatment of EBV-associated nodular sclerosing Hodgkin lymphoma in a patient with ataxia telangiectasia with brentuximab vedotin and reduced COPP plus rituximab. Pediatr Blood Cancer. (2015) 62:2018–20. doi: 10.1002/pbc.25621
28. Beier R, Sykora KW, Woessmann W, Maecker-Kolhoff B, Sauer M, Kreipe HH, et al. Allogeneic-matched sibling stem cell transplantation in a 13-year-old boy with ataxia telangiectasia and EBV-positive non-Hodgkin lymphoma. Bone Marrow Transplant. (2016) 51:1271–4. doi: 10.1038/bmt.2016.93
29. Du S, Scuderi R, Malicki DM, Willert J, Bastian J, Weidner N. Hodgkin's and non-Hodgkin's lymphomas occurring in two brothers with Wiskott-Aldrich syndrome and review of the literature. Pediatr Devel Pathol. (2011) 14:64–70. doi: 10.2350/10-01-0787-CR.1
30. Pasic S, Vujic D, Djuricic S, Jevtic D, Grujic B. Burkitt lymphoma-induced ileocolic intussusception in Wiskott-Aldrich syndrome. J Pediatr Hematol. (2006) 28:48–9.
31. Palenzuela G, Bernard F, Gardiner Q, Mondain M. Malignant B cell non-Hodgkin's lymphoma of the larynx in children with Wiskott Aldrich syndrome. Int J Pediatr Otorhinolaryngol. (2003) 67:989–93. doi: 10.1016/S0165-5876(03)00155-1
32. Sasahara Y, Fujie H, Kumaki S, Ohashi Y, Minegishi M, Tsuchiya S. Epstein-Barr virus-associated Hodgkin's disease in a patient with Wiskott-Aldrich syndrome. Acta Paediatr. (2001) 90:1348–51. doi: 10.1111/j.1651-2227.2001.tb01589.x
33. Nakanishi M, Kikuta H, Tomizawa K, Kojima K, Ishizaka A, Okano M, et al. Distinct clonotypic Epstein-Barr virus-induced fatal lymphoproliferative disorder in a patient with Wiskott-Aldrich syndrome. Cancer. (1993) 72:1376–81. doi: 10.1002/1097-0142(19930815)72:4<1376::AID-CNCR2820720437>3.0.CO;2-Q
34. Gulley ML, Chen CL, Raab-Traub N. Epstein-Barr virus-related lymphomagenesis in a child with Wiskott-Aldrich syndrome. Hematol Oncol. (1993) 11:139–45. doi: 10.1002/hon.2900110304
35. Ramos JT, Lopez-Laso E, Ruiz-Contreras J, Giancaspro E, Madero S. B cell non-Hodgkin's lymphoma in a girl with the DiGeorge anomaly. Arch Dis Childhood. (1999) 81:444–5. doi: 10.1136/adc.81.5.444
36. Taskinen M, Ranki A, Pukkala E, Jeskanen L, Kaitila I, Makitie O. Extended follow-up of the Finnish cartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet Part A. (2008) 146a:2370–5. doi: 10.1002/ajmg.a.32478
37. Dembowska-Baginska B, Perek D, Brozyna A, Wakulinska A, Olczak-Kowalczyk D, Gladkowska-Dura M, et al. Non-Hodgkin lymphoma (NHL) in children with Nijmegen Breakage syndrome (NBS). Pediatr Blood Cancer. (2009) 52:186–90. doi: 10.1002/pbc.21789
38. Bienemann K, Burkhardt B, Modlich S, Meyer U, Moricke A, Bienemann K, et al. Promising therapy results for lymphoid malignancies in children with chromosomal breakage syndromes (Ataxia teleangiectasia or Nijmegen-breakage syndrome): a retrospective survey. Br J Haematol. (2011) 155:468–76. doi: 10.1111/j.1365-2141.2011.08863.x
39. Kruger L, Demuth I, Neitzel H, Varon R, Sperling K, Chrzanowska KH, et al. Cancer incidence in Nijmegen breakage syndrome is modulated by the amount of a variant NBS protein. Carcinogenesis. (2007) 28:107–11. doi: 10.1093/carcin/bgl126
40. Gladkowska-Dura M, Dzierzanowska-Fangrat K, Dura WT, van Krieken JH, Chrzanowska KH, van Dongen JJ, et al. Unique morphological spectrum of lymphomas in Nijmegen breakage syndrome (NBS) patients with high frequency of consecutive lymphoma formation. J Pathol. (2008) 216:337–44. doi: 10.1002/path.2418
41. Seidemann K, Tiemann M, Henze G, Sauerbrey A, Muller S, Reiter A. Therapy for non-Hodgkin lymphoma in children with primary immunodeficiency: analysis of 19 patients from the BFM trials. Medic Pediatr Oncol. (1999) 33:536–44. doi: 10.1002/(SICI)1096-911X(199912)33:6<536::AID-MPO3>3.0.CO;2-Z
42. Dumic M, Radman I, Krnic N, Nola M, Kusec R, Begovic D, et al. Successful treatment of diffuse large B-cell non-hodgkin lymphoma with modified CHOP (cyclophosphamide/doxorubicin/vincristine/prednisone) chemotherapy and rituximab in a patient with Nijmegen syndrome. Clin Lymph Myeloma. (2007) 7:590–3. doi: 10.3816/CLM.2007.n.046
43. Jovanovic A, Minic P, Scekic-Guc M, Djuricic S, Cirkovic S, Weemaes C, et al. Successful treatment of hodgkin lymphoma in nijmegen breakage syndrome. J Pediatr Hematol. (2009) 31:49–52. doi: 10.1097/MPH.0b013e318190d72c
44. Gregorek H, Chrzanowska KH, Dzierzanowska-Fangrat K, Wakulinska A, Pietrucha B, Zapasnik A, et al. Nijmegen breakage syndrome: Long-term monitoring of viral and immunological biomarkers in peripheral blood before development of malignancy. Clin Immunol. (2010) 135:440–7. doi: 10.1016/j.clim.2010.01.008
45. Wolska-Kusnierz B, Gregorek H, Chrzanowska K, Piatosa B, Pietrucha B, Heropolitanska-Pliszka E, et al. Nijmegen breakage syndrome: clinical and immunological features, long-term outcome and treatment options - a retrospective analysis. J Clin Immunol. (2015) 35:538–49. doi: 10.1007/s10875-015-0186-9
46. Baykal C, Kavak A, Gulcan P, Buyukbabani N. Dyskeratosis congenita associated with three malignancies. J Eur Acad Dermatol Venereol. (2003) 17:216–8. doi: 10.1046/j.1468-3083.2003.00585.x
47. Basiratnia M, Baradaran-Heravi A, Yavarian M, Geramizadeh B, Karimi M. Non-hodgkin lymphoma in a child with schimke immuno-osseous dysplasia. Iran J Med Sci. (2011) 36:222–5. Retrieved from: http://ijms.sums.ac.ir/index.php/IJMS/article/view/771/200
48. Baradaran-Heravi A, Raams A, Lubieniecka J, Cho KS, DeHaai KA, Basiratnia M, et al. SMARCAL1 deficiency predisposes to non-Hodgkin lymphoma and hypersensitivity to genotoxic agents in vivo. Am J Med Genet Part A. (2012) 158a:2204–13. doi: 10.1002/ajmg.a.35532
49. Taha D, Boerkoel CF, Balfe JW, Khalifah M, Sloan EA, Barbar M, et al. Fatal lymphoproliferative disorder in a child with Schimke immuno-osseous dysplasia. Am J Med Genet Part A. (2004) 131:194–9. doi: 10.1002/ajmg.a.30356
50. Fuentes-Páez G, Saornil MA, Herreras JM, Alonso-Ballesteros M, Sánchez PS, García-Tejeiro M. CHARGE association, hyper-immunoglobulin M syndrome, and conjunctival MALT lymphoma. Cornea. (2007) 26:864–7. doi: 10.1097/ICO.0b013e31806c77d6
51. Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V, et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase delta syndrome. J Allergy Clin Immunol. (2014) 134:233–6. doi: 10.1016/j.jaci.2014.02.020
52. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: a large patient cohort study. J Allergy Clin Immunol. (2017) 139:597–606.e4. doi: 10.1016/j.jaci.2016.06.021
53. Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. (2013) 342:866–71. doi: 10.1126/science.1243292
54. Mellemkjaer L, Hammarstrom L, Andersen V, Yuen J, Heilmann C, Barington T, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: a combined Danish and Swedish study. Clin Exp Immunol. (2002) 130:495–500. doi: 10.1046/j.1365-2249.2002.02004.x
55. Gelmann E, Anderson T, Jaffe E, Broder S. Chemotherapy for lymphoma in a patient with common variable immunodeficiency: case report, literature review, and recommendations for chemotherapy in immunodeficient patients. Arch Int Med. (1982) 142:90–2. doi: 10.1001/archinte.1982.00340140092017
56. Aghamohammadi A, Parvaneh N, Tirgari F, Mahjoob F, Movahedi M, Gharagozlou M, et al. Lymphoma of mucosa-associated lymphoid tissue in common variable immunodeficiency. Leukemia Lymph. (2006) 47:343–6. doi: 10.1080/10428190500285285
57. Spickett GP. Current perspectives on common variable immunodeficiency (CVID). Clin Exp Allergy. (2001) 31:536–42. doi: 10.1046/j.1365-2222.2001.01117.x
58. Filipovich AH, Heinitz KJ, Robison LL, Frizzera G. The immunodeficiency cancer registry. a research resource. Am J Pediatr Hematol Oncol. (1987) 9:183–4. doi: 10.1097/00043426-198722000-00017
59. Kinlen LJ, Webster AD, Bird AG, Haile R, Peto J, Soothill JF, et al. Prospective study of cancer in patients with hypogammaglobulinaemia. Lancet. (1985) 1:263–6. doi: 10.1016/S0140-6736(85)91037-2
60. Gompels MM, Hodges E, Lock RJ, Angus B, White H, Larkin A, et al. Lymphoproliferative disease in antibody deficiency: a multi-centre study. Clin Exp Immunol. (2003) 134:314–20. doi: 10.1046/j.1365-2249.2003.02253.x
61. Rael E, Rakszawski K, Koller K, Bayerl M, Butte M, Zheng H. Treatment with rituximab and brentuximab vedotin in a patient of common variable immune deficiency-associated classic Hodgkin lymphoma. Biomarker Res. (2016) 4:7. doi: 10.1186/s40364-016-0061-8
62. Fernández Romero SD, Juri M, Paolini M, Malbrán A. Inmunodeficiencia Común Variable: Epidemiología y Manifestaciones Clínicas en 69 Pacientes. Buenos Aires: Medicina (2013). p. 315–23.
63. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. (1999) 92:34–48. doi: 10.1006/clim.1999.4725
64. Cunningham-Rundles C, Cooper DL, Duffy TP, Strauchen J. Lymphomas of mucosal-associated lymphoid tissue in common variable immunodeficiency. Am J Hematol. (2002) 69:171–8. doi: 10.1002/ajh.10050
65. Cunningham-Rundles C, Lieberman P, Hellman G, Chaganti RS. Non-Hodgkin lymphoma in common variable immunodeficiency. Am J Hematol. (1991) 37:69–74. doi: 10.1002/ajh.2830370202
66. Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol. (1987) 7:294–9. doi: 10.1007/BF00915550
67. Lamers CB, Wagener T, Assmann KJ, van Tongeren JH. Jejunal lymphoma in a patient with primary adult-onset hypogammaglobulinemia and nodular lymphoid hyperplasia of the small intestine. Digest Dis Sci. (1980) 25:553–7. doi: 10.1007/BF01315216
68. Gonzalez-Vitale JC, Gomez LG, Goldblum RM, Goldman AS, Patterson M. Immunoblastic lymphoma of small intestine complicating late-onset immunodeficiency. Cancer. (1982) 49:445–9. doi: 10.1002/1097-0142(19820201)49:3<445::AID-CNCR2820490309>3.0.CO;2-1
69. Castellano G, Moreno D, Galvao O, Ballestin C, Colina F, Mollejo M, et al. Malignant lymphoma of jejunum with common variable hypogammaglobulinemia and diffuse nodular hyperplasia of the small intestine. A case study and literature review. J Clin Gastroenterol. (1992) 15:128–35. doi: 10.1097/00004836-199209000-00010
70. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. (2007) 27:308–16. doi: 10.1007/s10875-007-9075-1
71. Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Quart J Med. (1993) 86:31–42.
72. Reichenberger F, Wyser C, Gonon M, Cathomas G, Tamm M. Pulmonary mucosa-associated lymphoid tissue lymphoma in a patient with common variable immunodeficiency syndrome. Respiration. (2001) 68:109–12. doi: 10.1159/000050475
73. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112:277–86. doi: 10.1182/blood-2007-11-124545
74. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. (2012) 119:1650–7. doi: 10.1182/blood-2011-09-377945
75. Wobser M, Kerstan A, Kneitz H, Goebeler M, Kunzmann V, Rosenwald A, et al. Primary cutaneous marginal zone lymphoma with sequential development of nodal marginal zone lymphoma in a patient with selective immunoglobulin A deficiency. J Cutaneous Pathol. (2013) 40:1035–41. doi: 10.1111/cup.12230
76. Pace R, Vinh DC. Autoimmune lymphoproliferative syndrome and epstein-barr virus-associated lymphoma: an adjunctive diagnostic role for monitoring EBV viremia? Case Rep Immunol. (2013) 2013:245893. doi: 10.1155/2013/245893
77. Sharapova SO, Fedorova AS, Pashchenko OE, Vahliarskaya SS, Guryanova IE, Migas AA, et al. Novel mutations in SH2D1A gene in X-linked lymphoproliferative syndrome, diagnosed after B-cell non-hodgkin lymphoma. J Pediatr Hematol Oncol. (2017) 39:e203–6. doi: 10.1097/MPH.0000000000000815
78. Hugle B, Astigarraga I, Henter JI, Porwit-MacDonald A, Meindl A, Schuster V. Simultaneous manifestation of fulminant infectious mononucleosis with haemophagocytic syndrome and B-cell lymphoma in X-linked lymphoproliferative disease. Eur J Pediatr. (2007) 166:589–93. doi: 10.1007/s00431-006-0290-1
79. Egeler RM, de Kraker J, Slater R, Purtilo DT. Documentation of Burkitt lymphoma with t(8;14) (q24;q32) in X-linked lymphoproliferative disease. Cancer. (1992) 70:683–7. doi: 10.1002/1097-0142(19920801)70:3<683::AID-CNCR2820700324>3.0.CO;2-C
80. Alme C, Satwani P, Alobeid B, Bhagat G, Kelly KM. Atypical clinical course in pediatric hodgkin lymphoma: association with germline mutations in interleukin-2-inducible T-cell kinase. J Pediatr Hematol Oncol. (2015) 37:507–8. doi: 10.1097/MPH.0000000000000366
81. Cipe FE, Aydogmus C, Serwas NK, Tugcu D, Demirkaya M, Biçici FA, et al. ITK deficiency: how can EBV be treated before lymphoma? Pediatr Blood Cancer. (2015) 62:2247–8. doi: 10.1002/pbc.25648
82. Shamriz O, Vilk SR, Wolf DG, Ta-Shma A, Averbuch D, Weintraub M, et al. Hematopoietic stem cell transplantation conditioning with use of rituximab in EBV related lymphoproliferative disorders. Clin Immunol. (2014) 151:79–83. doi: 10.1016/j.clim.2014.01.007
83. Linka RM, Risse SL, Bienemann K, Werner M, Linka Y, Krux F, et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia. (2012) 26:963–71. doi: 10.1038/leu.2011.371
84. Stepensky P, Weintraub M, Yanir A, Revel-Vilk S, Krux F, Huck K, et al. IL-2-inducible T-cell kinase deficiency: clinical presentation and therapeutic approach. Haematologica. (2011) 96:472–6. doi: 10.3324/haematol.2010.033910
85. Huck K, Feyen O, Niehues T, Ruschendorf F, Hubner N, Laws HJ, et al. Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Investig. (2009) 119:1350–8. doi: 10.1172/JCI37901
86. Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, et al. Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J Exp Med. (2017) 214:73–89. doi: 10.1084/jem.20160784
87. Abolhassani H, Edwards ES, Ikinciogullari A, Jing H, Borte S, Buggert M, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. (2017) 214:91–106. doi: 10.1084/jem.20160849
88. Alkhairy OK, Perez-Becker R, Driessen GJ, Abolhassani H, van Montfrans J, Borte S, et al. Novel mutations in TNFRSF7/CD27: Clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. (2015) 136:703–12.e10. doi: 10.1016/j.jaci.2015.02.022
89. Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ. XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus. Blood. (2014) 123:2148–52. doi: 10.1182/blood-2013-11-538686
90. Chaigne-Delalande B, Li FY, O'Connor GM, Lukacs MJ, Jiang P, Zheng L, et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science. (2013) 341:186–91. doi: 10.1126/science.1240094
91. Dhalla F, Murray S, Sadler R, Chaigne-Delalande B, Sadaoka T, Soilleux E, et al. Identification of a novel mutation in MAGT1 and progressive multifocal leucoencephalopathy in a 58-year-old man with XMEN disease. J Clin Immunol. (2015) 35:112–8. doi: 10.1007/s10875-014-0116-2
92. Patiroglu T, Haluk Akar H, Gilmour K, Unal E, Akif Ozdemir M, Bibi S, et al. A case of XMEN syndrome presented with severe auto-immune disorders mimicking autoimmune lymphoproliferative disease. Clin Immunol. (2015) 159:58–62. doi: 10.1016/j.clim.2015.04.015
93. Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature. (2014) 510:288–92. doi: 10.1038/nature13386
94. Neven B, Mamessier E, Bruneau J, Kaltenbach S, Kotlarz D, Suarez F, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood. (2013) 122:3713–22. doi: 10.1182/blood-2013-06-508267
95. Chae KM, Ertle JO, Tharp MD. B-cell lymphoma in a patient with WHIM syndrome. J Am Acad Dermatol. (2001) 44:124–8. doi: 10.1067/mjd.2001.111337
96. Imashuku S, Miyagawa A, Chiyonobu T, Ishida H, Yoshihara T, Teramura T, et al. Epstein-Barr virus-associated T-lymphoproliferative disease with hemophagocytic syndrome, followed by fatal intestinal B lymphoma in a young adult female with WHIM syndrome. Warts, hypogammaglobulinemia, infections, and myelokathexis. Ann Hematol. (2002) 81:470–3. doi: 10.1007/s00277-002-0489-9
97. Durham JC, Stephens DS, Rimland D, Nassar VH, Spira TJ. Common variable hypogammaglobulinemia complicated by an unusual T-suppressor/cytotoxic cell lymphoma. Cancer. (1987) 59:271–6. doi: 10.1002/1097-0142(19870115)59:2<271::AID-CNCR2820590216>3.0.CO;2-H
98. Kim JH, Bedrosian CL, Jain R, Schlossman DM. Peripheral T-cell lymphoma complicating common variable hypogammaglobulinemia. Am J Med. (1988) 85:123–5. doi: 10.1016/0002-9343(88)90519-0
99. Gottesman SR, Haas D, Ladanyi M, Amorosi EL. Peripheral T cell lymphoma in a patient with common variable immunodeficiency disease: case report and literature review. Leukemia Lymphoma. (1999) 32:589–95. doi: 10.3109/10428199909058418
100. Ott MM, Ott G, Klinker H, Trunk MJ, Katzenberger T, Muller-Hermelink HK. Abdominal T-cell non-Hodgkin's lymphoma of the gamma/delta type in a patient with selective immunoglobulin A deficiency. Am J Surg Pathol. (1998) 22:500–6. doi: 10.1097/00000478-199804000-00017
101. Kanavaros P, Rontogianni D, Hrissovergi D, Efthimiadoy A, Argyrakos T, Mastoris K, et al. Extranodal cytotoxic T-cell lymphoma in a patient with X-linked agammaglobulinaemia. Leukemia Lymphoma. (2001) 42:235–8. doi: 10.3109/10428190109097697
102. Gammon B, Robson A, Deonizio J, Arkin L, Guitart J. CD8(+) granulomatous cutaneous T-cell lymphoma: a potential association with immunodeficiency. J Am Acad Dermatol. (2014) 71:555–60. doi: 10.1016/j.jaad.2014.03.028
103. Pasic S, Vujic D, Fiorini M, Notarangelo LD. T-cell lymphoblastic leukemia/lymphoma in Nijmegen breakage syndrome. Haematologica. (2004) 89:Ecr27. Retreived from: http://www.haematologica.org/content/89/8/ECR27
104. Salzer E, Daschkey S, Choo S, Gombert M, Santos-Valente E, Ginzel S, et al. Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica. (2013) 98:473–8. doi: 10.3324/haematol.2012.068791
105. Hirschhorn R, Nicknam MN, Eng F, Yang DR, Borkowsky W. Novel deletion and a new missense mutation (Glu 217 Lys) at the catalytic site in two adenosine deaminase alleles of a patient with neonatal onset adenosine deaminase- severe combined immunodeficiency. J Immunol. (1992) 149:3107–12.
106. Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. (2014) 134:116–26. doi: 10.1016/j.jaci.2013.12.1077
107. Abbotts R, Thompson N, Madhusudan S. DNA repair in cancer: emerging targets for personalized therapy. Cancer Manage Res. (2014) 6:77–92. doi: 10.2147/CMAR.S50497
108. Albert MH, Bittner TC, Nonoyama S, Notarangelo LD, Burns S, Imai K, et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood. (2010) 115:3231–8. doi: 10.1182/blood-2009-09-239087
109. Cannons JL, Schwartzberg PL. SAP and lessons learned from a primary immunodeficiency. J Immunol. (2017) 199:1531–3. doi: 10.4049/jimmunol.1701007
110. Shouval DS, Ebens CL, Murchie R, McCann K, Rabah R, Klein C, et al. Large B-cell lymphoma in an adolescent patient with interleukin-10 receptor deficiency and history of infantile inflammatory bowel disease. J Pediatr Gastroenterol Nutr. (2016) 63:e15–7. doi: 10.1097/MPG.0000000000000532
111. Oft M. No immunosurveillance in human IL-10R deficiency. Blood. (2013) 122:3702–3. doi: 10.1182/blood-2013-10-531657
112. Hauck F, Voss R, Urban C, Seidel MG. Intrinsic and extrinsic causes of malignancies in patients with primary immunodeficiency disorders. J Aller Clin Immunol. (2018) 141:59–68.e4. doi: 10.1016/j.jaci.2017.06.009
113. de Miranda NF, Bjorkman A, Pan-Hammarstrom Q. DNA repair: the link between primary immunodeficiency and cancer. Ann NY Acad Sci. (2011) 1246:50–63. doi: 10.1111/j.1749-6632.2011.06322.x
114. Keijzers G, Bakula D, Scheibye-Knudsen M. Monogenic diseases of DNA repair. N Engl J Med. (2017) 377:1868–76. doi: 10.1056/NEJMra1703366
115. Rezaei N, Hedayat M, Aghamohammadi A, Nichols KE. Primary immunodeficiency diseases associated with increased susceptibility to viral infections and malignancies. J Allergy Clin Immunol. (2011) 127:1329–41.e2; quiz 42–3. doi: 10.1016/j.jaci.2011.02.047
116. Shabani M, Nichols KE, Rezaei N. Primary immunodeficiencies associated with EBV-Induced lymphoproliferative disorders. Crit Rev Oncol Hematol. (2016) 108:109–27. doi: 10.1016/j.critrevonc.2016.10.014
117. Latour S, Winter S. Inherited immunodeficiencies with high predisposition to Epstein-Barr virus-driven lymphoproliferative diseases. Front Immunol. (2018) 9:1103. doi: 10.3389/fimmu.2018.01103
118. Zuniga EI, Harker JA. T-cell exhaustion due to persistent antigen: quantity not quality? Eur J Immunol. (2012) 42:2285–9. doi: 10.1002/eji.201242852
119. Disis ML. Mechanism of action of immunotherapy. Semin Oncol. (2014) 41(Suppl. 5):S3–13. doi: 10.1053/j.seminoncol.2014.09.004
120. Johnson DB, Chandra S, Sosman JA. Immune checkpoint inhibitor toxicity in 2018. JAMA. (2018) 320:1702–3. doi: 10.1001/jama.2018.13995
121. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res. (2013) 19:3462–73. doi: 10.1158/1078-0432.CCR-13-0855
122. Yoon H, Park S, Ju H, Ha SY, Sohn I, Jo J, et al. Integrated copy number and gene expression profiling analysis of Epstein-Barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer. (2015) 54:383–96. doi: 10.1002/gcc.22249
123. Al-Humood S, AlQallaf A, Al-Shemmari S, Al-Faris L, Al-Ayadhy B. Genetic and immunohistochemical characterization of Epstein-Barr virus-associated diffuse large B-cell lymphoma. Acta Haematol. (2014) 131:1–10. doi: 10.1159/000350493
124. Uldrick TS, Little RF. How I treat classical Hodgkin lymphoma in patients infected with human immunodeficiency virus. Blood. (2015) 125:1226–35; quiz 355. doi: 10.1182/blood-2014-08-551598
125. Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. (2010) 126:602–10.e1–11. doi: 10.1016/j.jaci.2010.06.015
126. Wehr C, Gennery AR, Lindemans C, Schulz A, Hoenig M, Marks R, et al. Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol. (2015) 135:988–97.e6. doi: 10.1016/j.jaci.2014.11.029
127. Pai S-Y, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. (2014) 371:434–46. doi: 10.1056/NEJMoa1401177
128. Ngwube A, Hanson IC, Orange J, Rider NL, Seeborg F, Shearer W, et al. Outcomes after allogeneic transplant in patients with Wiskott-Aldrich syndrome. Biol Blood Marrow Transplant. (2018) 24:537–41. doi: 10.1016/j.bbmt.2017.11.019
129. Fox TA, Chakraverty R, Burns S, Carpenter B, Thomson K, Lowe D, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood. (2018) 131:917–31. doi: 10.1182/blood-2017-09-807487
130. Verhoeven D, Stoppelenburg AJ, Meyer-Wentrup F, Boes M. Increased risk of hematologic malignancies in primary immunodeficiency disorders: opportunities for immunotherapy. Clin Immunol. (2018) 190:22–31. doi: 10.1016/j.clim.2018.02.007
131. Bomken S, van der Werff Ten Bosch J, Attarbaschi A, Bacon CM, Borkhardt A, Boztug K, et al. Current understanding and future research priorities in malignancy associated with inborn errors of immunity and DNA repair disorders: the perspective of an interdisciplinary working group. Front Immunol. (2018) 9:2912. doi: 10.3389/fimmu.2018.02912
Keywords: primary immunodeficiencies, B cell lymphoma, T cell lymphoma, systematic (literature) reviews, immunodeficiency
Citation: Riaz IB, Faridi W, Patnaik MM and Abraham RS (2019) A Systematic Review on Predisposition to Lymphoid (B and T cell) Neoplasias in Patients With Primary Immunodeficiencies and Immune Dysregulatory Disorders (Inborn Errors of Immunity). Front. Immunol. 10:777. doi: 10.3389/fimmu.2019.00777
Received: 07 November 2018; Accepted: 25 March 2019;
Published: 16 April 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Fabian Hauck, LMU Munich, GermanyQiang Pan-Hammarström, Karolinska Institute (KI), Sweden
Eleonora Gambineri, University of Florence, Italy
Copyright © 2019 Riaz, Faridi, Patnaik and Abraham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roshini S. Abraham, cm9zaGluaS5hYnJhaGFtQG5hdGlvbndpZGVjaGlsZHJlbnMub3Jn