 Xi Zen Yap
Xi Zen Yap Lucie S. P. Hustin
Lucie S. P. Hustin Robert W. Sauerwein1
Robert W. Sauerwein1- 1Department of Medical Microbiology, RadboudUMC Centre for Infectious Diseases, Nijmegen, Netherlands
- 2Institut Curie, PSL Research University, CNRS UMR168, Paris, France
Humoral immunity is a critical effector arm for protection against malaria but develops only slowly after repeated infections. T cell-mediated regulatory dynamics affect the development of antibody responses to Plasmodium parasites. Here, we hypothesize that T follicular helper cell (TFH) polarization generated by repeated Plasmodium asexual blood-stage infections delays the onset of protective humoral responses. IFN-γ production promotes polarization toward TFH1 and increased generation of regulatory follicular helper cells (TFR). Delineating the mechanisms that drive TH1 polarization will provide clues for appropriate induction of lasting, protective immunity against malaria.
Naturally-Acquired Immunity in Malaria
Only after years of continued exposure to Plasmodium parasites do individuals from malaria endemic regions develop clinical immunity (CI), that protects against clinical disease but not from parasitaemia (1). This protection is mediated through both cellular and humoral immune effector mechanisms. In particular, humoral immunity (HI) apparently plays a pivotal role against blood-stages, which are responsible for pathology and disease. Seminal findings demonstrate that IgG transfer from malaria-immune adults to children with acute malaria can indeed reduce symptoms and parasite load (2).
Effective HI induction requires B cells to be activated by antigen-presenting cells (APCs), predominantly dendritic cells (DCs). Sustained “help” from cognate CD4+ T cells is subsequently required for B cell proliferation, affinity maturation, and Ig class-switching. T follicular helper cells (TFH), which co-localize with B cells in the germinal centers (GCs), are crucial for both naïve B cell activation during primary infections and reactivation of memory B cells (MBC) in secondary infections. TFH and other CD4+ helper T cells (TH) can drive naive B cells to differentiate into high-antibody-producing plasma cells (PC) or MBC, which rapidly reactivate and produce specific Abs during secondary infections.
While typically taking a number of years to develop fully, clinical malaria immunity is of relatively short duration and rapidly wanes in the absence of re-infection (3, 4). Antibody efficacy and specific MBC counts increase gradually with age and cumulative exposure, resulting in a strong TH1 (IFN-γ-producing) immune response (5–9). The origins of the relatively slow acquisition of clinical immunity, however, remain elusive.
Here we hypothesize that T cell responses generated by repeated blood-stage malaria infection may in fact delay the onset of potent humoral responses. We contextualize the role of TH and TFH polarization surrounding the B cell response in malaria, and suggest that excessive polarization toward the IFN-γ producing TH1 phenotype reduces the longevity of antibody responses.
B-cells and Plasma Cells Are Deregulated in Malaria
Potent humoral responses are characterized by the generation of specific and high-affinity long-lived PCs (LLPCs) and MBCs in the GCs. Yet both adults and children in malaria endemic areas show a delay in the development of MBC and short-lived antibodies targeting P. falciparum blood-stage antigens (10). Accordingly, antibodies generated during one acute malaria season are undetectable by the next (10). Similar delays in CI onset are found in malaria-naïve immigrants to Papua New Guinea (11).
Sustained parasitaemia may be a key factor affecting B cell differentiation. Recent studies have provided valuable insights into B cell subset dynamics and antibody kinetics in the context of Plasmodium infection. While it is clear that IgG+ MBCs are key effectors in long-term memory, high levels of non-IgG+ anti-P. falciparum MBCs may have a role in early protection (12). Frequent exposure to asexual parasites, as experienced in highly malaria-endemic regions, is associated with the development of MBCs with reduced memory function, known as atypical memory B-cells (AMBC). While the presence of AMBCs may contribute to the delayed and short-lived nature of HI to malaria (13), their presence may also be symptomatic of a more broadly deregulated humoral response.
Frequent parasite exposure seems to be a driving factor in AMBC development. AMBC frequency increases proportionate to transmission intensity, age, and cumulative malaria exposure (13–19), and AMBC proportions increase after each acute malaria episode (20). Conversely, the percentage of AMBCs declines in the absence of parasite exposure, inducing stable populations of malaria-specific classical MBCs (17, 19, 21, 22). This may be the result of direct B cell interactions with Plasmodium parasites, or indirectly generated by the pro-inflammatory environment (23, 24), or by a combination of the two, i.e., AMBCs as a product of persistent antigen engagement by B cells within a highly inflammatory environment of chronic malaria exposure, driven by TH1 cells (25).
Hence, inappropriate IFN-γ production may be a reflection of inadequate T cell help caused by frequent exposure to blood-stage P. falciparum.
Blood-Stage Infection Induces Changes in T Cell Phenotypes and Populations
Malaria parasites typically induce human T cells with high surface expression of PD-1 and LAG3 and high production of both IFN-γ and IL-10 (26–28). Hence, CD4+ T cells in the malarial environment frequently display a phenotype associated with immunosuppression. Furthermore, the malarial environment polarizes CD4+ T cells toward the IFN-γ-producing TH1-like phenotype, consequently reducing B-cell responses by suppressing antibody-inducing TH2 and TFH lineages. While this may be beneficial for containing parasite-mediated pathology, it may contribute to immunopathology and limit reactivation of long-lived MBC. Modeling analyses by Lonnberg et al indicate that monocytes in particular have a role in regulating the T cell response, producing cytokines which skew naïve cells away from the TFH lineage and toward a TH1 phenotype (26).
The Impact of TFH Cells on Humoral Immunity
The TFH subset is particularly crucial for B cell development in the GC and the subsequent generation of a functional memory B cell compartment. TFH responses are widely hypothesized to be disrupted in malaria, as reflected by the relatively high frequency of autoreactive AMBCs and classical MBCs (29).
Due to the challenges of obtaining secondary lymphoid tissue, human research on TFH cells has primarily concentrated on circulatory CD4+CXCR5+ TFH (30). These circulatory TFH cells share functional characteristics with GC TFH cells including IL-21 production and the ability to induce B cell differentiation in vitro (31). They also have properties of a central memory-like TFH population (26, 31–34). In contrast to GC-resident TFH, however, circulatory TFH cells lack BCL6 expression, which is required for survival and induction of secondary antibody responses (31, 35–38). BCL6 re-expression can be induced by re-challenge with cognate MBC (39), indicating that sustained antigen presence is required for TFH function.
In the last decade, circulatory TFH subsets equivalent to TH1, TH2, TH17, and TREG have been characterized in mice and humans (40, 41). TH1-like TFH cells (TFH1) show reduced potential to provide adequate help during antibody maturation ex vivo compared to TH2-like TFH cells (TFH2) (33, 35, 42). The concept that TFH subset imbalance may affect development of antimalarial immunity has gained more traction due to TFH subsets' potential roles in other chronic diseases, such as HIV (43). In parallel, polarization toward TH1-like responses has been well-documented in malaria and causes fundamental changes in multiple cell subtypes, such as induction of Th1-like regulatory cells (TREG1) (6, 28, 44).
Thus, dysfunctional GC processes and inappropriate TFH reactions are a likely consequence of malaria infection. Indeed, polarization of TFH is observed in Malian children, with more activated TFH1, more TH1-like cytokine responses, and less prominent TH2 polarization (26, 34, 45–47). This TH1-like cytokine response may lead to decreased GC reactions and therefore reduced generation and reactivation of T cell-dependent antibody responses (Figure 1).
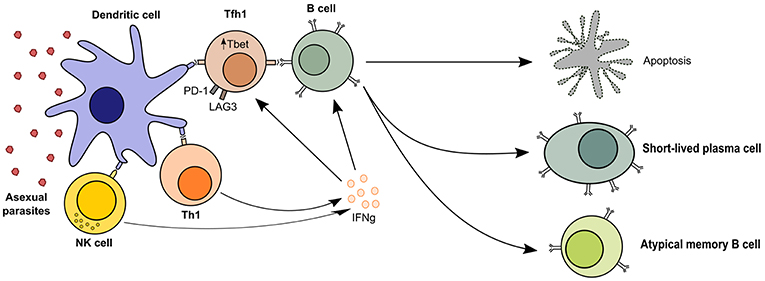
Figure 1. TH1-like T cell responses in malaria. Follicular T helper cells are required for B cell activation and the generation of humoral immunity, but malaria profoundly affects T cell polarization and leads to short-lived antibody responses. The presence of asexual parasitaemia promotes activation of TH1 and NK cells, which produce high levels of IFN-γ. This microenvironment promotes cellular upregulation of exhaustion markers like LAG3 and PD-1 and TFH differentiation into TFH1 cells, which are less effective at activating B cells. Quality of the T cell help in malaria-driven inflammation is therefore reduced, leading to B cell apoptosis or differentiation into short-lived plasma cells and atypical memory B cells, which are poor contributors to the long-term maintenance of humoral immunity.
Murine data suggest that circulatory TFH may represent pre-TFH generated from partly committed TFH lineage cells rather than mature memory GC-derived TFH cells (45). In murine malaria models, frequency of pre-TFH expressing the TH1-associated transcription factor Tbet increases after a single P. bergei ANKA infection (46). It will be important to clarify whether malaria-induced circulating TFH1 are simply pre-TFH generated in the periphery after a single exposure without entering the GC, and if circulating TFH2 therefore represent the mature TFH memory pool. This may explain the differential functionality of these two TFH subtypes in malaria. A proper understanding of the relationship between circulating- and GC TFH will be essential to delineate their particular role in the development of HI.
How Is the TH1-like Signature and TFH1-like Polarization Realized?
Studies with transgenic murine P. yoelii parasites suggest a positive feedback loop induced by Type I interferon and IL-2; TH1 cytokines secreted during Plasmodium infection increase CD4+ T cell responsiveness by up-regulating Tbet and BLIMP-1 (44, 47). Consequently, CD4+ T cells gain an increased predisposition to become TH1 cells.
Deregulation of humoral malaria immunity may be the result of an increased TFH1:TFH2 ratio in combination with the efficacy of the individual responses. Sustained polarization toward a TFH1 response after a single infection may affect an individual's ability to respond to subsequent malaria episodes. Frequencies of CXCR3+CCR6− TFH1s increase transiently but significantly during acute malaria, while CXCR3−CCR6− TFH2 frequencies decrease long-term in response to multiple malaria parasite exposures (48). In addition, T cell co-receptors may play a role in regulating TFH activation, as shown in P. yoelli-infected mice, where activation of OX40 leads to up-regulation of IFN-γ (49), resulting in activation of the inhibitory PD-1 pathway. Consequently, TFH help will shut down, resulting in dysfunctional B cell responses including the generation of AMBCs (25) and decreased parasite clearance due to lower specific IgM and IgG titres (49, 50). Therefore, CXCR3+ over-activation may be an important albeit not exclusive factor that limits T cell-dependent antibody responses to Plasmodium.
Co-infection with other pathogens can also impact humoral immunity to malaria. Multiple murine studies demonstrated that co-infection with murine Epstein-Barr virus analog MHV68 during P. yoelii XNL infection led to very high mortality from symptoms of malaria (51, 52). The latter study indicated that mortality was due to loss of humoral immunity by the MHV68 virus via induction of host IL-10 (52). Host factors involved in parasite sensing can also have a role: humanized mice engineered to express a single MHCII haplotype, HLA-DR4 (0401), had higher rates of parasitaemia and morbidity to P. yoelii 17XNL infection than mice engineered to express alternate haplotypes. The loss of parasite control was due to downregulation of humoral immunity by overproliferating TREGs (53).
Other Checkpoint Factors Influencing T Cell Differentiation in Malaria
Regulatory T cell subtypes are likely key modulators of HI. The recently characterized regulatory follicular helper T cell (TFR) subset is especially relevant for HI regulation. Contrary to TR1, which arise from TH1, TFR are a FOXP3+ subclass derived directly from TREG which express both BCL-6 and BLIMP-1 (54). Crucially, TFR can directly suppress both TFH and B cells in GC reactions and therefore directly affect GC formation (55–59).
TFR have not yet been studied in the context of malaria, even though their importance is indicated by their key role in controlling antibody production in HIV (60). TFR cell functionality is assumed to be determined by their ratio with TFH. As the proportion of TFR increases with age, similarly to TREGs (57), we hypothesize that TFR have the potential to play a role in the delayed onset of NAI. Murine studies show that the TFR fraction increases with age while the TFH proportion remains constant (60). TFR may therefore progressively regulate the TH1 driven over-activation of DCs, T cells and B-cells.
Conversely, a higher TFR:TFH ratio may inhibit TFH activation and proliferation, as suggested by TFR-induced downregulation of the proliferation marker Ki67 in TFH cells in vitro, dampening TFH1 activation (61, 62). However, TFR also downregulate the TH2-associated cytokines IL-21 and IL-4 in in vitro murine studies, potentially leading to marked defects in GC formation, and B cell affinity maturation (61, 63–65). Changes in the TFR:TFH ratio may therefore redirect GC B cells toward becoming extra-follicular MBCs and short-lived PCs, therefore further decreasing generation of long-lived high-affinity antibodies (58, 62).
Summary, Conclusions, and Outlook
Malaria infection induces TH1 polarization characterized by the production of IFN-γ. Overproduction of IFN-γ may be central to poor acquisition of HI by polarizing TFH toward TFH1 and causing a positive feedback loop of TH1 polarization. It will be crucial to understand the specific parasite components responsible for TH1 polarization so that we can better target parasite antigens which catalyze TH1 polarization.
Malaria-naïve adults and children from low-transmission regions tend to generate strong pro-inflammatory responses: TH1 cytokines IFN-γ and TNFα, and other pro-inflammatory cytokines such as IL-1β and IL-6, are produced, which may favor generation of TH1-like responses. However, children with sustained parasitaemia develop a cytokine signature consisting of IFN-γ, Type I IFN, and regulatory cytokines IL-10 and TGF-β (9, 66, 67). It is unclear whether this is related to parasite density, incidence of infections, or both. Parasite burden and transmission intensity could affect TFH polarization through systemic cytokine-mediated effects.
Dendritic cells and NK cells may be responsible for maintaining TH1 polarization. Malaria could affect early T cell polarization by disrupting dendritic cell function (68, 69), and DCs co-incubated with blood-stage parasites in vitro are shown to polarize naïve T cells toward a TH1-like phenotype that produces IFN-γ and TNFα (70, 71). Furthermore, DCs are required for NK cell activation to blood-stage parasites (72). NK cells are major producers of IFN-γ, and rapid reactivation of NK cells in response to blood-stage infection could lead to the formation of a TH1 cytokine signature, thereby inhibiting development of positive HI-forming responses. The presence of memory-like responses (trained immunity) from NK cells upon re-encountering pRBCs in vitro (73) suggests that NK cell activation in response to malaria may occur rapidly after the first infection, increasing early tendencies toward Th1-like responses. Moreover, NK cell cross-talk with dendritic cells is important for CD4 T cell priming in murine malaria models (74, 75), suggesting that NK cells may bias TH1 polarization through multiple pathways.
However, it is unclear whether the blood-derived TFH differ functionally from their GC counterparts. Better models of TFH will be required to study these differences and assess the functional relationship between TFH subsets and the generation of humoral immunity more thoroughly: what phenotypes are generated by B-cells co-stimulated by TFH1, the quality of the antibody response, and whether their ability to differentiate into LLPCs or classical MBCs is impacted by malaria-generated TFH1s. A culture system to induce TFH or novel systems such as humanized mice which could generate larger quantities of TFH and even allow for isolation of tissue-resident TFH would permit further, in-depth study of these cells. This would also permit mechanistic studies into how TFH1 polarization occurs.
In summary, malaria infection, especially repeated infection with high parasitaemia, may generate “inappropriate” TH1-like T cell responses that fail to provide the adequate environment for long-lasting HI. This may be due to (i) compromised TFH help, reducing the generation of functional GC and development of typical memory B-cells, leading to a loss of HI longevity; (ii) increased proliferation of regulatory subsets such as TFR which may further inhibit HI by decreasing TFH activation and proliferation; (iii) a strong TH1-like immune signature characterized by high production of IFN-γ, illustrated by the increased fraction of TH1 and other TH1-like cells, including the TFH1 subset. To break the cycle, we need improved methods to study TFH and understand the underlying mechanisms of TH1 polarization in malaria.
Author Contributions
XZY and LH wrote the first draft of the manuscript, which was reviewed by RWS. All authors have approved the publication of the final manuscript.
Funding
XZY is supported by funding from the Bill and Melinda Gates Foundation (Grant numbers. OPP1091355 and OPP1080385).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Doolan DL, Dobaño C, Baird JK. Acquired immunity to malaria. Clin Microbiol Rev. (2009) 22:13–36. doi: 10.1128/CMR.00025-08
2. Cohen S, McGregor IA, Carrington S. Gamma-globulin and acquired immunity to human malaria. Nature. (1961) 192:733–7. doi: 10.1038/192733a0
3. Kinyanjui SM, Conway DJ, Lanar DE, Marsh K. IgG antibody responses to plasmodium falciparum merozoite antigens in Kenyan children have a short half-life. Malar J. (2007) 6:82. doi: 10.1186/1475-2875-6-82
4. Marsh K, Kinyanjui S. Immune effector mechanisms in malaria. Parasite Immunol. (2006) 28:51–60. doi: 10.1111/j.1365-3024.2006.00808.x
5. Jagannathan P, Kim CC, Greenhouse B, Nankya F, Bowen K, Eccles-James I, et al. Loss and dysfunction of V 2+ T cells are associated with clinical tolerance to malaria. Sci Transl Med. (2014) 6:251ra117. doi: 10.1126/scitranslmed.3009793
6. Portugal S, Moebius J, Skinner J, Doumbo S, Doumtabe D, Kone Y, et al. Exposure-dependent control of malaria-induced inflammation in children. PLoS Pathog. (2014) 10:e1004079. doi: 10.1371/journal.ppat.1004079
7. Langhorne J, Ndungu FMM, Sponaas A-M, Marsh K. Immunity to malaria: more questions than answers. Nat Immunol. (2008) 9:725–32. doi: 10.1038/ni.f.205
8. Scholzen A, Sauerwein RW. Immune activation and induction of memory: lessons learned from controlled human malaria infection with Plasmodium falciparum. Parasitology. (2016) 143:224–35. doi: 10.1017/S0031182015000761
9. Montes de Oca M, Good MF, McCarthy JS, Engwerda CR. The impact of established immunoregulatory networks on vaccine efficacy and the development of immunity to malaria. J Immunol. (2016) 197:4518–26. doi: 10.4049/jimmunol.1600619
10. Weiss GE, Traore B, Kayentao K, Ongoiba A, Doumbo S, Doumtabe D, et al. The plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. (2010) 6:e1000912. doi: 10.1371/journal.ppat.1000912
11. Iqbal J, Perlmann P, Berzins K. Serological diversity of antigens expressed on the surface of erythrocytes infected with Plasmodium falciparum. Trans R Soc Trop Med Hyg. (1993) 87:583–8. doi: 10.1016/0035-9203(93)90097-A
12. Krishnamurty AT, Thouvenel CD, Portugal S, Keitany GJ, Kim KS, Holder A, et al. Somatically hypermutated plasmodium-specific IgM+ memory B cells Are rapid, plastic, early responders upon malaria rechallenge. Immunity. (2016) 45:402–14. doi: 10.1016/j.immuni.2016.06.014
13. Weiss GE, Crompton PD, Li S, Walsh LA, Moir S, Traore B, et al. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J Immunol. (2009) 183:2176–82. doi: 10.4049/jimmunol.0901297
14. Asito AS, Moormann AM, Kiprotich C, Ng'ang'a ZW, Ploutz-Snyder R, Rochford R. Alterations on peripheral B cell subsets following an acute uncomplicated clinical malaria infection in children. Malar J. (2008) 7:238. doi: 10.1186/1475-2875-7-238
15. Illingworth J, Butler NS, Roetynck S, Mwacharo J, Pierce SK, Bejon P, et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J Immunol. (2013) 190:1038–47. doi: 10.4049/jimmunol.1202438
16. Portugal S, Tipton CM, Sohn H, Kone Y, Wang J, Li S, et al. Malaria-associated atypical memory B cells exhibit markedly reduced B cell receptor signaling and effector function. Elife. (2015) 4:1–21. doi: 10.7554/eLife.07218
17. Portugal S, Doumtabe D, Traore B, Miller LH, Troye-Blomberg M, Doumbo OK, et al. B cell analysis of ethnic groups in mali with differential susceptibility to malaria. Malar J. (2012) 11:162. doi: 10.1186/1475-2875-11-162
18. Muellenbeck MF, Ueberheide B, Amulic B, Epp A, Fenyo D, Busse CE, et al. Atypical and classical memory B cells produce Plasmodium falciparum neutralizing antibodies. J Exp Med. (2013) 210:389–99. doi: 10.1084/jem.20121970
19. Nogaro SI, Hafalla JC, Walther B, Remarque EJ, Tetteh KKA, Conway DJ, et al. The breadth, but not the magnitude, of circulating memory B cell responses to P. falciparum increases with age/exposure in an area of low transmission. PLoS ONE. (2011) 6:e25582. doi: 10.1371/journal.pone.0025582
20. Sullivan RT, Ssewanyana I, Wamala S, Nankya F, Jagannathan P, Tappero JW, et al. B cell sub-types following acute malaria and associations with clinical immunity. Malar J. (2016) 15:139. doi: 10.1186/s12936-016-1190-0
21. Ndungu FM, Olotu A, Mwacharo J, Nyonda M, Apfeld J, Mramba LK, et al. Memory B cells are a more reliable archive for historical antimalarial responses than plasma antibodies in no-longer exposed children. Proc Natl Acad Sci USA. (2012) 109:8247–52. doi: 10.1073/pnas.1200472109
22. Ayieko C, Maue AC, Jura WGZO, Noland GS, Ayodo G, Rochford R, et al. Changes in B cell populations and merozoite surface protein-1-specific memory B cell responses after prolonged absence of detectable P. falciparum infection. PLoS ONE. (2013) 8:e67230. doi: 10.1371/journal.pone.0067230
23. Donati D, Zhang LP, Chen Q, Chêne A, Flick K, Nyström M, et al. Identification of a polyclonal B-cell activator in Plasmodium falciparum. Infect Immun. (2004) 72:5412–8. doi: 10.1128/IAI.72.9.5412-5418.2004
24. Donati D, Mok B, Chene A, Xu H, Thangarajh M, Glas R, et al. Increased B cell survival and preferential activation of the memory compartment by a malaria polyclonal B cell activator. J Immunol. (2006) 177:3035–44. doi: 10.4049/jimmunol.177.5.3035
25. Obeng-Adjei N, Portugal S, Holla P, Li S, Sohn H, et al. Malaria-induced interferon-γ drives the expansion of tbethi atypical memory B cells. PLOS Pathog. (2017) 13:e1006576. doi: 10.1371/journal.ppat.1006576
26. Lönnberg T, Svensson V, James KR, Fernandez-Ruiz D, Sebina I, Montandon R, et al. Single-cell RNA-seq and computational analysis using temporal mixture modeling resolves TH1/TFH fate bifurcation in malaria. Sci Immunol. (2017) 2:eaal2192. doi: 10.1126/sciimmunol.aal2192
27. Villegas-Mendez A, Inkson CA, Shaw TN, Strangward P, Couper KN. Long-Lived CD4 + IFN-γ + T cells rather than short-lived CD4 + IFN-γ + IL-10 + T cells initiate rapid IL-10 production to suppress anamnestic T cell responses during secondary malaria infection. J Immunol. (2016) 197:3152–64. doi: 10.4049/jimmunol.1600968
28. Jagannathan P, Eccles-James I, Bowen K, Nankya F, Auma A, Wamala S, et al. IFNγ/IL-10 Co-producing cells dominate the CD4 response to malaria in highly exposed children. PLoS Pathog. (2014) 10:e1003864. doi: 10.1371/journal.ppat.1003864
29. Hart GT, Akkaya M, Chida AS, Wei C, Jenks SA, Tipton C, et al. The regulation of inherently autoreactive VH4-34–expressing B cells in individuals living in a malaria-endemic area of West Africa. J Immunol. (2016) 197:3841–9. doi: 10.4049/jimmunol.1600491
30. Schmitt N, Ueno H. Blood Tfh cells come with colors. Immunity. (2013) 39:629–30. doi: 10.1016/j.immuni.2013.09.011
31. Chevalier N, Jarrossay D, Ho E, Avery DT, Ma CS, Yu D, et al. CXCR5 expressing human central memory CD4 T cells and their relevance for humoral immune responses. J Immunol. (2011) 186:5556–68. doi: 10.4049/jimmunol.1002828
32. Schmitt N, Bentebibel S-E, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol. (2014) 35:436–42. doi: 10.1016/j.it.2014.06.002
33. Locci M, Havenar-Daughton C, Landais E, Wu J, Kroenke MA, Arlehamn CL, et al. Human circulating PD-1+CXCR3–CXCR5+ memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity. (2013) 39:758–69. doi: 10.1016/j.immuni.2013.08.031
34. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. (2000) 192:1545–52. doi: 10.1084/jem.192.11.1545
35. Morita R, Schmitt N, Bentebibel S-E, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5+CD4+ T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. (2011) 34:108–21. doi: 10.1016/j.immuni.2010.12.012
36. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science. (2013) 341:673–7. doi: 10.1126/science.1241680
37. Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, et al. Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity. (2011) 34:961–72. doi: 10.1016/j.immuni.2011.03.025
38. Kroenke MA, Eto D, Locci M, Cho M, Davidson T, Haddad EK, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol. (2012) 188:3734–44. doi: 10.4049/jimmunol.1103246
39. Ise W, Inoue T, McLachlan JB, Kometani K, Kubo M, Okada T, et al. Memory B cells contribute to rapid Bcl6 expression by memory follicular helper T cells. Proc Natl Acad Sci USA. (2014) 111:11792–7. doi: 10.1073/pnas.1404671111
40. Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. (2011) 208:1377–88. doi: 10.1084/jem.20102065
41. Ioannidis LJ, Nie CQ, Ly A, Ryg-Cornejo V, Chiu CY, Hansen DS. Monocyte- and neutrophil-derived CXCL10 impairs efficient control of blood-stage malaria infection and promotes severe disease. J Immunol. (2016) 196:1227–38. doi: 10.4049/jimmunol.1501562
42. He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, et al. Circulating precursor CCR7loPD-1hi CXCR5+ CD4+ T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. (2013) 39:770–81. doi: 10.1016/j.immuni.2013.09.007
43. Baiyegunhi O, Ndlovu B, Ogunshola F, Ismail N, Walker BD, Ndung'u T, et al. Frequencies of circulating Th1-biased T follicular helper cells in acute HIV-1 infection correlate with the development of HIV-specific antibody responses and lower set point viral load. J Virol. (2018) 92:e00659–18. doi: 10.1128/JVI.00659-18
44. Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJJ, et al. Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLOS Pathog. (2016) 12:e1005945. doi: 10.1371/journal.ppat.1005945
45. Linterman MA, Rigby RJ, Wong RK, Yu D, Brink R, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med. (2009) 206:561–76. doi: 10.1084/jem.20081886
46. Ryg-Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL, et al. Severe malaria infections impair germinal center responses by inhibiting T follicular helper cell differentiation. Cell Rep. (2016) 14:68–81. doi: 10.1016/j.celrep.2015.12.006
47. Zander RA, Vijay R, Pack AD, Guthmiller JJ, Graham AC, Lindner SE, et al. Th1-like plasmodium -specific memory CD4 + T cells support humoral immunity. Cell Rep. (2017) 21:1839–52. doi: 10.1016/j.celrep.2017.10.077
48. Figueiredo MM, Costa PAC, Diniz SQ, Henriques PM, Kano FS, Tada MS, et al. T follicular helper cells regulate the activation of B lymphocytes and antibody production during plasmodium vivax infection. PLOS Pathog. (2017) 13:e1006484. doi: 10.1371/journal.ppat.1006484
49. Zander RA, Obeng-Adjei N, Guthmiller JJ, Kulu DI, Li J, Ongoiba A, et al. PD-1 co-inhibitory and OX40 co-stimulatory crosstalk regulates helper T cell differentiation and anti-plasmodium humoral immunity. Cell Host Microbe. (2015) 17:628–41. doi: 10.1016/j.chom.2015.03.007
50. Fernandes AAM, de Moura Carvalho LJ, Zanini GM, da Silva Ventura AMR, Souza JM, Cotias PM, et al. Similar cytokine responses and degrees of anemia in patients with Plasmodium falciparum and Plasmodium vivax infections in the brazilian amazon region. Clin Vaccine Immunol. (2008) 15:650–8. doi: 10.1128/CVI.00475-07
51. Haque A, Rachinel N, Quddus MR, Haque S, Kasper LH, Usherwood E. Co-infection of malaria and γ-herpesvirus: exacerbated lung inflammation or cross-protection depends on the stage of viral infection. Clin Exp Immunol. (2004) 138:396–404. doi: 10.1111/j.1365-2249.2004.02652.x
52. Matar CG, Anthony NR, O'Flaherty BM, Jacobs NT, Priyamvada L, Engwerda CR, et al. Gammaherpesvirus co-infection with malaria suppresses anti-parasitic humoral immunity. PLoS Pathog. (2015) 11:1–23. doi: 10.1371/journal.ppat.1004858
53. Wijayalath W, Danner R, Kleschenko Y, Majji S, Villasante EF, Richie TL, et al. HLA class II (DR0401) molecules induce Foxp3+ regulatory T cell suppression of B cells in plasmodium yoelii strain 17XNL malaria. Infect Immun. (2014) 82:286–97. doi: 10.1128/IAI.00272-13
54. Eivazi S, Bagheri S, Hashemzadeh MS, Ghalavand M, Qamsari ES, Dorostkar R, et al. Development of T follicular helper cells and their role in disease and immune system. Biomed Pharmacother. (2016) 84:1668–78. doi: 10.1016/j.biopha.2016.10.083
55. Wollenberg I, Agua-Doce A, Hernandez A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol. (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
56. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. (2011) 17:975–82. doi: 10.1038/nm.2425
57. Sage PT, Tan CL, Freeman GJ, Haigis M, Sharpe AH. Defective TFH cell function and increased TFR cells contribute to defective antibody production in aging. Cell Rep. (2015) 12:163–71. doi: 10.1016/j.celrep.2015.06.015
58. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. (2011) 17:983–8. doi: 10.1038/nm.2426
59. Alexander C-M, Tygrett LT, Boyden AW, Wolniak KL, Legge KL, Waldschmidt TJ. T regulatory cells participate in the control of germinal centre reactions. Immunology. (2011) 133:452–68. doi: 10.1111/j.1365-2567.2011.03456.x
60. Sage PT, Sharpe AH. T follicular regulatory cells. Immunol Rev. (2016) 271:246–59. doi: 10.1111/imr.12411
61. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory-like properties. J Clin Invest. (2014) 124:5191–204. doi: 10.1172/JCI76861
62. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity. (2014) 41:1026–39. doi: 10.1016/j.immuni.2014.12.005
63. Metwali A, Elliott D, Blum AM, Li J, Sandor M, Lynch R, et al. The granulomatous response in murine schistosomiasis mansoni does not switch to Th1 in IL-4-deficient C57BL/6 mice. J Immunol. (1996) 157:4546–53. Available at: http://www.ncbi.nlm.nih.gov/pubmed/8906833
64. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. (2010) 207:353–63. doi: 10.1084/jem.20091738
65. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity. (2008) 29:127–37. doi: 10.1016/j.immuni.2008.06.001
66. Wipasa J, Okell L, Sakkhachornphop S, Suphavilai C, Chawansuntati K, Liewsaree W, et al. Short-lived IFN-γ effector responses, but long-lived IL-10 memory responses, to malaria in an area of low malaria endemicity. PLoS Pathog. (2011) 7:e1001281. doi: 10.1371/journal.ppat.1001281
67. Bejon P, Mwacharo J, Kai O, Todryk S, Keating S, Lowe B, et al. The induction and persistence of T cell IFN-gamma responses after vaccination or natural exposure is suppressed by Plasmodium falciparum. J Immunol. (2007) 179:4193–201. doi: 10.4049/jimmunol.179.6.4193
68. Urban BC, Mwangi T, Ross A, Kinyanjui S, Mosobo M, Kai O, et al. Peripheral blood dendritic cells in children with acute Plasmodium falciparum malaria. Blood. (2001) 98:2859–61. doi: 10.1182/blood.V98.9.2859
69. Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F, et al. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect Immun. (2007) 75:3621–32. doi: 10.1128/IAI.00095-07
70. Götz A, Tang MS, Ty MC, Arama C, Ongoiba A, Doumtabe D, et al. Atypical activation of dendritic cells by Plasmodium falciparum. Proc Natl Acad Sci USA. (2017) 114:E10568–77. doi: 10.1073/pnas.1708383114
71. Clemente AM, Fadigati G, Caporale R, Marchese DG, Castronovo G, Sannella AR, et al. Modulation of the immune and inflammatory responses by Plasmodium falciparum. schizont extracts: role of myeloid dendritic cells in effector and regulatory functions of CD4+ lymphocytes. Infect Immun. (2013) 81:1842–51. doi: 10.1128/IAI.01226-12
72. Newman KC, Korbel DS, Hafalla JC, Riley EM. Cross-talk with myeloid accessory cells regulates human natural killer cell interferon-gamma responses to malaria. PLoS Pathog. (2006) 2:e118. doi: 10.1371/journal.ppat.0020118
73. McCall MBB, Roestenberg M, Ploemen I, Teirlinck A, Hopman J, De Mast Q, et al. Memory-like IFN-γ response by NK cells following malaria infection reveals the crucial role of T cells in NK cell activation by P. falciparum. Eur J Immunol. (2010) 40:3472–7. doi: 10.1002/eji.201040587
74. Ing R, Stevenson MM. Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun. (2009) 77:770–82. doi: 10.1128/IAI.00994-08
75. Ryg-Cornejo V, Nie CQ, Bernard NJ, Lundie RJ, Evans KJ, Crabb BS, et al. NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology. (2013) 218:263–71. doi: 10.1016/j.imbio.2012.05.018
Keywords: TH1, TFH1, IFN-γ, follicular T helper cells, B cells, malaria, humoral immunity
Citation: Yap XZ, Hustin LSP and Sauerwein RW (2019) TH1-Polarized TFH Cells Delay Naturally-Acquired Immunity to Malaria. Front. Immunol. 10:1096. doi: 10.3389/fimmu.2019.01096
Received: 26 December 2018; Accepted: 30 April 2019;
Published: 17 May 2019.
Edited by:
Christoph Hölscher, Forschungszentrum Borstel (LG), GermanyReviewed by:
Thomas Jacobs, Bernhard-Nocht-Institut für Tropenmedizin (BMITM), GermanyKylie Renee James, Wellcome Trust Sanger Institute (WT), United Kingdom
Copyright © 2019 Yap, Hustin and Sauerwein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Zen Yap, emVuLnlhcEByYWRib3VkdW1jLm5s
†These authors have contributed equally to this work