 Vivienne Woo
Vivienne Woo Emily M. Eshleman
Emily M. Eshleman Taylor Rice
Taylor Rice Jordan Whitt
Jordan Whitt Bruce A. Vallance
Bruce A. Vallance Theresa Alenghat
Theresa Alenghat- 1Division of Immunobiology and Center for Inflammation and Tolerance, Cincinnati Children's Hospital Medical Center, Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH, United States
- 2Division of Gastroenterology, Department of Pediatrics, BC Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada
Numerous bacterial pathogens infect the mammalian host by initially associating with epithelial cells that line the intestinal lumen. Recent work has revealed that commensal bacteria that reside in the intestine promote defense against pathogenic infection, however whether the microbiota direct host pathways that alter pathogen adherence is not well-understood. Here, by comparing germ-free mice, we identify that the microbiota decrease bacterial pathogen adherence and dampen epithelial expression of the cell surface glycoprotein C-type lectin 2e (Clec2e). Functional studies revealed that overexpression of this lectin promotes adherence of intestinal bacterial pathogens to mammalian cells. Interestingly, microbiota-sensitive downregulation of Clec2e corresponds with decreased histone acetylation of the Clec2e gene in intestinal epithelial cells. Histone deacetylation and transcriptional regulation of Clec2e depends on expression and recruitment of the histone deacetylase HDAC3. Thus, commensal bacteria epigenetically instruct epithelial cells to decrease expression of a C-type lectin that promotes pathogen adherence, revealing a novel mechanism for how the microbiota promote innate defense against infection.
Introduction
Infections of the gastrointestinal tract are a major cause of morbidity and mortality worldwide. Specifically, enteric infections caused by bacterial pathogens account for well over 200 million individual cases of enteritis resulting in an estimated 5 million deaths annually (1, 2). In addition to local intestinal infections, the gastrointestinal tract is the initial site of adhesion and entry for several pathogens that disseminate to cause systemic disease (3). Thus, adherence and invasion are critical steps in the pathogenesis of both enteric and systemic bacterial infections. In order to establish disease, pathogens can interact with host cells by expressing adhesin molecules which recognize various components such as extracellular matrix proteins, integral membrane adhesion receptors, and cell membrane associated glycoproteins (4). These interactions between bacterial pathogens and host cells are not only critical for initiating infection, but also direct tissue tropism, species specificity, and host susceptibility to infection (4–7). Therefore, understanding how pathogenic adherence is mediated is critical for directing effective approaches that prevent and treat enteric infections.
In addition to pathogenic bacteria, the mammalian gastrointestinal tract harbors trillions of innocuous commensal bacteria. These commensal microbes, collectively termed the microbiota, are required for healthy intestinal development and immune cell activation (8). Importantly, the presence of the microbiota has also been consistently shown to be essential for host defense against pathogenic infections (8, 9). While several mechanisms have been proposed to account for microbiota-dependent protection against infection, many pathways indicate that commensal bacteria can potentiate host-cell intrinsic defenses (10–12). Intestinal epithelial cells (IECs) reside at the direct interface between the microbiota and underlying host immune cells and are in constant contact with both beneficial as well as invading bacteria. Thus, IECs are a key cell type to which enteric pathogens often directly associate with in order to infect and invade the host. In addition to mediating binding and sensing of microbial components, these critically located cells can actively respond to pathogenic challenges by secreting antimicrobial peptides, mucins, and cytokines that prime and regulate innate and adaptive immune cell compartments (13–16). However, it is not clear whether the microbiota restrict enteric infection by regulating pathogen binding to the intestinal epithelium.
In mammalian cells, DNA is packaged around histone proteins that are condensed into a higher order structure called chromatin. In general, chromatin structure itself restricts access of transcriptional machinery to the genome thereby repressing gene expression. However, covalent modifications of the amino-terminal tails of histones, specifically, acetylation, methylation, phosphorylation, SUMOylation, and ubiquitination are associated with conformational changes in the chromatin landscape. For example, histone acetylation is known to generate an open chromatin structure that contributes to active transcription (17–19). These modifications are mediated by epigenetic modifying enzymes such as histone acetyltransferases and histone deacetylases (HDACs). The balance and pattern of these modifications on specific histone tails regulate chromatin reorganization and direct transcriptional machinery. Thus, epigenetic modifications enable environmental signals to trigger transcriptional changes without altering underlying DNA sequence (20–22).
In this study, we aimed to test whether the microbiota affect IEC-intrinsic pathways that alter the ability of pathogens to adhere to the IECs. Citrobacter rodentium, a murine enteric pathogen with a similar pathogenesis to enteropathogenic E. coli in humans, infects the host by initially adhering to IECs. By employing germ-free (GF) mice, we identified that the microbiota reduce pathogen colonization with C. rodentium during infection and instruct decreased IEC interactions with the pathogen. Global gene expression analyses revealed that the microbiota highly suppressed IEC expression of the cell-surface C-type lectin 2e (Clec2e). Interestingly, functional studies showed that overexpression of Clec2e enhanced pathogen bacterial binding to the mammalian cell membrane. Furthermore, microbiota-dependent transcriptional suppression of Clec2e in IECs correlated with decreased histone acetylation and recruitment of the histone deacetylase, HDAC3. Collectively, these data demonstrate a novel mechanism by which commensal bacteria in the intestine epigenetically regulate expression of a pathogen-binding glycoprotein to promote host defense against infection.
Materials and Methods
Mice and in vivo Infections
Conventionally-housed C57Bl/6J mice were purchased from Jackson Laboratories and maintained in our specific-pathogen free colony at CCHMC. Germ-free (GF) mice were maintained in plastic isolators in the CCHMC Gnotobiotic Mouse Facility, fed autoclaved feed and water, and monitored to ensure absence of microbes. HDAC3FF mice (23) were crossed to C57Bl/6J mice expressing Cre recombinase under control of the IEC-specific villin promoter (24) to generate HDAC3ΔIEC mice (25). Mice were housed up to 4 per cage in a ventilated cage system in a 12 h light/dark cycle, with free access to water and food. For C. rodentium infection, age- and gender- matched mice were orally inoculated with 109 colony forming units (CFUs) of C. rodentium (26, 27). To enumerate intestinal bacterial burdens, stool was collected in PBS and homogenized in a TissueLyser II at 30 Hz for 3 min. Homogenates were serially diluted and plated on MacConkey agar. CFUs were counted and normalized to stool weight after 18 h. All experiments were performed according to the animal guidelines upon approval of the Institutional Animal Care and Use Committee at CCHMC.
IEC Harvest, RNA Analyses, Western Blotting
IECs were harvested from mouse intestine as described previously (25, 27, 28). IECs from the small intestine were harvested from the most distal 12 cm section. RNA was isolated from cells using the RNeasy Kit (Qiagen) then subjected to reverse transcription with Verso reverse transcriptase (Thermo Fisher). Directional polyA RNA-seq for IECs from the small intestine was performed by the Sequencing Core at the University of Cincinnati (28). Sequence reads were aligned by using Illumina sequence analysis pipeline by the Laboratory for Statistical Genomics and Systems Biology at the University of Cincinnati. Real-time PCR was performed using SYBR (Applied Biosystems) and analyzed with a threshold in the linear range of amplification using primer sequences as follows: Clec2eF: 5′-AGCAAGGTTCACAGCTCTCC-3′; Clec2eR: 5′-GCTGCTATGGAGTGATCATGG-3′; RegIIIγF: 5′-TTCCTGTCCTCCATGATCAAA-3′; RegIIIγR: 5′-CATCCACCTCTGTTGGGTTC-3′; HPRTF: 5′-GATTAGCGATGAACCAGGT-3′; HPRTR: 5′-CCTCCCATCTCCTTCATGACA-3′. Expression analysis in IECs from large intestine of HDAC3FF and HDAC3ΔIEC mice by microarray was described previously (25). For western blot analyses, total cell lysates were probed with anti-histone H3 (Santa Cruz) and anti-DDK (FLAG) (Origene) and imaged using an Odyssey Fc imager (LICOR). Global expression data has been deposited in NCBI's Gene Expression Omnibus (GEO) and is accessible through accession number GSE128362.
ChIP-Sequencing
ChIP was performed as described previously with few modifications (29). Briefly, cells were fixed in 1% PFA for 10 min and quenched with glycine. Total cell extracts were sonicated using a Covaris S220 Focused-ultrasonicator and nuclear extracts were immunoprecipitated with rabbit anti-H3K9Ac (Millipore, 06-942) or rabbit anti-HDAC3 (Abcam, ab7030) using a SX-8G IP-STAR robot. Sequencing was performed using Illumina HiSeq 2500, mapped to mus musculus genome mm10 with Bowtie and peaks were identified with MACS (30, 31) and visualized in Biowardrobe (32). ChIP-qPCR was performed using SYBR (Applied Biosystems) and analyzed as fold difference normalized to an unaffected control gene. Reactions were run on a real-time PCR system (QuantStudio3; Applied Biosystems) with custom made primer pairs: Clec2e-ChIPF: 5′-ACACAAGATGCAGCGGAGAT-3′; Clec2e-ChIPR: 5′-GTGAAGGGGTTTTCACTAGGGG-3′; Insl-ChIPF: 5′-CAGAGACCATCAGCAAGCAG-3′; Insl-ChIPR: 5′-TTCTCCCTAAAGTCGCTGGA-3′; Albumin-ChIPF: 5′-AGAGCGATCTTTCTGCACACA-3′; Albumin-ChIPR: 5′-AGGAGAAAGGTTACCCACTTCATTT-3′. ChIP-seq data is accessible through GEO series accession numbers GSE50453 and GSE128369.
Cell Culture and Immunofluorescence
HEK293T cells were cultured in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C and 5% CO2. Cells were transiently transfected with pCMV6-Clec2e-myc-DDK (FLAG) vector (Origene, MR202134) using Lipofectamine 3000 (Thermo Fisher). Transfected cells were seeded onto Retronectin (Takara Bio) coated chamber slides (Ibidi) and infected with 106 CFUs of GFP-expressing C. rodentium for 6 h in antibiotic-free media (26, 27). Cells were washed in PBS 3 times and fixed in 4% paraformaldehyde for 20 min. Fixed cells were blocked with 2% BSA for 1 h at room temperature and stained in 0.5% BSA with 488-anti-GFP (Thermo Fisher, 1:300), Phalloidin (Invitrogen, 1:200) and DAPI (Invitrogen, 1:1,000) for 1 h at room temperature. Stained cells were visualized using Nikon A1R LUN-V inverted confocal microscope.
Intestinal Organoids
Murine organoids were generated from colonic crypts isolated from germ-free and conventionally-housed mice as previously described (33). Briefly, dissected colons were opened longitudinally, scrapped to remove intestinal contents and outer cells, washed repeatedly in ice-cold PBS, and cut into 1-cm pieces. Colonic pieces were incubated in chelation buffer (2 mM EDTA in PBS) for 30 min at 4°C with rotation. The tissue was transferred into a new tube containing Shaking Buffer (PBS, 43.3 mM sucrose, 54.9 mM sorbitol) and gently shaken by hand for 2–4 min. Colonic crypts were resuspended and plated in Matrigel (Corning) with organoid culture media (60% Advanced DMEM/F12 media supplemented with 10 mM HEPES, 2 mM L-glutamate, 40% L-WRN conditioned media, 1x N2 supplement, 1x B27 supplement, 50 ng/mL murine EGF, and 10 μM Y-27632 ROCK inhibitor) overlaid. Culture media was changed every 3–4 days. Organoid cultures were infected with GFP-C. rodentium at a concentration of 106 CFUs for 24 h. After incubation, organoids were washed 3 times in ice-cold PBS, dislodged from plate and Matrigel, and fixed in 1% PFA for 1 h at 4°C. GF organoids were stimulated with 10 ng/mL of E. coli LPS for 24 h.
Flow Cytometry
Cells were stained for flow cytometry using the following fluorescence-conjugated antibodies diluted in FACS Buffer (2% FBS, 0.01% Sodium Azide, PBS): PE anti-CD326 (EpCAM) (Clone: G8.8, eBioscience), BUV395 anti-CD45.2 (Clone: 104, BD Biosciences), 488 anti-GFP (Clone; FM264G, BioLegend). Dead cells were excluded with the Fixable Violet Dead Cell Stain Kit (Invitrogen). Samples were acquired on the BD LSRFortessa and analyzed with FlowJo Software (Treestar). The geometric mean fluorescence intensity (MFI) for GFP-C. rodentium expression was assessed and the background MFI determined in uninfected controls was subtracted from infected samples.
Bacterial Adhesion Assay
Adhesion of pathogenic bacteria to mammalian cells was determined as previously described (34). Briefly, HEK293T cells were seeded at 70–90% confluency and incubated with GFP-C. rodentium or wild-type Salmonella enterica serovar Typhimurium at a multiplicity of infection (MOI) of 5:1 (bacteria:cells) for 6 h in antibiotic-free media. Cells were washed 3 times with PBS and adherent bacteria were detached using a 1% triton-X 100 lysis buffer and plated onto MacConkey agar in 10-fold serial dilutions. Colony forming units (CFUs) were counted after 16 h.
Statistical Analyses
Results are expressed as mean ± SEM. Statistical significance was determined with the Student's t-test, with all data meeting the assumptions of the statistical test used. Results were considered significant at *p < 0.05; **p < 0.01; ***p < 0.001. Statistical significance was calculated using Prism version 7.0 (GraphPad Software).
Results
Microbiota Decrease Pathogen Adherence to Intestinal Epithelial Cells
Citrobacter rodentium (C. rodentium) is a murine bacterial pathogen with similar pathogenesis to enteropathogenic E. coli in humans. Germ-free (GF) mice infected with C. rodentium exhibited significantly higher pathogen burdens compared to conventionally-housed (CNV) mice (Figure 1A), indicating that the microbiota enhance defense against pathogenic colonization. Interestingly, microbiota-sensitive protection against infection was detected as early as day 4 post-infection, suggesting the presence of the microbiota affect the initial establishment of C. rodentium colonization. Intestinal epithelial cells (IECs) produce antimicrobial peptides and consistent with previous studies (35), the microbiota induced IEC expression of the antimicrobial peptide RegIIIγ that targets bacterial pathogens (Figure 1B) (36). In order to investigate how the microbiota induce epithelial-intrinsic defense, intestinal epithelial organoids that are devoid of immune cells were compared. Interestingly, intestinal epithelial organoids generated from CNV-housed mice expressed significantly reduced levels of RegIIIγ (Figure 1B). However, despite this impairment in RegIIIγ expression, organoids from CNV mice exhibited lower adherent GFP-expressing C. rodentium compared to GF organoids as measured by flow cytometry (Figure 1C). These data suggest that other mechanisms, aside from RegIIIγ, contribute to microbiota-sensitive IEC-intrinsic resistance against pathogenic bacterial adherence.
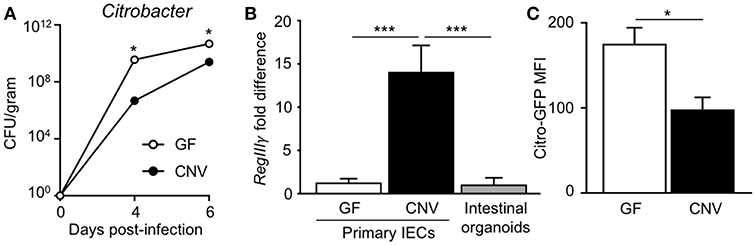
Figure 1. Microbiota exposure decreases pathogen adherence to intestinal epithelial cells. (A) C. rodentium colony forming units (CFUs) in stool of infected germ-free (GF) and conventionally-housed (CNV) mice during early course of infection (day 4–6). Data are representative of at least 3 independent experiments with 3–4 mice per group. (B) Relative RegIIIγ mRNA expression in IECs isolated from the large intestine of GF and CNV mice compared to intestinal organoid cultures. (C) MFI of GFP-C. rodentium infected intestinal organoids derived from GF and CNV mice. Data represent two independent experiments with 3–4 mice per group. Results are mean ± SEM. *p < 0.05, ***p < 0.001.
Microbiota Exposure Downregulates C-Type Lectin 2e Expression in Intestinal Epithelial Cells
In order to identify potential mediators of pathogen adherence that are regulated by the microbiota, we compared genes expressed in IECs harvested from the intestine of GF and CNV mice by RNA-sequencing. These analyses identified C-type lectin 2 member e (Clec2e; Clr-a) as one of the most significantly downregulated genes in IECs in response to microbial exposure (Figure 2A). Clec2e expression was confirmed to be decreased by quantitative PCR (qPCR) in IECs from independent cohorts of GF and CNV mice in both the small intestine (Figure 2B) and large intestine (Figure 2C). To investigate whether microbiota-suppressed Clec2e expression was maintained in the absence of immune cells or persistent microbial stimulation, intestinal organoid cultures were generated from colonic crypts isolated from GF and CNV mice (Figure 2D). Consistent with IECs in vivo, Clec2e expression was repressed in organoids derived from CNV mice (Figure 2E), suggesting prior exposure to the microbiota led to sustained downregulation of Clec2e. To determine how the microbiota may suppress Clec2e expression, intestinal organoids derived from GF mice were incubated with LPS (Figure 2F). LPS reduced Clec2e expression in IECs, although less than observed in CNV organoids, suggesting that microbial-derived components may collectively regulate epithelial Clec2e expression.
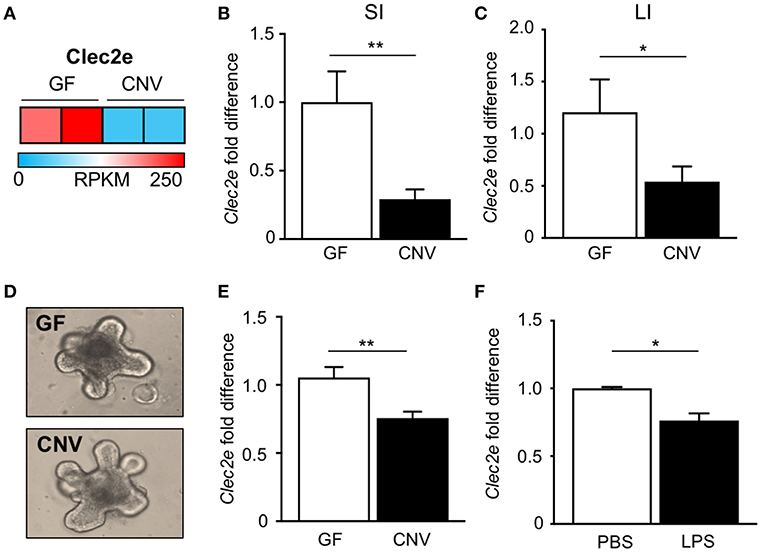
Figure 2. Microbiota downregulate intestinal epithelial expression of C-type lectin Clec2e. (A) Relative Clec2e expression levels in IECs harvested from small intestine of GF and CNV mice, represented as reads per kilobase per million mapped reads (RPKM). (B,C) Real-time quantitative PCR analysis of Clec2e in IECs from GF and CNV mice isolated from (B) small intestine (SI) or (C) large intestine (LI). (D) Intestinal organoids derived from GF and CNV mice. (E) Clec2e mRNA expression in GF and CNV organoids. (F) Clec2e mRNA expression in GF intestinal organoids in the absence or presence of LPS. Data represent two independent experiments with 3–4 mice per group. Results are mean ± SEM. *p < 0.05, **p < 0.01.
Expression of Clec2e Increases Cellular Adherence of Enteric Bacterial Pathogens
Clec2e is a homodimeric cell surface glycoprotein expressed in the intestinal epithelium that shares homology with other C-type lectins (37, 38). However, unlike other CLEC2 family members, Clec2e does not interact with Nkrp1 receptors (37, 39). C-type lectin receptor family members contain extracellular carbohydrate binding domains that associate with common pathogen-associated molecular patterns including mannose, fucose, and β-glycan (40, 41), provoking the hypothesis that Clec2e may facilitate bacterial adhesion to host cells. To test whether Clec2e plays a role in bacterial adhesion to mammalian cells, Clec2e-FLAG was overexpressed in HEK293T cells followed by incubation with either Salmonella or C. rodentium, enteric bacterial pathogens that directly bind to IECs. Expression of transfected Clec2e was confirmed by Western analyses (Figure 3A). Interestingly, Clec2e-expressing cells exhibited significantly greater adherence of Salmonella (Figure 3B) and C. rodentium (Figure 3C) compared to negative control cells. Bacterial adherence of GFP-expressing C. rodentium to Clec2e-expressing cells was confirmed at the cellular level by immunofluorescence (Figure 3D) and flow cytometry (Figure 3E). Together, these data indicate that Clec2e expression promotes adherence of bacterial pathogens to mammalian cells.
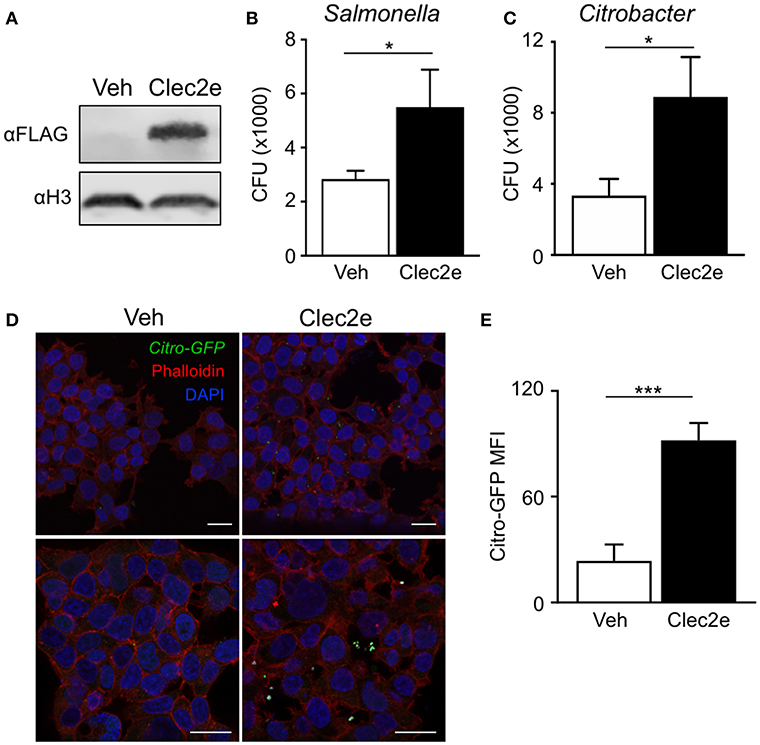
Figure 3. Expression of Clec2e enables increased adherence of bacterial pathogens. Clec2e overexpressed in HEK293T cells using a FLAG-tagged Clec2e expression vector. (A) Western blot analysis of Clec2e-FLAG expression in vehicle or Clec2e-FLAG transfected HEK293T cell lysates. (B,C) In vitro bacterial adhesion assay of infected HEK293T cells, expressed as adherent CFUs 6 h post infection. (D) Immunofluorescent staining of HEK293T cells infected with GFP-expressing C. rodentium (green). Mammalian cell membrane and nuclei are stained with Phalloidin (red) and DAPI (blue), respectively. Scale bars, 25 μm. (E) MFI of GFP-C. rodentium bound to vehicle-treated or Clec2e-transfected HEK293T cells. Data represent 2 independent experiments with n = 3 per group. Results are mean ± SEM. *p < 0.05, ***p < 0.001.
Microbiota Induce Loss of Histone Acetylation and HDAC3 Recruitment Within Regulatory Regions of Clec2e
Environmental factors can regulate mammalian gene expression through epigenetic modifications of the chromatin, such as DNA methylation and histone acetylation. Consistent with this, recent studies have revealed that epigenetic pathways may be essential in mediating host-microbe dynamics (17, 42, 43). Therefore, to determine whether the microbiota epigenetically modify chromatin at the Clec2e gene, histone acetylation was compared in primary IECs harvested from GF and CNV mice. For these analyses, chromatin immunoprecipitation (ChIP)-sequencing (seq) was performed for the histone mark H3K9Ac, which is associated with permissive and actively transcribed chromatin (17). Remarkably, ChIP-seq analyses revealed that H3K9Ac levels were significantly decreased at multiple sites within the Clec2e locus in IECs isolated from CNV mice compared to GF controls (Figure 4A). This loss of histone acetylation in regulatory regions of Clec2e due to the microbiota was confirmed by ChIP-qPCR for H3K9Ac (Figure 4B). Previous studies have demonstrated that histone acetylation in IECs can be regulated by epigenetic-modifying enzymes called histone deacetylase (HDACs) (44, 45). The class I histone deacetylase 3 (HDAC3) deacetylates histone H3K9Ac and mediates microbiota-dependent regulation of epithelial gene expression (25, 27). Thus, to determine whether HDAC3 epigenetically regulates Clec2e, HDAC3 recruitment was first examined by ChIP. HDAC3 was enriched at the site of differential H3K9Ac in Clec2e (Figure 4B) relative to a negative non-HDAC3 target (Figure 4C), supporting that Clec2e is a direct target of HDAC3. Interestingly, IECs from CNV mice exhibited significantly higher HDAC3 recruitment to Clec2e compared to IECs from GF mice (Figure 4D). Collectively, these data indicate that the microbiota direct epigenetic regulation of Clec2e through HDAC3.
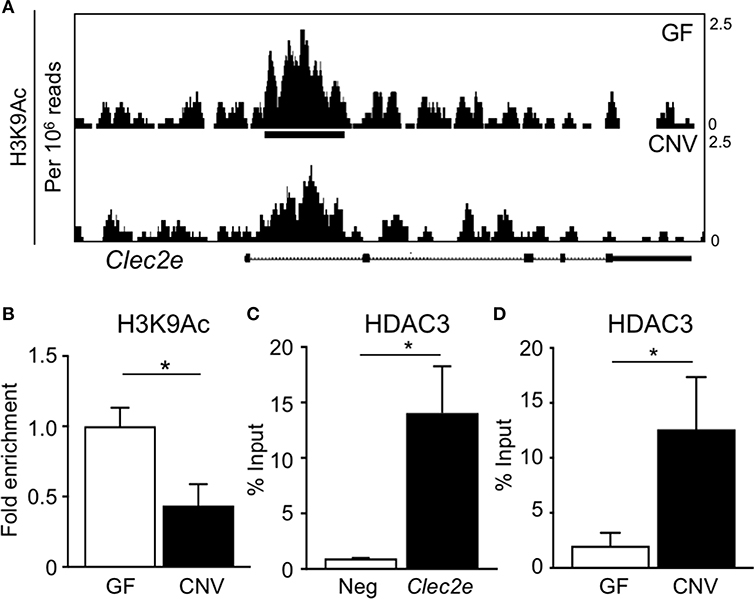
Figure 4. Microbiota induce histone deacetylation and HDAC3 enrichment at Clec2e gene. (A) ChIP-seq for H3K9Ac in primary IECs isolated from the small intestine of GF and CNV mice. (B) ChIP-qPCR for H3K9Ac in Clec2e from IECs. (C) ChIP-qPCR for HDAC3 in Clec2e as percent of input, relative to a negative control gene (Insulin 1). (D) HDAC3 ChIP-qPCR in Clec2e from GF and CNV IECs. Data represent two independent experiments with 3-4 mice per group. Results are mean ± SEM. *p < 0.05.
HDAC3 Regulates Epithelial Clec2e Expression and Pathogen Adherence
The microbiota-dependent increase in HDAC3 recruitment to Clec2e suggests that loss of H3K9Ac and decreased expression of Clec2e in response to the microbiota could be mediated by HDAC3. Thus, to directly test whether IEC-intrinsic HDAC3 regulates histone acetylation within the Clec2e gene, ChIP-seq for H3K9Ac was performed in IECs harvested from mice that lack HDAC3 expression specifically in IECs (HDAC3ΔIEC) compared to floxed HDAC3FF control mice (25). Consistent with histone deacetylation by HDAC3, IECs harvested from the large intestine of mice lacking IEC-HDAC3 (HDAC3ΔIEC) exhibited significantly higher levels of H3K9Ac in Clec2e compared to IECs from HDAC3FF mice (Figure 5A). Increased H3K9Ac enrichment within the microbiota-sensitive regulatory region in the Clec2e gene was also identified in IECs from the small intestine of HDAC3ΔIEC mice (Figure 5B), indicating that Clec2e histone acetylation is regulated by epithelial HDAC3. HDAC3 recruitment is associated with transcriptional repression of bound genes. Thus, to test whether elevated H3K9Ac with HDAC3 depletion corresponds with altered expression, Clec2e mRNA expression was measured in IECs from HDAC3FF and HDAC3ΔIEC mice. Consistent with the role of HDAC3 as a transcriptional repressor of direct targets, these analyses revealed significantly increased Clec2e expression in IECs harvested from the small and large intestine of HDAC3ΔIEC mice compared to IECs from HDAC3FF controls (Figure 5C), indicating that Clec2e expression in HDAC3-deficient IECs results from impaired HDAC3-mediated deacetylation within the Clec2e gene. Collectively, these studies demonstrate that epigenetic and transcriptional regulation of Clec2e broadly depends on epithelial HDAC3 expression in the intestine.
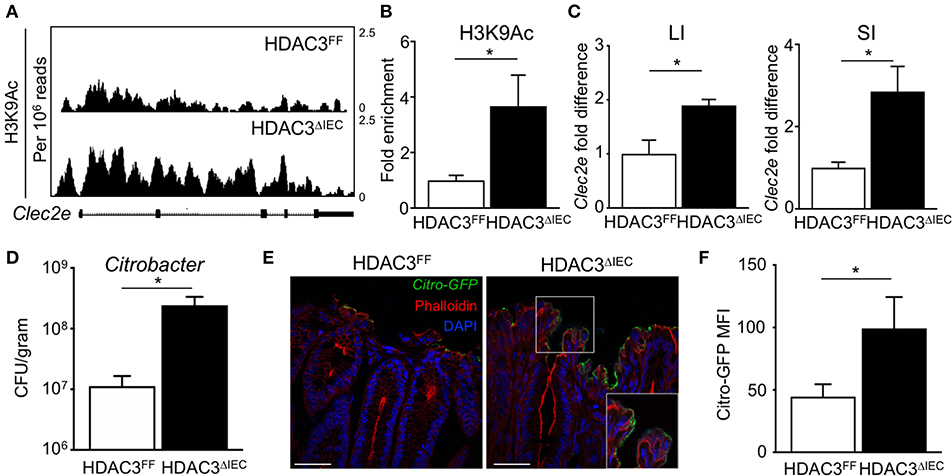
Figure 5. HDAC3 regulates epithelial Clec2e expression and pathogen adherence. (A) ChIP-seq for H3K9Ac in primary IECs isolated from the large intestine of HDAC3FF and HDAC3ΔIEC mice. (B) ChIP-qPCR for H3K9Ac in Clec2e in IECs from the small intestine. (C) Clec2e expression in IECs from large and small intestine of HDAC3FF and HDAC3ΔIEC mice. (D) C. rodentium CFUs in stool at day 6 post infection. (E) Fluorescence microscopy of colon from HDAC3FF and HDAC3ΔIEC mice infected with GFP-C. rodentium. (Green: GFP-C. rodentium, Red: Phalloidin, Blue: DAPI). Scale bars, 25 μm. (F) MFI of GFP-C. rodentium infected intestinal organoids derived from HDAC3FF and HDAC3ΔIEC mice. Data are representative of at least 2 independent experiments with 3–4 mice per group. Results are mean ± SEM. *p < 0.05.
To next test whether HDAC3-dependent regulation of IECs affects pathogen adhesion, HDAC3FF and HDAC3ΔIEC mice were infected with C. rodentium. Interestingly, C. rodentium-infected HDAC3ΔIEC mice exhibited higher pathogen burden (Figure 5D) and increased GFP-C. rodentium adherence to IECs (Figure 5E) relative to infected HDAC3FF control mice, confirming increased pathogen adhesion in HDAC3ΔIEC mice. Further, to test this in the absence of immune cells, intestinal organoids were generated from the colon of control HDACFF mice and mice lacking HDAC3 in IECs. Consistent with the in vivo findings, HDAC3ΔIEC organoids exhibited elevated GFP-C. rodentium binding compared to cells from floxed controls (Figure 5F). Taken together, these data indicate that regulation of Clec2e expression in IECs by HDAC3-mediated histone deacetylation can promote decreased bacterial pathogen adherence to the intestinal epithelium.
Discussion
The intestinal epithelium not only maintains intestinal homeostasis to innocuous commensals, but it also defends against invading pathogens (13, 46). Our data indicate that the microbiota can promote epithelial defense by epigenetically suppressing Clec2e-mediated pathogen adherence. Consistent with previously published data (10, 27), we show that GF mice are more susceptible to enteric infection relative to microbiota-replete mice. Previous studies have focused on investigating how the microbiota impact immune cell activation and antibacterial immunity (11, 47, 48), however we observed very early susceptibility to C. rodentium infection in GF mice suggesting an important role for innate responses. By employing intestinal organoid cultures, we determined that the microbiota directly impact IEC-intrinsic defense and identified that Clec2e downregulation by the microbiota can reduce pathogen colonization. The microbiota influence several aspects of IEC biology and microbiota-sensitive alterations in IEC composition can also impact host resistance to enteric infection. However, Clec2e is expressed throughout the intestinal epithelium including progenitor and differentiated epithelial cells (37), suggesting that differences in epithelial composition induced by the microbiota are unlikely to be a main mechanism regulating Clec2e expression.
Consistent with our data, studies using GF mice and mouse models that are deficient for pattern recognition receptor mediators have revealed that mucins and antimicrobial peptides require microbial stimulation for expression (35, 49–52). These proteins work in concert to prevent intestinal infection by restricting bacterial adhesion and invasion, limiting microbial growth and colonization, and directly killing bacteria. For example, mucins function by forming a protective barrier that limits access of microbes to underlying IECs and can bind to several enteric pathogens including C. rodentium to prevent adhesion (26, 53). In addition, expression of cathelicidin-related antimicrobial peptide by IECs plays an important and non-redundant role in preventing C. rodentium adhesion and colonization, especially in early stages of infection (54). Another member of the C-type lectin family with structural similarity to Clec2e, RegIIIγ, binds intestinal bacteria via interactions with peptidoglycan carbohydrates and directly lyses bacteria (35). These studies, combined with our Clec2e data, demonstrate that the microbiota direct multiple IEC-intrinsic host defenses that alter bacterial access to IECs and limit adhesion and colonization.
Similar to enteropathogenic E. coli and enterohemorrhagic E. coli, C. rodentium employs a type 3 secretion system and other virulence strategies to attach to the apical plasma membrane of IECs (55). Salmonella is also equipped with a type 3 secretion system and several fimbriae proteins that enable adherence and invasion to colonic IECs (3, 4, 56). Genetic deletion of type 3 secretion systems or fimbriae molecules drastically reduces bacterial colonization, confirming the necessity of these molecules for pathogenesis (26, 55, 56). Fimbriae and other filopodia-like extensions that enable bacterial adhesion often interact with host plasma membrane associated proteins. Salmonella fimbriae bind to a specific glycosylated moiety that is abundantly expressed in the intestinal epithelium (56). Interestingly, Clec2e is a heavily glycosylated protein whose expression is restricted to the intestinal epithelium and is downregulated with LPS or Poly:(IC) stimulation (37), suggesting it may play a functional role in regulating intestinal host defense. Future investigation will require GF and CNV Clec2eΔIEC knockout models in combination with mono-association studies to dissect the contribution of specific commensal microbes or microbial components that influence in vivo regulation of pathogen control by Clec2e.
Through global RNA-sequencing analysis we identified a drastic reduction in the expression of the C-type lectin protein, Clec2e, in IECs isolated from CNV mice compared to GF controls. Clec2e (Clr-a) is an orphan C-type lectin molecule that closely resembles the natural killer (NK) gene complex receptor, Clec2h (Clr-f). However, unlike Clec2h, Clec2e does not bind any known NK cell receptors (37, 39, 57). In addition to being signaling partners for NK cell receptors, C-type lectin molecules play a crucial role in recognition of conserved pathogen-associated molecular patterns. Specifically, C-type lectin receptors bind carbohydrate structures commonly associated with microbial cell wall components including mannose, fucose, and β-glucans (35, 40, 41). Further, expression of Clec2e is restricted to the intestinal epithelium and is downregulated with LPS and Poly:(IC) in a TLR3-dependent manner (37). Here, we demonstrate that overexpression of Clec2e is sufficient to promote bacterial adherence to mammalian cells. While the ligand of Clec2e remains unknown, its structural similarities to Dectin-1 and RegIII microbial binding proteins, along with our bacterial adhesion data, suggests Clec2e may bind conserved microbial cell wall components or bacterial pili and fimbria necessary for cellular adherence. Although expanded studies are needed to fully interrogate how Clec2e interacts with commensal bacterial populations and pathogens beyond Salmonella and C. rodentium, a lack of Clec2e suppression may contribute to heightened susceptibility of GF or antibiotic-treated mice to infection (48).
Epigenetic modifications enable host cells to alter gene expression without modifying the genetic sequence and changes in the host epigenome occur downstream of external environmental signals (20–22). Recent studies, focused predominantly on immune cells types, have supported that the microbiota may imprint or epigenetically prime genes in the host through enzymes such as DNA methyltransferases (58), histone methyltransferases (29, 59), and HDACs (25, 60–63). In addition to HDAC3, other class I HDACs are expressed in IECs (44, 45) and the role of these HDACs as well as other regulatory proteins may further alter epigenetic regulation of the Clec2e gene in IECs. IECs are equipped to sense and respond to common microbial moieties such as LPS, and previous studies showed TLR4 expression in IECs was epigenetically primed by the microbiota (64). Specifically, DNA methylation of TLR4 was decreased in GF mice compared to CNV controls, consistent with reduced TLR4 expression with microbial exposure (64). Histone acetylation is associated with permissive and actively transcribed chromatin. Our data using intestinal organoids revealed that the microbiota mediate durable changes in IECs that remain even after microbial stimulation has been removed. In the presence of the microbiota, H3K9Ac was reduced in the Clec2e gene which directly corresponds with reduced expression of Clec2e in IECs from CNV mice, indicating that the microbiota epigenetically regulate Clec2e expression. This study further demonstrates that H3K9Ac in Clec2e is regulated by HDAC3 as loss of HDAC3 expression leads to increased histone acetylation and loss of transcriptional repression of Clec2e. Taken together, our data demonstrate a novel mechanism by which the microbiota promote host defense through suppression of IEC-intrinsic pathways that are coopted for pathogen adherence and highlights that epigenetic regulation of innate pathways in IECs may represent a potent, long-lasting mechanism by which the microbiota prime host defense.
Data Availability
The datasets generated for this study can be found in Gene Expression Omnibus (GEO), GSE128362, GSE50453, GSE128369.
Ethics Statement
All experiments were performed according to the animal guidelines upon approval of the Institutional Animal Care and Use Committee at CCHMC.
Author Contributions
TA, VW, and EE designed the studies and analyzed the data. VW, EE, TR, and JW carried out experiments. BV provided bacterial strains. TA, VW, and EE wrote the manuscript.
Funding
This research is supported by the National Institutes of Health (DK093784, DK114123, DK116868 to TA), a Crohn's and Colitis Foundation/Janssen/AGA award to TA, and the Arnold W. Strauss Fellowship to EE. TA holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund and is a Pew Scholar in the Biomedical Sciences, supported by the Pew Charitable Trust. BV holds the CH.I.L.D. Foundation Chair in Pediatric Gastroenterology. This project is supported in part by PHS grant P30 DK078392 and the CCHMC Trustee Award and Procter Scholar's Program.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the members of the Alenghat lab for critical reading of the manuscript. We also thank CCHMC Veterinary Services, Research Flow Cytometry Core, Confocal Imaging Core and the University of Cincinnati Genomics, Epigenomics and Sequencing Core for services and technical assistance.
References
1. Dupont HL. Acute infectious diarrhea in immunocompetent adults. N Engl J Med. (2014) 370:1532–40. doi: 10.1056/NEJMra1301069
3. Cossart P, Sansonetti PJ. Bacterial invasion : the paradigms of enteroinvasive pathogens. Science. (2004) 304:242–9. doi: 10.1126/science.1090124
4. Pizarro-Cerdá J, Cossart P. Bacterial adhesion and entry into host cells. Cell. (2006) 124:715–27. doi: 10.1016/j.cell.2006.02.012
5. Fitzhenry RJ, Reece S, Trabulsi LR, Heuschkel R, Murch S, Thomson M, et al. Tissue tropism of enteropathogenic Escherichia coli strains belonging to the O55 serogroup. Infect Immun. (2002) 70:4362–8. doi: 10.1128/IAI.70.8.4362
6. Flores J, Okhuysen PC. Genetics of susceptibility to infection with enteric pathogens. Curr Opin Infect Dis. (2009) 22:471–6. doi: 10.1097/QCO.0b013e3283304eb6.Genetics
7. Baumler A, Fang FC. Host specificity of bacterial pathogens. Cold Spring Harb Perscpectives Med. (2013) 3:a010041. doi: 10.1101/cshperspect.a010041
8. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. (2014) 157:121–41. doi: 10.1016/j.cell.2014.03.011
9. Libertucci J, Young VB. The role of the microbiota in infectious diseases. Nat Microbiol. (2019) 4:35–45. doi: 10.1038/s41564-018-0278-4
10. Kamada N, Kim YG, Sham HP, Vallance BA, Puente JL, Martens EC, et al. Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science. (2012) 336:1325–30. doi: 10.1126/science.1222195
11. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
12. Ganal SC, Sanos SL, Kallfass C, Oberle K, Johner C, Kirschning C, et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity. (2012) 37:171–86. doi: 10.1016/j.immuni.2012.05.020
13. Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. (2014) 14:141–53. doi: 10.1038/nri3608
14. Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. (2012) 12:503–16. doi: 10.1038/nri3228
15. Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, et al. Epithelial-intrinsic IKKα expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. (2015) 212:1513–28. doi: 10.1084/jem.20141831
16. Ramanan D, Cadwell K. Intrinsic defense mechanisms of the intestinal epithelium. Cell Host Microbe. (2016) 19:434–41. doi: 10.1016/j.chom.2016.03.003
17. Woo V, Alenghat T. Host – microbiota interactions : epigenomic regulation. Curr Opin Immunol. (2017) 44:52–60. doi: 10.1016/j.coi.2016.12.001
18. Alenghat T. Epigenomics and the microbiota. Toxicol Pathol. (2015) 43:101–6. doi: 10.1177/0192623314553805
19. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. (1997) 389:349–52. doi: 10.1038/38664
20. Renz H, von Mutius E, Brandtzaeg P, Cookson WO, Autenrieth IB, Haller D. Gene-environment interactions in chronic inflammatory disease. Nat Immunol. (2011) 12:273–7. doi: 10.1038/ni0411-273
21. Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–3. doi: 10.1101/gad.1787609.Copyright
22. Happel N, Doenecke D. Histone H1 and its isoforms: contribution to chromatin structure and function. Gene. (2009) 431:1–12. doi: 10.1016/j.gene.2008.11.003
23. Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev. (2011) 25:2480–8. doi: 10.1101/gad.175950.111
24. Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem. (2002) 277:33275–83. doi: 10.1074/jbc.M204935200
25. Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. (2013) 504:153–7. doi: 10.1038/nature12687
26. Bergstrom KSB, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. (2010) 6:e1000902. doi: 10.1371/journal.ppat.1000902
27. Navabi N, Whitt J, Wu S, Woo V, Moncivaiz J, Jordan MB, et al. Epithelial histone deacetylase 3 instructs intestinal immunity by coordinating local lymphocyte activation. Cell Rep. (2017) 19:1165–75. doi: 10.1016/j.celrep.2017.04.046
28. Whitt J, Woo V, Lee P, Moncivaiz J, Haberman Y, Denson L, et al. Disruption of epithelial HDAC3 in intestine prevents diet-induced obesity in mice. Gastroenterology. (2018) 155:501–13. doi: 10.1053/j.gastro.2018.04.017
29. Kelly D, Kotliar M, Woo V, Jagannathan S, Whitt J, Moncivaiz J, et al. Microbiota-sensitive epigenetic signature predicts inflammation in Crohn's disease. JCI Insight. (2018) 3:122104. doi: 10.1172/jci.insight.122104
30. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. (2009) 10:R25. doi: 10.1186/gb-2009-10-3-r25
31. Zhang Y, Liu T, Ca M, Eeckhoute J, Berstein B, Nusbaum C, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. (2008) 9:R137. doi: 10.1186/gb-2008-9-9-r137
32. Kartashov AV, Barski A. Biowardrobe: an integrated platform for analysis of epigenomics and transcriptomics data. Genome Biol. (2015) 2015:16. doi: 10.1186/s13059-015-0720-3
33. Waddell A, Vallance JE, Hummel A, Alenghat T, Rosen MJ. IL-33 induces murine intestinal goblet cell differentiation indirectly via innate lymphoid cell IL-13 secretion. J Immunol. (2018) 2018:ji1800292. doi: 10.4049/jimmunol.1800292
34. Letourneau J, Levesque C, Berthiaume F, Jacques M, Mourez M. In vitro assay of bacterial adhesion onto mammalian epithelial cells. J Vis Exp. (2011) 1:3–6. doi: 10.3791/2783
35. Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. (2006) 1126–30. doi: 10.1126/science.1127119.Symbiotic
36. Mukherjee S, Zheng H, Derebe MG, Callenberg KM, Partch C, Rollins D, et al. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature. (2014) 505:103–7. doi: 10.1038/nature12729
37. Rutkowski E, Leibelt S, Born C, Friede ME, Bauer S, Weil S, et al. Clr-a: a novel immune-related C-type lectin-like molecule exclusively expressed by mouse gut epithelium. J Immunol. (2017) 198:916–26. doi: 10.4049/jimmunol.1600666
38. Miki T, Holst O, Hardt WD. The bactericidal activity of the C-type lectin RegIIIβ against gram-negative bacteria involves binding to lipid a.pdf. J Biol Chem. (2012) 287:34844–55. doi: 10.1074/jbc.M112.399998
39. Zhang Q, Rahim MMA, Allan DSJ, Tu MM, Belanger S, Abou-Samra E, et al. Mouse Nkrp1-Clr gene cluster sequence and expression analyses reveal conservation of tissue-specific MHC-independent immunosurveillance. PLoS ONE. (2012) 7:e50561. doi: 10.1371/journal.pone.0050561
40. Osorio F, Reis e Sousa C. Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity. (2011) 34:651–64. doi: 10.1016/j.immuni.2011.05.001
41. Hoving JC, Wilson GJ, Brown GD, Town C, Africa S. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol. (2014) 16:185–94. doi: 10.1111/cmi.12249
42. Obata Y, Furusawa Y, Hase K. Epigenetic modifications of the immune system in health and disease. Immunol Cell Biol. (2015) 93:226–32. doi: 10.1038/icb.2014.114
43. Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. (2016) 535:65–74. doi: 10.1038/nature18847
44. Wilson EM, Rotwein P. Control of MyoD function during initiation of muscle differentiation by an autocrine signaling pathway activated by insulin-like growth factor-II. J Biol Chem. (2006) 281:29962–71. doi: 10.1074/jbc.M605445200
45. Turgeon N, Blais M, Gagné JM, Tardif V, Boudreau F, Perreault N, et al. HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE. (2013) 8:e73785. doi: 10.1371/journal.pone.0073785
46. Pott J, Hornef M. Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep. (2012) 13:684–98. doi: 10.1038/embor.2012.96
47. Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity. (2012) 37:158–70. doi: 10.1016/j.immuni.2012.04.011
48. Abt MC, Pamer EG. Commensal bacteria mediated defenses against pathogens. Curr Opin Immunol. (2014) 29:16–22. doi: 10.1016/j.coi.2014.03.003
49. Mukherjee S, Hooper L V. Antimicrobial defense of the intestine. Immunity. (2015) 42:28–39. doi: 10.1016/j.immuni.2014.12.028
50. Johansson MEV, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. (2015) 18:582–92. doi: 10.1016/j.chom.2015.10.007
51. Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun. (2002) 70:953–63. doi: 10.1128/IAI.70.2.953-963.2002
52. Natividad JMM, Hayes CL, Motta JP, Jury J, Galipeau HJ, Philip V, et al. Differential induction of antimicrobial REGIII by the intestinal microbiota and Bifidobacterium breve NCC2950. Appl Environ Microbiol. (2013) 79:7745–54. doi: 10.1128/AEM.02470-13
53. Florin THJ, Mcguckin MA, Linde SK. Mucin dynamics in intestinal bacterial infection. PLoS ONE. (2008) 3:e3952. doi: 10.1371/journal.pone.0003952
54. Iimura M, Gallo RL, Hase K, Miyamoto Y, Eckmann L, Kagnoff MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J Immunol. (2005) 174:4901–7. doi: 10.4049/jimmunol.174.8.4901
55. Mundy R, MacDonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. (2005) 7:1697–706. doi: 10.1111/j.1462-5822.2005.00625.x
56. Chessa D, Winter MG, Jakomin M, Bäumler AJ. Salmonella enterica serotype Typhimurium Std fimbriae bind terminal α(1,2)fucose residues in the cecal mucosa. Mol Microbiol. (2009) 71:864–75. doi: 10.1111/j.1365-2958.2008.06566.x
57. Leibelt S, Friede ME, Rohe C, Gütle D, Rutkowski E, Weigert A, et al. Dedicated immunosensing of the mouse intestinal epithelium facilitated by a pair of genetically coupled lectin-like receptors. Mucosal Immunol. (2015) 8:232–42. doi: 10.1038/mi.2014.60
58. Yu D-H, Gadkari M, Zhou Q, Yu S, Gao N, Guan Y, et al. Postnatal epigenetic regulation of intestinal stem cells requires DNA methylation and is guided by the microbiome. Genome Biol. (2015) 16:211. doi: 10.1186/s13059-015-0763-5
59. Burgess SL, Saleh M, Cowardin CA, Buonomo E, Noor Z, Watanabe K, et al. Role of serum amyloid A, GM-CSF and bone marrow granulocyte-monocyte precursor expansion in segmented filamentous bacteria-mediated protection from Entamoeba histolytica. Infect Immun. (2016) 84:IAI.00316–16. doi: 10.1128/IAI.00316-16
60. Schulthess J, Pandey S, Capitani M, Rue-Albrecht K, Arnold I, Franchini F, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. (2019) 50:432–45. doi: 10.1016/J.IMMUNI.2018.12.018
61. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
62. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
63. Park J, Kim M, Kang SG, Jannasch AH, Cooper B, Patterson J, et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. (2015) 8:80–93. doi: 10.1038/mi.2014.44
Keywords: microbiota, HDAC, CLEC, intestine epithelial cells, citrobacter, epigenetic
Citation: Woo V, Eshleman EM, Rice T, Whitt J, Vallance BA and Alenghat T (2019) Microbiota Inhibit Epithelial Pathogen Adherence by Epigenetically Regulating C-Type Lectin Expression. Front. Immunol. 10:928. doi: 10.3389/fimmu.2019.00928
Received: 02 February 2019; Accepted: 11 April 2019;
Published: 07 May 2019.
Edited by:
Sudhanshu Shekhar, University of Oslo, NorwayReviewed by:
Elena Verdu, McMaster University, CanadaKyoko Takahashi, Nihon University, Japan
Masato Tsuda, Nihon University, Japan
Copyright © 2019 Woo, Eshleman, Rice, Whitt, Vallance and Alenghat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Theresa Alenghat, dGhlcmVzYS5hbGVuZ2hhdEBjY2htYy5vcmc=
†These authors have contributed equally to this work