 Lilly Stahl
Lilly Stahl Anna Duenkel1†
Anna Duenkel1†- 1Immune Tolerance Unit, Fraunhofer Institute of Cell Therapy and Immunology, Leipzig, Germany
- 2Max-Bürger Research Center, Institute for Clinical Immunology, University of Leipzig Medical Center, Leipzig, Germany
T cell modulation in the clinical background of autoimmune diseases or allogeneic cell and organ transplantations with concurrent preservation of their natural immunological functions (e.g., pathogen defense) is the major obstacle in immunology. An anti-human CD4 antibody (MAX.16H5) was applied intravenously in clinical trials for the treatment of autoimmune diseases (e.g., rheumatoid arthritis) and acute late-onset rejection after transplantation of a renal allograft. The response rates were remarkable and no critical allergic problems or side effects were obtained. During the treatment of autoimmune diseases with the murine MAX.16H5 IgG1 antibody its effector mechanisms with effects on lymphocytes, cytokines, laboratory and clinical parameters, adverse effects as well as pharmacodynamics and kinetics were studied in detail. However, as the possibility of developing immune reactions against the murine IgG1 Fc-part remains, the murine antibody was chimerized, inheriting CD4-directed variable domains of the MAX.16H5 IgG1 connected to a human IgG4 backbone. Both antibodies were studied in vitro and in specific humanized mouse transplantation models in vivo with a new scope. By ex vivo incubation of an allogeneic immune cell transplant with MAX.16H5 a new therapy strategy has emerged for the first time enabling both the preservation of the graft-vs.-leukemia (GVL) effect and the permanent suppression of the acute graft-vs.-host disease (aGVHD) without conventional immunosuppression. In this review, we especially focus on experimental data and clinical trials obtained from the treatment of autoimmune diseases with the murine MAX.16H5 IgG1 antibody. Insights gained from these trials have paved the way to better understand the effects with the chimerized MAX.16H5 IgG4 as novel therapeutic approach in the context of GVHD prevention.
Introduction
Besides the T cell receptor (TCR) and the CD3 antigen, other molecules are expressed on T cells but are also present on other hematopoietic cells (1). Monoclonal antibodies targeting such antigens (other than the TCR or CD3) can therefore bind to several antigen-expressing cell types. The CD4 molecule is expressed on T cells, monocytes and macrophages and contains four immunoglobulin-like domains (D1–D4) (2). It acts as a co-receptor during antigen presentation and associates with the TCR upon major histocompatibility complex (MHC) binding (1, 2). In the past, several therapeutic strategies using CD4-directed antibodies were investigated for the treatment of several autoimmune diseases [reviewed in Wofsy (3) and Burmester et al. (4)]. In this context, the human CD4+ T-cell clone 2C11 was generated (5) for immunization of BALB/c mice to produce the monoclonal anti-human CD4 antibody MAX.16H5 (initial name 30F16H5) (6, 7). Therefore, splenocytes of the immunized mice were fused with X63-Ag8.653 mouse myeloma cells to generate hybridoma cells (6–8). To examine their binding properties, antibody-containing hybridoma supernatants were incubated with CD4+ T cells prepared from peripheral blood (PB) which were subsequently analyzed using enzyme-linked immunosorbent assays (ELISA) and cytofluorometric analyses (6). For the development as a therapeutic antibody, MAX.16H5 IgG1 was selected because of its high affinity to CD4 (9). In this study, researchers compared 225 different CD4-directed antibodies regarding their CD4 binding properties and kinetics showing that MAX.16H5 IgG1 shared some fine specificities with gp120 with regard to the recognition of different mutated CD4 versions (9). At the same time, experiments were performed to obtain information about the binding properties of gp120 and MAX.16H5 IgG1 to the CD4 molecule by using peptides (10). The peptide TbYICbEbVEDQKAcEE was reported to inhibit CD4 binding of both gp120 and MAX.16H5 IgG1 (10). During the early years, the antibody was tested in several different assays thereby obtaining solid information not only about antigen-binding properties, but also about antibody-mediated effector mechanisms. Throughout the clinical development of the murine MAX16.H5 IgG1, pharmacodynamic and pharmacokinetic data were collected. Upon administration in patients, the mode-of-action of the antibody, the induced CD4 and immunomodulation were studied intensively. Since MAX.16H5 IgG1 was applied systemically, clinical data implementing cytokine profiles, acute-phase-reactant evaluation and side effects were obtained and documented. This review summarizes the clinical development of the therapeutic use of MAX.16H5 IgG1 for the treatment of autoimmune diseases toward a promising treatment option for hematopoietic stem cell transplantation (HSCT)-related GVHD.
MAX.16H5 IgG1 in the Treatment of Autoimmune Diseases
In total, 47 patients have been treated with the murine wild type antibody MAX.16H5 IgG1 (7, 11–24). The individuals suffered from varying diseases or conditions: rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), inflammatory bowel disease (IBD), or acute late-onset rejection after transplantation of a renal allograft (Table 1). Thereby, RA was the most studied disease. Besides the studies focusing on the applicability of MAX.16H5 IgG1 as a promising therapeutic antibody format, several studies were published about the use of 99mTc-labeled MAX.16H5 IgG1 in RA patients to report the localization/accumulation, the pharmacokinetics, and the elimination process of the antibody (7, 11, 23, 24). A broad variety of parameters was obtained during these clinical trials to evaluate the safety and efficacy of the systemic therapeutic administration of MAX.16H5 IgG1. Table 1 provides an overview of the studies performed in humans.
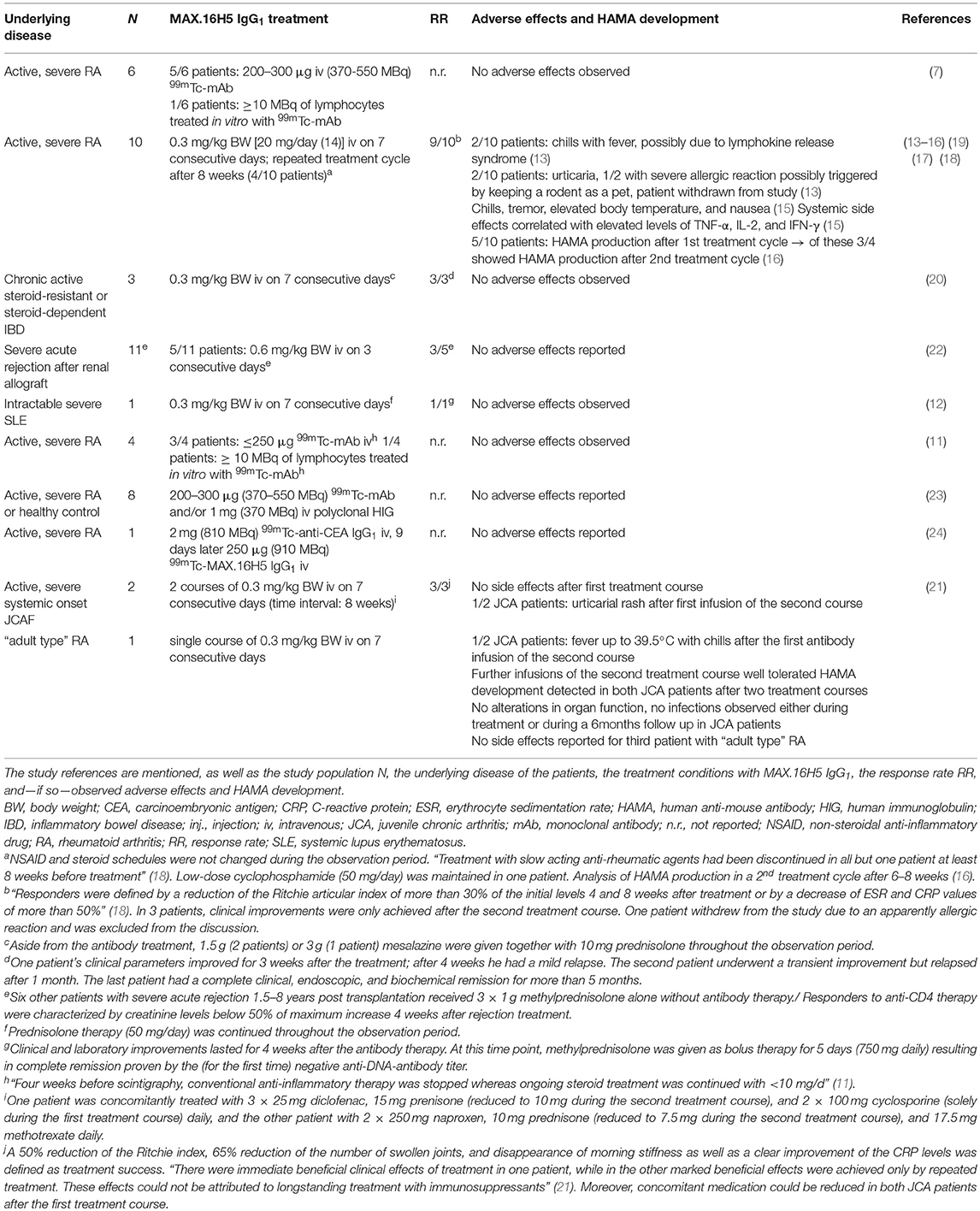
Table 1. Results of human studies using MAX.16H5 IgG1.
Since it is known that murine antibodies can cause immunological reactions in humans, the administration of the murine MAX.16H5 IgG1 was particularly examined. Immunological reactions against the Fc-part of the murine antibody were expected, and in three different studies that administered MAX.16H5 IgG1 in the background of RA the production of human anti-mouse antibodies (HAMAs) was investigated (13, 16, 21). Based on the obtained datasets authors concluded that HAMA production followed by MAX.16H5 IgG1 administration was rather low and that HAMA activities are directed against specific determinants of the antibody, including anti-idiotypic reactivity (16). Furthermore, it was shown that MAX.16H5 IgG1 F(ab)2 directed HAMA (IgG) levels did not exceed levels higher than 0.7 mg/l after the first and 1.7 mg/l after the second course of therapy (13). Compared to HAMA activities exceeding 100 mg/l measured in other studies using monoclonal murine antibodies against cancer antigens (25, 26) in immunocompetent patients, the HAMA amounts directed against MAX.16H5 IgG1 were rather low, but detectable. Overall, even in studies using the murine IgG1 isotype of MAX.16H5, only low HAMA amounts were detected which allowed for further treatment cycles without loss of efficacy (16).
MAX.16H5 IgG1 Mediated Effector Mechanisms
Antibodies can mediate effector mechanisms by both binding the antigen via the Fab domain and binding Fc receptors (FcRs) expressed on effector cells through the Fc part. Mouse IgG1 is known to bind two different murine Fc receptors, mFcγRIIb and mFcγRIII (27). The mFcγRIIb is an immunoreceptor tyrosine-based inhibitory motif (ITIM)-carrying receptor, which is highly expressed on murine B cells, granulocytes, macrophages, monocytes and dendritic cells (27). In contrast, mFcγRIII is absent on B cells but highly expressed on monocytes, macrophages, dendritic cells, and granulocytes thereby mediating activating signals via an immunoreceptor tyrosine-based activation motif (ITAM) [reviewed in Bruhns (27)]. Mice produce three different IgG subclasses which do not only differ in their FcR binding specificity but also bear diverse capacity to activate the complement system (27–30). Based on serum bactericidal activity measurements the following hierarchy of murine IgG induced complement activity was proposed: IgG3 > IgG2 > IgG1 (30). It has to be noted that in these assays human serum was used as a source of complement (30).
To this date, no MAX.16H5 IgG1 mediated complement-dependent cytotoxicity (CDC) (using rabbit serum as a source of complement) or antibody-dependent cell-mediated cytotoxicity (ADCC) using granulocytes or peripheral blood monocytes as effector cells was obtained in in vitro assays (13, 31).
Effects on Lymphocytes
In general, the MAX.16H5 IgG1 treatment results in a decrease of CD4+ cells in RA patients and therefore to an overall reduction of CD3+ cells (13, 14). Neither CD8+ nor B-cell values were changed in RA patients (13, 14). Immune cells from RA patients treated with MAX.16H5 IgG1 showed reduced proliferation to various stimulatory agents 1 h post injection (p.i.) (13). However, in four out of nine patients, increased mitogen responses were induced after 8 days, which was indicative for unaltered clinical effects in these patients (13). Blood samples from IBD patients treated with MAX.16H5 IgG1 showed a reduced lymphocyte proliferation after stimulation with mitogens and recall antigens (20), too.
Immortalized and interleukin (IL)-2-dependent CD4+ T cells revealed reduced mitotic activity (not increased apoptosis) after incubation with MAX.16H5 IgG1 or its F(ab')2 (32). The same effect was observed with the Fab of MAX.16H5 and gp120 of HIV which could be prevented by high concentrations of IL-2 (32). The authors showed that this effect was connected to a decreased amount of lymphocyte-specific protein tyrosine kinase (Lck) bound to the intracellular domain of CD4 (32).
For further investigation of intracellular signaling pathways, the calcium release after TCR stimulation was examined in peripheral blood mononuclear cells (PBMCs) of RA patients or healthy donors treated with MAX.16H5 IgG1 (33, 34). In one study, samples from healthy donors showed reduced intracellular calcium levels after MAX.16H5 IgG1 incubation and TCR stimulation in vitro, but only if MAX.16H5 IgG1 was still bound to the CD4 molecule (33). In a second study, the intracellular calcium concentration did not increase after solely incubation with MAX.16H5 IgG1 (34). Only after cross-linking of CD3 and CD4 by anti-mouse goat serum an increased intracellular calcium signaling was obtained if MAX.16H5 incubation was performed for a maximum of 5 min (34). For longer incubation periods, the calcium signal decreased again indicating that full T cell activation by CD3 occurs rapidly within a short time (34). These data indicate a strong dependence of preincubation time. Furthermore, the increased calcium release only after cross-linking of CD3 and CD4 leads to the following speculation: the T cell activation was impaired due to transient but asynchronous activity of different kinases in T cells and intercellular cross-talk between T cells and monocytes was required (34).
At the time the studies were conducted, regulatory T cells (Tregs) were not yet identified as important targets to follow in clinical GVHD research: By 2000, Tregs were identified as suppressors of autoimmunity in vitro and in mouse models [reviewed in Shevach (35) and Sakaguchi (36)]. However, the flow cytometric identification of Tregs remained difficult until they were specified as positive for CD4, CD25, and forkhead box protein P3 (FoxP3) in 2003 (37–39). Therefore, the clinical data on MAX.16H5 IgG1 lack information on the Treg population.
Effects on B-Cell Crosstalk With T-Helper Cells and Immunoglobulin Secretion
PBMCs of healthy individuals were assessed for effects of MAX.16H5 IgG1 incubation on B-cell differentiation and resulting IgG and IgM production (40). It was found that incubation with MAX.16H5 IgG1 inhibited B-cell differentiation and following immunoglobulin (Ig) production (40). Even in the presence of mitogens and IL-2 or IL-4, MAX.16H5 IgG1 addition reduced Ig secretion (40). Moreover, the production of IL-2 and IL-4 by T-helper (Th) cells was minimally influenced by MAX.16H5 IgG1 under various stimulating conditions (40). Thus, cytokines were not responsible for lower Ig secretion after MAX.16H5 incubation. More likely, the reduction of direct cellular contacts between Th and B cells by MAX.16H5 IgG1 and its F(ab')2 lead to reduced crosstalk between the two cell types causing reduced Ig secretion indicating that CD4-blockade by MAX.16H5 interferes with early T-B cell collaboration (40). In RA patients, Ig reduction was observed after MAX.16H5 IgG1 treatment, especially rheumatoid factor (RF) production, indicating that this effect is also present in vivo (13).
Effects on Monocytes
In the treatment of RA with MAX.16H5 IgG1, CD14+ monocytes in the PB were reduced one hour after infusion of MAX.16H5 IgG1 (13). Continuing the MAX.16H5 IgG1 treatment kept monocyte levels in normal ranges (13). The authors offer two possible explanations: either the monocyte/macrophage system is responsible for the depletion of antibody-coated T cells or the MAX.16H5 IgG1 bound to the CD4 molecule present on a monocyte subset results in temporary monocyte reduction in the PB (13). On the other hand, reduced crosstalk between Th cells and monocytes may play a role in the observed reduction of monocyte activation (13). In different studies, the same RA patients were monitored for monocyte activation indicated by heightened neopterin serum values, MHC class II expression, monocyte counts, and IL-1 production prior to MAX.16H5 IgG1 application (14). These parameters could be reduced after MAX.16H5 IgG1 treatment (14). Moreover, elevated levels of soluble CD14 (sCD14) detected in five patients prior to MAX.16H5 IgG1 treatment were reduced in three patients after antibody application (18). IL-1 and IL-6 serum levels correlated to sCD14 concentrations in RA patients (18). A comparison between therapy responders and non-responders revealed reduced monocyte and Th cell counts in the responder group, whereas both values increased again in the non-responder group after 1 week (18).
Cytokine Release
In chronic inflammatory diseases such as RA and SLE, the release of pro-inflammatory cytokines plays a crucial role in disease progression. Elevated levels of cytokines produced by CD4+ cells including tumor necrosis factor (TNF)-α, IL-1, IL-6, and IL-17 favor disease pathogenesis [reviewed in Lourenço and La Cava (41) and McInnes and Schett (42)]. Monoclonal antibodies (mAbs) against these molecules or their respective receptors are therapeutic options to treat patients with autoimmune diseases (43). Therefore, the effect of the treatment with MAX.16H5 IgG1 targeting human CD4+ cells regarding cytokine release was studied in detail in vivo and in vitro.
IL-6 is known as the most important inducer and regulator of acute-phase response (44). Elevated IL-6 levels were measured in most RA patients before MAX.16H5 treatment (18). The IL-6 levels rapidly declined in four patients during the treatment course, which was observed in parallel with substantial clinical and laboratory improvement (18). On the other hand, one patient showed a slight increase of IL-6 during first treatment course and did not respond to treatment (18). One individual demonstrated a considerable increase of IL-6 and underwent an allergic skin reaction after the first injection (18). In that special patient, the IL-6 levels decreased to the pretreatment values after the treatment was stopped (18).
Besides the positive effect of IL-6 reduction, cytokine release due to MAX.16H5 IgG1 application was analyzed as potential side effect. During the therapy of RA patients with MAX.16H5 IgG1, symptomatic patients showed elevated serum levels of TNF-α, IFN-γ, and/or IL-2 (15). Comparison of the modulation efficacy of CD4+ T cells induced by MAX.16H5 treatment did not reveal any difference between patients without clinical adverse effects and those developing systemic side effects (15). The authors hypothesized that the clinical adverse effects were likely a result of lymphocyte activation and/or a monocyte/macrophage interaction with lymphocytes (15). A comparable side effect profile was described for mAb OKT3 treatment (45, 46) but MAX.16H5 IgG1 induced effects were milder and of much shorter duration which made a further treatment of the patient unnecessary (15). It has to be noted, that only a small cohort of patients systemically received MAX.16H5 IgG1 therapy for autoimmune disease treatment, which complicates drawing solid conclusions.
The cytokine production in SLE patients was analyzed in vitro as well (47). Spontaneous IL-6 secretion was heightened in blood cell cultures from patients with active SLE compared to cultures from inactive SLE patients and healthy controls (47). After incubation with MAX.16H5 IgG1, cell cultures of active SLE patients demonstrated reduced IL-6 levels, whereas TNF-α levels were not significantly altered (47). When samples from healthy volunteers were stimulated either with phytohemagglutinin (PHA) or lipopolysaccharide (LPS) MAX.16H5 IgG1 induced IL-6 decrease was found to be antibody dose-dependent (47). In control wells it was shown that the addition of methylprednisolone to the cell cultures of stimulated healthy volunteer samples, stimulated inactive SLE samples and unstimulated active SLE samples not only reduced IL-6 but also TNF-α secretion markedly (47). Summarized it was shown that MAX.16H5 antibody incubation altered stimulated IL-6 secretion of in vitro blood cell cultures obtained from SLE patients and healthy individuals. The decrease of stimulated IL-6 secretion was dose-dependent. Other than methylprednisolone MAX.16H5 IgG1 incubation did not influence TNF-α levels in these assays (47).
Laboratory and Clinical Parameters
In contrast to other anti-CD4 antibodies, MAX.16H5 was the only one improving not only clinical but also laboratory parameters in RA patients [reviewed in Burmester and Emmrich (48)]. Additionally, MAX.16H5 application showed an effect on parameters which were associated with monocyte/macrophage activation (14). In general, a significant decrease of laboratory [erythrocyte sedimentation rate (ESR), RF titer, C-reactive protein (CRP) levels] and clinical parameters (Ritchie articular index and swollen joints) was observed (13). In one of the patients, no impact on ESR and CRP levels was observed during the first cycle of MAX.16H5 infusion (17). Four years before the treatment with MAX.16H5 IgG1, the patient was diagnosed with RA. Due to a trauma, he underwent splenectomy earlier in life (17). After the second course of MAX.16H5 therapy, ESR and CRP levels were reduced, possibly followed after decreased IL-6 serum values (17). Since the change in laboratory variables did not translate into an improvement of clinical parameters, low dose chlorambucil was implemented into the treatment regimen. The combination of CD4 directed antibody therapy together with chemotherapeutic medication resulted in clinical improvements which also translated in continued reduced levels of certain inflammatory parameters (ESR and CRP) (17). Overall, no adverse effects (especially infections) were observed (17).
The clinical parameters of two children were assessed in another study where MAX.16H5 was given i.v. for treatment of refractory juvenile chronic arthritis (JCA) (21). One patient benefited from the antibody therapy clinically within 1 week after the first antibody application. A second antibody application showed an even more improved response compared to the first course of treatment and symptoms like fever and rash were reduced for around 2 months (21). In the second patient, two cycles of treatment were needed to obtain notable improvement of clinical symptoms. Also, these juvenile patients did not show any signs of adverse side effects caused by the MAX.16H5 IgG1 antibody treatment (21).
Patients suffering from severe acute rejection after kidney transplantation also benefitted from the therapy with MAX.16H5 IgG1 (22). Histological signs of acute rejection (if present) disappeared as a response to the MAX.16H5 IgG1 treatment. All patients showed rapid decreasing serum creatinine levels within the first 3 days post injection. However, graft function was impaired in two patients 3–4 weeks after therapy and one patient experienced transplant rejection again after 10 weeks (22). The authors observed a rapid effect of the MAX.16H5 antibody in the treatment of acute rejection after kidney transplantation and concluded that CD4+ T cells seem to play an important role in the rejection process. They further suggested to implement the antibody therapy in established immunosuppression treatment protocols to improve therapeutic efficacy (22).
MAX.16H5 IgG1 application was also shown to be effective in the treatment of inflammatory bowel disease (IBD) (20). Especially, when other treatment options are exhausted and conventional therapeutics are ineffective MAX.16H5 IgG1 can be used as a treatment option in IBD (20). As also discussed in the treatment of autoimmune diseases earlier, single cycle administration of MAX.16H5 was insufficient to reach persistent therapeutic success (20).
Other Effects
Since the exact mechanism of the MAX.16H5 IgG1 induced effects were not sufficiently explained neither in vitro nor in vivo, researchers focused on the intracellular signaling after antibody binding to its antigen. By using U937 target cells, the activation of complex inositol polyphosphate responses and Ca2+ increase after MAX.16H5 IgG1 antibody treatment was investigated in vitro independently from TCR signaling (49). The authors showed, that MAX.16H5 IgG1 incubation alone was not sufficient to induce Ca2+ increase in CD4-expressing cells (PB-monocytes and the monocyte cell line U937) (49). When goat anti-mouse antiserum was added, clear crosslinking of MAX.16H5 IgG1 was obtained leading to heightened Ca2+ levels (49). The outcome of experiments in U937 cells using F(ab)2 fragments of MAX.16H5 together with F(ab)2 crosslinking agents were not applicable to observations made with whole antibodies (49). The authors concluded that in U937 cells only “[…] crosslinking of CD4 and FcγR, but not cross-linking of CD4 alone specifically activates the inositol polyphosphate/Ca2+ signal transduction pathway” (49).
Pharmacokinetics and Pharmacodynamics in Humans
Pharmacokinetic and pharmacodynamic studies with 99mTc-labeled MAX.16H5 IgG1 were performed in RA patients (7, 11, 23, 24). In one study, patients received either i.v. injection of 99mTc-labeled MAX.16H5 IgG1 antibody or ex vivo 99mTc-labeled MAX.16H5 IgG1 incubated PB-lymphocytes (7). Following images/scans were taken using a gamma camera (7). The use of the CD4-directed MAX.16H5 IgG1 antibody for medical imaging in order to obtain information about disease progression in RA or also for diagnosis was promising since techniques used at that time, e.g., 99mTc-early methylene diphosphonate (MDP) bone scans, were rather unsatisfactory (7). The authors concluded that 99mTc-labeled MAX.16H5 imaged the localization of disease joints more precisely than MDP scans, which made it a promising tool for the diagnosis of autoimmune arthritis (7).
Organ Activity Distribution and Kinetics
In context with their participation in a therapeutic trial for RA [preliminary data after 2 patients enrolled evaluated in (50)], 4 patients received either radio-labeled MAX.16.H5 IgG1 antibody i.v. or PB-lymphocytes labeled with the antibody [1 patient] (11). The “[…] study was mainly concerned with the evaluation of the kinetic behavior of the antibody-labeled cells in the patients” (11). In general, the maximum of activity [100%] of the antibody or the antibody-labeled lymphocytes was reached within a few minutes in the heart and lung (ca. 4.5 min), whereas the maximum of radio-labeled activity was obtained after ca. 12 min in the spleen and about 19 min in the liver (11). However, 90 min after the injection, radiolabeled MAX.16H5 antibody activity reincreased in the patients' hip joints after a first injection peak (11). Interestingly, organ kinetic curves were comparable between patients receiving 99mTc-labeled MAX.16H5 IgG1 and the patient who received ex vivo antibody-labeled lymphocytes (11). Additionally, study examined the whole-body radioactivity distribution at two different time points [4 and 24 h p.i.] (11). “The splenic uptake decreased by about 39% from 4 h […] to 24 h p.i. […]” (11). A moderate increase of activity measured in the liver was recorded (11). Nevertheless, at both time points, approximately 50% of radioactivity was measured in the bone marrow. Joints overall showed a rather low activity with 0.5 ± 0.09% for not-diseased joints vs. 2 (after 4 h) to 2.5% (after 24 h) for a single affected joint (11).
Another study (23) evaluated the kinetic differences between labeled MAX.16H5 IgG1 and polyclonal human immunoglobulin (HIG) in RA patients or healthy controls to exclude non-specific accumulation of immunoglobulin. Compared to HIG, 99mTc-labeled MAX.16H5 IgG1 showed a higher uptake in the liver and in the spleen of RA patients at 24 h p.i. (23). Since the MAX.16H5 IgG1 “[…] showed a higher target-to-background ratio in arthritic knee and elbow joints in comparison to polyclonal HIG used for conventional imaging […]” the authors discussed a potential beneficial application of the antibody in the “[…] detection of inflammatory infiltrates rich in CD4-positive cells” (23).
Adverse Effects
The adverse effects observed in studies with human patients or healthy volunteers are summarized in Table 1. In different trials using the 99mTc-labeled MAX.16H5 IgG1, no adverse reaction was observed after the intravenous application (7, 11, 23, 24).
Several studies examined the treatment of RA patients with MAX.16H5 IgG1. In general, only occasional and minor side effects were observed which probably resulted from short lived cytokine peaks (15). The infusions were well-tolerated [reviewed in Burmester and Emmrich (48)]. Immediate adverse effects were allergic reactions on rare occasion as well as nausea and fever being symptomatic for the development of a mild to moderate cytokine release syndrome (15). A long-term effect on the topic of laboratory parameters was the development of HAMAs (13, 16, 21). Approximately 25% of the HAMA activity was directed against idiotypic determinants (16). Significant HAMA concentrations were measured between 2 and 12 weeks p.i. (16). Still, in contrast to monoclonal antibodies directed against other T cell epitopes, the amounts of these antibodies were low and never exceeded 2.0 (after 1 cycle) or 2.2 mg/l (after 2 cycles) (16). Thus, patients could be retreated without loss of efficacy (13) as similarly shown with other anti-human CD4 antibodies (51).
The reduced CD4+ T cell numbers did not result in infectious problems in any study (48). Together with an unaltered or even elevated T cell reactivity in vitro [4 patients showed heightened T cell reactivity to common antigens and mitogens when CD4+ cell numbers were still reduced (13)], this observation points to low numbers of CD4+ Th cells being sufficient to maintain the function of the cellular immune system (13, 48).
In the trial using MAX.16H5 IgG1 in the treatment of refractory JCA in two patients the first treatment course was tolerated without side effects (21). Application of MAX.16H5 IgG1 in the treatment of either SLE or IBD patients did not show any side or adverse effect (12, 20). In the therapy of acute rejection in long-term renal allograft recipients, no adverse effects due to the treatment were mentioned in the original article (22).
Although not all trials showed the development of HAMAs as side effect, the possibility of developing immune reactions against the murine IgG1 Fc-part remains. This risk was considered to be reduced by the development of a chimerized, humanized version of the MAX.16H5 IgG4 antibody.
Development of a Chimerized MAX.16H5 IgG4 Monoclonal Antibody
The use of murine antibody formats for therapeutic interventions was shown to be connected to the development of several side effects, e.g., antibody responses (HAMA) or allergic reactions, which led to the development of optimization protocols in antibody design using recombinant DNA tools already in the 1980's (52, 53). Morrison et al. described a process called “chimerization” where heavy and light chain variable DNA sequences of a murine antibody were connected to DNA sequences encoding human IgG1/2 and sequences encoding for the human kappa light chain, respectively (53). Morrison and colleagues discussed the potential of such “near-human” antibody formats with respect to reduced side effect profiles when administered in vivo (53). In 1997, the approval of rituximab, a chimeric CD20-directed IgG1 antibody for the treatment of lymphoma eventually paved the way for modified antibody formats (54). In December 2018, 75 antibodies as therapeutics were approved for the treatment of a variety of diseases including 9 antibodies carrying human Fc domains and murine variable sequence motifs and are therefore defined as chimeric (43). The aforementioned CD20-directed antibody rituximab is listed as well as e.g., obiltoxaximab, which reached US-approval more recently (2016) and is used for the treatment and prophylaxis of inhalational anthrax (55). The chimerization of MAX.16H5 was promoted in order to reduce immunogenicity of the antibody for potential clinical applications. The MAX.16H5 chimerization process was started in 2007. A CD4-directed murine IgG1 antibody-expressing hybridoma clone was used as starting material. By combining cloning and sequencing techniques together with in silico modeling, variable regions of light and heavy chains were extracted, analyzed, modified and connected to human constant regions as commissioned (Figure 1) (56). Mammalian cells were used for the production of the chimeric antibody (56). Binding profiles of MAX.16H5 IgG4 were comparable with the murine MAX.16H5 IgG1 proving that the correct variable regions were chosen to generate the chimeric antibody (56).
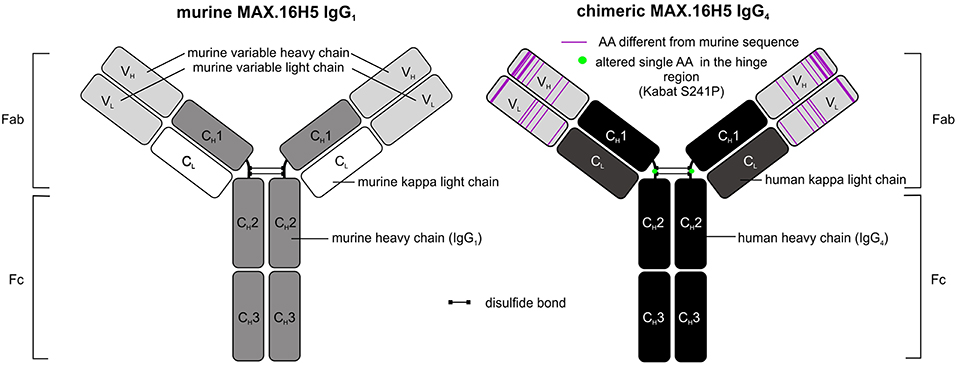
Figure 1. Schematic representation of murine MAX.16H5 IgG1 and chimeric MAX.16H5 IgG4 antibodies.
In general, Fc parts of antibodies are known to mediate effector mechanisms which include their interaction with both certain Fc receptors expressed on effector cells and the activation of the complement system (57). Depending on their Fc-binding capacities and mediated effector mechanisms, therapeutic antibodies can be used for different therapeutic purposes. In the setting of HSCT or GVHD, an Fc-mediated depletion of CD4+ cells was not our desired therapeutic approach. Therefore, MAX.16H5 variable domains were specifically connected to human IgG4 and not to human IgG1 constant domains since it is known that IgG4 is a weak activator of ADCC and CDC (58) [reviewed in Davies and Sutton (59)]. Because of the functional features of the IgG4 isotype, its translation into the clinical application in the field of immune checkpoint blockade was quite successful. To date, pembrolizumab (MK-3475) and nivolumab (MDX-1106), two antibodies harboring IgG4 backbones, are approved for immune checkpoint blockade in the USA and the EU (43). The molecules effectively inhibited programmed cell death protein 1/programmed cell death 1 ligand 1 (PD-1/PD-L1) interactions but were shown to be inactive eliciting Fc-mediated effector functions [reviewed in Topalian et al. (60)]. Another challenge in the development of IgG4-based therapeutic antibodies was the isotype's feature of the so-called Fab-arm exchange where single heavy chain-light chain dimers can form bivalent antibodies with other single heavy chain-light chain dimers (61). From a biological point of view it was discussed that the bivalency of IgG4 molecules may reduce their “pathological potential” (61). For certain applications, therapeutic antibody manufacturing can be challenging due to the Fab-arm exchange of IgG4. This led to the development of several antibody structure optimization strategies in the past (59, 61). A study published in 2009 showed that bispecific IgG4 antibodies were detectable in blood samples from patients who received an unmodified IgG4-based therapeutic antibody (62). The antibody formed half-molecules in vivo and furthermore, assembled to bispecific antibodies with patient-specific endogenous IgG4 (62). The group introduced a S228P amino acid substitution in the hinge-region of the antibody and showed the prevention of Fab-arm exchange impressively (62). The MAX.16H5 IgG4 sequence was optimized in the same manner in order to prevent Fab-arm exchange (Figure 1) (56), to ensure the stability of the antibody, and the reliability of the manufacturing process during clinical development and GMP production.
Non-clinical Development of the Chimerized Anti-human Anti-CD4 Antibody—Studies of MAX.16H5 IgG1 and IgG4 in Murine GVHD Models
It was sought to examine the properties of the antibody's influence on the immune system using mouse models. To study the effect of the MAX.16H5 antibody against human CD4 expressing cells in mice, a genetically modified murine model with a C56BL/6 background was developed, which functionally expressed human CD4 on T cells while the murine CD4 gene was knocked-out (63–65). Additionally to the murine MHC II the T cells expressed human HLA-DR17 (63–65). In first experiments, spleen and lymph node samples of triple transgenic (TTG) mice previously immunized with tetanus toxoid (TT) showed reduced immune response after MAX.16H5 IgG1 treatment and re-stimulation with TT ex vivo (66). The same effect emerged in vivo after injection with 15 μg/g body weight (BW) MAX.16H5 IgG1 (67) and the F(ab)2 of MAX.16H5 was as potent as the whole antibody (65). Surprisingly, unresponsiveness was preserved after the mice underwent another antigen boost without prior antibody administration, thus indicating a long-lasting but not depleting effect of MAX.16H5, which was moreover antigen-specific and dependent on the ability to form the immunological synapse between CD4 and HLA-DR (65). Therefore, we speculate, that MAX.16H5 does not induce a general immune suppression but only to antigens present simultaneously with or shortly after antibody treatment, most likely while the antibody is still bound to its ligand when the HLA-DR molecule encounters the TCR. This observation led to the idea that MAX.16H5 may conquer an old challenge in immunology: induction of specific tolerance and influencing both host-vs.-graft and graft-vs.-host reactions. The latter severely limit the application of allogeneic hematopoietic stem cell (HSC) and immune cell transplantations for the treatment of, e.g., autoimmune diseases or hematopoietic cancers such as leukemia.
Acute GVHD is the main complication of allogeneic hematopoietic stem cell transplantation (HSCT) and the main reason for early transplantation-associated mortality (68). Conventional immunosuppressive drugs such as corticosteroids are not specific and suppress the entire immune system (69). So far, no therapy manages HSCT or donor lymphocyte infusion without the need for conventional systemic immunosuppressive drugs. Promising in vivo data were obtained regarding the feasibility and effectivity of ex vivo graft incubation with MAX.16H5 IgG1 and the antibody's influence on GVHD down modulation after allogeneic full-mismatch immune stem cell transplantation if the graft from TTG mice was preincubated with the antibody (70–72). Of note, removing unbound antibody molecules from the graft did not reduce its effectiveness (70–72). The graft's unresponsiveness to allogeneic BALB/cwt antigens was even preserved if immune and stem cells from transplanted GVHD-free mice were transferred to new BALB/cwt mice without MAX.16H5 preincubation—a phenomenon called “infectious tolerance” (71). This phenomenon is possibly achieved by heightened levels of Tregs present in mice receiving MAX.16H5-preincubated grafts suggesting a possible role for long-term unresponsiveness in vivo (71). Two studies did not only focus on accelerated GVHD but also on the maintained GVL effect mediated by the transplanted antibody incubated immune cell graft. To investigate the GVL effect mediated by the transplanted graft, BALB/cwt animals did not only receive TTG immune cell grafts but also P815 cells, a murine mastocytoma cell line (70). It was shown that graft preincubation with the murine MAX.16H5 IgG1 antibody did not influence the GVL effect which was induced by the transplanted immune cells (70, 71). In another murine model, the murine MAX.16H5 IgG1 also prolonged the survival of recipient C3H/HeN mice (receiving a TTG immune cell graft to induce GVHD) even if they were co-transplanted with myeloblast-like murine cell line 32D Clone 3 (32D) expressing human Fms like tyrosine kinase 3 with the internal tandem repeat duplication (FLT3ITD), which constitutes an aggressive acute myeloid leukemia (AML) cell line model (72).
The ex vivo graft incubation with MAX.16H5 together with a subsequent washing protocol was found to be an attractive and promising therapeutic setting in HSCT (31, 70, 71). It got even more into focus when patients, who received a therapeutic antibody, suffered from side-effects which were mainly caused by a systemic inflammatory response as a response to systemic application (73). In 2006, unpredicted side-effects emerged during a phase-I (first-in-man) clinical trial with a CD28-directed monoclonal antibody called TGN1412 (73, 74). The antibody was developed to modulate Treg expansion and was praised as “a promising novel tool for the treatment of human autoimmune diseases” (75). As of today, the cytokine release syndrome is a known side-effect of different immunotherapeutic interventions such as therapeutic antibodies (e.g., CD20-directed mAbs, bispecific T cell engagers, and immune checkpoint inhibitors) or chimeric antigen receptor (CAR) T cells [reviewed in Shimabukuro-Vornhagen et al. (76)]. Due to safety considerations, the development and optimization of the MAX.16H5 antibody format for the treatment of GVHD was shifted to ex vivo graft incubation rather than systemic administration of the therapeutic antibody.
After the chimerized MAX.16H5 IgG4 was available, its effectiveness in preventing GVHD was evaluated using a NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse model (31). Here, mice received a xenogeneic human PBMC transplant to initiate acute GVHD (31). Both variants, the murine IgG1 and the chimeric IgG4 of the MAX.16H5 antibody, were compared regarding their ability to down regulate GVHD development. Unspecific isotype antibodies were used as controls in these experiments (31). The data were published in 2016 and showed that both the murine IgG1 and the chimerized IgG4 were able to prevent GVHD in this experimental setting (31). The mice received immune cell grafts which were ex vivo incubated with the respective antibodies making a systemic application of MAX.16H5 unnecessary. After 2 h of MAX.16H5 IgG1 or -IgG4 preincubation, the grafts were washed with PBS to remove excess, unbound antibodies. Finally 2 × 107 MAX.16H5 or isotype control treated graft cells were diluted in 150 μl 0.9% NaCl and were injected into the tail vein of the animals (31). Mice receiving MAX.16H5 murine IgG1 or chimeric IgG4 antibody incubated grafts showed a significant prolonged survival in comparison to mice receiving grafts incubated with isotype control antibodies (31). During these animal experiments, data were obtained collecting several parameters such as general health status (e.g., fur, weight, behavior, mobility), immune cell reconstitution (white blood cell counts, flow cytometric analyses of human T cell, B cell, and lymphocyte markers) and histological data (apoptotic cells in the gut of the mice, TUNEL) (31). Importantly, MAX.16H5 IgG1 (murine) or IgG4 (chimeric) incubation of immune cell grafts comparably weakened GVHD development significantly but did not impair the engraftment of the transplanted cells (31).
Concluding Remarks
It was early known that even the murine IgG1 istoype of MAX.16H5 does not stimulate CDC or ADCC (13), but the mechanism of action is still not understood. Over the years of development, many theories have been arosen on its mechanism of action to induce tolerance. Despite being of interest, most data were not published as they showed negative results. Due to the quick responses observed in clinical trials, it was speculated that the antibody's mechanism was independent of antigen recognition which would take longer but was more likely caused by inhibition of preactivated IL-2-dependent T cells (32). However, this theory cannot explain why the antibody is effective in GVHD prevention after an ex vivo incubation, i.e., before the cells of the transplant had antigen contact. Moreover, the monocyte/macrophage system was thought to be involved, but this idea too was in conflict with reported data (13). Rising Treg amounts in a murine model (71) are probably a mediator of tolerance but do not explain the mechanism how tolerance is achieved.
The majority of the studies performed with the anti-human CD4 antibody MAX.16H5 focused on the treatment of autoimmune diseases such as RA, SLE, IDB and JCA and revealed striking results that identified the MAX.16H5 antibody as a promising alternative for conventional therapeutics. Following initial systemic application of MAX.16H5, a new strategy was developed leading to similar success in therapy and improved safety of patients: graft CD4+ cells were incubated ex vivo with the MAX.16H5 antibody and re-infused into the patient (Figure 2). This innovative approach extended the scope from the treatment of autoimmune diseases to hematological malignancies. By ex vivo incubation of an allogeneic immune- and stem cell transplant with the epitope-specific anti-human CD4 antibody MAX.16H5, a new therapy strategy has emerged for the first time enabling both the preservation of the GVL effect of the transplant and the permanent suppression of GVHD without the need for conventional immunosuppression. The following essential benefits of this innovative therapeutic approach can be expected: First, the treatment of a human immune cell transplant does not deteriorate the anti-tumor effectiveness with regard to different leukemia types. Second, it is expected that an allogeneic HSC graft treated with MAX.16H5 anti-human CD4 antibodies leads to a general improvement of the survival due to suppression of the GVHD. Furthermore, the dosage and amount of conventional immunosuppressive drugs (toxicity, side effects, and duration of the treatment) can be reduced and the patients' quality of life could be improved. Due to the suppression of the GVHD, patients lacking a suitable donor will be applicable to receive donor cells whose transplantation normally would be associated with a higher risk for GVHD (e.g., more HLA mismatches). Thus, immune cell therapies will become applicable to cure other diseases (e.g., autoimmune diseases and primary immunodeficiencies) whose curative treatment regimen does currently not include this form of therapy because of a high risk for GVHD development. Finally yet importantly, the incubation of the allogeneic HSCT grafts with the epitope-specific anti-human CD4 antibody MAX.16H5 can be performed outside of the body which reduces side effects and therapy costs, antibody amounts as well as improves the safety of the transplantation remarkably.
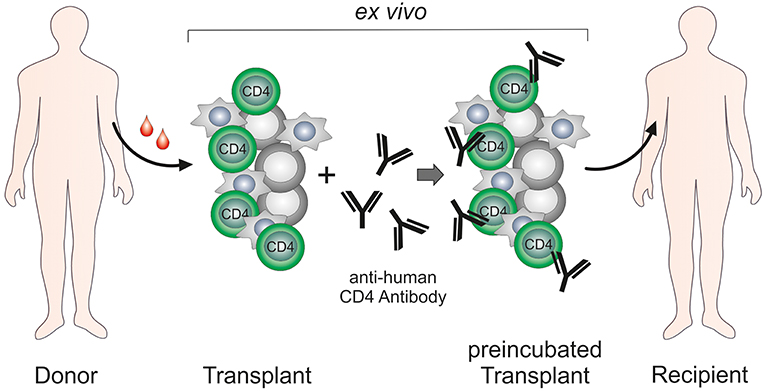
Figure 2. Ex vivo treatment of hematopoietic stem cell/immune cell grafts by anti-human CD4 antibody MAX.16H5.
Author Contributions
LS, AD, NH, UT, and SF analyzed the publications, created the figures and tables, and wrote the paper.
Funding
This study was supported by the Fraunhofer Society, the Fraunhofer Cluster of Immune Mediated Diseases (CIMD), and the Leistungs- und Transferzentrum Chemie- und Biosystemtechnik which is supported by Sächsische Aufbaubank (SAB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the colleagues from the Fraunhofer Cluster of Immune Mediated Diseases (CIMD) for their helpful discussion and proof reading of the manuscript. Furthermore, we thank Prof. Dr. F. Emmrich for his valuable notes regarding the MAX.16H5 anti-human CD4 antibody. Study design, collection of data, analysis, decision to publish and preparation of the manuscript were not influenced.
Abbreviations
AA, amino acid; ADCC, antibody dependent cellular cytotoxicity; aGVHD, acute graft-vs.-host disease; AML, acute myeloid leukemia; BW, body weight; CDC, complement dependent cytolysis; CEA, carcinoembryonic antigen; CRP, C-reactive protein; ELISA, enzyme-linked immunosorbent assay; ESR, erythrocyte sedimentation rate; F(ab)2, fragment antigen binding region; Fc, fragment crystallizable region; FcR, Fc receptor; FLT3, Fms like tyrosine kinase 3; FoxP3, forkhead box protein P3; GVHD, graft-vs.-host disease; GVL effect, graft-vs.-leukemia effect; HAMA, human anti-mouse antibody; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; HSC, hematopoietic stem cell; HSCT, hematopoietic stem cell transplantation; IBD, inflammatory bowel disease; Ig, immunoglobulin; IL, interleukin; ITD, internal tandem duplication; ITAM, immunoreceptor tyrosine-based activating motif; ITIM, immunoreceptor tyrosine-based inhibitory motif; i.v., intravenous; JCA, juvenile chronic arthritis; Lck, lymphocyte-specific protein tyrosine kinase; mAb, monoclonal antibody; LPS, lipopolysaccharide; MHC, major histocompatibility complex; NSG, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; PB, peripheral blood; PBMC, peripheral blood mononuclear cell; PD-1, programmed cell death protein 1; PD-L1, programmed cell death 1 ligand 1; PHA, phytohemagglutinin; p.i., post injection; RA, rheumatoid arthritis; RF, rheumatoid factor; sCD14, soluble CD14; SLE, systemic lupus erythematosus; TCR, T-cell receptor; Th cell, T-helper cell; Treg, regulatory T cell; TT, tetanus toxoid.
References
1. Knapp W, editor. Leucocyte Typing IV: White Cell Differentiation Antigens. Oxford: Oxford University Press (1989). p. 1182.
2. Sweet RW, Truneh A, Hendrickson WA. CD4: Its structure, role in immune function and AIDS pathogenesis, and potential as a pharmacological target. Curr Opin Biotechnol. (1991) 2:622–33. doi: 10.1016/0958-1669(91)90089-N
3. Wofsy D. Strategies for treating autoimmune disease with monoclonal antibodies. West J Med. (1985) 143:804–9.
4. Burmester GR, Horneff G, Emmrich F. Management of early inflammatory arthritis. Intervention with immunomodulatory agents: monoclonal antibody therapy. Baillieres Clin Rheumatol. (1992) 6:415–34.
5. Emmrich F, Kaufmann SH. Human T-cell clones with reactivity to Mycobacterium leprae as tools for the characterization of potential vaccines against leprosy. Infect Immun. (1986) 51:879–83.
6. Emmrich F, Hunsmann G, Lüke W. Monoclonal Antibody for Inhibiting Cell Infection by HIV Viruses (WO1990002199 A1). Google Patents (1989). Available online at: http://www.google.com/patents/WO1990002199A1?cl=en (accessed July 4, 2016).
7. Becker W, Emmrich F, Horneff G, Burmester G, Seiler F, Schwarz A, et al. Imaging rheumatoid arthritis specifically with technetium 99m CD4-specific (T-helper lymphocytes) antibodies. Eur J Nucl Med. (1990) 17:156–9.
8. Kearney JF, Radbruch A, Liesegang B, Rajewsky K. A new mouse myeloma cell line that has lost immunoglobulin expression but permits the construction of antibody-secreting hybrid cell lines. J Immunol. (1979)123:1548–50.
9. Davis SJ, Schockmel GA, Somoza C, Buck DW, Healey DG, Rieber EP, et al. Antibody and HIV-1 gp120 recognition of CD4 undermines the concept of mimicry between antibodies and receptors. Nature. (1992) 358:76–9. doi: 10.1038/358076a0
10. Repke H, Gabuzda D, Palu G, Emmrich F, Sodroski J. Effects of CD4 synthetic peptides on HIV type I envelope glycoprotein function. J Immunol. (1992) 149:1809–16.
11. Becker W, Horneff G, Emmrich F, Burmester G, Kalden J, Wolf F. Kinetics of 99mTc-labelled antibodies against CD4 (T-helper) lymphocytes in man. Nuklearmedizin. (1992) 31:84–90.
12. Hiepe F, Volk HD, Apostoloff E, Baehr R von, Emmrich F. Treatment of severe systemic lupus erythematosus with anti-CD4 monoclonal antibody. Lancet. (1991) 338:1529–30.
13. Horneff G, Burmester GR, Emmrich F, Kalden JR. Treatment of rheumatoid arthritis with an anti-CD4 monoclonal antibody. Arthritis Rheum. (1991) 34:129–40.
14. Horneff G, Emmrich F, Kalden JR, Burmester GR. Suppression of monocyte-macrophage activation by anti-CD4 therapy in patients with chronic polyarthritis. Immun Infekt. (1991) 19:55–6.
15. Horneff G, Krause A, Emmrich F, Kalden JR, Burmester GR. Elevated levels of circulating tumor necrosis factor-alpha, interferon-gamma, and interleukin-2 in systemic reactions induced by anti-CD4 therapy in patients with rheumatoid arthritis. Cytokine. (1991) 3:266–7.
16. Horneff G, Winkler T, Kalden JR, Emmrich F, Burmester GR. Human anti-mouse antibody response induced by anti-CD4 monoclonal antibody therapy in patients with rheumatoid arthritis. Clin Immunol Immunopathol. (1991) 59:89–103.
17. Horneff G, Emmrich F, Reiter C, Kalden JR, Burmester GR. Persistent depletion of CD4+ T cells and inversion of the CD4/CD8 T cell ratio induced by anti-CD4 therapy. J Rheumatol. (1992) 19:1845–50.
18. Horneff G, Sack U, Kalden JR, Emmrich F, Burmester GR. Reduction of monocyte-macrophage activation markers upon anti-CD4 treatment. Decreased levels of IL-1, IL-6, neopterin and soluble CD14 in patients with rheumatoid arthritis. Clin Exp Immunol. (1993) 91:207–13.
19. Emmrich F, Horneff G, Becker W, Luke W, Potocnik A, Kanzy U, et al. An anti-CD4 antibody for treatment of chronic inflammatory arthritis. Agents Actions Suppl. (1991) 32:165–70.
20. Emmrich J, Seyfarth M, Fleig WE, Emmrich F. Treatment of inflammatory bowel disease with anti-CD4 monoclonal antibody. Lancet. (1991) 338:570–1.
21. Horneff G, Dirksen U, Schulze-Koops H, Emmrich F, Wahn V. Treatment of refractory juvenile chronic arthritis by monoclonal CD4 antibodies: a pilot study in two children. Ann Rheum Dis. (1995) 54:846–9.
22. Reinke P, Volk HD, Miller H, Neuhaus K, Fietze E, Herberger J, et al. Anti-CD4 therapy of acute rejection in long-term renal allograft recipients. Lancet. (1991) 338:702–3.
23. Kinne RW, Becker W, Schwab J, Horneff G, Schwarz A, Kalden JR, et al. Comparison of 99Tcm-labelled specific murine anti-CD4 monoclonal antibodies and non-specific human immunoglobulin for imaging inflamed joints in rheumatoid arthritis. Nucl Med Commun. (1993) 14:667–75.
24. Kinne RW, Becker W, Schwab J, Schwarz A, Kalden JR, Emmrich F, et al. Imaging rheumatoid arthritis joints with technetium-99m labelled specific anti-CD4- and non-specific monoclonal antibodies. Eur J Nucl Med. (1994) 21:176–80.
25. Courtenay-Luck NS, Epenetos AA, Moore R, Larche M, Pectasides D, Dhokia B, et al. Development of primary and secondary immune responses to mouse monoclonal antibodies used in the diagnosis and therapy of malignant neoplasms. Cancer Res. (1986) 46(12 Pt 1):6489–93.
26. Schroff RW, Foon KA, Beatty SM, Oldham RK, Morgan AC, JR . Human anti-murine immunoglobulin responses in patients receiving monoclonal antibody therapy. Cancer Res. (1985) 45:879–85.
27. Bruhns P. Properties of mouse and human IgG receptors and their contribution to disease models. Blood. (2012) 119:5640–9. doi: 10.1182/blood-2012-01-380121
28. Neuberger MS, Rajewsky K. Activation of mouse complement by monoclonal mouse antibodies. Eur J Immunol. (1981) 11:1012–6. doi: 10.1002/eji.1830111212
29. Ey PL, Russell-Jones GJ, Jenkin CR. Isotypes of mouse IgG—I. Evidence for ‘non-complement-fixing' IgG1 antibodies and characterization of their capacity to interfere with IgG2, sensitization of target red blood cells for lysis by complement. Mol Immunol. (1980) 17:699–710. doi: 10.1016/0161-5890(80)90139-X
30. Michaelsen TE, Kolberg J, Aase A, Herstad TK, Høiby EA. The four mouse IgG isotypes differ extensively in bactericidal and opsonophagocytic activity when reacting with the P1.16 epitope on the outer membrane PorA protein of Neisseria meningitidis. Scand J Immunol. (2004) 59:34–9. doi: 10.1111/j.0300-9475.2004.01362.x
31. Hilger N, Glaser J, Muller C, Halbich C, Muller A, Schwertassek U, et al. Attenuation of graft-versus-host-disease in NOD scid IL-2Rgamma-/- (NSG) mice by ex vivo modulation of human CD4+ T cells. Cytometry A. (2016) 89:803–15. doi: 10.1002/cyto.a.22930
32. Bröker BM, Yu A, Tsygankov AY, Fickenscher H, Chitaev NA, Müller-Fleckenstein I, et al. Engagement of the CD4 receptor inhibits the interleukin-2-dependent proliferation of human T cells transformed byHerpesvirus saimiri. Eur. J. Immunol. (1994) 24:843–50. doi: 10.1002/eji.1830240411
33. Horneff G, Guse AH, Schulze-Koops H, Kalden JR, Burmester GR, Emmrich F. Human CD4 modulation in vivo induced by antibody treatment. Clin Immunol Immunopathol. (1993) 66:80–90.
34. Guse AH, Tsygankov A, Broker BM, Roth E, Emmrich F. Signal transduction in T lymphocytes and monocytes: effects of the anti-CD4 antibody MAX.16H5. Year Immunol. (1993) 7:175–81.
35. Shevach EM. Regulatory T cells in autoimmmunity*. Annu. Rev. Immunol. (2000) 18:423–49. doi: 10.1146/annurev.immunol.18.1.423
37. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
38. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
39. Khattri R, Cox T, Yasayko S-A, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. (2003) 4:337. doi: 10.1038/ni909
40. Wang J, Yan T, Simmer B, Emmrich F. The effect of anti-CD4 on helper function of CD4,45RA+ versus CD4,45RO+ T cells. Clin Exp Immunol. (1994) 95:128–34.
41. Lourenço EV, La Cava A. Cytokines in systemic lupus erythematosus. Curr Mol Med. (2009) 9:242–54.
42. McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. (2007) 7:429–42. doi: 10.1038/nri2094
43. The Antibody Society. Approved Antibodies. (2015) Available online at: https://www.antibodysociety.org/news/approved-antibodies/ (accessed March 19, 2018).
44. Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. (1990) 265:621–36.
45. Chatenoud L, Ferran C, Reuter A, Legendre C, Gevaert Y, Kreis H, et al. Systemic reaction to the anti-T-cell monoclonal antibody OKT3 in relation to serum levels of tumor necrosis factor and interferon-gamma corrected. N Engl J Med. (1989) 320:1420–1. doi: 10.1056/NEJM198905253202117
46. Suthanthiran M, Fotino M, Riggio RR, Cheigh JS, Stenzel KH. OKT3-associated adverse reactions: mechanistic basis and therapeutic options. Am J Kidney Dis. (1989) 14(5 Suppl. 2):39–44.
47. Brink I, Thiele B, Burmester GR, Trebeljahr G, Emmrich F, Hiepe F. Effects of anti-CD4 antibodies on the release of IL-6 and TNF-alpha in whole blood samples from patients with systemic lupus erythematosus. Lupus. (1999) 8:723–30.
48. Burmester GR, Emmrich F. Anti-CD4 therapy in rheumatoid arthritis. Clin Exp Rheumatol. (1993) 11(Suppl. 8):45.
49. Guse AH, Roth E, Broker BM, Emmrich F. Complex inositol polyphosphate response induced by co-cross-linking of CD4 and Fc gamma receptors in the human monocytoid cell line U937. J Immunol. (1992) 149:2452–8.
50. Horneff G, Burmester GR, Strobel G, Gramatzki M, Kalden JR, Emmrich F. Therapie der chronischen Polyarthritis mit einem monoklonalen Antikörper gegen das CD4-Antigen auf T-Helferzellen. Aktuelle Rheumatol. (1989) 1989:232.
51. Reiter C, Kakavand B, Rieber EP, Schattenkirchner M, Riethmuller G, Kruger K. Treatment of rheumatoid arthritis with monoclonal CD4 antibody M-T151. Clinical results and immunopharmacologic effects in an open study, including repeated administration. Arthritis Rheum. (1991) 34:525–36.
52. Nadler LM, Stashenko P, Hardy R, Kaplan WD, Button LN, Kufe DW, et al. Serotherapy of a patient with a monoclonal antibody directed against a human lymphoma-associated antigen. Cancer Res. (1980) 40:3147–54.
53. Morrison SL, Johnson MJ, Herzenberg LA, Oi VT. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc Natl Acad Sci. (1984) 81:6851–5.
54. McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol. (1998) 16:2825–33. doi: 10.1200/JCO.1998.16.8.2825
55. Greig SL. Obiltoxaximab: first global approval. Drugs. (2016) 76:823–30. doi: 10.1007/s40265-016-0577-0
56. Fricke S, Emmrich F, Hilger N. Anti-CD4 Antibodies to Prevent in Particular GRAFT-versus-Host Disease (GvHD): US Patent (US 9,745,552 B2) (2011).
57. Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. (2017) 9:63–73. doi: 10.1007/s13238-017-0473-8
58. Brüggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. (1987) 166:1351–61.
59. Davies AM, Sutton BJ. Human IgG4: a structural perspective. Immunol Rev. (2015) 268:139–59. doi: 10.1111/imr.12349
60. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. (2012) 24:207–12. doi: 10.1016/j.coi.2011.12.009
61. Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. (2002) 105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x
62. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. (2009) 27:767–71. doi: 10.1038/nbt.1553
63. Laub R, Dorsch M, Wedekind D, Meyer D, Schröder S, Ermann J, et al. Replacement of murine by human CD4 and introduction of HLA-DR17 in mice: a triple-transgenic animal model to study human MHC II-CD4 interaction in situ. J Exp Anim Sci. (1999) 39:122–35.
64. Meyer D, Laub R, Dorsch M, Thamm B, Ermann J, Lehmann J, et al. Replacement of murine by human CD4 and introduction of HLA-DR in mice: a triple-transgenic animal model to study human MHC II-CD4 interaction in situ. In: 28th Annual Meeting of the Deutsche Gesellschaft für Immunologie. September 24-27, 1997. Abstracts. Elsevier GmbH (1997). p. 153.
65. Laub R, Brecht R, Dorsch M, Valey U, Wenk K, Emmrich F. Anti-human CD4 induces peripheral tolerance in a human CD4+, murine CD4-, HLA-DR+ advanced transgenic mouse model. J Immunol. (2002) 169:2947–55. doi: 10.4049/jimmunol.169.6.2947
66. Laub R, Dorsch M, Meyer D, Ermann J, Hedrich HJ, Emmrich F. A multiple transgenic mouse model with a partially humanized activation pathway for helper T cell responses. J Immunol Methods. (2000) 246:37–50. doi: 10.1016/S0022-1759(00)00288-X
67. Laub R, Dorsch M, Wenk K, Emmrich F. Induction of immunologic tolerance to tetanus toxoid by anti-human CD4 in HLA-DR3(+)/human CD4(+)/murine CD4(-) multiple transgenic mice. Transplant Proc. (2001) 33:2182–3. doi: 10.1016/S0041-1345(01)01934-0
68. Holtan SG, Pasquini M, Weisdorf DJ. Acute graft-versus-host disease: a bench-to-bedside update. Blood. (2014) 124:363–73. doi: 10.1182/blood-2014-01-514786
69. Garnett C, Apperley JF, Pavlu J. Treatment and management of graft-versus-host disease: improving response and survival. Ther Adv Hematol. (2013) 4:366–78. doi: 10.1177/2040620713489842
70. Schmidt F, Hilger N, Oelkrug C, Svanidze E, Ruschpler P, Eichler W, et al. Flow cytometric analysis of the graft-versus-Leukemia-effect after hematopoietic stem cell transplantation in mice. Cytometry A. (2015) 87:334–45. doi: 10.1002/cyto.a.22619
71. Fricke S, Hilger N, Fricke C, Schonfelder U, Behre G, Ruschpler P, et al. Prevention of graft-versus-host-disease with preserved graft-versus-leukemia-effect by ex vivo and in vivo modulation of CD4(+) T-cells. Cell Mol Life Sci. (2014) 71:2135–48. doi: 10.1007/s00018-013-1476-0
72. Hilger N, Mueller C, Stahl L, Mueller AM, Zoennchen B, Dluczek S, et al. Incubation of immune cell grafts with MAX.16H5 IgG1 anti-human CD4 antibody prolonged survival after hematopoieticstem cell transplantation in a mouse model for Fms like tyrosine kinase 3 positive acute myeloid leukemia. Front. Immunol. (2018) 9:1131. doi: 10.3389/fimmu.2018.02408
73. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
74. Pearson H. Tragic drug trial spotlights potent molecule. Nature. (2006). doi: 10.1038/news060313-17
75. Beyersdorf N, Hanke T, Kerkau T, Hünig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev. (2006) 5:40–5. doi: 10.1016/j.autrev.2005.06.001
Keywords: T cell modulation, anti-human CD4 antibody, MAX.16H5, autoimmune disease, graft-vs.-host disease, graft-vs.-leukemia effect
Citation: Stahl L, Duenkel A, Hilger N, Tretbar US and Fricke S (2019) The Epitope-Specific Anti-human CD4 Antibody MAX.16H5 and Its Role in Immune Tolerance. Front. Immunol. 10:1035. doi: 10.3389/fimmu.2019.01035
Received: 30 November 2018; Accepted: 23 April 2019;
Published: 24 May 2019.
Edited by:
Everett Meyer, Stanford University, United StatesReviewed by:
Suparna Dutt, Stanford University, United StatesAlexander Steinkasserer, University Hospital Erlangen, Germany
Copyright © 2019 Stahl, Duenkel, Hilger, Tretbar and Fricke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lilly Stahl, bGlsbHkuc3RhaGxAaXppLmZyYXVuaG9mZXIuZGU=
†These authors have contributed equally to this work
‡These authors shared last authorship