 Ning Song
Ning Song Friedrich Thaiss1*
Friedrich Thaiss1* Linlin Guo
Linlin Guo- 1III. Department of Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Department of Critical Care Medicine, The First Affiliated Hospital of Harbin Medical University, Harbin, China
The global burden of chronic kidney disease will increase during the next century. As NFκB, first described more than 30 years ago, plays a major role in immune and non-immune-mediated diseases and in inflammatory and metabolic disorders, this review article summarizes current knowledge on the role of NFκB in in vivo kidney injury and describes the new and so far not completely understood crosstalk between canonical and non-canonical NFκB pathways in T-lymphocyte activation in renal disease.
Introduction
More than 30 years ago, a protein binding to a specific conserved DNA sequence in nuclear extracts of activated B lymphocyte was identified by Sen and Baltimore (1) and called nuclear factor binding near the κ light-chain gene in B cells (NFκB), although it is not B cell specific, as is now known. NFκB is a protein family consisting of 5 dimers, RelA (p65), RelB, c-Rel, p50 (generated from p105), and p52 (generated from p100), which can form a variety of homodimers or heterodimers. All the subsets share a Rel homology domain that can bind DNA, dimerize and translocate into the nuclear compartment. However, only RelA, RelB and c-Rel have the necessary transcriptional activation domain for target gene expression (2). Normally, NFκB dimers are inactivated through interacting with the inhibitor of κB (IκB) (3). The IκB proteins, including IκBα, IκBβ, and IκBε, preferentially act on different c-Rel and p65 dimers (4), and sometimes, p100 that is not processed to p52 can act as an IκB protein, termed IκBδ (5).
NFκB is regulated by two pathways: the canonical (NEMO-dependent) and the non-canonical (NEMO-independent) pathway. The canonical NFκB pathway always responds rapidly and is mediated by a kinase complex comprising IKKα, IKKβ, and regulatory NEMO (IKKγ). Immune signals, including antigens, Toll-like receptor (TLR) ligand and inflammatory cytokines, such as tumor necrosis factor (TNF) and interleukin 1β (IL-1β), can lead to phosphorylation-dependent activation of this kinase complex. The activated complex will phosphorylate the IκB bound to NFκB dimers (such as p50-p65 and p50-c-Rel), leading to ubiquitination and subsequent proteasome-induced degradation of IκB (6). The degradation releases the NFκB dimers, allowing them to bind κB site-containing DNA and rapidly accumulate in the nuclear compartment. After activation, the synthesized IκB proteins bind to and inhibit NFκB activity and traffic it back to form a negative feedback loop (6).
The non-canonical pathway can be stimulated by members of the TNF cytokine family (such as CD40 ligand, TWEAK and lymphotoxin-β) (7, 8) and membrane attack complexes (MACs) (9). Compared with the constitutive processing of p105 in the canonical pathway, the activation of the non-canonical pathway is based on the processing of p100. This process is accomplished via phosphorylation and subsequent ubiquitination of p100. An important protein, NFκB-inducing kinase (NIK, or MAP3K14), plays a role in phosphorylation through activation of kinase IKKα (10, 11). Once activated, IKKα will trigger the generation of p50-RelB via proteolytic processing of p100 and binding to RelB. The activated NFκB dimers translocate into the nucleus, bind to the DNA and have a major effect on lymphoid organogenesis (Figure 1).
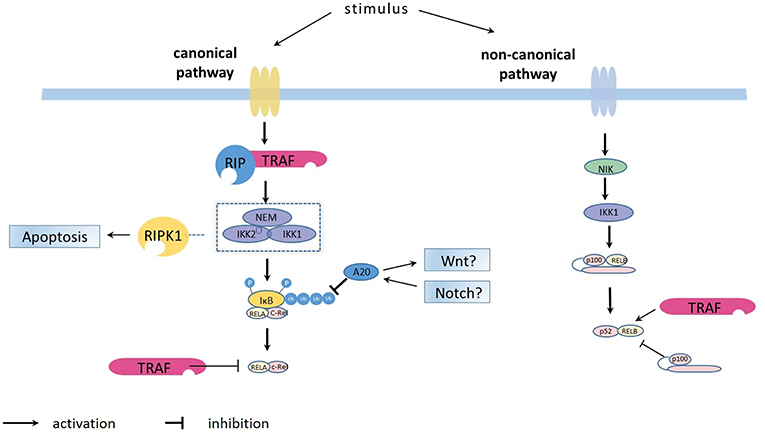
Figure 1. Overview on the principles of canonical and non-canonical NFκB pathways activation. The canonical NFκB pathway is activated by the kinase complex comprising IKKα (IKK1), IKKβ (IKK2) and regulatory NEMO which are for example activated via the TRAF/ RIP complexes (https://doi.org/10.3389/fimmu.2019.00326). The activated IKK1/IKK2/NEMO complex phosphorylates the IκB bound to NFκB dimers (such as p50-p65 and p50-c-Rel), leading to ubiquitination and subsequent proteasome-induced degradation of IκB. The degradation releases the NFκB dimers, allowing them to bind κB site-containing DNA and rapidly accumulate in the nuclear compartment. After activation, the synthesized IκB proteins and also TRAF bind to and inhibit NFκB activity and traffic it back to form a negative feedback loop. The ubiquitin-editing enzyme A20, regulated by the WNT and Notch pathways, represses NFκB function by deubiquitinating of IκB. The non-canonical NFκB pathway is activated through NFκB-inducing kinase (NIK, or MAP3K14), which phosphorylates IKKα (IKK1) which triggers the generation of p50-RelB via proteolytic processing of p100 and binding to RelB.
Ubiquitination plays a major role in controlling many transcription factors, such as NFκB. As a reversible and dynamic event, ubiquitination is involved in the upstream activation of both the canonical and non-canonical NFκB pathways (12). However, post-translational modifications, including ubiquitination, are also critical in regulating the NFκB transcriptional response. Stimulus-dependent induction of these modifications can affect the ability of NFκB dimers to bind DNA, interact with IκB proteins, recruit essential co-activators and alter the stability of the proteins. For example, after stimulation of TLRs, the linear ubiquitin assembly complex (LUBAC), which is composed of heme-oxidized IRP2 (HOIL-1L), HOIL-interacting protein (HOIP) and SHANK-associated RH domain-interacting protein (SHARPIN), is recruited by TRAF6 and target NEMO to activate the NFκB pathway (13, 14). However, it is now apparent that ubiquitination and proteasomal degradation of NFκB subunits also represents a major limiting factor in the NFκB transcriptional response (12, 15) (see also below).
Upon activation, NFκB dimers are transported to the nuclear compartment and regulate gene expression by binding to enhancer elements and recruit chromatin remodeling complexes to enhance transcription (16, 17). RelA, for example, recruits acetyltransferases or histone deacetylases after phosphorylation to form the p300/CBP complex (18). c-Rel dimers may remove repressive H3K9 dimethyl modifications from gene promoters to promote pro-inflammatory gene expression. RelA at the same promoter may drive divergent phenotypes by modifying transcriptional noise by targeting chromatin changes (19). NFκB may also synergistically regulate transcription with other transcription factors (12). Recent studies have shown that during activation of key inflammatory genes, NFκB and IRF3 show a gene-specific cooperation in the transcriptional regulation of these genes (20, 21).
In the canonical pathway, IκBs inhibit NFκB dimers and restore the resting state. There are several IκB proteins, including IκBα, IκBβ, and the inhibitory regions on both p100 and p105. As the predominant inhibitory protein, IκB can not only bind NFκB dimers in the cytoplasm, but can also translocate into the nucleus and halt transcription through binding with dimers. On the other hand, dimers that only contain p50 or p52 without transactivation domains repress active dimer binding (22). Upstream of the IκBs, the IRAK-M and several NLRs (NLRP12, NLRX1, and NLRC3) can also negatively regulate NFκB by inhibiting key components in the pathways (23). In the process of posttranscriptional regulation, some micro-RNAs (miRNAs), for example, miR-146a and, miR-302b, and some RNA-binding proteins, such as tristeraprolin (TTP), Regnase-1 and Roquin, regulate the stability or translation of mRNAs encoding specific signaling adaptors of the NFκB pathway (24).
In the negative regulation of NFκB, deubiquitylation plays also a vital role. The ubiquitin-editing enzyme A20 (TNFAIP3) represses NFκB function by deubiquitinating several intermediate NFκB signaling molecules, such as RIP1, TRAF6, and MALT1 (25). Other DUBs, such as CYLD, OTULIN, and cellular zinc finger anti-NFκB (Cezanne) (23), also negatively regulate the NFκB pathway in specific cells. More recently, a novel function for IKKα has been described: it nucleates the ubiquitin-editing enzyme A20 complex, thus enhancing the negative regulatory function in canonical NFκB signaling (26).
The NFκB pathway, especially the canonical one, dynamically regulates the response to external stimulation. In the presence of IκBs and the A20 negative feedback loop, activated NFκB shuttles between the nucleus and the cytoplasm and causes the oscillation of transcription activation and inactivation. For example, different intervals of pulsatile TNFα stimulation particularly effect the transcription of late genes. On the other hand, stimulation with LPS causes only slow and non-oscillatory NFκB activation (27, 28). Playing a key role in this oscillation, newly synthesized IκB is restored in a distant location in the cytoplasm and acts as a pool for resetting NFκB (29). Moreover, the export of IκB mRNA from the nucleus regulates the persistency of the oscillation, and importation of IκB protein effects the frequency of oscillation (30, 31). Body temperature and the circadian clock also regulate the oscillation of NFκB through both IκBα and A20 (32, 33).
In a single cell population, various stimuli and target genes contribute to different dynamics, but the dynamics of NFκB in an identical population of cells are even more complex and differentially regulated considering the susceptibility and inherent stochasticity of the system in each single cell (34–36). Although NFκB plays a vital role in the activation and development of B cells, it is also of fundamental importance in every cell type, such as in innate immune cells and lymphocytes (37), as well as renal (38), and liver (39) parenchymal cells. NFκB has an effect on multiple diseases, for instance, on inflammatory, metabolic, and autoimmune diseases (24, 40) and cancer (41–43). In this review, we focus on the role NFκB may play in in vivo kidney diseases.
Renal Disease—the Perspective of the Global Burden of Chronic Kidney Injury
Kidney diseases can be induced by numerous causes, such as infection, immune and non-immune injury or autoimmune diseases. Most patients who do not repair from their kidney injury will progress to chronic kidney disease (CKD) and finally develop renal failure. In 2002, CDK was the 17th worldwide leading cause of death. The annual mortality rate due to CKD has increased from 11.6 to 15.8 per 100,000 population from 1990 to 2013. It is projected that by 2030, the global burden of kidney diseases will become the 13th leading cause of death worldwide (44). No matter where CKD originates from, inflammation and the immune system play a vital role in the induction of renal injury.
The pathophysiology of kidney diseases is complex, and some of the most recent relevant mechanisms described are briefly mentioned here. The innate immune system includes pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) induced on innate immune cells, for example, macrophages, neutrophils and dendritic cells. DAMP and PAMP are recognized by pattern-recognition receptors (PRR) expressed on these cells. Toll-like receptors (TLRs) are members of the PRR family and can be found either on the surface or in the intracellular compartment of cells. TLRs can activate the canonical NFκB pathway, which is responsible for the transcription of pro-inflammatory cytokines and chemokines and may result in the progression of kidney disease (45–47). Adaptive immune components, including various T helper (Th) cells, such as Th1, Th2, Th17, and T follicular (Tfh) cells, are derived from naïve CD4 T cells. Th1 and Th17 and the pro-inflammatory cytokines secreted by these cells are involved in many inflammatory renal diseases (48–51). NFκB functions not only in the transcription of pro-inflammation genes in Th1 and Th17 cells but also promotes their differentiation from naïve CD4 T cells through the interaction with innate immune cells (52). In addition to immune cells, some renal cells, for instance mesangial and tubular epithelial cells, can also promote inflammation through the TLRs-NFκB axis (53–55).
The complement system is also involved in the pathogenesis of kidney diseases (56, 57), in kidney transplantation (58, 59) and in T-cell mediated immune injury (60, 61). The classical pathway induced by IgG1, IgG3 and IgM activates C1q, resulting in the activation of C3 through C2 and C4. The mannose-binding lectin (MBL) pathway activate C2 and C4, but not C1, and the alternate pathway activates C3 directly. All three pathways proceed through C3 and C5, causing the production of C5a and C5b. C5a augments inflammation through attracting the inflammation cells, and by contrast, C5b generates the terminal membrane attach complex (C5b-9) (62, 63). The NFκB pathway plays an important role in renal damage mediated by enhanced complement activation (64).
Renal Disease—Glomerulonephritis
Glomerulonephritis (GN) includes various forms, such as postinfectious GN, lupus nephritis, IgA nephropathy, anti-glomerular basement membrane (GBM) disease and primary and idiopathic membranous nephropathy (iMN), which result from the interaction between renal cells and the immune system with and without immune complex formation. Antigen-Ig antibody complexes (IC) emerge through circulating immune complexes trapped in the glomerulus or from immune deposits with exogenous or endogenous antigens in situ (65). For example, in iMN, most patients, nearly 70%, have antibodies directed against the podocyte membrane antigen phospholipase A2 receptor1 (PLA2R) (66, 67). As another important part of the adaptive immune system in glomerulonephritis, T cells, especially diverse T helper (Th) cells, play a vital role in the induction and maintenance of renal diseases. Accumulating evidence indicates that Th17-cells and their characteristic cytokine IL-17 accelerate the induction of nephrotoxic nephritis (NTN), anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis and lupus nephritis (48).
The role of NFκB in in vivo models of immune glomerular injury is less clear. TANK-binding kinase 1 (TBK1) can suppress the production of IgA in B cells by accelerating the degradation of NIK, which plays an important role in the NFκB non-canonical pathway (68, 69). Our data showed that in crescentic GN, an inhibitor of IKK2, unexpectedly accelerated the disease, which may have resulted from Treg cell impairment (70). In another experimental model of crescentic GN, deficiency in NFκB1 (p50) increased the severity of acute glomerular injury but NFκB1 did not influence the chronic, fibrotic phase (71). In the podocyte, NEMO can activate proinflammatory signaling and aggravate GN (72). In podocyte-specific NEMO knockout mice, glomerular lesion can resolve faster by changing the podocyte cytoskeletal dynamic independently of NFκB (73). In mesangial cells, for example, in lupus nephritis, inhibition of NFκB blocks cell proliferation through the cyclin D1 pathway (74).
Ubiquitin C-terminal hydrolase L1 (UCH-L1) is a deubiquitinating enzyme that influences the inflammatory response by transcription through the ubiquitin-proteasome system (UPS). NFκB increases UCH-L1 expression in podocytes, which plays a role in immune complex-mediated, but not in non-immune complex-mediated, GN (75, 76). UCH-L1 knockout mice have an exacerbation of kidney injury, probably through the decline of proteasomal activity and the increase in oxidative-modified and polyubiquitinated proteins (77). Disruption of ABIN1, another deubiquitinating enzyme, increases the severity of GN through the increased activation of NFκB in podocytes (78). NFκB also induces the expression of periostin and accelerates kidney injury in GN through the activation of integrin-β3 (79).
In patients with immune-mediated glomerular injury, the activation of NFκB has been shown by immunohistochemistry. NFκB is overexpressed in IgA nephropathy, especially in patients with high proteinuria and reduced renal function (80, 81). In patients with VEGF-targeted therapy, increased expression of NFκB (RelA) can repress the content of c-mip in renal thrombotic microangiopathy (TMA) (82). The immunohistochemical examinations in patients with glomerulonephritis demonstrate RelA expression, while other NFκB proteins such as RelB, p52, or p50 have rarely been described. All these morphological examinations in human biopsy only describe the correlation of NFκB expression and clinical outcome parameters in these patients; more evidence about the direct causal relationship of NFκB and disease is still lacking. Single-nucleotide polymorphisms of NFκB, NFκB-inhibitors and NFκB-kinases have also been associated with glomerular kidney diseases (83, 84).
Renal Disease—Acute Kidney Injury
Acute kidney injury (AKI) is a syndrome defined by a rapid decrease in glomerular filtration, which may be induced by sepsis, ischemia/reperfusion injury (IRI) or nephrotoxic drugs. It is associated with substantial mortality and morbidity in high-income and in low- to middle-income areas (85). Importantly, AKI not only contributes to short-term adverse outcomes, but the survivors may suffer from chronic kidney (CKD) and even end-stage renal disease (ESRD).
As one of the most important components of the pathogenesis, systematic inhibition of NFκB affects the severity of AKI. In a disease model induced by folic acid, inhibition of NFκB mitigates AKI-injury by reduction of RelA and NFκB2 activation (86). In another experimental model of AKI, inhibition of IκB kinase ameliorates injury and improves kidney function even if started 24 h after initiation of AKI (87). NFκB plays its role through immune and epithelial cells in AKI, linking the coordination between inflammation and cell death (88). In innate immune cells, suppression of NFκB in macrophages through blockade of CD38 alleviates LPS-induced AKI (89). In addition, activation of NFκB can increase the infiltration of M1 macrophages in AKI, which may involve the pattern recognition receptor Mincle (90). There is evidence that inhibition of NFκB, specifically in T cells using IκBαΔN-Tg mice to express a transdominant form of IκBα that constitutively represses the activity of multiple NFκB/Rel proteins, protects kidneys from ischemia/reperfusion injury in mice (91). However, by contrast, data from our group showed that in a similar animal model, lymphocyte-specific knockout of IKK2 or NEMO aggravates the damage through the increased infiltration of Th17 cells (92). This discrepancy may result from differences in T cell subpopulations infiltrating the kidneys or from different NFκB subunit activation.
In NFκB tubular epithelial-specific knockout mice, the decrease in NFκB activation protects mice from ischemic AKI through reduced apoptosis and chemokine expression (93). In response to ischemia- and cisplatin-induced AKI, the anti-inflammatory effect of KIM-1 expression is due to the interaction of KIM-1 with p85 and subsequent PI3K-dependent downmodulation of NFκB, suggesting that KIM-1-mediated epithelial cell phagocytosis of apoptotic cells may protect the kidneys after acute injury by downregulating NFκB (94). NFκB, however, may also have anti-inflammatory properties (95). In the IRI mice model, preactivation of NFκB by LPS alleviates subsequent kidney injury, which is accomplished by the HIF-2α-regulated nitric oxide production (96). However, NFκB activation in podocytes is dispensable in the pathogenesis of IRI (97).
As for the function of NFκB in the long-term outcome of AKI, inhibiting IκB kinase showed that AKI induced fibrosis was reduced within 28 days (87). Another group has shown that the disease severity of unilateral ureteric obstruction (UUO) in NFκB1−/− (p50) animals was equivalent with that in wild-type animals (71). Tumor necrosis factor-like weak inducer of apoptosis (TWEAK), which promotes inflammation via NFκB/STAT3 pathways, may also play a major role in the long-term outcome of AKI by activating fibroblast growth factor-inducible-14 (Fn14) receptor, as has been reviewed recently (98). Single-nucleotide polymorphisms of NFκB and NFκB kinases have also been associated with AKI (99).
Renal Disease—Metabolic Kidney Injury
Obesity is intensely related to type 2 diabetes mellitus (T2D), cardiovascular disease, fatty liver disease and obesity-associated cancers. These diseases are assembled as “metabolic syndrome”. The pathology of all these diseases emerges from insulin resistance and the constantly chronic, low-grade inflammation that is caused by the activation of various inflammatory signals, such as NFκB, JNK, and inflammasomes (100). The kidney is susceptible to diabetes and obesity, with functional impairment including increased proteinuria and glomerular hyperfiltration (101). Therefore, treating chronic, low-grade inflammation by an anti-inflammatory therapy may be a good strategy for T2D-associated kidney injury.
A role of celastrol, an NFκB inhibitor, on insulin resistance and renal function has been demonstrated in diabetic db/db mice (102). In these experiments, celastrol treatment reduced the creatinine levels, albuminuria, glomerular matrix expansion as well as renal lipids and insulin resistance (102). In another study, peritoneal injection of miR-451 improved renal function in diabetic nephropathy, and the effect was accomplished by the suppression of the LMP7/NFκB pathway-mediated proinflammatory molecule expression (103). Downregulation of NFκB can also ameliorate kidney injury through the reduction of the glomerular NLRP3 inflammasome in diabetic nephropathy (DN), which has been demonstrated in experiments exploring the mechanism of pioglitazone in DN (104). In addition, it was found that in T2D, enhanced NFκB activation impaired vascular function, including myogenic tone, vascular reactivity and inflammatory response, through the detection of PARP-1, SP-1, and COX-2 activity (105). Resolvins, naturally occurring polyunsaturated fatty acids, have anti-inflammatory actions in several tissues, including the kidneys. Resolvins exert this anti-inflammatory effect in diabetes by suppressing inflammatory responses via at least five molecular mechanisms: inhibition of the nucleotide-binding oligomerization domain protein 3 inflammasome, inhibition of nuclear factor κB molecular pathways, improvement of oxidative stress, modulation of nitric oxide synthesis/release and prevention of local and systemic leukocytosis (106). In addition, sirtuin-1 (SIRT1) has a protective role in diabetic kidney disease through the deacetylation of transcription factors such as p53, FOXO, RelA/p65NFκB, STAT3, and PGC1α/PPARγ (107). Taken together, these studies show that NFκB may play a dominant role in the pathogenesis of diabetic nephropathy and metabolic renal diseases (108).
Renal Disease—Autoimmune Kidney Injury
Autoimmune diseases result from the loss of tolerance to endogenous self-antigens, leading to a varied range of chronic inflammatory conditions that result in substantial morbidity and mortality. Immunosuppressive therapy only improves outcomes in some of the patients with autoimmune diseases and may be accompanied by undesirable treatment side effects. To maintain immune self-tolerance, thymic T-cell selection, Treg cell homeostasis and the elimination of autoreactive B-cell clones are essential, and in all these processes, NFκB subsets play a vital role (109). There is no NFκB inhibitor used for the treatment of autoimmune diseases yet, but various promising approaches targeting diverse stages of NFκB, including IKK, IκB, the ubiquitin-proteasome system, DNA binding, and posttranscriptional and posttranslational modifications are being investigated (109, 110).
A20, A20-binding inhibitor of NFκB (ABIN1), and their genes TNFAIP3 and TNIP1, respectively, are the focus of recent promising therapies. In an experimental model of lupus nephritis (LN), knock-in mice expressing an inactive form of ABIN1 developed progressive GN related to class III and IV LN in humans (83). In addition, ABIN1-deficient mice developed a progressive, lupus-like inflammatory disease characterized by expansion of myeloid cells and leukocyte infiltrations in different parenchymatous organs. These results demonstrated that ABIN1 is an essential anti-inflammatory component of TLR-signaling pathways that controls C/EBPβ and NFκB activity (111).
NFκB may also affect inflammatory autoimmune diseases through the regulation of Tregs. The NFκB family of transcription factors, in particular the canonical pathway subunits, c-Rel and p65, are crucial for the development, maintenance, and function of Tregs. These observations highlight canonical NFκB signaling as a master regulator of Treg development and function, and demonstrate the discrete functions of individual NFκB subunits in Treg-dependent immune tolerance (112–114). The role of the alternative, non-canonical NFκB pathway components, p100 and RelB, in Treg biology was less clear until recently (115). In the latter study, conditional deletion of the p100 gene, nfkb2, in Tregs, resulted in massive inflammation because of impaired suppressive function of nfkb2-deficient Tregs. Surprisingly, deletion of both relb and nfkb2 rescued the inflammatory phenotype, demonstrating an essential role for p100 as an inhibitor of RelB in Tregs (115). In another experiment about the alternative NFκB pathway, NIK SMI1 was shown to be a potent small-molecular inhibitor of NIK, which selectively repressed the non-canonical NFκB pathway and mitigated SLE in mice (116).
Analyses of renal biopsies from patients with different types of lupus nephritis showed increased expression of the transcription factor Nrf2. In an experimental model, Nrf2−/− mice suffered from greater renal damage and more severe pathological alterations in the kidney after the induction of a lupus-like disease. Knockdown of Nrf2 expression aggravated the nuclear factor-kappa B (NFκB) pathway and increased the levels of reactive oxygen species, iNOS, TGFβ1, and fibronectin. These results suggest that Nrf2 improved lupus nephritis by neutralizing reactive oxygen species and by negatively regulating the NFκB and TGFβ1 signaling pathways (117). In an experimental model of ANCA-induced GN, the activation of p50/p65 heterodimers in endothelial cells stimulated by ANCA-primed neutrophils promoted the development of GN (118). In an investigation of SNPs in patients with LN, polymorphisms of TNIP1 were related to LN in patients of European ancestry and in African-Americans (see above) (83). Biallelic loss-of-function alleles accounts for many early-onset and severe immune deficiencies, but some less severe immune disorders with heterozygous mutation are less well-understood. A recent clinical study showed that heterozygous mutation of IKBKB, which encodes IKK2, led to combined T and B lymphocyte deficiency in patients with acquired immune deficiency (119).
Renal Disease—Kidney Transplantation
Ischemia-reperfusion injury is inevitable in organ transplantation and may cause renal allograft dysfunction. During cold storage and the inflammatory injury following reperfusion, NFκB is activated and plays an important role. In a mouse model of kidney transplantation, knockdown of RelB by local intra-arterial perfusion of the donor kidneys with siRNA relieved IRI in transplanted kidneys by attenuating inflammatory cytokines and cell apoptosis (120). Dexmedetomidine, a sedative drug used in the clinic, reduced kidney injury after transplantation through the inhibition of the TLR4/NFκB pathway in rats (121).
The long-term outcome of renal allografts is reduced by chronic rejection, presenting as allograft vasculopathy (AV). AV is an intriguing process that involves not only adaptive but also innate immunity, such as NK cells, neutrophils and endothelial cells. Deposition of MAC enhances graft immunogenicity instead of causing cell death directly through activation of the non-canonical NFκB pathway (122). Calcineurin inhibitors (CNI) are used for preventing rejection of transplanted kidneys and eventually are one of the major reasons of chronic allograft dysfunction. There is evidence that suppression of NFκB through neutralizing high-mobility group box1 (HMGB1) in mice mitigates chronic cyclosporine nephrotoxicity (123).
In humans, all the components of the NFκB pathway were shown to be expressed in renal allografts with acute rejection and chronic renal allograft injury through qPCR, but there is no clear functional relationship between the expression of NFκB components and allograft rejection or long-term outcome (124). Spontaneous, clinical operational tolerance, characterized as well-functioning allograft without rejection after recession of immunosuppression, has been associated with lower c-Rel expression and a higher proportion of Tregs in allograft biopsies (125). Higher frequencies of mutant genotypes of tag-SNPs of FOXP3 and NFκB1 occurred in patients with end-stage renal disease and with acute rejection episodes (126). These studies indicate a significant relationship between NFκB canonical pathway activation and rejection episodes, but further information on the role of the non-canonical NFκB pathway in human kidney transplantation is needed.
Renal Disease—NFκB Pathway Crosstalk
NFκB has comprehensive physiological and pathophysiological effects. However, it does not exist as an isolated signaling pathway. There is intensive crosstalk with many other parallel signaling pathways (127–130). This review will only briefly mention the topic of crosstalk between the NFκB pathways and other signaling pathways but focus on what is currently known on the possible interactions between the canonical and non-canonical NFκB pathways in renal disease.
Tumor necrosis factor receptor-associated factors (TRAFs) and receptor-interacting proteins (RIPs), serving as the signals between NFκB receptors and IKK complexes, are critical not only for the IKK activation but also other pathways as the AP-1 pathway (131). IKKα is involved in tumorigenesis in an NFκB-independent way, as has been shown in colorectal cancer (CRC) (132). In CRC cells, mutant BRAF activates the phosphorylation of the proteolytic fragment of IKKα (p45-IKKα) instead of activation of NFκB and contributes to the CRC proliferation (132). In mice, RIPK1 kinase-dependent cell death can be prevented by the IKKα/IKKβ complex, but not by IKKα nor IKKβ alone, during TNF signaling in an NFκB-independent way. RIPK1 in the TNFR1 complex is phosphorylated directly by IKKα/IKKβ instead of promoting cell death by binding to FADD/caspase-8 (133). Subsequently, this anti-cell death function of IKKα/IKKβ was detected in human cells (134). In research about the effect of temperature on NFκB dynamics, a strong down-regulation of AXIN2, a key factor of the Wnt pathway, is observed, which suggests Wnt/NFκB crosstalk (32). There also exists a distinct activation mechanism of the non-canonical pathway in the defense against pathogens. The complement membrane attach complexes (MAC) form an endosome-based signaling complex with the kinases AKT and NIK through phosphorylation and mediate NIK stabilization instead of a TRAF3-dependent pathway (9). Both the Notch pathway and NFκB effect the development of Tregs, and the Notch signaling pathway crosstalks with the NFκB pathway. In the tumor microenvironment, hyperactive Notch3 enhances the canonical NFκB pathway, regulates the developmental aspects and activity of Tregs, and changes the response to tumors (112). The crosstalk between the NFκB and other parallel signaling pathways has not been addressed in renal diseases in detail (135).
For now, NFκB has two main activation pathways, the canonical and non-canonical pathways, but the two pathways are not completely separate. A diverse crosstalk occurs at the levels of gene transcription, subunit dimerization, IKK complexes, and upstream signaling and stimulation. For example, the TNF-induced canonical NFκB pathway induces the expression of RelB and inhibits the expression of the CXCL12 gene mediated by the non-canonical pathway (136). Intriguingly, subunits from the two pathways may dimerize and modulate gene expression. For example, in T cells, p52 from the non-canonical pathway cooperates with c-Rel from the canonical pathway to induce the expression of GM-CSF (9). IKKα can activate both the canonical pathway with support from IKKβ and IKKγ and the non-canonical pathway by interacting with NIK. There must exist some unknown mechanisms that activate IKKα in each pathway specifically. Recent results show that the surface structure of IKKα determines the interaction with NIK and the processing of p100 in the non-canonical pathway (137). TNF-like weak inducer of apoptosis (TWEAK) activates both the canonical and non-canonical pathways in different kinds of kidney cells. TWEAK can activate the non-canonical NFκB pathway and promote the expression of CCL21 in podocytes independent of the canonical pathway (138). TRAF2, a common upstream signal in the activation of the canonical and non-canonical pathways, is involved in the protection of cardiac injury through the crosstalk of these pathways by upregulation of RelB as well as diminishing canonical NFκB signaling (139). Crosstalk between the canonical and non-canonical NFκB pathways is essential for medullary thymic epithelial cells to induce central immune tolerance and in Tregs (140). The key function of p100 in Tregs is to be a negative regulator of p52-independent, RelB-containing complexes. These data therefore provide further insight into how constitutive activation of the canonical and alternative NFκB pathways might drive pathology in chronic inflammation and autoimmune disease and demonstrate that the adequate balance of NFκB activity, regulated by NFκB2 p100, is essential for maintaining optimal Treg function and immune tolerance (115). We have recently shown in two experimental models of renal injury that the non-canonical NFκB signaling pathway is critical for Th17 cell induction and that the Th17 cell immune function depends on the fine-tuning of canonical NFκB signaling (92, 141). The crosstalk between canonical and non-canonical NFκB signaling in the complex regulation of Th17 cells and Tregs warrants further investigation to identify potent candidate genes for the manipulation of pathogenicity of Th17 cells without affecting nonpathogenic Th17 and Treg cells that may be critical for tissue homeostasis in renal diseases (109, 142, 143) (Figure 2).
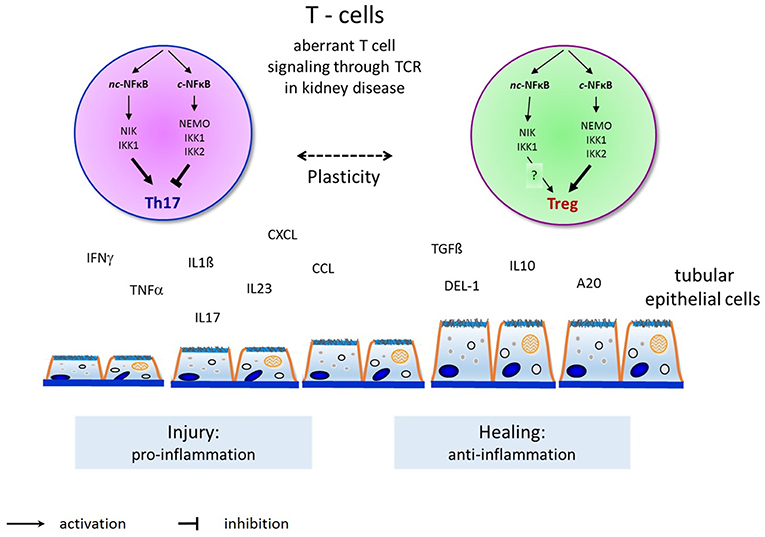
Figure 2. Overview and principles of canonical and non-canonical NFκB pathway activation in T-helper cell differentiation to induce Th17-cells and Tregs and their interaction with endogenous renal cells (such as renal tubular epithelial or glomerular endothelial or mesangial cells) in renal diseases. Th17-cells activation by the non-canonical NFκB pathway induce renal injury (as shown here by tubular epithelial cell damage) through proinflammatory cytokines as shown on the left. Tregs activation by the canonical NFκB pathway permits renal healing (as shown by tubular epithelial cell healing) through anti-inflammatory cytokines on the right.
Conclusion
As a critical regulator of inflammation and cell survival, the NFκB pathway is a promising target for diagnosing and treating kidney diseases. For modulation of the NFκB pathway in the clinic, a number of molecules can effectively inhibit NFκB signaling by targeting the receptors, associated adaptors, IKKs, IκBs and transcriptional regulators (144). There is further clinical evidence on small-molecule inhibitors of IKKα and NIK from recent trials on anti-cancer therapies (145). These clinical trials showed that the cancer-selective pharmacodynamic response of DTP3, the co-inhibitor of the JNK-activating kinase MKK7, and the NFκB-regulated antiapoptotic factor GADD45β could be of clinical value through GADD45B expression in multiple myeloma, which was confirmed to be effective in a subsequent clinical trial (146). However, considering the complexity of the NFκB pathways and the crosstalk between the pathways, there are numerous NFκB-independent side effects that are unrelated to the on-target effects of the inhibitors (147). Therefore, a more specific regulation of NFκB in the targeted disease must be cell-specific.
In addition, as the NFκB system has a broad impact on all kinds of tissues, modulation of NFκB in diseased cells without effecting normal cells must be guaranteed. Therefore, we also need to focus on an appropriate delivery system that will transfer small-molecular inhibitors or genes to specific cells through cell-specific recognition. For example, in macrophages, the acidic lysosomes, the liposomes, the glucan shell microparticles, and the oligopeptide complex have been used in the treatment of metabolic diseases (148). There are also new and emerging therapeutic opportunities with nanoparticles. For example, in hyperglycemia, gold nanoparticles reduce inflammation and apoptosis through tuberin-mTOR/ NFκB pathway inhibition in macrophages (149). In cancer cells, Zn-CuO nanoparticles can induce autophagy and apoptosis through the NFκB pathway (150). Nanoparticles can target a particular cell type as a carrier when packaged with specific receptors (148). More recently, approaches of targeted therapies for the treatment of inflammatory diseases have been highlighted in kidney disease and other diseases, as well through regulation of the NFκB/NLRP3 axis, regulation of the NFκB signaling pathway by miRNAs to cell-specifically suppress NFκB, and activation of the PI3K/Akt/eNOS pathways or by Bcl3 expression (151–162).
Although it has been known for more than 30 years, much remains to be learned about the NFκB pathway, the crosstalk between the canonical and non-canonical NFκB pathways, the interaction of the NFκB pathway with other signal transduction pathways, and the NIK-, IKK-, and NEMO-dependent but NFκB-proteins independent effects in the gene regulation of renal diseases. However, new and exciting NFκB-specific strategies are now being explored for cancer and metabolic diseases and will open new therapeutic opportunities for disease, including cell-specific and phase-specific oscillatory inhibition of NFκB in kidney injury. A complete understanding of these molecular pathways is important, as this may lead to the development of new, effective therapeutic strategies in renal diseases.
Author Contributions
FT and NS contributed to the conception and design of the study. NS wrote the first draft of the manuscript. All of the authors contributed to manuscript revision and read and approved the submitted version. The first author, NS takes primary responsibility for communication with the journal and editorial office during the submission process, throughout peer review and during publication. The first author is also responsible for ensuring that the submission adheres to all of the journal's requirements, including, but not exclusive to, the details of authorship, study ethics and ethics approval, clinical trial registration documents and conflict of interest declaration. The first and corresponding author will be available post-publication to respond to any queries or critiques.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the support by grants from the Deutsche Forschungsgemeinschaft (DFG Th 343/12-2).
Abbreviations
AKI, acute kidney injury; ANCA, Antineutrophil cytoplasmic antibodies; AV, allograft vasculopathy; CKD, chronic kidney disease; CNI, calcineurin inhibitor; CXCL12, C-X-C Motif Chemokine Ligand 12; DAMP, damage-associated molecular patterns; ESRD, end-stage renal disease; GBM, anti-glomerular basement membrane; GN, glomerulonephritis; Imn, idiopathic membranous nephropathy; IRAK, Interleukin-1 receptor-associated kinase; IRI, ischemia/ reperfusion injury; LP, lupus nephritis; MAC, membrane attach complexes; NLRs, NOD-like receptors; NTN, nephrotoxic nephritis; PAMP, pathogen-associated molecular patterns; PRR, pattern-recognition receptors; RIP, receptor-interacting proteins; T2D, type 2 diabetes mellitus; TBK, TANK-binding kinase; Th, T helper; TLRs, Toll-like receptors; TMA, renal thrombotic microangiopathy; TMA, thrombotic microangiopathy; TRAF, Tumor necrosis factor receptor-associated factors; TTP, tristeraprolin; UUO, unilateral ureteric obstruction.
References
1. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. (1986) 46:705–16.
2. Wan F, Lenardo MJ. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harbor Perspect Biol. (2009) 1:a000067. doi: 10.1101/cshperspect.a000067
3. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. (2006) 25:6680–4. doi: 10.1038/sj.onc.1209954
4. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspect Biol. (2009) 1:a000034. doi: 10.1101/cshperspect.a000034
5. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med. (2016) 8:227–41. doi: 10.1002/wsbm.1331
6. Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. (2002) 298:1241–5. doi: 10.1126/science.1071914
7. Gerondakis S, Fulford TS, Messina NL, Grumont RJ. NF-kappaB control of T cell development. Nat Immunol. (2014) 15:15–25. doi: 10.1038/ni.2785
8. Lindsay SA, Wasserman SA. Conventional and non-conventional drosophila toll signaling. Dev Comp Immunol. (2014) 42:16–24. doi: 10.1016/j.dci.2013.04.011
9. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
10. Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. (2001) 7:401–9. doi: 10.1016/S1097-2765(01)00187-3
11. Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. (2001) 293:1495–9. doi: 10.1126/science.1062677
12. Collins PE, Mitxitorena I, Carmody RJ. The ubiquitination of NF-κB subunits in the control of transcription. Cells. (2016) 5:23. doi: 10.3390/cells5020023
13. Courtois G, Fauvarque MO. The many roles of ubiquitin in NF-kappaB Signaling. Biomedicines. (2018) 6:E43. doi: 10.3390/biomedicines6020043
14. Zhang J, Hu MM, Wang YY, Shu HB. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem. (2012) 287:28646–55. doi: 10.1074/jbc.M112.362608
15. Lork M, Staal J, Beyaert R. Ubiquitination and phosphorylation of the CARD11-BCL10-MALT1 signalosome in T cells. Cell Immunol. (2018). doi: 10.1016/j.cellimm.2018.11.001. [Epub ahead of print].
16. Harhaj EW, Dixit VM. Regulation of NF-kappaB by deubiquitinases. Immunol Rev. (2012) 246:107–24. doi: 10.1111/j.1600-065X.2012.01100.x
17. Mitchell JP, Carmody RJ. NF-kappaB and the transcriptional control of inflammation. Int Rev Cell Mol Biol. (2018) 335:41–84. doi: 10.1016/bs.ircmb.2017.07.007
18. Aksentijevich I, Zhou Q. NF-kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol. (2017) 8:399. doi: 10.3389/fimmu.2017.00399
19. Natoli G, Chiocca S. Nuclear ubiquitin ligases, NF-kappaB degradation, and the control of inflammation. Sci Signal. (2008) 1:pe1. doi: 10.1126/stke.11pe1
20. Natoli G. From the beauty of genomic landscapes to the strength of transcriptional mechanisms. Cell. (2016) 165:18–9. doi: 10.1016/j.cell.2016.03.011
21. Tong AJ, Liu X, Thomas BJ, Lissner MM, Baker MR, Senagolage MD, et al. A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell. (2016) 165:165–79. doi: 10.1016/j.cell.2016.01.020
22. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. (2016) 7:160. doi: 10.3389/fimmu.2016.00160
23. Rothschild DE, McDaniel DK, Ringel-Scaia VM, Allen IC. Modulating inflammation through the negative regulation of N-κB signaling. J Leukoc Biol. (2018) 103:1131–50. (2018). doi: 10.1002/jlb.3mir0817-346rrr
24. Afonina IS, Zhong Z, Karin M, Beyaert R. Limiting inflammation-the negative regulation of NF-kappaB and the NLRP3 inflammasome. Nat Immunol. (2017) 18:861–9. doi: 10.1038/ni.3772
25. Zhang M, Peng LL, Wang Y, Wang JS, Liu J, Liu MM, et al. Roles of A20 in autoimmune diseases. Immunol Res. (2016) 64:337–44. doi: 10.1007/s12026-015-8677-6
26. Pujari R, Hunte R, Khan WN, Shembade N. A20-mediated negative regulation of canonical NF-kappaB signaling pathway. Immunol Res. (2013) 57:166–71. doi: 10.1007/s12026-013-8463-2
27. Kobayashi T, Kageyama R. Dynamic advances in NF-kappaB signaling analysis. Sci Signal. (2009) 2:pe47. doi: 10.1126/scisignal.281pe47
28. Ashall L, Horton CA, Nelson DE, Paszek P, Harper CV, Sillitoe K, et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. (2009) 324:242–6. doi: 10.1126/science.1164860
29. Ohshima D, Inoue J, Ichikawa K. Roles of spatial parameters on the oscillation of nuclear NF-kappaB: computer simulations of a 3D spherical cell. PLoS ONE. (2012) 7:e46911. doi: 10.1371/journal.pone.0046911
30. Ohshima D, Ichikawa K. Regulation of NF-kappaB oscillation by nuclear transport: mechanisms determining the persistency and frequency of oscillation. PLoS ONE. (2015) 10:e0127633. doi: 10.1371/journal.pone.0127633
31. Inoue K, Shinohara H, Behar M, Yumoto N, Tanaka G, Hoffmann A, et al. Oscillation dynamics underlie functional switching of NF-kappaB for B-cell activation. NPJ Syst Biol Appl. (2016) 2:16024. doi: 10.1038/npjsba.2016.24
32. Harper CV, Woodcock DJ, Lam C, Garcia-Albornoz M, Adamson A, Ashall L, et al. Temperature regulates NF-kappaB dynamics and function through timing of A20 transcription. Proc Natl Acad Sci USA. (2018) 115:E5243–e9. doi: 10.1073/pnas.1803609115
33. Wang X, Yu W, Zheng L. The dynamics of NF-kappaB pathway regulated by circadian clock. Math Biosci. (2015) 260:47–53. doi: 10.1016/j.mbs.2014.07.012
34. Colombo F, Zambrano S, Agresti A. NF-kappaB, the importance of being dynamic: role and insights in cancer. Biomedicines. (2018) 6:E45. doi: 10.3390/biomedicines6020045
35. Nelson RH, Nelson DE. Signal Distortion: How intracellular pathogens alter host cell fate by modulating NF-kappaB dynamics. Front Immunol. (2018) 9:2962. doi: 10.3389/fimmu.2018.02962
36. Callegari A, Sieben C, Benke A, Suter DM, Fierz B, Mazza D, et al. Single-molecule dynamics and genome-wide transcriptomics reveal that NF-kB (p65)-DNA binding times can be decoupled from transcriptional activation. PLoS Genet. (2019) 15:e1007891. doi: 10.1371/journal.pgen.1007891
37. Ben-Neriah Y. Regulatory functions of ubiquitination in the immune system. Nat Immunol. (2002) 3:20–6. doi: 10.1038/ni0102-20
38. Sanz AB, Sanchez-Nino MD, Ramos AM, Moreno JA, Santamaria B, Ruiz-Ortega M, et al. NF-kappaB in renal inflammation. J Am Soc Nephrol. (2010) 21:1254–62. doi: 10.1681/asn.2010020218
39. Luedde T, Beraza N, Kotsikoris V, van Loo G, Nenci A, De Vos R, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. (2007) 11:119–32. doi: 10.1016/j.ccr.2006.12.016
40. Kondylis V, Kumari S, Vlantis K, Pasparakis M. The interplay of IKK, NF-kappaB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol Rev. (2017) 277:113–27. doi: 10.1111/imr.12550
41. Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, et al. NF-kappaB c-Rel Is crucial for the regulatory T cell immune checkpoint in cancer. Cell. (2017) 170:1096–108 e13. doi: 10.1016/j.cell.2017.08.004
42. Shi JH, Sun SC. Tumor necrosis factor receptor-associated factor regulation of nuclear factor kappaB and mitogen-activated protein kinase pathways. Front Immunol. (2018) 9:1849. doi: 10.3389/fimmu.2018.01849
43. Tegowski M, Baldwin A. Noncanonical NF-kappaB in Cancer. Biomedicines. (2018) 6:66. doi: 10.3390/biomedicines6020066
44. Li PK, Ma TK. Global impact of nephropathies. Nephrology. (2017) 22 (Suppl 4):9–13. doi: 10.1111/nep.13146
45. Knauf F, Brewer JR, Flavell RA. Immunity, microbiota and kidney disease. Nat Rev Nephrol. (2019). doi: 10.1038/s41581-019-0118-7. [Epub ahead of print]
46. Xia H, Bao W, Shi S. Innate immune activity in glomerular podocytes. Front Immunol. (2017) 8:122. doi: 10.3389/fimmu.2017.00122
47. Anders HJ. Of Inflammasomes and Alarmins: IL-1beta and IL-1alpha in Kidney Disease. J Am Soc Nephrol. (2016) 27:2564–75. doi: 10.1681/asn.2016020177
48. Krebs CF, Schmidt T, Riedel JH, Panzer U. T helper type 17 cells in immune-mediated glomerular disease. Nat Rev Nephrol. 13:647–659. doi: 10.1038/nrneph.2017.112
49. Yang C, Huang XR, Fung E, Liu HF, Lan HY. The Regulatory T-cell transcription factor Foxp3 protects against crescentic glomerulonephritis. Sci Rep. (2017) 7:1481. doi: 10.1038/s41598-017-01515-8
50. Schmidt T, Luebbe J, Paust HJ, Panzer U. Mechanisms and functions of IL-17 signaling in renal autoimmune diseases. Mol Immunol. (2018) 104:90–9. doi: 10.1016/j.molimm.2018.09.005
51. Krebs CF, Paust HJ, Krohn S, Koyro T, Brix SR, Riedel JH, et al. Autoimmune renal disease is exacerbated by S1P-receptor-1-dependent intestinal Th17 cell migration to the kidney. Immunity. (2016) 45:1078–92. doi: 10.1016/j.immuni.2016.10.020
52. Zhang H, Sun SC. NF-kappaB in inflammation and renal diseases. Cell Biosci. (2015) 5:63. doi: 10.1186/s13578-015-0056-4
53. Nishikawa H, Taniguchi Y, Matsumoto T, Arima N, Masaki M, Shimamura Y, et al. Knockout of the interleukin-36 receptor protects against renal ischemia-reperfusion injury by reduction of proinflammatory cytokines. Kidney Int. (2018) 93:599–614. doi: 10.1016/j.kint.2017.09.017
54. Liu H, Xiong J, He T, Xiao T, Li Y, Yu Y, et al. High uric acid-induced epithelial-mesenchymal transition of renal tubular epithelial cells via the TLR4/NF-kB signaling pathway. Am J Nephrol. (2017) 46:333–42. doi: 10.1159/000481668
55. Li Y, Li X, He K, Li B, Liu K, Qi J, et al. C-peptide prevents NF-kappaB from recruiting p300 and binding to the inos promoter in diabetic nephropathy. FASEB J. (2018) 32:2269–79. doi: 10.1096/fj.201700891R
56. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nature Rev Nephrol. (2018) 14:26–47. doi: 10.1038/nrneph.2017.156
57. Angeletti A, Reyes-Bahamonde J, Cravedi P, Campbell KN. Complement in non-antibody-mediated kidney diseases. Front Med. (2017) 4:99. doi: 10.3389/fmed.2017.00099
58. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-kappaB signaling in endothelial cells. Circulation. (2013) 128:2504–16. doi: 10.1161/circulationaha.113.002972
59. Qin L, Li G, Kirkiles-Smith N, Clark P, Fang C, Wang Y, et al. Complement C5 inhibition reduces T cell-mediated allograft vasculopathy caused by both alloantibody and ischemia reperfusion injury in humanized mice. Am J Transpl. (2016) 16:2865–76. doi: 10.1111/ajt.13834
60. Arbore G, Kemper C, Kolev M. Intracellular complement - the complosome - in immune cell regulation. Mol Immunol. (2017) 89:2–9. doi: 10.1016/j.molimm.2017.05.012
61. Reichhardt MP, Meri S. Intracellular complement activation-an alarm raising mechanism? Semin Immunol. (2018) 38:54–62. doi: 10.1016/j.smim.2018.03.003
62. Manthiram K, Zhou Q, Aksentijevich I, Kastner DL. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol. (2017) 18:832–42. doi: 10.1038/ni.3777
63. Jane-wit D, Surovtseva YV, Qin L, Li G, Liu R, Clark P, et al. Complement membrane attack complexes activate noncanonical NF-kappaB by forming an Akt+ NIK+ signalosome on Rab5+ endosomes. Proc Natl Acad Sci USA. (2015) 112:9686–91. doi: 10.1073/pnas.1503535112
64. Liu M, Wang H, Zhang J, Yang X, Li B, Wu C, et al. NF-kappaB signaling pathway-enhanced complement activation mediates renal injury in trichloroethylene-sensitized mice. J Immunotoxicol. (2018) 15:63–72. doi: 10.1080/1547691x.2017.1420712
65. Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. J Am Soc Nephrol. (2012) 23:381–99. doi: 10.1681/asn.2011030304
66. Couser WG. Primary membranous nephropathy. Clin J Am Soc Nephrol. (2017) 12:983–97. doi: 10.2215/cjn.11761116
67. Hoxha E, Stahl RAK. Translational Aspects of Primary Membranous Nephropathy. Semin Nephrol. 37:436–46. doi: 10.1016/j.semnephrol.2017.05.017
68. Bram RJ. TBK1 suppression of IgA in the NIK of time. Nat Immunol. (2012) 13:1027–9. doi: 10.1038/ni.2451
69. Jin J, Xiao Y, Chang JH, Yu J, Hu H, Starr R, et al. The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-kappaB signaling. Nat Immunol. (2012) 13:1101–9. doi: 10.1038/ni.2423
70. Gotot J, Piotrowski E, Otte MS, Tittel AP, Linlin G, Yao C, et al. Inhibitor of NFkappaB Kinase Subunit 2 blockade hinders the initiation but aggravates the progression of crescentic GN. J Am Soc Nephrol. (2016) 27:1917–24. doi: 10.1681/asn.2015060699
71. Fearn A, Situmorang GR, Fox C, Oakley F, Howarth R, Wilson CL, et al. The NF-kappaB1 is a key regulator of acute but not chronic renal injury. Cell Death Dis. (2017) 8:e2883. doi: 10.1038/cddis.2017.233
72. Brahler S, Ising C, Hagmann H, Rasmus M, Hoehne M, Kurschat C, et al. Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria. Am J Physiol Renal Physiol. (2012) 303:F1473–85. doi: 10.1152/ajprenal.00031.2012
73. Brahler S, Ising C, Barrera Aranda B, Hohne M, Schermer B, Benzing T, et al. The NF-kappaB essential modulator (NEMO) controls podocyte cytoskeletal dynamics independently of NF-kappaB. Am J Physiol Renal Physiol. (2015) 309:F617–26. doi: 10.1152/ajprenal.00059.2015
74. Feng XJ, Liu SX, Wu C, Kang PP, Liu QJ, Hao J, et al. The PTEN/PI3K/Akt signaling pathway mediates HMGB1-induced cell proliferation by regulating the NF-kappaB/cyclin D1 pathway in mouse mesangial cells. Am J Physiol Cell Physiol. (2014) 306:C1119–28. doi: 10.1152/ajpcell.00385.2013
75. Zhang H, Mao X, Sun Y, Hu R, Luo W, Zhao Z, et al. NF-kappaB upregulates ubiquitin C-terminal hydrolase 1 in diseased podocytes in glomerulonephritis. Mol Med Rep. (2015) 12:2893–901. doi: 10.3892/mmr.2015.3780
76. Zhang H, Sun Y, Hu R, Luo W, Mao X, Zhao Z, et al. The regulation of the UCH-L1 gene by transcription factor NF-kappaB in podocytes. Cell Signal. (2013) 25:1574–85. doi: 10.1016/j.cellsig.2013.03.018
77. Radon V, Czesla M, Reichelt J, Fehlert J, Hammel A, Rosendahl A, et al. Ubiquitin C-terminal hydrolase L1 is required for regulated protein degradation through the ubiquitin proteasome system in kidney. Kidney Int. (2018) 93:110–27. doi: 10.1016/j.kint.2017.05.016
78. Korte EA, Caster DJ, Barati MT, Tan M, Zheng S, Berthier CC, et al. ABIN1 Determines severity of glomerulonephritis via activation of intrinsic glomerular inflammation. Am J Pathol. (2017) 187:2799–810. doi: 10.1016/j.ajpath.2017.08.018
79. Prakoura N, Kavvadas P, Kormann R, Dussaule JC, Chadjichristos CE, Chatziantoniou C. NFkappaB-induced periostin activates integrin-beta3 signaling to promote renal injury in GN. J Am Soc Nephrol. (2017) 28:1475–90. doi: 10.1681/asn.2016070709
80. Danilewicz M, Wagrowska-Danilewicz M. Tubular NF-kB is overexpressed in proteinuric patients with IgA nephropathy. Folia Histochem et Cytobiol. (2012) 50:93–8. doi: 10.2478/18702
81. Silva GE, Costa RS, Ravinal RC, Ramalho LZ, Dos Reis MA, Coimbra TM, et al. NF-kB expression in IgA nephropathy outcome. Dis Markers. (2011) 31:9–15. doi: 10.3233/dma-2011-0795
82. Izzedine H, Mangier M, Ory V, Zhang SY, Sendeyo K, Bouachi K, et al. Expression patterns of RelA and c-mip are associated with different glomerular diseases following anti-VEGF therapy. Kidney Int. (2014) 85:457–70. doi: 10.1038/ki.2013.344
83. Caster DJ, Korte EA, Nanda SK, McLeish KR, Oliver RK, G'Sell R T, et al. ABIN1 dysfunction as a genetic basis for lupus nephritis. J Am Soc Nephrol. (2013) 24:1743–54. doi: 10.1681/asn.2013020148
84. Zhang JY, Wang M, Tian L, Genovese G, Yan P, Wilson JG, et al. UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci USA. (2018) 115:3446–51. doi: 10.1073/pnas.1716113115
85. Hoste EAJ, Kellum JA, Selby NM, Zarbock A, Palevsky PM, Bagshaw SM, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrology. (2018) 14:607–25. doi: 10.1038/s41581-018-0052-0
86. Kumar D, Singla SK, Puri V, Puri S. The restrained expression of NF-kB in renal tissue ameliorates folic acid induced acute kidney injury in mice. PLoS ONE. (2015) 10:e115947. doi: 10.1371/journal.pone.0115947
87. Johnson FL, Patel NSA, Purvis GSD, Chiazza F, Chen J, Sordi R, et al. Inhibition of IκB kinase at 24 hours after acute kidney injury improves recovery of renal function and attenuates fibrosis. J Am Heart Assoc. (2017) 6:e005092. doi: 10.1161/jaha.116.005092
88. Kezic A, Stajic N, Thaiss F. Innate immune response in kidney ischemia/reperfusion injury: potential target for therapy. J Immunol Res. (2017) 2017:6305439. doi: 10.1155/2017/6305439
89. Shu B, Feng Y, Gui Y, Lu Q, Wei W, Xue X, et al. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signall. (2018) 42:249–58. doi: 10.1016/j.cellsig.2017.10.014
90. Lv LL, Tang PM, Li CJ, You YK, Li J, Huang XR, et al. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. (2017) 91:587–602. doi: 10.1016/j.kint.2016.10.020
91. Xue C, Liu Y, Li C, Li Y, Yang T, Xie L, et al. Powerful protection against renal ischemia reperfusion injury by T cell-specific NF-kappaB inhibition. Transplantation. (2014) 97:391–6. doi: 10.1097/01.tp.0000438622.89310.95
92. Guo L, Lee HH, Noriega ML, Paust HJ, Zahner G, Thaiss F. Lymphocyte-specific deletion of IKK2 or NEMO mediates an increase in intrarenal Th17 cells and accelerates renal damage in an ischemia-reperfusion injury mouse model. Am J Physiol Renal Physiol. (2016) 311:F1005–f14. doi: 10.1152/ajprenal.00242.2016
93. Marko L, Vigolo E, Hinze C, Park JK, Roel G, Balogh A, et al. Tubular epithelial NF-kappaB activity regulates ischemic AKI. J Am Soc Nephrol. (2016) 27:2658–69. doi: 10.1681/asn.2015070748
94. Yang L, Brooks CR, Xiao S, Sabbisetti V, Yeung MY, Hsiao LL, et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Investigat. (2015) 125:1620–36. doi: 10.1172/jci75417
95. Lawrence T, Fong C. The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int J Biochem Cell Biol. (2010) 42:519–23. doi: 10.1016/j.biocel.2009.12.016
96. He K, Chen X, Han C, Xu L, Zhang J, Zhang M, et al. Lipopolysaccharide-induced cross-tolerance against renal ischemia-reperfusion injury is mediated by hypoxia-inducible factor-2alpha-regulated nitric oxide production. Kidney Int. (2014) 85:276–88. doi: 10.1038/ki.2013.342
97. Yamashita M, Yoshida T, Hayashi M. Podocyte NF-κB is dispensable for the pathogenesis of renal ischemia-reperfusion injury. Physiol Rep. (2016) 4:e12912. doi: 10.14814/phy2.12912
98. Sanz AB, Ruiz-Andres O, Sanchez-Nino MD, Ruiz-Ortega M, Ramos AM, Ortiz A. Out of the TWEAKlight: Elucidating the Role of Fn14 and TWEAK in Acute Kidney Injury. Semin Nephrol. (2016) 36:189–98. doi: 10.1016/j.semnephrol.2016.03.006
99. Bhatraju P, Hsu C, Mukherjee P, Glavan BJ, Burt A, Mikacenic C, et al. Associations between single nucleotide polymorphisms in the FAS pathway and acute kidney injury. Crit Care. (2015) 19:368. doi: 10.1186/s13054-015-1084-5
100. Catrysse L, van Loo G. Inflammation and the metabolic syndrome: the tissue-specific functions of NF-kappaB. Trends Cell Biol. (2017) 27:417–29. doi: 10.1016/j.tcb.2017.01.006
101. Decleves AE, Sharma K. Novel targets of antifibrotic and anti-inflammatory treatment in CKD. Nat Rev Nephrol. (2014) 10:257–67. doi: 10.1038/nrneph.2014.31
102. Kim JE, Lee MH, Nam DH, Song HK, Kang YS, Lee JE, et al. Celastrol, an NF-kappaB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS ONE. (2013) 8:e62068doi: 10.1371/journal.pone.0062068
103. Sun Y, Peng R, Peng H, Liu H, Wen L, Wu T, et al. miR-451 suppresses the NF-kappaB-mediated proinflammatory molecules expression through inhibiting LMP7 in diabetic nephropathy. Mol Cel Endocrinol. (2016) 433:75–86. doi: 10.1016/j.mce.2016.06.004
104. Wang Y, Yu B, Wang L, Yang M, Xia Z, Wei W, et al. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLoS ONE. (2017) 12:e0181248. doi: 10.1371/journal.pone.0181248
105. Kassan M, Choi SK, Galan M, Bishop A, Umezawa K, Trebak M, et al. Enhanced NF-kappaB activity impairs vascular function through PARP-1-, SP-1-, and COX-2-dependent mechanisms in type 2 diabetes. Diabetes. (2013) 62:2078–87. doi: 10.2337/db12-1374
106. Yaribeygi H, Atkin SL, Simental-Mendia LE, Barreto GE, Sahebkar. Anti-inflammatory effects of resolvins in diabeticnephropathy: mechanistic pathways. J Cell Physiol. (2019) 1–10. doi: 10.1002/jcp.28315. [Epub ahead of print].
107. Zhong Y, Lee K, He JC. SIRT1 Is a potential drug target for treatment of diabetic kidney disease. Front Endocrinol. (2018) 9:624. doi: 10.3389/fendo.2018.00624
108. Artunc F, Schleicher E, Weigert C, Fritsche A, Stefan N, Haring HU. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol. (2016) 12:721–37. doi: 10.1038/nrneph.2016.145
109. Miraghazadeh B, Cook MC. Nuclear factor-kappaB in autoimmunity: man and mouse. Front Immunol. (2018) 9:613. doi: 10.3389/fimmu.2018.00613
110. Herrington FD, Carmody RJ, Goodyear CS. Modulation of NF-kappaB signaling as a therapeutic target in autoimmunity. J Biomol Screen. (2016) 21:223–42. doi: 10.1177/1087057115617456
111. Zhou J, Wu R, High AA, Slaughter CA, Finkelstein D, Rehg JE, et al. A20-binding inhibitor of NF-kappaB (ABIN1) controls Toll-like receptor-mediated CCAAT/enhancer-binding protein beta activation and protects from inflammatory disease. Proc Natl Acad Sci USA. (2011) 108:E998–1006. doi: 10.1073/pnas.1106232108
112. Ferrandino F, Grazioli P, Bellavia D, Campese AF, Screpanti I, Felli MP. Notch and NF-kappaB: coach and players of regulatory T-cell response in cancer. Front Immunol. (2018) 9:2165. doi: 10.3389/fimmu.2018.02165
113. Oh H, Ghosh S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. (2013) 252:41–51. doi: 10.1111/imr.12033
114. van der Veeken J, Arvey A, Rudensky A. Transcriptional control of regulatory T-cell differentiation. Cold Spring Harbor Symposia Quant Biol. (2013) 78:215–22. doi: 10.1101/sqb.2013.78.020289
115. Grinberg-Bleyer Y, Caron R, Seeley JJ, De Silva NS, Schindler CW, Hayden MS, et al. The alternative NF-kappaB pathway in regulatory T cell homeostasis and suppressive function. J Immunol. (2018) 200:2362–71. doi: 10.4049/jimmunol.1800042
116. Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol EB, et al. NF-kappaB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat Commun. (2018) 9:179. doi: 10.1038/s41467-017-02672-0
117. Jiang T, Tian F, Zheng H, Whitman SA, Lin Y, Zhang Z, et al. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-kappaB-mediated inflammatory response. Kidney Int. (2014) 85:333–43. doi: 10.1038/ki.2013.343
118. Choi M, Schreiber A, Eulenberg-Gustavus C, Scheidereit C, Kamps J, Kettritz R. Endothelial NF-kappaB blockade abrogates ANCA-induced GN. J Am Soc Nephrol. (2017) 28:3191–204. doi: 10.1681/asn.2016060690
119. Cardinez C, Miraghazadeh B, Tanita K, da Silva E, Hoshino A, Okada S, et al. Gain-of-function IKBKB mutation causes human combined immune deficiency. J Exp Med. (2018) 215:2715–24. doi: 10.1084/jem.20180639
120. Zheng X, Zang G, Jiang J, He W, Johnston NJ, Ling H, et al. Attenuating ischemia-reperfusion injury in kidney transplantation by perfusing donor organs with siRNA cocktail solution. Transplantation. (2016) 100:743–52. doi: 10.1097/tp.0000000000000960
121. Yao H, Chi X, Jin Y, Wang Y, Huang P, Wu S, et al. Dexmedetomidine Inhibits TLR4/NF-kappaB activation and reduces acute kidney injury after orthotopic autologous liver transplantation in rats. Sci Rep. (2015) 5:16849. doi: 10.1038/srep16849
122. Merola J, Jane-Wit DD, Pober JS. Recent advances in allograft vasculopathy. Curr Opin Organ Transplant. (2017) 22:1–7. doi: 10.1097/mot.0000000000000370
123. Park HS, Kim EN, Kim MY, Lim JH, Kim HW, Park CW, et al. The protective effect of neutralizing high-mobility group box1 against chronic cyclosporine nephrotoxicity in mice. Transplant Immunol. (2016) 34:42–9. doi: 10.1016/j.trim.2015.11.001
124. Seeger H, Lindenmeyer MT, Cohen CD, Jaeckel C, Nelson PJ, Chen J, et al. Lymphotoxin expression in human and murine renal allografts. PLoS ONE. (2018) 13:e0189396. doi: 10.1371/journal.pone.0189396
125. Becker LE, de Oliveira Biazotto F, Conrad H, Schaier M, Kihm LP, Gross-Weissmann ML, et al. Cellular infiltrates and NFkappaB subunit c-Rel signaling in kidney allografts of patients with clinical operational tolerance. Transplantation. (2012) 94:729–37. doi: 10.1097/TP.0b013e31826032be
126. Misra MK, Mishra A, Pandey SK, Kapoor R, Sharma RK, Agrawal S. Association of functional genetic variants of transcription factor Forkhead Box P3 and Nuclear Factor-kappaB with end-stage renal disease and renal allograft outcome. Gene. (2016) 581:57–65. doi: 10.1016/j.gene.2016.01.028
127. Baizabal-Aguirre VM. Editorial: Cross-talk mechanisms of Wnt/beta-catenin signaling components with TLR-activated signaling molecules in the inflammatory response. Front Immunol. (2017) 8:1396. doi: 10.3389/fimmu.2017.01396
128. Ghosh S, Dass JF. Study of pathway cross-talk interactions with NF-kappaB leading to its activation via ubiquitination or phosphorylation: a brief review. Gene. (2016) 584:97–109. doi: 10.1016/j.gene.2016.03.008
129. Hoffmann A. Immune response signaling: combinatorial and dynamic control. Trends Immunol. (2016) 37:570–2. doi: 10.1016/j.it.2016.07.003
130. Ghosh S, Febin Prabhu Dass J. Non-canonical pathway network modelling and ubiquitination site prediction through homology modelling of NF-kappaB. Gene. (2016) 581:48–56. doi: 10.1016/j.gene.2016.01.025
131. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. (2011) 12:695–708. doi: 10.1038/ni.2065
132. Margalef P, Colomer C, Villanueva A, Montagut C, Iglesias M, Bellosillo B, et al. BRAF-induced tumorigenesis is IKKalpha-dependent but NF-kappaB-independent. Sci Signal. (2015) 8:ra38. doi: 10.1126/scisignal.2005886
133. Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell. (2015) 60:63–76. doi: 10.1016/j.molcel.2015.07.032
134. Slotta C, Storm J, Pfisterer N, Henkel E, Kleinwachter S, Pieper M, et al. IKK1/2 protect human cells from TNF-mediated RIPK1-dependent apoptosis in an NF-kappaB-independent manner. Biochim et Biophy Acta. (2018) 1865:1025–33. doi: 10.1016/j.bbamcr.2018.04.003
135. Sheng X, Zuo X, Liu X, Zhou Y, Sun X. Crosstalk between TLR4 and Notch1 signaling in the IgA nephropathy during inflammatory response. Int Urol Nephrol. (2018) 50:779–85. doi: 10.1007/s11255-017-1760-2
136. Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. (2012) 246:125–40. doi: 10.1111/j.1600-065X.2011.01088.x
137. Polley S, Passos DO, Huang DB, Mulero MC, Mazumder A, Biswas T, et al. Structural Basis for the Activation of IKK1/alpha. Cell Rep. (2016) 17:1907–14. doi: 10.1016/j.celrep.2016.10.067
138. Valino-Rivas L, Gonzalez-Lafuente L, Sanz AB, Ruiz-Ortega M, Ortiz A, Sanchez-Nino MD. Non-canonical NFkappaB activation promotes chemokine expression in podocytes. Sci Rep. (2016) 6:28857. doi: 10.1038/srep28857
139. Evans S, Tzeng HP, Veis DJ, Matkovich S, Weinheimer C, Kovacs A, et al. TNF receptor-activated factor 2 mediates cardiac protection through noncanonical NF-kappaB signaling. JCI Insight. (2018) 3:98278. doi: 10.1172/jci.insight.98278
140. Riemann M, Andreas N, Fedoseeva M, Meier E, Weih D, Freytag H, et al. Central immune tolerance depends on crosstalk between the classical and alternative NF-kappaB pathways in medullary thymic epithelial cells. J Autoimmun. (2017) 81:56–67. doi: 10.1016/j.jaut.2017.03.007
141. Guo L, Huang J, Chen M, Piotrowski E, Song N, Zahner G, et al. T-lymphocyte-specific knockout of IKK-2 or NEMO induces Th17 cells in an experimental nephrotoxic nephritis mouse model. FASEB J. (2018): 33:2359–71. doi: 10.1096/fj.201800485RR
142. Stein T, Wollschlegel A, Te H, Weiss J, Joshi K, Kinzel B, et al. Interferon regulatory factor 5 and nuclear factor κ-B exhibit cooperating but also divergent roles in the regulation of pro-inflammatory cytokines important for the development of TH1 and TH17 responses. FEBS J. (2018). 285:3097–113. doi: 10.1111/febs.14600
143. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 ligation in human T regulatory cells enhances IL2-induced cell proliferation through the non-canonical NF-kappaB pathway. Sci Rep. (2018) 8:12079. doi: 10.1038/s41598-018-30621-4
144. Durand JK, Baldwin AS. Targeting IKK and NF-kappaB for Therapy. Adv Protein Chem Struct Biol. (2017) 107:77–115. doi: 10.1016/bs.apcsb.2016.11.006
145. Paul A, Edwards J, Pepper C, Mackay S. Inhibitory-kappaB Kinase (IKK) alpha and nuclear factor-kappaB (NFkappaB)-Inducing Kinase (NIK) as anti-cancer drug targets. Cells. (2018) 7. doi: 10.3390/cells7100176
146. Tornatore L, Capece D, D'Andrea D, Begalli F, Verzella D, Bennett J, et al. Clinical proof of concept for a safe and effective NF-κB-targeting strategy in multiple myeloma. Br J Haematol. (2018). doi: 10.1111/bjh.15569. [Epub ahead of print].
147. Cahill KE, Morshed RA, Yamini B. Nuclear factor-kappaB in glioblastoma: insights into regulators and targeted therapy. Neuro-oncology. (2016) 18:329–39. doi: 10.1093/neuonc/nov265
148. Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-targeted therapeutics for metabolic disease. Trends Pharmacol Sci. (2018) 39:536–46. doi: 10.1016/j.tips.2018.03.001
149. Rizwan H, Mohanta J, Si S, Pal A. Gold nanoparticles reduce high glucose-induced oxidative-nitrosative stress regulated inflammation and apoptosis via tuberin-mTOR/NF-kappaB pathways in macrophages. Int J Nanomed. (2017) 12:5841–62. doi: 10.2147/ijn.s141839
150. Xu H, Yuan R, Liu X, Li X, Qiao G, Li C, et al. Zn-doped CuO nanocomposites inhibit tumor growth by NF-?B pathway cross-linked autophagy and apoptosis. Nanomedicine (Lond.). (2019) 14:131–49. doi: 10.2217/nnm-2018-0366
151. Poveda J, Sanz AB, Carrasco S, Ruiz-Ortega M, Cannata-Ortiz P, Sanchez-Nino MD, et al. Bcl3: a regulator of NF-kappaB inducible by TWEAK in acute kidney injury with anti-inflammatory and antiapoptotic properties in tubular cells. Exp Mol Med. (2017) 49:e352. doi: 10.1038/emm.2017.89
152. Wang S, Lin Y, Yuan X, Li F, Guo L, Wu B. REV-ERBalpha integrates colon clock with experimental colitis through regulation of NF-kappaB/NLRP3 axis. Nat Commun. (2018) 9:4246. doi: 10.1038/s41467-018-06568-5
153. Zhu L, Xu H, Lv W, He Z, Ye P, Wang Y, et al. miR-199b-5p regulates immune-mediated allograft rejection after lung transplantation through the GSK3beta and NF-kappaB Pathways. Inflammation. (2018) 41:1524–35. doi: 10.1007/s10753-018-0799-2
154. Bennett J, Capece D, Begalli F, Verzella D, D'Andrea D, Tornatore L, et al. NF-kappaB in the crosshairs: Rethinking an old riddle. Int J Biochem Cell Biol. (2018) 95:108–12. doi: 10.1016/j.biocel.2017.12.020
155. Prescott JA, Cook SJ. Targeting IKKbeta in cancer: challenges and opportunities for the therapeutic utilisation of IKKbeta inhibitors. Cells. (2018) 7:E115. doi: 10.3390/cells7090115
156. Wu J, Ding J, Yang J, Guo X, Zheng Y. MicroRNA roles in the nuclear factor kappa B signaling pathway in cancer. Front Immunol. (2018) 9:546. doi: 10.3389/fimmu.2018.00546
157. Li H, Jiang T, Li MQ, Zheng XL, Zhao GJ. Transcriptional regulation of macrophages polarization by MicroRNAs. Front Immunol. (2018) 9:1175. doi: 10.3389/fimmu.2018.01175
158. Zhong L, Simard MJ, Huot J. Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation. FASEB J. (2018) 32:4070–84. doi: 10.1096/fj.201701536R
159. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
160. Pichler R, Afkarian M, Dieter BP, Tuttle KR. Immunity and inflammation in diabetic kidney disease: translating mechanisms to biomarkers and treatment targets. Am J Physiol Renal Physiol. (2017) 312:F716–f31. doi: 10.1152/ajprenal.00314.2016
161. Ye H, Su B, Ni H, Li L, Chen X, You X, et al. microRNA-199a may be involved in the pathogenesis of lupus nephritis via modulating the activation of NF-kappaB by targeting Klotho. Mol Immunol. (2018) 103:235–42. doi: 10.1016/j.molimm.2018.10.003
162. Liu L, Pang XL, Shang WJ, Xie HC, Wang JX, Feng GW. Over-expressed microRNA-181a reduces glomerular sclerosis and renal tubular epithelial injury in rats with chronic kidney disease via down-regulation of the TLR/NF-kappaB pathway by binding to CRY1. Mol Med. (2018) 24:49. doi: 10.1186/s10020-018-0045-2
Keywords: NFκB, kidney disease, glomerulonephritis, AKI, autoimmune disease, kidney transplantation, NFκB pathway crosstalk
Citation: Song N, Thaiss F and Guo L (2019) NFκB and Kidney Injury. Front. Immunol. 10:815. doi: 10.3389/fimmu.2019.00815
Received: 18 December 2018; Accepted: 27 March 2019;
Published: 16 April 2019.
Edited by:
Valentin A. Pavlov, Northwell Health, United StatesReviewed by:
Liudmila V. Spirina, Russian Academy of Sciences, RussiaMichael May, University of Pennsylvania, United States
David Charles Budd, GlaxoSmithKline, United Kingdom
Copyright © 2019 Song, Thaiss and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedrich Thaiss, dGhhaXNzQHVrZS5kZQ==