 Husni Elbahesh
Husni Elbahesh Thomas Gerlach
Thomas Gerlach Giulietta Saletti
Giulietta Saletti Guus F. Rimmelzwaan
Guus F. Rimmelzwaan- Research Center for Emerging Infections and Zoonoses, University of Veterinary Medicine (TiHo), Hanover, Germany
Despite causing pandemics and yearly epidemics that result in significant morbidity and mortality, our arsenal of options to treat influenza A virus (IAV) infections remains limited and is challenged by the virus itself. While vaccination is the preferred intervention strategy against influenza, its efficacy is reduced in the elderly and infants who are most susceptible to severe and/or fatal infections. In addition, antigenic variation of IAV complicates the production of efficacious vaccines. Similarly, effectiveness of currently used antiviral drugs is jeopardized by the development of resistance to these drugs. Like many viruses, IAV is reliant on host factors and signaling-pathways for its replication, which could potentially offer alternative options to treat infections. While host-factors have long been recognized as attractive therapeutic candidates against other viruses, only recently they have been targeted for development as IAV antivirals. Future strategies to combat IAV infections will most likely include approaches that alter host-virus interactions on the one hand or dampen harmful host immune responses on the other, with the use of biological response modifiers (BRMs). In principle, BRMs are biologically active agents including antibodies, small peptides, and/or other (small) molecules that can influence the immune response. BRMs are already being used in the clinic to treat malignancies and autoimmune diseases. Repurposing such agents would allow for accelerated use against severe and potentially fatal IAV infections. In this review, we will address the potential therapeutic use of different BRM classes to modulate the immune response induced after IAV infections.
Introduction
Influenza viruses (IVs) are responsible for significant morbidity and mortality in the human population with ~500,000 annual deaths worldwide. IVs can cause severe acute respiratory disease especially in high-risk populations like children, the elderly and the immunocompromised. While both influenza A and B viruses (IAV and IBV, respectively) cause annual epidemics, the majority of severe human infections are caused by IAV.
IVs have segmented negative-sense single-stranded RNA genomes. The lack of proof-reading activity of the viral RNA-dependent RNA polymerase (RdRp) and successive replication can lead to the accumulation of nucleotide mutations which drive antigenic drift. In addition, the segmented nature of their genome allows genetic reassortment between IV's to take place, which can produce novel strains that have acquired alternative antigenically distinct hemagglutinin, also known as antigenic shift. Both antigenic drift and antigenic shift contribute to the IV's ability to evade pre-existing host immunity induced by previous infections.
Early recognition and responses to IV infection are largely mediated by innate immune sensors expressed by its primary target, the alveolar epithelial cells (1, 2). Recognition of IVs is mediated by pattern recognition receptors (PRRs) that include Toll like receptors (TLRs), retinoinc acid inducible gene-I (RIG-I), and nucleotide oligomerization domain (NOD)-like receptor family pyrin domain containing 3 (NLRP3); all of which can recognize viral RNAs during various stages of the infection cycle (3–5). Activation of these sensors triggers signaling cascades that lead to the production of interferons as well as pro-inflammatory cytokines and chemokines ultimately resulting in an antiviral state within the surrounding cells/tissue (6). Accordingly, IVs have multiple mechanisms to evade these responses mediated by the viral nonstructural 1 protein (NS1), polymerase basic 1 protein (PB1), polymerase basic 2 protein (PB2), polymerase acidic (PA) and nucleoprotein (NP) [reviewed in van de Sandt et al. (1) and Chen et al. (2)].
In otherwise healthy individuals, IAV infections are mild and the ensuing pro- and anti-inflammatory responses are balanced. In contrast, a “cytokine storm” is typically associated with severe infections including those caused by highly pathogenic IV strains. During a cytokine storm, chemokine and cytokine responses are dysregulated in both intensity and kinetics resulting in excessive damage to the host due to infiltration of inflammatory immune cells. Acute lung injury (ALI) caused by this inflammatory response is typically characterized by significant damage or destruction of the respiratory epithelium leading to acute respiratory distress syndrome (ARDS) (7, 8).
Clinical treatment options for severe influenza virus infections remain limited and relying heavily on the administration of antiviral neuraminidase inhibitors (NAIs) and supportive critical care (9). However, NAIs have not been effective in patients with severe H7N9 or H5N1 infections and there is evidence that fatal outcomes are associated with development of antiviral resistance in patients (10–12). While virus-targeted therapies remain the standard approach, IV's mutability and adaptation to current antivirals has highlighted the need for new therapeutic options that target host factors that regulate IV infections and resulting immune responses. In either approach, the focus is to prevent or limit damage to the lung epithelium due to exaggerated or dysregulated immune cell responses. Biological response modifiers (BRMs) can alter the immune response thereby offering an additional therapeutic approach to treating severe infections. In this review, we highlight several studies that have shown the viability of BRMs as potential treatment options. For clarity, BRMs are categorized based on the type of biological agent (Table 1).
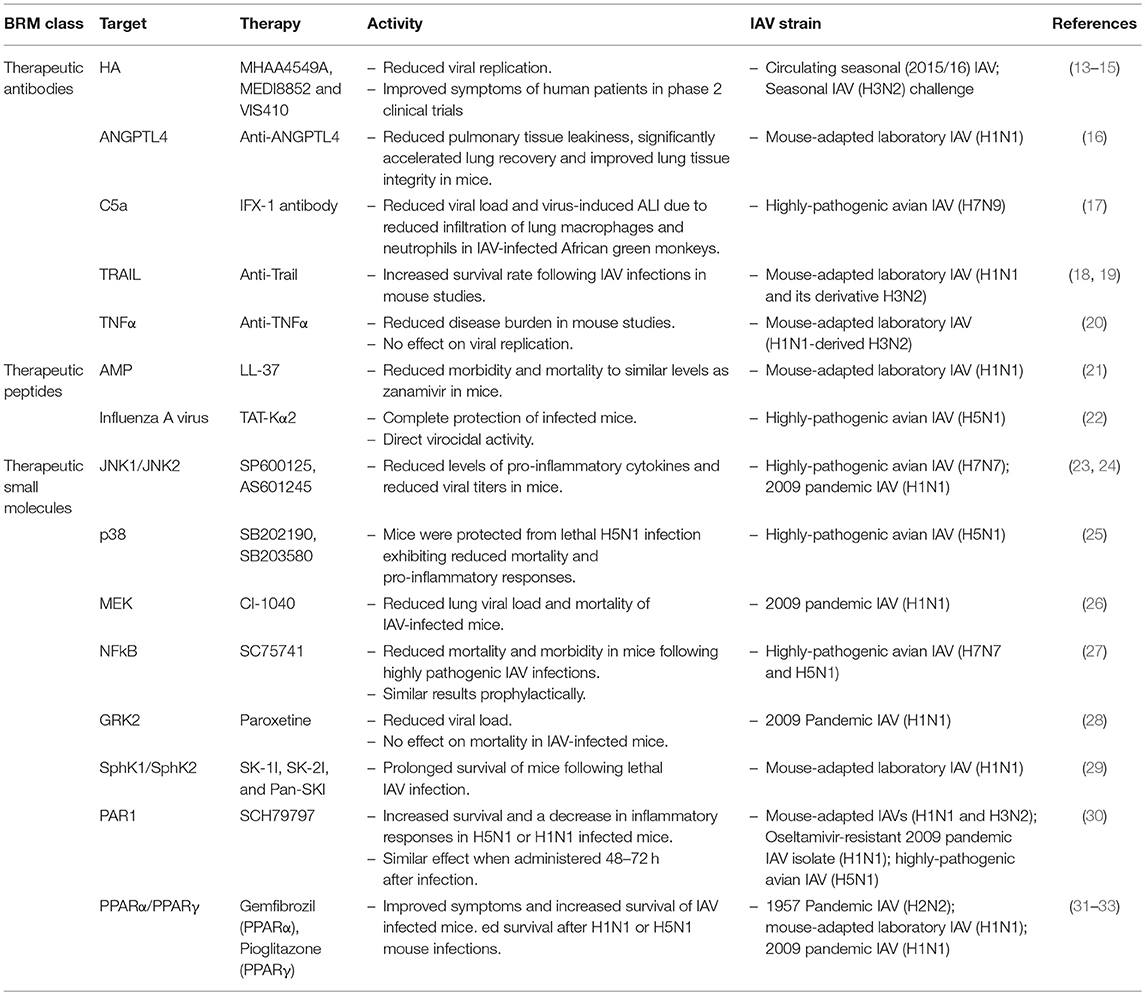
Table 1. Biological response modifiers discussed.
Biological Response Modifiers
Therapeutic Antibodies
IAV infections and some vaccines elicit broadly-neutralizing antibodies (Abs) that target the viral HA-stem. However, their abundance and immune-subdominance is overshadowed by Abs targeting the HA-head domain. The effectiveness of these HA-stem Abs against a broad range of IAV subtypes, makes them an attractive target not only for vaccine development but also as antivirals. Indeed, several HA-stem specific human monoclonal Abs are now being evaluated in clinical trials [reviewed in Davidson (34)]. MHAA4549A, MEDI8852, and VIS410 are human monoclonal Abs that have been shown to control viral replication and improve symptoms of human patients in phase 2 clinical trials (13–15).
While virus-specific Abs aim to reduce antigenic load, Abs to host targets aim at limiting the secondary wave of cytokines and reduce prolonged damaging cellular infiltration during severe infections. Host-target directed antibodies have been utilized to target key regulators of this inflammatory wave and could potentially be used to dampen these overt responses.
Angiopoietin-like 4 (ANGPTL4) is a soluble angiogenic-regulating protein. Following proteolytic cleavage, the C-terminal portion (cANGPTL4) is involved in integrin-dependent wound repair and can regulate vascular permeability (35, 36). ANGPTL4 was significantly elevated in lung biopsies from IAV-induced pneumonia patients (16). In mouse studies, neutralizing anti-ANGPTL4 Abs reduced pulmonary tissue leakiness significantly accelerating lung recovery and improved lung tissue integrity (16).
Neutrophil infiltration into the alveolar space occurs within 1 day following IAV infections (37). Neutrophil extracellular traps (NETs) released during IAV-induced pneumonia into the alveolar space caused alveolar damage (38). The complement protein C5a was shown to induce NETs release and administration of anti-C5a Abs (IFX-1) reduced H7N9-induced ALI due to reduced infiltration of lung macrophages and neutrophils as well as reduction of viral load in African green monkeys (17, 39).
Tumor necrosis factor alpha (TNFα) is a key cytokine for controlling severe IAV infections. It regulates two main antiviral functions: the induction of (i) the NFkB pathway, which ultimately controls expression of several inflammatory cytokines and (ii) apoptosis through multiple signaling cascades (40, 41). TNF upregulation during IAV infections correlates with infection severity, especially following highly pathogenic IAV-infections (42–44). Mice treated with anti-TNF Abs showed reduced disease burden; however, the authors of that study reported no effect on viral replication (20).
TNF-related apoptosis inducing ligand (TRAIL) can trigger apoptosis in IAV-infected cells. IAV-infected human epithelial cells are sensitized to TRAIL-mediated apoptosis while peripheral blood mononuclear cells upregulate TRAIL expression. Moreover, administration of monoclonal Abs against TRAIL increases survival rate following IAV infections in mouse studies (18, 19).
Therapeutic Peptides
Antimicrobial peptides (AMPs) are host proteins that have direct antibacterial and antiviral activities and can modulate immune responses to infections. While the literature is largely focused on the antibacterial aspects of AMPs, several studies have highlighted the antiviral potential of AMPs against several viruses including IVs [reviewed in Hsieh and Hartshorn (45) and Albericio and Kruger (46)]. LL-37 is a human cathelicidin derived AMP that is found predominantly in neutrophils and its expression can also be induced in epithelial cells and macrophages (47). Aerosol administration of either human LL-37 or its mouse counterpart mCRAMP led to reduced morbidity and mortality to similar levels as the neuraminidase inhibitor zanamivir that is used for the treatment of human influenza patients (21).
Both cellular and viral FADD-like IL-1β-converting enzyme-inhibitory protein (cFLIP and vFLIP, respectively) protect cells from death receptor mediated apoptosis. Kα2 is a vFLIP-derived peptide that consists of 10 amino acids from the α2 helix of the Kaposi's sarcoma herpes virus (KSHV) death effector domain 1 protein. A synthetic version of this peptide, TAT-Kα2, was generated by fusing Kα2 to a portion of the HIV TAT protein (22, 48). In mouse challenge studies, intranasal administration of TAT-Kα2 at the time of infection with highly pathogenic avian H5N1 virus resulted in protection of the treated mice. No replicating virus was detected in the lungs at either 3 or 5 days after infection suggesting complete protection from infection (22). It should be noted that this effect is largely due to direct destabilization of the virions by the TAT-Kα2 peptide and it is likely that infection in treated mice was not established; the efficacy of this AMP has not been determined during an established infection and warrants further investigation.
Therapeutic Small Molecules
Host kinases regulate not only IAV entry and replication but also initiate antiviral signaling cascades that regulate expression of pro-inflammatory chemokines and cytokines during infections and present viable targets for intervention (24, 49–58).
IAV infection has been shown to upregulate c-Jun N-terminal kinases 1 and 2 (JNK1/JNK2). These kinases directly regulate the induction of pro-inflammatory responses. IAV-induced JNK1/JNK2 activation mediates production of chemokines and cytokines including TNF-α, interferon β (IFN-β), and interleukin 6 (IL-6) (24). In vivo inhibition of JNK1/JNK2 resulted in reduced levels of pro-inflammatory cytokines and reduced viral titers (23, 24).
The mitogen activated protein kinase (MAPK), p38, regulates viral entry and replication (55, 59). Furthermore, p38 regulates IFN stimulated gene (ISG) gene expression and ultimately cytokine production via STAT1 phosphorylation (25). Using either of two specific p38 inhibitors (SB 202190 or SB 203580), mice were protected from lethal H5N1 infection exhibiting reduced mortality and pro-inflammatory responses (25). Activation of another MAPK, MEK, is required for efficient IAV replication and its inhibition results in viral ribonucleoprotein (vRNP) retention and reduced titers of progeny virus (26, 60, 61). Importantly, treatment of mice with the clinically approved MEK inhibitor (CI-1040) showed reduced lung viral load and mortality of mice following infection with a lethal dose of pandemic H1N1 IAV; interestingly, this inhibitor significantly out-performed the clinically recommended oseltamivir in these studies (26).
Another central regulator of immune responses at the epithelium as well as immune cells is the NF-κB signaling pathway. Accordingly, IAV has evolved several mechanisms to modulate this pathway to counteract antiviral responses including directly targeting the IkB kinase (IKK) (62, 63). SC75741 is a potent NFkB inhibitor that functions by reducing the ability of the p65 subunit of the NFkB complex to bind DNA; thereby limiting its transcription-regulating functions (64, 65). In vivo administration of SC75741 at 4 days after lethal infection with either H5N1 or H7N7 avian viruses resulted in significant protection with most mice surviving and showing little to no clinical symptoms; similar results were obtained by prophylactic administration (27).
G-protein coupled receptor kinase 2 (GRK2) is best known for its phosphorylation of GPCRs in cardiac tissue resulting in recruitment of β-arrestin to facilitate rapid receptor internalization and lysozomal degradation (66). Recent phosphoproteomic studies identified GRK2 as a potentially proviral host protein for IAV that plays a major role in virion uncoating (28). Although in vivo inhibition of GRK2 using paroxetine led to a significant reduction in upper respiratory tract viral load and to a modest reduction in lower respiratory tract titers at 4 days post infection, this inhibition was not protective from lethal infections (28). However, it is possible that the route of administration (intraperitoneal vs. intranasal) and dosing regimen influenced the results.
Sphingosin kinases (SphK) are lipid kinases that mediate conversion of sphingosine to bioactive lipid sphingosine 1-phosphate (S1P) (67), a known modulator of central apoptotic pathways (68). IAV infections leads to increased expression and activation of SphK1 and SphK2 (29) and in vitro inhibition of SphK1 was shown to decrease IAV RNA synthesis via suppression of NFkB activation (69). Treatment of mice with specific inhibitors to either SphK1 or SphK2 or a pan-SphK inhibitor led to prolonged survival of mice following lethal IAV infection (29).
Peroxisome proliferator-activated receptors (PPARα, PPARβ, and PPARγ) regulate metabolic homeostasis and are important mediators of the inflammatory response. Several PPAR agonists have been investigated for efficacy during IAV infections with varying results. Gemfibrozil (PPARα agonist) not only improved symptoms when administered 4 days after infections with an H2N2 virus, but also increased survival of IAV infected mice (31). Prophylactic treatment of H1N1-infected mice with pioglitazone (PPARγ agonist) resulted in increased survival (32). Combined activation of PPARγ and its downstream target AMPK improved survival of mice infected with pandemic IAV strains (33).
Protease activated receptor (PARs) link protease activity to inflammatory cellular responses (70). PAR1 expression is upregulated in the mouse airways following IAV infections (71). Intranasal administration of a PAR1 antagonist (SCH79797) at the time of infection with various IAV strains including highly pathogenic avian H5N1 and pandemic H1N1 viruses led to increased survival and a decrease in inflammatory responses. Moreover, this effect was also observed when SCH79797 was administered 48–72 h after infection (30).
The use of statins, angiotensin II receptor blockers (ARBs) and angiotensin converting enzyme inhibitors (ACEi) has been proposed to regulate the IAV-induced cytokine storm in severe infections (72, 73). Retrospective studies conducted separately in Mexico, Netherlands, UK and USA reported an association of reduced IAV-related pneumonia and lower case fatality due to lower respiratory tract IAV infections with statin treatment (74–77). However, this association was contested in two additional studies that found no benefit of statin treatment on IAV-induced disease burden (78, 79). This uncertainty regarding the IAV therapeutic potential of these widely used compounds warrants further investigations at the basic science level and in clinical trials.
Perspectives and Future Directions
The continuous accumulation of adaptive mutations and the introduction of novel viruses in the human population continue to pose a threat to public health, especially to individuals at high risk to influenza. The emergence of strains resistant to existing classes of antiviral drugs and reduced vaccine effectiveness highlights the need for the development of alternative intervention strategies. Therefore, therapeutic approaches that can diminish the potential for drug-resistance while being effective against multiple IAV subtypes/strains are highly desirable. Targeting host cell factors meets these criteria and is more likely to avoid overtly robust immune responses thereby reducing disease severity and improve patient outcome (Figure 1).
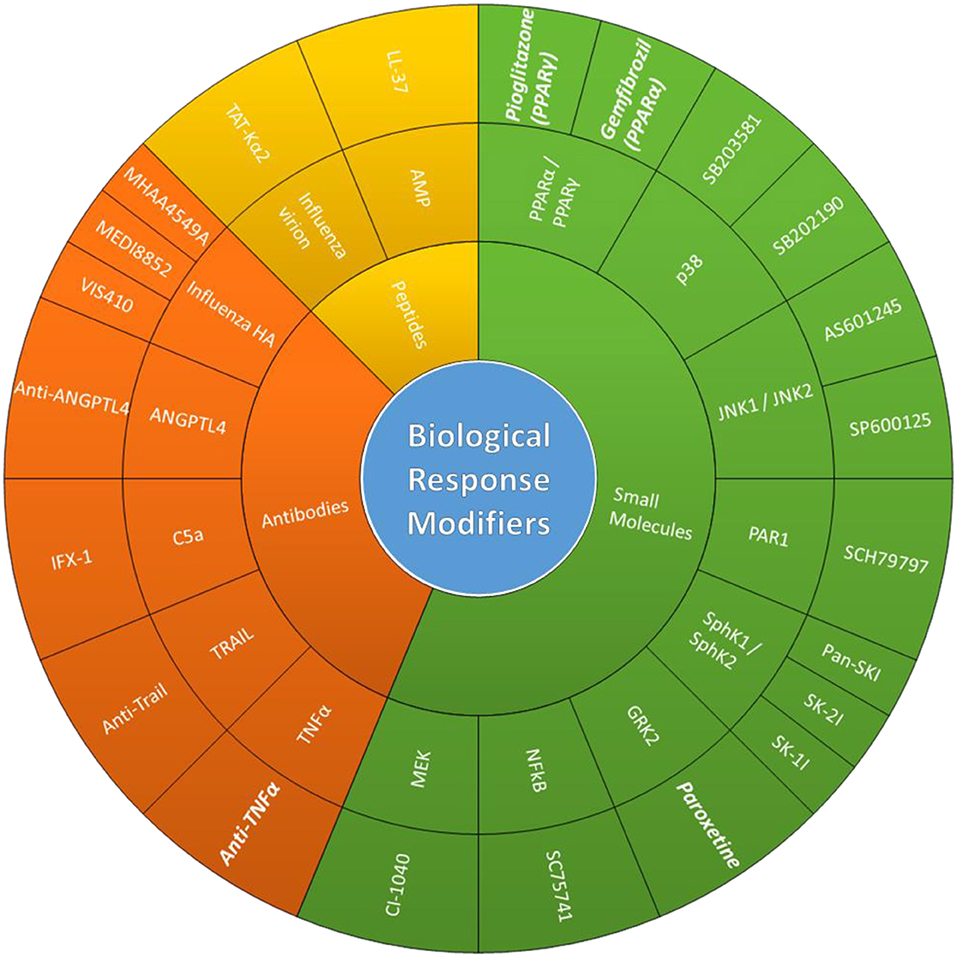
Figure 1. Biological response modifiers (BRMs). Potentially therapeutic BRMs that have shown antiviral/immunomodulatory effects during in vivo IV infections. Schematic organizing BRMs based on BRM class, host/virus targets, compounds used in cited studies (from innermost to outermost ring). FDA-approved BRMs cited in this review are in bold and italics.
A large effort has been made in recent years to identify host proteins to serve as intervention targets against IV infections. Several genetic and proteomic screens have identified several promising hits with potential roles in the IV replication cycle (80–90). In addition to these genome-wide screens, viral and host protein interactions can be mapped into networks that can also be used to identify host factors critical for IV replication (91, 92). Interestingly, meta-analysis of some these studies shows limited overlap in the genes/proteins identified as required host factors (87, 93–95). This is likely due to study-specific variations in IV types/strains and cell-lines used, inclusion/exclusion criteria, limited hit-validations and methods used to “knock-down/out” these genes.
Local microenvironment within a given tissue can dictate the quality and intensity of an immune response. Inhibition or activation of critical signaling pathways expressed in both respiratory tract epithelial and immune cells by BRMs can have opposite and unintended consequences. As discussed above, TRAIL regulates immune cell-mediated apoptosis of infected cells and several studies have shown that blocking TRAIL signaling by genomic deletion or depletion by monoclonal antibody administration can improve infection outcome in IAV-infected mice. Indeed inhibition of TRAIL signaling in alveolar macrophages and other monocytes limits their ability to induce apoptosis in alveolar cells, prevents lung tissue damage and promotes survival (19, 96, 97). However, CD8+ T cells from TRAIL−/− mice are less able to protect mice from severe infections, consistent with impaired TRAIL-mediated effector functions of CD8+ T cells (18). Similarly, opposing beneficial and detrimental outcomes have also been observed in studies using Bcl-2 inhibitors to treat IAV infections (98, 99).
BRM delivery should be guided by immune system “compartmentalization” to ensure they elicit balanced immune responses. Ideally, mucosal delivery deposits BRMs that reduce viral titers at the site of IAV replication; however, systemic delivery of certain BRMs might be required to dampen dysregulated responses. This not only depends on the BRMs used but also on the timing of their administration. Moreover, the duration of treatment with BRMs must be considered because sustained inhibition of certain inflammatory responses can result in an immune status that increases susceptibility to secondary opportunistic infections.
Repurposing of clinically approved drugs could potentially be used as BRMs for the treatment of severe IAV infectious and should be explored (86, 89, 90). Considering that susceptibility to severe IAV infections is influenced by host genetics and host-specific immune responses, selection of therapeutic BRMs should be carried out using in vivo model systems that are representative of the immune status spectrum and underlying conditions of high-risk influenza patients (young, immunocompromised, non-naive, obese, pregnant, or aged). Using these model systems will increase the likelihood of identifying BRMs with clinically relevant antiviral and immunomodulatory potentials.
Author Contributions
HE, TG, GS, and GR conceptualized and composed the manuscript. GR and HE oversaw all aspects of the manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the Alexander von Humboldt Foundation in the framework of the Alexander von Humboldt Professorship endowed by the German Federal Ministry of Education and Research. We apologize to any investigators whose relevant work was not included due to space limitations.
References
1. van de Sandt CE, Kreijtz JH, Rimmelzwaan GF. Evasion of influenza A viruses from innate and adaptive immune responses. Viruses. (2012) 4:1438–76. doi: 10.3390/v4091438
2. Chen X, Liu S, Goraya MU, Maarouf M, Huang S, Chen JL. Host immune response to influenza A virus infection. Front Immunol. (2018) 9:320. doi: 10.3389/fimmu.2018.00320
3. Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. (2009) 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x
4. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. (2010) 32:305–15. doi: 10.1016/j.immuni.2010.03.012
5. Pang IK, Iwasaki A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. (2011) 32:34–41. doi: 10.1016/j.it.2010.11.004
6. Sanders CJ, Doherty PC, Thomas PG. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res. (2011) 343:13–21. doi: 10.1007/s00441-010-1043-z
7. Kuiken T, Riteau B, Fouchier RA, Rimmelzwaan GF. Pathogenesis of influenza virus infections: the good, the bad and the ugly. Curr Opin Virol. (2012) 2:276–86. doi: 10.1016/j.coviro.2012.02.013
8. Short KR, Kroeze E, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis. (2014) 14:57–69. doi: 10.1016/S1473-3099(13)70286-X
9. Zambon M. Developments in the treatment of severe influenza: lessons from the pandemic of 2009 and new prospects for therapy. Curr Opin Infect Dis. (2014) 27:560–5. doi: 10.1097/QCO.0000000000000113
10. de Jong MD, Thanh TT, Khanh TH, Hien VM, Smith GJD, Chau NV, et al. Brief report - Oseltamivir resistance during treatment of influenza A (H5N1) infection. N Engl J Med. (2005) 353:2667–72. doi: 10.1056/NEJMoa054512
11. Chan PK, Lee N, Zaman M, Adisasmito W, Coker R, Hanshaoworakul W, et al. Determinants of antiviral effectiveness in influenza virus A subtype H5N1. J Infect Dis. (2012) 206:1359–66. doi: 10.1093/infdis/jis509
12. Hu YW, Lu SH, Song ZG, Wang W, Hao P, Li JH, et al. Association between adverse clinical outcome in human disease caused by novel influenza A H7N9 virus and sustained viral shedding and emergence of antiviral resistance. Lancet. (2013) 381:2273–9. doi: 10.1016/S0140-6736(13)61125-3
13. Wollacott AM, Boni MF, Szretter KJ, Sloan SE, Yousofshahi M, Viswanathan K, et al. Safety and upper respiratory pharmacokinetics of the hemagglutinin stalk-binding antibody VIS410 support treatment and prophylaxis based on population modeling of seasonal influenza A outbreaks. EBioMedicine. (2016) 5:147–55. doi: 10.1016/j.ebiom.2016.02.021
14. Ali O, Takas T, Nyborg AC, Jensen KM, Dubovsky F, Mallory R. A phase 2a study to evaluate the safety of MEDI8852 in outpatient adults with acute, uncomplicated influenza, A. Open Forum Infect Dis. (2017). 4(Suppl. 1):S519. doi: 10.1093/ofid/ofx163.1352
15. McBride JM, Lim JJ, Burgess T, Deng R, Derby MA, Maia M, et al. Phase 2 randomized trial of the safety and efficacy of MHAA4549A, a broadly neutralizing monoclonal antibody, in a human influenza A virus challenge model. Antimicrob Agents Chemother. (2017) 61:e01154-17. doi: 10.1128/AAC.01154-17
16. Li L, Chong HC, Ng SY, Kwok KW, Teo Z, Tan EH, et al. Angiopoietin-like 4 increases pulmonary tissue leakiness and damage during influenza pneumonia. Cell Rep. (2015) 10:654–63. doi: 10.1016/j.celrep.2015.01.011
17. Sun S, Zhao G, Liu C, Fan W, Zhou X, Zeng L, et al. Treatment with anti-C5a antibody improves the outcome of H7N9 virus infection in African green monkeys. Clin Infect Dis. (2015) 60:586–95. doi: 10.1093/cid/ciu887
18. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol. (2008) 181:4918–25. doi: 10.4049/jimmunol.181.7.4918
19. Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. (2008) 205:3065–77. doi: 10.1084/jem.20080201
20. Hussell T, Pennycook A, Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. (2001) 31:2566–73. doi: 10.1002/1521-4141(200109)31:9<2566::AID-IMMU2566>3.0.CO;2-L
21. Barlow PG, Svoboda P, Mackellar A, Nash AA, York IA, Pohl J, et al. Antiviral activity and increased host defense against influenza infection elicited by the human cathelicidin LL-37. PLoS ONE. (2011) 6:e25333. doi: 10.1371/journal.pone.0025333
22. Moon HJ, Nikapitiya C, Lee HC, Park ME, Kim JH, Kim TH, et al. Inhibition of highly pathogenic avian influenza (HPAI) virus by a peptide derived from vFLIP through its direct destabilization of viruses. Sci Rep. (2017) 7:4875. doi: 10.1038/s41598-017-04777-4
23. Nacken W, Ehrhardt C, Ludwig S. Small molecule inhibitors of the c-Jun N-terminal kinase (JNK) possess antiviral activity against highly pathogenic avian and human pandemic influenza A viruses. Biol Chem. (2012) 393:525–34. doi: 10.1515/hsz-2011-0270
24. Xie J, Zhang S, Hu Y, Li D, Cui J, Xue J, et al. Regulatory roles of c-jun in H5N1 influenza virus replication and host inflammation. Biochim Biophys Acta. (2014). 1842(12 Pt A):2479–88. doi: 10.1016/j.bbadis.2014.04.017
25. Borgeling Y, Schmolke M, Viemann D, Nordhoff C, Roth J, Ludwig S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J Biol Chem. (2014) 289:13–27. doi: 10.1074/jbc.M113.469239
26. Haasbach E, Muller C, Ehrhardt C, Schreiber A, Pleschka S, Ludwig S, et al. The MEK-inhibitor CI-1040 displays a broad anti-influenza virus activity in vitro and provides a prolonged treatment window compared to standard of care in vivo. Antiviral Res. (2017) 142:178–84. doi: 10.1016/j.antiviral.2017.03.024
27. Haasbach E, Reiling SJ, Ehrhardt C, Droebner K, Ruckle A, Hrincius ER, et al. The NF-kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza A virus. Antiviral Res. (2013) 99:336–44. doi: 10.1016/j.antiviral.2013.06.008
28. Yanguez E, Hunziker A, Dobay MP, Yildiz S, Schading S, Elshina E, et al. Phosphoproteomic-based kinase profiling early in influenza virus infection identifies GRK2 as antiviral drug target. Nat Commun. (2018) 9:3679. doi: 10.1038/s41467-018-06119-y
29. Xia C, Seo YJ, Studstill CJ, Vijayan M, Wolf JJ, Hahm B. Transient inhibition of sphingosine kinases confers protection to influenza A virus infected mice. Antiviral Res. (2018) 158:171–7. doi: 10.1016/j.antiviral.2018.08.010
30. Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, et al. PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest. (2013) 123:206–14. doi: 10.1172/JCI61667
31. Budd A, Alleva L, Alsharifi M, Koskinen A, Smythe V, Mullbacher A, et al. Increased survival after gemfibrozil treatment of severe mouse influenza. Antimicrob Agents Chemother. (2007) 51:2965–8. doi: 10.1128/AAC.00219-07
32. Aldridge JR Jr Moseley CE, Boltz DA, Negovetich NJ, Reynolds C, Franks J, et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci USA. (2009) 106:5306–11. doi: 10.1073/pnas.0900655106
33. Moseley CE, Webster RG, Aldridge JR. Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influenza Other Respir Viruses. (2010) 4:307–11. doi: 10.1111/j.1750-2659.2010.00155.x
34. Davidson S. Treating influenza infection, from now and into the future. Front Immunol. (2018) 9:1946. doi: 10.3389/fimmu.2018.01946
35. Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L, et al. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J Biol Chem. (2010) 285:32999–3009. doi: 10.1074/jbc.M110.108175
36. Guo L, Li SY, Ji FY, Zhao YF, Zhong Y, Lv XJ, et al. Role of Angptl4 in vascular permeability and inflammation. Inflamm Res. (2014) 63:13–22. doi: 10.1007/s00011-013-0678-0
37. Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. (2008) 4:e1000115. doi: 10.1371/journal.ppat.1000115
38. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
39. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
40. Julkunen I, Sareneva T, Pirhonen J, Ronni T, Melen K, Matikainen S. Molecular pathogenesis of influenza A virus infection and virus-induced regulation of cytokine gene expression. Cytokine Growth Factor Rev. (2001) 12:171–80. doi: 10.1016/S1359-6101(00)00026-5
41. Iwai A, Shiozaki T, Miyazaki T. Relevance of signaling molecules for apoptosis induction on influenza A virus replication. Biochem Biophys Res Commun. (2013) 441:531–7. doi: 10.1016/j.bbrc.2013.10.100
42. Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. (2007) 81:2736–44. doi: 10.1128/JVI.02336-06
43. Peiris JS, Cheung CY, Leung CY, Nicholls JM. Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol. (2009) 30:574–84. doi: 10.1016/j.it.2009.09.004
44. Guo XJ, Thomas PG. New fronts emerge in the influenza cytokine storm. Semin Immunopathol. (2017) 39:541–50. doi: 10.1007/s00281-017-0636-y
45. Hsieh IN, Hartshorn KL. The role of antimicrobial peptides in influenza virus infection and their potential as antiviral and immunomodulatory therapy. Pharmaceuticals. (2016) 9:53. doi: 10.3390/ph9030053
46. Albericio F, Kruger HG. Therapeutic peptides. Future Med Chem. (2012) 4:1527–31. doi: 10.4155/fmc.12.94
47. Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. (2004) 75:39–48. doi: 10.1189/jlb.0403147
48. Lee JS, Li QL, Lee JY, Lee SH, Jeong JH, Lee HR, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. (2009) 11:1355–U1225. doi: 10.1038/ncb1980
49. Ohmichi M, Pang L, Ribon V, Gazit A, Levitzki A, Saltiel ARJB. The tyrosine kinase inhibitor tyrphostin blocks the cellular actions of nerve growth factor. Biochemistry. (1993) 32:4650–8. doi: 10.1021/bi00068a024
50. Sieczkarski SB, Brown HA, Whittaker GR. Role of protein kinase C betaII in influenza virus entry via late endosomes. J Virol. (2003) 77:460–9. doi: 10.1128/JVI.77.1.460-469.2003
51. Ehrhardt C. From virus entry to release: the diverse functions of PI3K during RNA virus infections. Future Virol. (2011) 6:1225–39. doi: 10.2217/fvl.11.90
52. Marjuki H, Gornitzky A, Marathe BM, Ilyushina NA, Aldridge JR, Desai G, et al. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell Microbiol. (2011) 13:587–601. doi: 10.1111/j.1462-5822.2010.01556.x
53. Planz O. Development of cellular signaling pathway inhibitors as new antivirals against influenza. Antiviral Res. (2013) 98:457–68. doi: 10.1016/j.antiviral.2013.04.008
54. Elbahesh H, Cline T, Baranovich T, Govorkova EA, Schultz-Cherry S, Russell CJ. Novel roles of focal adhesion kinase in cytoplasmic entry and replication of influenza A viruses. J Virol. (2014) 88:6714–28. doi: 10.1128/JVI.00530-14
55. Choi MS, Heo J, Yi CM, Ban J, Lee NJ, Lee NR, et al. A novel p38 mitogen activated protein kinase (MAPK) specific inhibitor suppresses respiratory syncytial virus and influenza A virus replication by inhibiting virus-induced p38 MAPK activation. Biochem Biophys Res Commun. (2016) 477:311–6. doi: 10.1016/j.bbrc.2016.06.111
56. Elbahesh H, Bergmann S, Russell CJ. Focal adhesion kinase (FAK) regulates polymerase activity of multiple influenza A virus subtypes. Virology. (2016) 499:369–74. doi: 10.1016/j.virol.2016.10.002
57. Zhang J, Ruan T, Sheng T, Wang J, Sun J, Wang J, et al. Role of c-Jun terminal kinase (JNK) activation in influenza A virus-induced autophagy and replication. Virology. (2018) 526:1–12. doi: 10.1016/j.virol.2018.09.020
58. Meineke R, Rimmelzwaan GF, Elbahesh H. Influenza virus infections and cellular kinases. Viruses. (2019) 11:171. doi: 10.3390/v11020171
59. Marchant D, Singhera GK, Utokaparch S, Hackett TL, Boyd JH, Luo Z, et al. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J Virol. (2010) 84:11359–73. doi: 10.1128/JVI.00804-10
60. Droebner K, Pleschka S, Ludwig S, Planz O. Antiviral activity of the MEK-inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo. Antiviral Res. (2011) 92:195–203. doi: 10.1016/j.antiviral.2011.08.002
61. Haasbach E, Hartmayer C, Planz O. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro. Antiviral Res. (2013) 98:319–24. doi: 10.1016/j.antiviral.2013.03.006
62. Chu WM, Ostertag D, Li ZW, Chang L, Chen Y, Hu Y, et al. JNK2 and IKKbeta are required for activating the innate response to viral infection. Immunity. (1999) 11:721–31. doi: 10.1016/S1074-7613(00)80146-6
63. Gao S, Song L, Li J, Zhang Z, Peng H, Jiang W, et al. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell Microbiol. (2012) 14:1849–66. doi: 10.1111/cmi.12005
64. Leban J, Baierl M, Mies J, Trentinaglia V, Rath S, Kronthaler K, et al. A novel class of potent NF-kappaB signaling inhibitors. Bioorg Med Chem Lett. (2007) 17:5858–62. doi: 10.1016/j.bmcl.2007.08.022
65. Ehrhardt C, Ruckle A, Hrincius ER, Haasbach E, Anhlan D, Ahmann K, et al. The NF-kappaB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol. (2013) 15:1198–211. doi: 10.1111/cmi.12108
66. Huang ZM, Gao E, Chuprun JK, Koch WJ. GRK2 in the heart: a GPCR kinase and beyond. Antioxid Redox Signal. (2014) 21:2032–43. doi: 10.1089/ars.2014.5876
67. Pitson SM. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem Sci. (2011) 36:97–107. doi: 10.1016/j.tibs.2010.08.001
68. Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochimica et Biophysica Acta (BBA) Biomembranes. (2006) 1758:2016–26. doi: 10.1016/j.bbamem.2006.08.007
69. Seo YJ, Pritzl CJ, Vijayan M, Bomb K, McClain ME, Alexander S, et al. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral RNA synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PLoS ONE. (2013) 8:e75005. doi: 10.1371/journal.pone.0075005
70. Coughlin SR, Camerer E. PARticipation in inflammation. J Clin Invest. (2003) 111:25–7. doi: 10.1172/JCI17564
71. Lan RS, Stewart GA, Goldie RG, Henry PJ. Altered expression and in vivo lung function of protease-activated receptors during influenza A virus infection in mice. Am J Physiol Lung Cell Mol Physiol. (2004) 286:L388–398. doi: 10.1152/ajplung.00286.2003
72. Fedson DS. Pandemic influenza: a potential role for statins in treatment and prophylaxis. Clin Infect Dis. (2006) 43:199–205. doi: 10.1086/505116
73. Fedson DS. Treating influenza with statins and other immunomodulatory agents. Antiviral Res. (2013) 99:417–35. doi: 10.1016/j.antiviral.2013.06.018
74. Enserink M. Infectious disease. Old drugs losing effectiveness against flu; could statins fill gap? Science. (2005) 309:1976–7. doi: 10.1126/science.309.5743.1976a
75. Frost FJ, Petersen H, Tollestrup K, Skipper B. Influenza and COPD mortality protection as pleiotropic, dose-dependent effects of statins. Chest. (2007) 131:1006–12. doi: 10.1378/chest.06-1997
76. Carrillo-Esper R, Sosa-Garcia JO, Arch-Tirado E. [Experience in the management of the severe form of human influenza A H1N1 pneumonia in an intensive care unit]. Cir Cir. (2011) 79:409–16.
77. Vandermeer ML, Thomas AR, Kamimoto L, Reingold A, Gershman K, Meek J, et al. Association between use of statins and mortality among patients hospitalized with laboratory-confirmed influenza virus infections: a multistate study. J Infect Dis. (2012) 205:13–9. doi: 10.1093/infdis/jir695
78. Kwong JC, Li P, Redelmeier DA. Influenza morbidity and mortality in elderly patients receiving statins: a cohort study. PLoS ONE. (2009) 4:e8087. doi: 10.1371/journal.pone.0008087
79. Fleming DM, Verlander NQ, Elliot AJ, Zhao H, Gelb D, Jehring D, et al. An assessment of the effect of statin use on the incidence of acute respiratory infections in England during winters 1998-1999 to 2005-2006. Epidemiol Infect. (2010) 138:1281–8. doi: 10.1017/S0950268810000105
80. Hao L, Sakurai A, Watanabe T, Sorensen E, Nidom CA, Newton MA, et al. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. (2008) 454:890–3. doi: 10.1038/nature07151
81. Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. (2009) 139:1243–54. doi: 10.1016/j.cell.2009.12.017
82. Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. (2009) 139:1255–67. doi: 10.1016/j.cell.2009.12.018
83. Sui BQ, Bamba D, Weng K, Ung H, Chang SJ, Van Dyke J, et al. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology. (2009) 387:473–81. doi: 10.1016/j.virol.2009.02.046
84. Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. (2010) 463:818–22. doi: 10.1038/nature08760
85. Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, et al. Human host factors required for influenza virus replication. Nature. (2010) 463:813–7. doi: 10.1038/nature08699
86. Tripp RA, Mark Tompkins S. Antiviral effects of inhibiting host gene expression. Curr Top Microbiol Immunol. (2015) 386:459–77. doi: 10.1007/82_2014_409
87. Watanabe T, Kawaoka Y. Influenza virus-host interactomes as a basis for antiviral drug development. Curr Opin Virol. (2015) 14:71–8. doi: 10.1016/j.coviro.2015.08.008
88. Han J, Perez JT, Chen C, Li Y, Benitez A, Kandasamy M, et al. Genome-wide CRISPR/Cas9 screen identifies host factors essential for influenza virus replication. Cell Rep. (2018) 23:596–607. doi: 10.1016/j.celrep.2018.03.045
89. Li CC, Wang XJ, Wang HR. Repurposing host-based therapeutics to control coronavirus and influenza virus. Drug Discov Today. (2019) 24:726–36. doi: 10.1016/j.drudis.2019.01.018
90. Pizzorno A, Terrier O, Nicolas de Lamballerie C, Julien T, Padey B, Traversier A, et al. Repurposing of drugs as novel influenza inhibitors from clinical gene expression infection signatures. Front Immunol. (2019) 10:60. doi: 10.3389/fimmu.2019.00060
91. Wang L, Fu B, Li W, Patil G, Liu L, Dorf ME, et al. Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat Commun. (2017) 8:13876. doi: 10.1038/ncomms13876
92. Ackerman EE, Kawakami E, Katoh M, Watanabe T, Watanabe S, Tomita Y, et al. Network-guided discovery of influenza virus replication host factors. MBio. (2018) 9:e02002-18. doi: 10.1128/mBio.02002-18
93. Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe. (2010) 7:427–39. doi: 10.1016/j.chom.2010.05.008
94. de Chassey B, Meyniel-Schicklin L, Aublin-Gex A, Andre P, Lotteau V. Genetic screens for the control of influenza virus replication: from meta-analysis to drug discovery. Mol Biosyst. (2012) 8:1297–303. doi: 10.1039/c2mb05416g
95. Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, et al. Meta- and orthogonal integration of influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe. (2015) 18:723–35. doi: 10.1016/j.chom.2015.11.002
96. Brincks EL, Gurung P, Langlois RA, Hemann EA, Legge KL, Griffith TS. The magnitude of the T cell response to a clinically significant dose of influenza virus is regulated by TRAIL. J Immunol. (2011) 187:4581–8. doi: 10.4049/jimmunol.1002241
97. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun. (2014) 5:3864. doi: 10.1038/ncomms4864
98. Kakkola L, Denisova OV, Tynell J, Viiliainen J, Ysenbaert T, Matos RC, et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. (2013) 4:e742. doi: 10.1038/cddis.2013.267
Keywords: influenza, treatment, response modifiers, antiviral, immune response, immunomodulators
Citation: Elbahesh H, Gerlach T, Saletti G and Rimmelzwaan GF (2019) Response Modifiers: Tweaking the Immune Response Against Influenza A Virus. Front. Immunol. 10:809. doi: 10.3389/fimmu.2019.00809
Received: 10 January 2019; Accepted: 26 March 2019;
Published: 12 April 2019.
Edited by:
Abhijeet Anil Bakre, University of Georgia, United StatesReviewed by:
Elisa Vicenzi, San Raffaele Hospital (IRCCS), ItalyGiulia Fabozzi, National Institute of Allergy and Infectious Diseases (NIAID), United States
Copyright © 2019 Elbahesh, Gerlach, Saletti and Rimmelzwaan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Husni Elbahesh, aHVzbmkuZWxiYWhlc2hAdGloby1oYW5ub3Zlci5kZQ==
Guus F. Rimmelzwaan, Z3V1cy5yaW1tZWx6d2FhbkB0aWhvLWhhbm5vdmVyLmRl