 Ole Petter Rekvig
Ole Petter Rekvig- Department of Medical Biology, Faculty of Health Sciences, University of Tromsø, Tromsø, Norway
This study aims to understand what lupus nephritis is, its origin, clinical context, and its pathogenesis. Truly, we encounter many conceptual and immanent tribulations in our attempts to search for the pathogenesis of this disease—and how to explain its assumed link to SLE. Central in the present landscape stay a short history of the early studies that substantiated the structures of isolated or chromatin-assembled mammalian dsDNA, and its assumed, highly controversial role in induction of anti-dsDNA antibodies. Arguments discussed here may provoke the view that anti-dsDNA antibodies are not what we think they are, as they may be antibodies operational in quite different biological contexts, although they bind dsDNA by chance. This may not mean that these antibodies are not pathogenic but they do not inform how they are so. This theoretical study centers the content around the origin and impact of extra-cellular DNA, and if dsDNA has an effect on the adaptive immune system. The pathogenic potential of chromatin-anti-dsDNA antibody interactions is limited to incite lupus nephritis and dermatitis which may be linked in a common pathogenic process. These are major criteria in SLE classification systems but are not shared with other defined manifestations in SLE, which may mean that they are their own disease entities, and not integrated in SLE. Today, the models thought to explain lupus nephritis are divergent and inconsistent. We miss a comprehensive perspective to try the different models against each other. To do this, we need to take all elements of the syndrome SLE into account. This can only be achieved by concentrating on the interactions between autoimmunity, immunopathology, deviant cell death and necrotic chromatin in context of elements of system science. System science provides a framework where data generated by experts can be compared, and tested against each other. This approach open for consensus on central elements making up “lupus nephritis” to separate what we agree on and how to understand the basis for conflicting models. This has not been done yet in a systematic context.
Introduction
In this critical review, different aspects of pathogenic processes suspected or proven to be involved in lupus nephritis are discussed; (i) The exposure of dsDNA, and the impact of its surface structure and net charge exposed in pure dsDNA vs. DNA in chromatin; (ii) Anti-dsDNA antibodies, whether homologous or heterologous depending on whether instigated by DNA or non-DNA structures, and what they recognize in glomeruli; (iii) If lupus nephritis in a critical sense is an intrinsic part of SLE; and as a direct consequence of the last question; (iv) Whether SLE is an abstraction without a clear definition, which may allow us to regard lupus nephritis as a single disease entity; and (v) Whether production of anti-dsDNA antibodies induce the same pathogenic processes in non-SLE (like in cancer) patients as they do in SLE. In other words, can lupus nephritis etiologically be regarded as an integrated part of SLE—or can it stand alone? These dilemmas may not center around a clinical diagnosis, but around processes that may describe the molecular and cellular events that in sum define lupus nephritis. In this context, it is important to discuss factors that prime the inflammatory processes in lupus nephritis, and not secondary inflammatory mediators like complement activation, cytokines or their receptors, because the initiators of lupus nephritis inherit the principle, while inflammatory pathways are secondary responses instigated by the principal inducers of lupus nephritis—like type II or type III immune mediated tissue inflammation. In fact, if we summarize data over the last decades, both type II and type III have been claimed to account for lupus nephritis. One tribulation is whether type II immune mediated nephritis is more like Goodpasture syndrome (1, 2) than like lupus nephritis. However, there are many more problems that need to be solved before we can develop a true pathogenic model of lupus nephritis (see below). These problems represent the focus of this study.
The dsDNA: Structure, Autoimmune Inducer, and Target—Status and a Short Scientific History
In two foregoing studies, an historical and contemporary overview of anti-dsDNA antibodies (3) and a condensed history of the evolution of our contemporary opinions on SLE (4) have been published. These two studies aimed at a central understanding of the role of dsDNA and how it is involved in lupus nephritis. On the other hand, it is possible that dsDNA plays a bystander role in the disease, if e.g., anti-dsDNA antibodies recognize different obligate glomerular structures (see below). In that sense it is essential to approach historical and contemporary studies and hypotheses as backdrops to understand how paradigms related to SLE and anti-dsDNA antibodies have evolved over time. In other words, history is also in this context important to consider in order to understand contemporary paradigms. Ludvik Fleck once said: “For the current state of knowledge remains vague when history is not considered, just as history remains vague without substantive knowledge of the current state” [(5), cited in (4)].
Whether the antibodies described in 1957 in SLE (6–9) were specific for dsDNA and not for other DNA structures like ssDNA can be discussed in terms of history of science on dsDNA. The scientific history of DNA originates from studies performed during the 19th century. DNA was first identified as a unique substance in the late 1860s by the Swiss chemist Friedrich Miescher [(10), see also the biographical presentation of Miescher by (11)]. In the aftermath of Miescher's discovery, studies revealed fundamental details about the DNA molecule. This resulted in important discoveries describing the chemical composition of DNA, including its primary chemical components and the ways in which chains joined with one each other. Central scientists were Phoebus Levene, who provided evidence that different forms of nucleic acids existed—DNA and RNA, and he also determined that DNA contained adenine, guanine, thymine, cytosine, deoxyribose, and a phosphate group (12); and Erwin Chargaff, who was the first to present evidence that the DNA structure exists as a double helix constituted by two complementary single-strand DNA molecules (13, 14) (see below). With these important and pioneering studies, the enigma of inheritance started to be revealed.
A central researcher aiming to solve this scientific puzzle was Rosalind Elsie Franklin, a British chemist [see the comprehensive biography on Rosalind Franklin by Brenda Maddox, (15)]. Franklin contributed significantly to the discovery of the three-dimensional structure of dsDNA by X-ray crystallography (16). In these studies, she precisely described the double helix of DNA, a discovery that placed her in the first row of those days biochemical scientists aimed to describe the nature, structure and function, basically of Miescher's “nuclein” (11) and transformed it into dsDNA! Watson and Crick and their studies that were published in 1953 stated that the DNA molecule exists in the form of a three-dimensional double helix (17). Their conclusions were based particularly on Franklin's analyses and her interpretations, but also on results of the studies performed by Levene and Chargaff. One may consider if Watson and Crick at all should be in the first line of candidates to receive the Nobel price. Levene, Chargaff and Franklin presented all the elements to describe dsDNA as a double helix three-dimensional DNA structure.
The central work of Chargaff, Levene, and Franklin were remodeled into the paradigms now called The Chargaff's rules. These paradigms state that any DNA from cells of any species have a 1:1 ratio (base Pair Rule) of pyrimidine and purine bases. They stated that the amount of guanine equals the amount of cytosine, and that the amount of adenine equals the amount of thymine. This double helix pattern of DNA is equal in DNA from all species and provides evidence that we all evolve from the same genetical principle (see Figure 1 and Table 1).
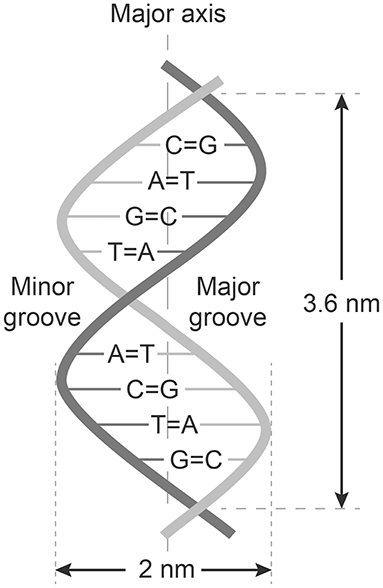
Figure 1. Structure of dsDNA and Chargaffs rules for a double-helix dsDNA. In this figure Chargaff's first rule demonstrates that DNA from any cell of all organisms have a 1:1 ratio (base Pair Rule) of pyrimidine and purine bases and, more specifically, that the amount of guanine is equal to cytosine and the amount of adenine is equal to thymine. This pattern is found in both strands of the DNA. The figure also demonstrates Chargaffs second rule saying that the proportion of A/T and C/G holds true for both strands.
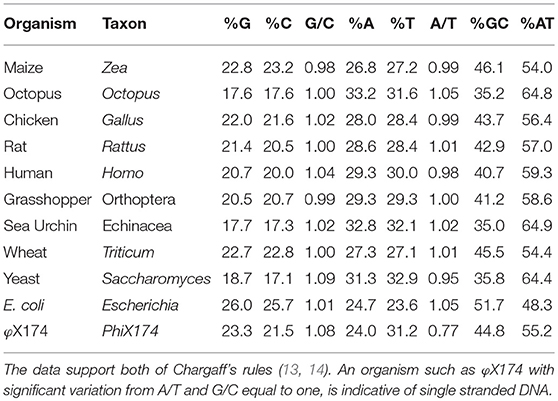
Table 1. This table is a representative sample of Chargaff's et al. (13) data, taken with slightly modified table published by Bansal (18), listing the base composition of DNA from various organisms.
Chargaffs Rules (13, 14)
Chargaff demonstrated that the double helix was created and stabilized by A–T and C–G interactions. The data of his experiments were organized and summarized as Chargaff's Two Rules (see Table 1 for examples including human dsDNA):
1. The number of Adenine bases is equal to the number of Thymine bases and the number of Cytosine is equal to Guanine bases: (nA = nT; A/T = 1; nC = nG; C/G = 1), and the sum of A, T, C, G, is always 100% in the DNA double helix molecule isolated from a cell.
2. The proportion of A/T and C/G holds true for both strands.
In sum: A/T = G/C = 1.
All antibodies that bind nucleic acids characterized by the ratio in the formula 1 given above must consequently bee defined as anti-dsDNA or anti-native dsDNA antibodies.
Considering the rough methods Chargaff, co-workers and successors used to purify nuclein (DNA), the double helix must have been very robust. We know that the DNA purified for the purpose of detecting anti-dsDNA antibodies was in fact dominantly dsDNA also without further active elimination of ssDNA [(19, 20), Rekvig, unpublished observations]. Thus, the dsDNA structure described above turned out to be the target for anti-dsDNA antibodies in SLE, a statement that also may be valid for the early 1938 detection of anti-DNA antibodies in context of infections (21–23), and that complexes between them had the potential to induce inflammation in SLE-related lupus nephritis [for review see e.g., (4) and below].
Anti-dsDNA Antibodies: How Are They Formed—and in Which Principal Clinical Contexts
To answer these questions, we have to rigorously define whether an anti-dsDNA antibody represents a response to exposed dsDNA or to a non-dsDNA/non-DNA structure by molecular mimicry (see below).
Anti-dsDNA Antibodies: Is dsDNA a Stable Structure That May Be Immunogenic in vivo?
Interpretation of the structure originally called nuclein, as a derivation from Chargaffs rules, the DNA was most probably used in the first assays in the form of the canonical double helix DNA. Since the A/T and G/C ratios were stable [Table 1, and Figure 1 (13, 17)] and since they in sum did not deviate toward an overrepresentation of any of the bases that could indicate presence of ssDNA domains (Table 1), we can in retrospect conclude that the autoantibodies observed in 1957 in SLE (6–9) recognized dsDNA as a stable structure—and that they cross-reacted with dsDNA from as different species as like viral, bacteria, and mammals. DNA is present in all nucleated cells. If exposed chromatin is potentially dangerous to the body, as discussed by e.g., Darrah and Andrade (24) and by others (25–29), this may illuminate how important it is to remove chromatin from dying cells in an abrupt and silent way.
However, in individuals with anti-dsDNA antibodies and impaired clearance of cell debris including necrotic chromatin, like in SLE (30–37), this may change the situation from a controlled removal of chromatin into a condition where chromatin debris remains exposed and may be a contributor to produce and or amplify anti-dsDNA antibodies by interaction with TLR9 and to promote inflammation. This may be caused by slow removal of extra-cellular dsDNA by e.g., silencing of DNase I or blocking of DNase I activity since binding of anti-dsDNA antibodies to DNA may inhibit the effect of the endonucleases [discussed in (29)]. In this situation, externalized DNA is further targeted by anti-dsDNA/anti-chromatin antibodies, and immune complexes may be formed both in situ and in circulation (3, 38–40) and, as a consequence, induce serious inflammation (see below).
Furthermore, once anti-dsDNA antibody production is initiated (irrespective mechanism), an anti-dsDNA antibody may bind dsDNA in the extra-cellular compartment. In a normal situation, in vivo autologous dsDNA is rapidly and completely digested by DNases. On the other hand, anti-dsDNA antibodies may be produced and form immune complexes with the consequence that autologous DNA-containing fragments are resistant to DNases, then they may bind the DNA-specific B cell receptor and is transported into endosomes/lysosomes where TLR9 is sensing unmethylated CpG dinucleotides (41). Stimulated TLR9 acts via MYD88 and TRAF6, leading to NF-kappa-B activation, cytokine secretion and the inflammatory response (42, 43). TLR9 promote in this way increased inflammation and amplification of anti-dsDNA antibodies [(44, 45), see a model in Figure 2].
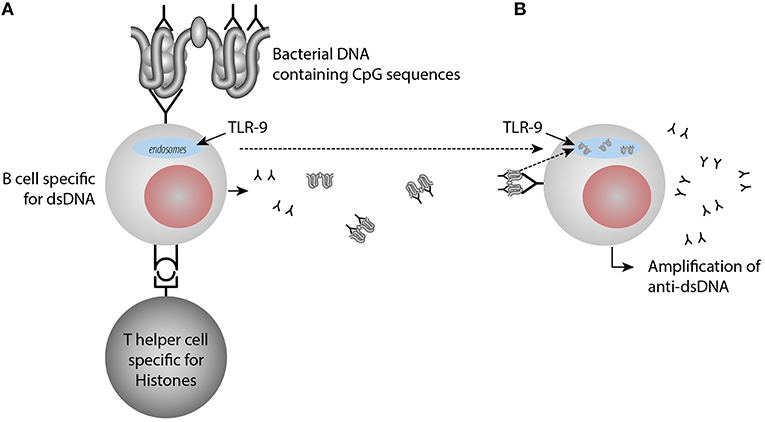
Figure 2. Amplification of anti-dsDNA antibody responses through activation of TLR9 by immune-complexes containing DNA-anti-DNA. In (A) anti-dsDNA antibodies are induced by a classical hapten-carrier complex, in which dsDNA in form of small chromatin complexes represent the hapten and histone peptides represent the carrier protein. These interactions transform B cells into anti-dsDNA antibody producing plasma cells and enter the extracellular space. Upon cell death, chromatin is degraded and removed in a fast and silent way by DNases. Anti-dsDNA antibodies bind these small chromatin fragments, make them resistant to DNase. Then they bind dsDNA through the dsDNA-specific B cell receptor and the dsDNA fragments enter into the endosomes, where TLR9 is sensing unmethylated CpG dinucleotides (B). Stimulated TLR9 promotes cytokine secretion and the inflammatory response and amplification of anti-dsDNA antibodies. TLR9 promote in this way increased inflammation and amplification of anti-dsDNA antibodies upon TLR9 sensing of CpG9.
In sum, the pioneers that described nuclein (11) as dsDNA (12, 13, 17), had a substantial influence on the discovery of anti-dsDNA antibodies in an autoimmune context in 1957 (6–9) and on the potential of dsDNA and anti-dsDNA antibody complexes to drive inflammation, as we see in e.g., lupus nephritis.
SLE: A Disease With High Rates of Infectivity and DNA-Specific Autoimmunity—Is the Latter Depending on the First?
Does the immune system need infection as a sine qua non-contributor to incite anti-chromatin/anti-dsDNA antibodies (see main hypothesis in Figure 3A)—and is this hypothesis a factual substantiation of the hapten-carrier system in which T cells recognize the infectious-derived chromatin-bound protein (exemplified by polyomavirus T antigen in Figure 3A) while B cells recognize hapten-like autologous chromatin structures as dsDNA, histones, transcription factors and other chromatin-associated proteins [Figure 3A (46–50), reviewed in (3, 51)].
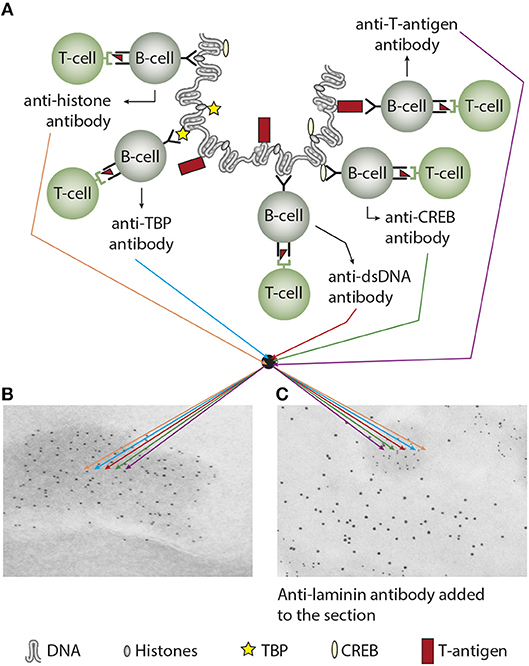
Figure 3. Experimental induction of anti-dsDNA antibodies and other chromatin autoantibodies by in vivo expression of a single viral dsDNA-binding protein. In (A), Injection of normal mice with plasmids encoding wild type polyomavirus DNA-binding T antigen in context of eukaryotic promoters predictively induced production of antibodies to T antigen and significant production of antibodies to mammalian dsDNA, histones, and to certain transcription factors like TATA-binding protein (TBP) and cAMP- responsive element-binding protein (CREB). All autologous chromatin-derived ligands physically linked to T antigen can therefore be rendered immunogenic to autoimmune B cells that present peptides derived from T antigen. Therefore, concerted production of autoantibodies specific for chromatin antigens, including dsDNA and histones, is not depending on a systemic lupus erythematosus background, but may appear also in quite healthy individuals. In (B) the group of chromatin autoantibodies notably including anti-dsDNA antibodies target exposed chromatin in kidneys. As demonstrated by immune electron microscopy, it is evident that the autoantibodies target electron dense structure (EDS), convincingly demonstrated to constitute chromatin fragments (the left immune electron microscopy in (B). These autoantibodies did not bind GBM structures or in the mesangial matrix (seen as clean membranes). However, anti-laminin antibodies added to the sections in vitro bound GBM (sees as 10 nm gold particle-labeled antibodies), and they did not co-localize with in vivo-bound anti-chromatin antibodies (seen as 5 nm gold particles) (C). These data argue for the fact that anti-dsDNA/anti-chromatin antibodies bound chromatin fragments, and they did not bind inherent, regular membrane components. (A) Is copied from Rekvig (4).
Over so many years, we have not succeeded in understanding what the immune system recognize and act upon in context of spontaneous production of anti-dsDNA antibodies in vivo. Since the 1980s, studies were concerned around the following problems; what instigated anti-dsDNA antibodies, what were their targets, dsDNA or non-DNA structures (52–54), why did they correlate with disease (SLE) [(55), reviewed in (4)], how to detect them in the most appropriate way, and what make them pathogenic (56–58)? This is a concentrate of the problems being in focus over the last 50 years—and still is. Do cell death in context of infection, and consequent release of hetero-complexes between host chromatin and infectious-derived ligands explain the whole repertoire of chromatin antibodies in SLE (see Figure 3A), known to be overrepresented with respect to infections and to factual production of anti-dsDNA antibodies [see e.g., references (59–72)]? For example, the Epstein-Barr nuclear antigen 1 (EBNA1) (50); the C-terminal DNA-binding domain of the human papillomavirus E2 protein (46); the Fus 1 peptide derived from Trypanosoma cruzi (73); or polyomavirus large T antigen (47, 48) have all the evident and predictive potential to render dsDNA/chromatin immunogenic in vivo upon complex formation. Infection and autoimmunity may therefore be linked together in many situations where infections tend to be chronic or recurrent, and cell death rates are high (see Figure 3A as a model to explain linked production of different chromatin antibodies when infectious-derived proteins bind chromatin fragments). This model is not restricted to chromatin autoimmunity, but also to other autologous proteins. For example, Dong et al. demonstrated that complexes of T antigen and the tumor suppressor protein p53 terminated tolerance to p53 (74, 75).
In this sense, one idea is that infectious DNA-binding proteins and DNA/chromatin fragments are walking hand in hand in their successful promotion of chromatin autoimmunity. In this picture B cells represent the autoimmune hand while infectious protein-specific T cells represent the immune hand—and upon contact they stimulate each other and transform the B cells to be autoantibody-producing plasma cells. This has been directly demonstrated in studies were T antigen-expressing plasmids were injected in experimental animals under control of eukaryotic transcription factors (47, 48). In this context, exposed chromatin and its different molecular structures can all be targeted by anti-dsDNA and anti-chromatin antibodies if induced by chromatin fragment-viral DNA-binding proteins (See Figures 3A,B for a model thinking). This model says that specters of chromatin antibodies which are induced by chromatin-peptide complexes all can target exposed chromatin in situ and provoke serious inflammation.
Cancer: A Group of Malignant Diseases With High Rates of Infectivity and DNA-Specific Autoimmunity—Is the Latter Depending on the First?
In this sense, cancer may represent a mirror image of autoimmunity in SLE with respect to infectivity rates and termination of tolerance for dsDNA and chromatin constituents. In line with this, anti-dsDNA antibodies are frequently detected in cancers [reviewed in (3, 4)]. In 1991, the Nobel prize winner zur Hausen suggested that most of all of human cancers worldwide are linked to viral infections, including human papillomaviruses, human T-cell leukemia viruses, hepatitis B virus, Epstein-Barr virus and polyomaviruses (76, 77). At the same time, several virus-associated cancer forms are connected with the production of autoantibodies against dsDNA [see e.g., (47, 48) reviewed in (3)]. The impact of viruses in different cancer forms, and if or how viruses influence the malignancy of tumor cells may, according to zur Hausen, need to be revised in light of new viruses that has been discovered in cancer forms since zur Hausen's data were discussed in his 1991 Science paper (76). Since cancer and SLE are largely segregated, the slight over-representation of cancer in SLE (78) does not reduce the arguments for the view that anti-dsDNA antibodies are generated independently, although possibly by similar molecular and cellular mechanisms (3) in the two different types of conditions.
In SLE, the antibodies may crossreact with and bind inherent renal antigens or chromatin fragments exposed in kidneys [(79), present study] and initiate nephritis, although the two binding profiles are principally different as one is type II and the other is type III immune mediated inflammation. On the other hand, inflammation in juxtaposition to a tumor may indicate that autoantibodies may target chromatin released from necrotic tumor cells and promote local inflammation in analogy to kidney inflammation caused by antibody-binding to chromatin exposed in glomeruli (80, 81). Implication of anti-dsDNA antibodies in tumor-associated tissue has not been directly investigated. Also in cancers, anti-dsDNA antibodies are from a principal concern not clinical epiphenomenons, although their genesis is still poorly understood (if not categorized as local infectious-driven autoantibodies as principally outlined in Figures 3A,B). One potent hypothesis may therefore be the impact in cancers of infections and anti-dsDNA antibodies that are induced by complexes of tumor-derived chromatin and DNA-binding infectious-derived peptides.
Genesis of the Anti-dsDNA Antibody in vivo: Closely Linked to Infections
The role of light chain editing to abolish and control anti-dsDNA reactivity is recently discussed in SLE [see reference (4) for a brief discussion]. This type of regulation can be impaired by SLE susceptibility factors, thereby allowing DNA-specific B cells to expand in SLE [see (4) and references herein].
Till now, no clear evidence have been presented that convincingly state that anti-dsDNA antibodies are initiated by sole exposed autologous DNA/chromatin [(3, 51, 73), discussed in a highly relevant way back in 1994 by (82)], irrespective whether they are exposed as native or cell death-associated modified chromatin structures [discussed in (3, 29, 51)]. However, infectious-derived DNA/chromatin-binding proteins in complex with chromatin fragments can provide strong T helper cell stimuli and promote transformation of autoimmune B cells into autoantibody-producing plasma cells (3). This brings to light that the infectivity state characteristic of SLE or of cancers is in intimate context with (auto-)immune competent cells both physically and functionally (47, 48). This was directly hinted on already at the time of the first discovery of anti-dsDNA antibodies in 1938–1939 in patients suffering from bacterial infections (21–23, 83). Other up today examples of the link between polyomavirus infection and anti-dsDNA antibodies was shown in small children with primary polyomavirus BK infections (84). These infected children produced antibodies to polyomavirus T antigen and transiently to mammalian dsDNA. T antigen is the BK virus' transcription factor and is therefore a DNA-binding protein that in a native situation binds both viral and host cell DNA [see above, reviewed in (85)]. In this situation T antigen was assumed to serve as a T helper cell-stimulating protein presented by DNA-specific B cells, once the T cells had been primed by dendritic cells presenting T antigen-derived peptides [discussed in Figure 3, reviewed in (3)]. Thus, both along the spontaneous BK virus infection (48, 84) and as a consequence of experimental expression of T antigen in vivo or other infectious agents, appearance of anti-dsDNA and other anti-chromatin antibodies is a predictive outcome [(46, 47), reviewed in (51)]. Then, why do children with primary BKV that produce anti-dsDNA antibodies not develop lupus-like nephritis or dermatitis? This may be explained by absence of exposed chromatin in glomeruli and in the dermal basement membrane zone of the skin due to the transient nature of the infection. This will be further discussed below.
Deviant Cell Death Events Promote Exposure of DNA/Chromatin—Immunogen or Target?
If exposed dsDNA in form of chromatin has the potential to induce anti-dsDNA antibodies remain as an attractive, although yet an unproven model (29). Chromatin released by cell death may be linked to aseptic inflammation, and to the role of disordered cell death processes like exposure of DNA-containing neutrophil extracellular traps (NETs), secondary necrotic chromatin, microparticles, and may be linked to reduced elimination of dead cell debris (whether of apoptotic or necrotic origin) (27, 33, 86). NETs were first observed by Brinkmann et al. (87). Still, however, their function as an assumed complex defense structure (88) is not fully resolved [see a thoughtful discussion by (89)]. On the other hand, NETS and secondary necrotic chromatin have in several studies been suspected to be involved in inflammatory processes (28, 35, 90–92), and is assumed to account for increased levels of anti-chromatin antibodies. The latter association does not, according to my understanding of the relevant literature, mean that NETS or apoptotic chromatin induce anti-dsDNA antibodies. This is thoroughly discussed by Gupta and Kaplan in their review (25) who reached the conclusion that “….many of the molecules externalized through NET formation are considered to be key autoantigens and might be involved in the generation or enhancement of autoimmune responses in predisposed individuals…..” However, they did not state that NETS had the potential to induce anti-dsDNA antibodies. Similarly, Pieterse and van der Vlag conclude in their study “…it can be concluded that increased apoptosis or NETosis on its own is not sufficient to break immunological tolerance to nuclear autoantigens in SLE, and additional factors are required to turn apoptotic material or NETs into danger triggers of autoimmunity.” (29). Still, we do not understand whether NETs, necrotic chromatin or microparticles have the potential to induce antibodies to dsDNA or to native histones, although it has been demonstrated that they may initiate antibodies against cell death-modified histones [discussed in (25)].
In support of these considerations, Radic and Dwivedi have recently published a comprehensive and critical review on controversies related to NETs, cell death and autoimmunity (93). They came to the same conclusion as presented here as they hesitate to accept that NETs promote humoral autoimmunity against native chromatin components, inclusive dsDNA. The autoimmune consequence of perturbed order of cell death and the impact on adaptive immunity is hard to comprehend. It is probably an abstraction and not proven by evidence that these processes have the potential to promote production of anti-dsDNA antibodies, although the same structures may drive innate immune-dependent inflammation in SLE (36, 90, 94). However, diminished removal of nuclear debris has been demonstrated to correlate with production of antibodies to cell death-induced structural changes of proteins in chromatin. This is in harmony with earlier observations that while histone H4 is non-immunogenic, triacetylated histone H4 is (95). Recently, Dieker et al. observed that autoantibodies against modified histone peptides in SLE patients were associated with disease activity and lupus nephritis (91).
Similarly, T cell responses to analogous modified structures do not allow us to interprete such (helper) T cells as activator of B cells and thus induce true, anti-native dsDNA autoantibodies [(26, 91, 94, 96–104), reviewed by Pieterse and van der Vlag (29)]. Data that demonstrate that deranged cell death debris can activate T and B cells specific for altered self chromatin are settled by solid experiments (26, 35, 91, 91, 92, 105). Whether antibodies or T cells against death-associated chromatin modifications have the potential to induce inflammation has not been thoroughly studied, but their recognition and binding to modified (homologous) chromatin structures exposed as NETs might well promote in situ formed immunocomplexes, and consequently inflammation. In harmony with these critical comments, Gordon et al. (96) demonstrated that NETs inhibition by different approaches, like genetically manipulated Nox-deficient mice, or by deletion of PADi4 or pharmacological inhibition of PAD4 activity hardly had any influence on nephritis, and NETs inhibition did not affect any aspects of nephritis, did not lead to loss of tolerance, nor to immune activation (96). Pharmacological inhibition of PAD activity did not affect the progression of nephritis into end-organ disease in inducible models of glomerulonephritis. The authors conclude that the data oppose the concept that NETs promote autoimmunity and target organ injury in SLE (96) in agreement with earlier observations by Campbell et al. (97).
Nevertheless, NETs may serve as in situ targets for the autoimmune responses and participate in evolution of organ injury in SLE. Thus, true anti-dsDNA antibodies may have the ability to sensitize NETs by forming immune complexes and to initiate inflammation since dsDNA in NETs may remain in their native state, and not modified during deviant cell death, as opposed to immunity to chromatin in secondary necrotic cells in which apoptotically modified autoantigens (dsDNA, high mobility group box 1 protein, apoptosis-associated chromatin modifications, e.g., histones H3-K27-me3; H2A/H4 AcK8,12, 16; and H2B-AcK12) are present (106).
Autoimmunity to dsDNA and native chromatin exists, but till now, their spontaneous appearance in a native context is still enigmatic. There is no solid evidence to say that native chromatin has immunogenic potential. However, native chromatin in complex with a DNA-binding viral protein (see above) is immunogenic because T cell tolerance, as is operative for native chromatin, is circumvented by the immunogenic infectious-derived carrier protein. There are yet no firm evidence stating that antibodies to native dsDNA are induced by perturbed cell death, although disorganized cell death may induce and enhance production of antibodies to chromatin-associated proteins modified in context of cell death (29, 93). Thus, although anti-dsDNA antibodies are easily detected in SLE, it is hard to explain why the antibodies materialize themselves and how they harm organs like the kidneys and skin in context of SLE (see below).
Pathogenic Potential of Anti-dsDNA Antibodies
Isolated dsDNA is negatively charged due to solvent phase exposed phosphate groups that makes up every nucleotide that consists of pentose, nitrogenous bases, and phosphate groups (see above). This makes it unlikely that isolated dsDNA binds directly to glomerulus basement membranes (GBM) in context of lupus nephritis because GBM is overall anionic and would therefore repel dsDNA. Rather, since mammalian dsDNA is part of chromatin, consisting of histone octamers, histone H1, and a large array of other non-histone proteins with various charges, dsDNA may indirectly bind to GBM through interaction of solvent phase cationic protein tails with anionic GBM structures. This forms the basis for formation of immune complexes between anti-dsDNA antibodies and dsDNA, and deposition of the complexes in situ along the GBM, and in the mesangial matrix of circulating dsDNA-containing immune complexes [reviewed in (3) and (81)]. By using surface plasmon resonance, we demonstrated that isolated dsDNA did not bind collagen or laminin, while chromatin fragments bound with relatively high avidity, irrespective presence or absence of complex-bound anti-dsDNA antibodies (107, 108). These data harmonize nicely with experiments performed in the Berden laboratory, where they demonstrated that immune complexes of anti-dsDNA antibodies and nucleosomes bound in glomeruli of perfused kidneys, while highly pure anti-dsDNA antibodies did not bind (109–111). Nevertheless, these data open for two ways how chromatin may promote inflammation; either by binding anti-dsDNA antibodies to chromatin exposed in situ, or by binding preformed chromatin-IgG complexes to GBM.
Anti-dsDNA Antibodies: Are They Induced by dsDNA or Non-dsDNA Structures in vivo?
On the other hand, anti-dsDNA antibodies have in many studies been proven to be instigated by non-DNA structures [discussed in e.g., (3, 112–114), see Table 2 for examples]. Therefore, anti-dsDNA antibodies may represent two principally different antibody populations; real anti-dsDNA antibodies induced by dsDNA, or (quasi) antibodies with potential to bind dsDNA although instigated by non-dsDNA structures. We are today not able to distinguish which is which. In context of the question if anti-dsDNA antibodies are induced by dsDNA or non-dsDNA structures in vivo, a logic issue would be if anti-dsDNA antibodies are pathogenic because they recognize dsDNA (homologous interaction) or non-dsDNA (heterologous interaction) in the kidneys.
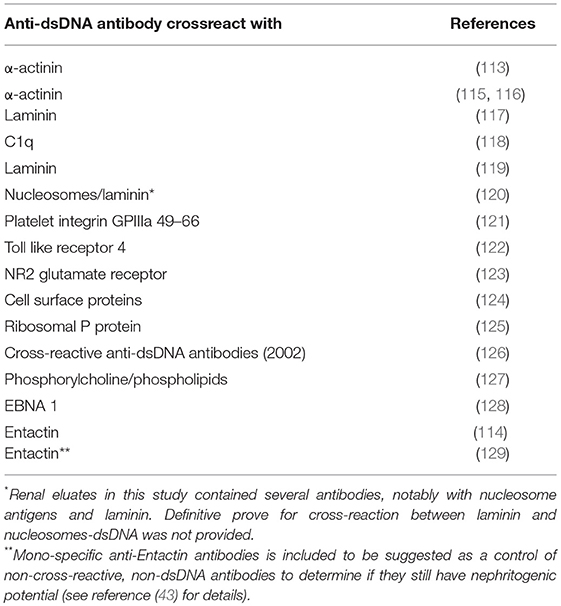
Table 2. Examples of anti-dsDNA antibodies that cross-react with non-DNA structures.
Thus, anti-dsDNA antibodies may exert a pathogenic process by direct binding to inherent cross-reactive renal structures. This demonstrates that anti-dsDNA antibodies may promote two principally opposite pathogenic processes; They either bind chromatin fragments that are exposed and associated with GBM structures [denoted in this context “the chromatin model” see models in Figures 3, 4 (80)] or, they bind directly to glomerular antigens like laminin, collagen, entactin, and others by cross-reaction (denoted “the cross-reaction model,” see relevant data in Table 2, and Figure 4). Two variants of the chromatin model exist. In one; chromatin fragments are exposed in membranes and matrices due to the fact that chromatin fragments bind membranes and matrices at high affinity. If anti-dsDNA antibodies bind this form of chromatin, the immune complexes are formed in situ (107). In the other variant, formation of IgG-chromatin immune complexes occurs in circulation. Such pre-formed IgG- chromatin fragment immune complexes may bind primarily in the mesangial matrix and in GBM [reviewed in (3) and (4)]. The second variant is experimentally demonstrated. Injection of immunologically normal mice with highly pure anti-dsDNA monoclonal antibodies (mAbs) resulted in deposition of chromatin-fragment–IgG complexes in the mesangium and GBM (131, 132). Concentration of circulating chromatin fragments was significantly reduced after infusion of the antibodies. Similarly, if (NZB × NZW) nephritic mice were injected with pure biotinylated mAbs, these antibodies were observed in immune complex deposits observed as electron dense structures (EDS) in the mesangial matrix and in GBM (132). These data demonstrate that the experimental mAbs bound chromatin fragments in vivo. However, these experiments did not allow us to conclude whether they formed immune complexes in situ or in circulation. In line with this, circulating DNA–anti-dsDNA antibody complexes have been described (133) and discussed (134) in the context of SLE. Whether circulating complexes were associated to a certain stage of nephritis was not determined in those studies.
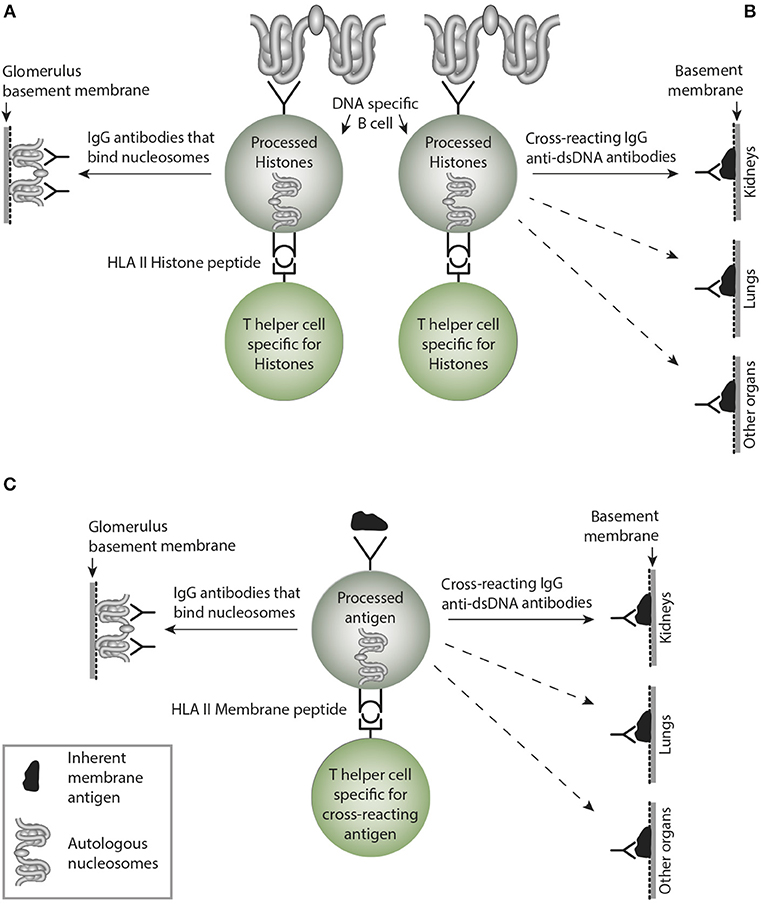
Figure 4. Non-cross-reacting and crossreacting IgG immune responses induced by homologous or cross-reacting antigens. In (A) the B cells are specific for DNA as presented in chromatin. In the left side, B cells recognize and produce antibodies that bind (dsDNA) chromatin, i.e., homologous recognition. These may target exposed chromatin and initiate lupus nephritis by a Type III immune mediated tissue inflammation. In (B) the B cells are specific for dsDNA, but may, however secrete cross-reacting antibodies not targeting solely dsDNA. Instead they bind non-dsDNA cross-reacting inherent glomerular basement membranes, like entactin, laminin or collagen (see text). This has not been entirely investigated, but these cross-reactive antibodies might well initiate Goodpasture analog kidney, skin or lung diseases. This has not been considered in the literature (see text). In (C), the B cells are specific for a (membrane)-component and cross-react with nucleosomes. Since the IgG antibodies may recognize membrane components in e.g., lung and other organs, they inherit the nature of collagen IV-like antibodies in Goodpasture syndrome. Although many such cross-reactions have been described, they have not drawn much attention in pathophysiological contexts. More studies are needed to explore these contexts. This figure is reprinted from Rekvig (130).
Combining data discussed above, mesangial nephritis, representing the early phase of lupus nephritis, may be instigated by circulating immune complexes (132), while progression of lupus nephritis into end stage organ disease is associated with silencing of the major renal endonuclease DNase I (see below). Loss of renal DNase I leaves chromatin from apoptotic cells undigested, and being retained in GBM. In situ formation of immune complexes by binding of circulating anti-dsDNA antibodies to the exposed chromatin fragments forms the basis for severe lupus nephritis (135). Thus, the chromatin model is in clear opposition to the cross-reactive model, and reflects the real pathogenic process of lupus nephritis (see below). It seems that we are far from reaching consensus on pathogenesis of lupus nephritis. Importantly, a proven cross-reactivity of an anti-dsDNA antibody will not provide information about which of the target antigens that binds the antibody in vivo.
Why Are Anti-dsDNA Antibodies Pathogenic—and Are They All Pathogenic?
A central question is if the pure existence of anti-dsDNA antibodies is pathogenic through binding cross-reactive, obligate renal structures in SLE, or if they are epiphenomenons in absence of exposed chromatin structures (3, 4, 114, 136, 137). Thus, we have to evaluate how and why they are harmful, and in which contexts (36, 90, 94, 114, 138). This dilemma relates to the pathogenic effect irrespective whether in SLE or in other diseases like different cancer forms (see above). There is no reason to assume that an anti-dsDNA antibody produced in context of SLE differ in pathogenic impact from those produced in context of infection or cancer. There are many reasons to argue for and against these paradigms. However, these antibodies are pathogenic, but only in presence of relevant target structures. In other words; all have pathogenic potential, but they do not always transform potentiality into activity—i.e., transformation depends on whether the targets are exposed and accessible in vivo.
Lupus Nephritis: Contexts and Pathogeneses
While in end 1930s, DNA without further structural distinction or knowledge was determined to be an acceptor for anti-dsDNA antibodies (21–23, 83, 139). Shortly after the presence of anti-dsDNA antibodies were confirmed, they were in 1957 described in SLE (6–9, 140). In the following text, exposed dsDNA, like in NETs, chromatin or microparticles will not be further discussed. Rather, specificity of nephritogenic antibodies will be the focus.
In context of the discovery of SLE-related anti-dsDNA antibodies, it was soon clear that these antibodies were associated with SLE and with lupus nephritis. This perception represented a conceptual advantage in our understanding of pathogenic processes in SLE, although Fu et al. (136) proposed that anti-dsDNA antibodies are not crucial nor necessary to cause lupus nephritis. Nevertheless, this concept derives from 5 facts: (1) DNA has been reported to bind collagen, a component of GBM (141); (2) chromatin fragments bind laminin and collagen (107); (3) the nephritogenic antibodies bind DNA (chromatin fragments) (38, 142); (4) anti-dsDNA antibodies can be purified from nephritic kidneys (38, 143, 144); (5) infusion of anti-dsDNA antibodies promote nephritis by binding glomerular structures (either GBM or exposed chromatin) in non-autoimmune mice (114, 122, 131, 132, 145, 146).
In a strict context, these facts involve recognition of DNA by antibodies linked to autoimmune inflammation in SLE, but do not necessarily provide information about which of the structures represent glomerular targets for the SLE-associated antibodies [chromatin or inherent glomerular structures, see e.g., the divergent interpretations by (130) and (40)]. The data only demonstrate that the pathogenic antibodies recognize at least dsDNA. As we will see, anti-dsDNA antibodies may even not by definition be denoted anti-dsDNA antibodies due to the vast number of cross-reactions/cross-stimulations with non-DNA/non-chromatin ligands or complex structures—like those in matrices and membranes (see the following discussion of this problem). Traditionally, we call this antibody family “anti-dsDNA” and/or “cross-reacting anti-dsDNA” antibodies. But these are merely biological abstractions as long as we are not able to explain their real initiators and targets in vivo.
Lupus Nephritis Pathogenesis: The Need to Distinguish and Validate Pathogenic Models
From what we know today, we may be forced to define a hierarchy of antibody specificities that are involved in the genesis of lupus nephritis. This may, surprisingly, not be a concise distinction: Maybe monospecific anti-dsDNA antibody is a fiction—indicating that all antibodies are oligospecific (multiple specificities)—or at least cross-reactive (dual specificity)? We have simply not yet sufficient information about this problem [see e.g., (147, 148)]. These somewhat naïve statements cannot rule out other non-DNA lupus nephritis-associated inflammatory factors, like antibodies that are dominantly specific for glomerular structures, as collagen (2, 149), laminin (115, 150), entactin (114), or the role of T cells [see e.g., discussions in (151, 152, 152–156)]. These may be relevant candidates to understand the inflammatory genesis of lupus nephritis.
A Hierarchy of Disparate Anti-dsDNA Antibodies Are Pathogenic in Lupus Nephritis
In this context, there is an imperative need to understand the biological and pathogenic meaning of these factual observations. Therefore, we have to dissect in vivo-bound antibodies and antibodies with potential to bind in vivo, into four categories:
• Antibodies specifically binding chromatin and DNA (51), and anti-dsDNA antibodies that may be formed as a consequence of somatic mutation, even though the reverted germ-line V regions did not show any measurable autoreactivity in the elegant study of Wellman et al. (157). Their results indicate that anti-dsDNA autoantibodies may even develop from non-autoreactive B-cells by somatic hypermutation (157);
• Antibodies that cross-react with DNA and non-DNA glomerular structures (see Table 2, for examples and corresponding references);
• Antibodies that bind native chromatin fragments but not dsDNA;
• Antibodies bound in vivo but have no specificity for chromatin structures, but for glomerular non-DNA structures exposed in the membranes, like entactin, laminin, and collagen [see Table 2 with relevant references, and the extensive review by (158)].
One Pathogenic Model Implies That Anti-dsDNA Antibodies Bind Glomerulus Membrane-Associated Chromatin Fragments
Co-localization immune electron microscopy (IEM) analyses demonstrated that the electron-dense structures in mesangial matrix and in GBM were targeted in vitro by antibodies to dsDNA, histones and transcription factors, whereas co-localization terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) IEM demonstrated that these structures contained high concentrations of in vivo-bound IgG and TUNEL-positive DNA in both murine (159) and human (159, 160) lupus nephritis. These and similar data (135) indicate that anti-dsDNA antibodies exert their nephritogenic effect by binding to exposed chromatin fragments in glomerular membranes and the mesangial matrices (143, 160, 161), which is consistent with the fact that antibodies eluted from nephritic kidneys show specificity for chromatin fragments, histones and DNA as common denominators, although several other specificities have been detected in such eluates [see above (38, 143, 144)]. The early phase of mesangial nephritis might indeed be initiated by circulating immune complexes consisting of chromatin fragments complexed with IgG (132). Notably, by high resolution IEM we never observed antibodies bound in vivo to native GBM itself, nor to the mesangial matrix surrounding EDS (see Figure 3B as example, where antibodies are confined to EDS leaving GBM unstained).
The Role of Renal DNase I in Progressive Lupus Nephritis
We have demonstrated that progressive lupus nephritis correlates with loss of the central renal Dnase I endonuclease mRNA, and DNase I endonuclease activity. This event coincided with significantly reduced fragmentation of chromatin, leaving large chromatin fragments that accumulate in situ in glomeruli. If this happens in glomeruli of a person that produce anti-dsDNA antibodies, complexes of these partners (IgG anti-dsDNA antibodies and retained chromatin fragments) exert deleterious inflammatory effects on the integrity and function of the kidneys. Although not proven by solid evidence, chromatin fragments in kidneys with selectively silenced DNase I gene expression may derive from kidneys themselves, at least in progressive disease (86, 135, 162, 163).
Chromatin Metabolism in Kidneys in the Course of Lupus Nephritis
From both theoretical considerations and the comprehensive sets of coherent data discussed above, it is fair to conclude that glomerular extracellular chromatin fragments play a direct role in lupus nephritis, where they serve as homologous targets for anti-dsDNA/anti-chromatin antibodies. This conclusion also implies that the antibodies do not have an a priori nephritogenic potential in absence of chromatin. However, when chromatin is exposed in glomeruli, the antibodies are rendered nephritic (132). That is, isolated presence of either of the factors—the antibody or chromatin—remain in the body as clinical epiphenomenons! Therefore, the core nature of both murine and human lupus nephritis is pointing at an acquired error of renal chromatin metabolism due to silenced DNase I gene expression as a key event in disease progression [reviewed in (3, 4)].
Another Pathogenic Model to Describe Lupus Nephritis Implies That Cross-Reactive Anti-dsDNA Antibodies Interact Directly With Glomerular Non-DNA Structures
The cross-reactive model inherits several problems that need to be described by experiments and analyses in lupus-prone mice and patients. The following questions need considerations.
Is the Cross-Reacting Immune Response Sustained Over Time—The Problem of Affinity Maturation?
This is a central problem in this model. Sustained immune B cell stimulation may open for a successive loss of the cross-reactive specificity while the homologous response may remain with increased avidity and titer. Considering a sustained stimulus of the dsDNA-specific B cells by dsDNA, they will increase avidity for dsDNA as a consequence of affinity mutations, and since these mutations are random, they will/may by chance mutate away from the cross-reactive specificity. Thus, over time the cross-reactive specificity may slowly die out due to sense mutations for the immunogen (dsDNA), and non-sense mutation for the crossreacting specificity that inevitably will die out (see model thinking as presented in Figure 5).
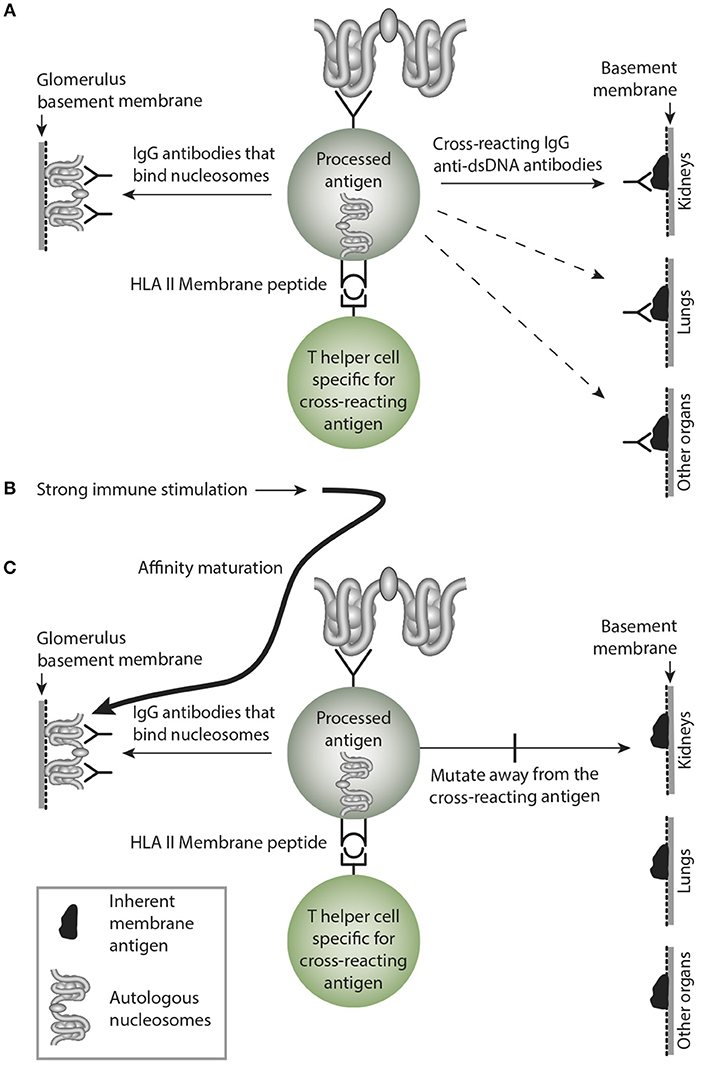
Figure 5. Affinity maturation may transform a cross-reacting antibody into a monospecific antibody. In (A) a B cell bind nucleosomes by its antigen receptor, process them and present nucleosomes-derived peptides in context of HLA class II to peptide-specific T helper cells. In this example, the B cell transform into antibody-secreting plasma cells, and the emerging cross-reacting IgG antibody recognize nucleosomes, and they cross-react with an inherent glomerular membrane antigen like e.g., laminin. Since laminin is part of membranes in different organs, the cross-reactive antibody may bind in glomeruli, lungs and in other organs, similar to the anti-collagen IV in Goodpasture Syndrome. In (B) a strong and possibly sustained stimulus recruits more somatically mutated IgG molecules. In (C) mutations are selected that promote increased affinity for the B cell antigen, while mutations diminish affinity for the cross-reacting antigen, since these antigens are not selected by nucleosomes. This is a mechanism that may transform oligospecific into mono-specific IgG antibodies. This model therefore indicates that the effect of the cross-reactive specificity may over time faint or die out.
Is the Avidity for a Cross-Reactive Antibody Similar for Both Ligands or Will the Highest Avidity Direct the Antibody to That Antigen?
When we started our studies on the pathogenesis on lupus nephritis, we foresaw this problem. Therefore, we developed highly sensitive electron microscopy (EM) variants that with relatively high precision could determine the nature of the glomerular targets for nephritogenic antibodies. This included transmission EM and IEM to trace binding of antibodies in vivo, co-localization IEM, where we added different experimental antibodies to the renal sections, like antibodies to dsDNA, histones, transcription factors and GBM ligands like laminin, in order to analyze which of the added antibodies co-localized with in vivo-bound IgG. In addition, we analyzed loci of in vivo bound antibodies by TUNEL-colocalized IEM where we observed that TUNEL-positive DNA co-localized with in vivo-bound IgG. All our results were consistent and demonstrated that IgGs that bound in vivo were exclusively seen in EM as part of electron dense structures (see details in Figure 3B). No binding to podocytes or to regular GBM structures were observed (79). The same was true when we translated these experimental analyses to human lupus nephritis (79, 159).
Will Antibodies Mono-Specific for a Non-DNA Cross-Reacting Antigen Bind in Glomeruli?
This question—and obvious deviating experiments—is in fact neglected in the literature in this context. We know that many anti-dsDNA antibodies cross-react with a large panel of non-dsDNA structures [See Table 2, and e.g., (112)]. By injecting cross-reacting (dsDNA-X) and non-cross-reacting non-dsDNA (X) antibodies into mice, may solve if one—or both specificities contribute to lupus nephritis.
Similarly, If Crossreaction of Anti-dsDNA Antibodies With Renal Antigens Is Instrumental in Initiating Lupus Nephritis, Then Why Does the Disease Start in the Mesangial Matrix?
This is exactly what we observe in the BW mouse after injection of purified anti-dsDNA antibodies (135), and linked to loss of renal DNaseI endonuclease, the disease expanded from mesangial nephritis to membranoproliferative nephritis with deposits of the antibodies in GBM where they co-localize with chromatin fragments. If the antibodies bound in vivo crossreacted with e.g., laminin or entactin, we expected they should bind simultaneously in the mesangium and GBM.
Are Cross-Reactive Antibodies Eluted From Nephritic Kidneys?
In search of the biological meaning of cross-reacting antibodies as essential in lupus nephritis, there are so far too few systematic analyses addressed to solve this problem. One clear exemption is the study of Deocharan et al. They analyzed anti-dsDNA antibodies that crossreacted with α-actinin and observed strong antibody activity in renal eluates (113). However, it is difficult from such observations to determine if the antibodies bound exposed chromatin or exposed α-actinin. More important, it would have been of strong interest if control injection with a non-cross-reacting (non-dsDNA) counterpart was performed. If they could be rescued from kidneys by elution, it would have been easier to make stronger conclusion on the impact of assumed pathogenic cross-reactive antibodies.
Concluding Remarks
This study discusses two central problems: Are antibodies binding dsDNA really anti-dsDNA antibodies, or do they recognize dsDNA after being instigated by a non-dsDNA (cross-reacting) antigen. Secondly, and in line with the first problem, are these antibodies nephritic because they bind chromatin exposed on glomerular membranes or are they nephritic because they recognize inherent glomerular membrane structures. These two models—the chromatin model and the cross-reactive model—are still not fully understood, have not been agreed on, and are still promoting controversies. Yet, the discussions and contradictions aimed to describe the pathogenesis of lupus nephritis characterize the contemporary situation. Thus, we still lack a coordinated and open minded approach to obtain a general and evidence-based perspective by not taking all aspects of the syndrome SLE into account. This can only be achieved by concentrating on the biological and pathophysiological interactions between its different disease-promoting elements. We need a framework in which dissection of published data generated by experts in different fields like immunology, pathology, immunopathology, and experimental animal research can be combined and confronted with each other simply in order to determine what we agree on (is the anti-dsDNA antibody important?), what must be done (study the impact of the other side of the cross-reacting anti-dsDNA antibody), and what the best strategy forward must be (to collaborate between the different schools of hypotheses). Whatever its nature and origin might be, anti-dsDNA antibodies are a strange and challenging phenomenon—so is lupus nephritis and SLE also. And do not forget the role of T cells in lupus nephritis! As a conclusion for now, we are producing increasing numbers of puzzle pieces connected to the eponym SLE. We are not, however, halting and concentrating on organizing the picture that may tell us why the puzzle pieces belong to each other. New phenomenons are not needed if we do not put them into a context leading to our understanding of SLE and how to treat it.
Disclosure
The author declares he has no competing interests, or other interests that might be perceived to influence the results and discussions reported in this manuscript.
Ethics Statement
The present manuscript is a review on murine and human SLE and lupus nephritis. All data are taken from original studies approved by relevant ethical committees.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This study was supported by the University of Tromsø as Milieu Support and a grant given by Norwegian Center for Molecular Medicine, University of Oslo. The publication charges for this article have been funded by a grant from the Publication Fund of UiT The Arctic University of Nor.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Dhivya Thiyagarajan, Ph.D., Institute of Medical Biology, University of Tromsø (IMB, UiT), for critical and eye-opening discussions, Gunnar Rekvig, Ph.D., Tokyo University of Foreign Studies, for critical reading and textual improvements, and Ugo Moens, IMB, UiT, for continuous support over years. I am thankful to Rod Wolstenholme, Section for Dissemination Services, Faculty of Health Sciences, for expert help to prepare the figures. His stamina has helped me a lot.
References
1. Borza DB, Bondar O, Colon S, Todd P, Sado Y, Neilson EG, et al. Goodpasture autoantibodies unmask cryptic epitopes by selectively dissociating autoantigen complexes lacking structural reinforcement: novel mechanisms for immune privilege and autoimmune pathogenesis. J Biol Chem. (2005) 280:27147–54. doi: 10.1074/jbc.M504050200
2. Borza DB, Neilson EG, Hudson BG. Pathogenesis of goodpasture syndrome: a molecular perspective. Semin Nephrol. (2003) 23:522–31. doi: 10.1053/S0270-9295(03)00131-1
3. Rekvig OP. The anti-DNA antibody: origin and impact, dogmas and controversies. Nat Rev Rheumatol. (2015)11:530–40. doi: 10.1038/nrrheum.2015.69
4. Rekvig OP. Systemic lupus erythematosus: definitions, contexts, conflicts, enigmas. Front Immunol. (2018) 9:387. doi: 10.3389/fimmu.2018.00387
5. Ludwik Fleck. Entstehung und Entwicklung Einer Wissenschaftlichen Tatsache. Einführung in die Lehre vom Denkstil und Denkkollektiv. Basel: Suhrkamp Taschenbuch Wissenschaft (1935).
6. Miescher P, Strassle R. New serological methods for the detection of the L.E. factor. Vox Sang. (1957) 2:283–7. doi: 10.1159/000478330
7. Robbins WC, Holman HR, Deicher H, Kunkel HG. Complement fixation with cell nuclei and DNA in lupus erythematosus. Proc Soc Exp Biol Med. (1957) 96:575–9. doi: 10.3181/00379727-96-23545
8. Ceppellini R, Polli E, Celada F. A DNA-reacting factor in serum of a patient with lupus erythematosus diffusus. Proc Soc Exp Biol Med. (1957) 96:572–4. doi: 10.3181/00379727-96-23544
9. Seligman M. [Serology-evidence in serum from patients with disseminated lupus erythermatosus of a substance determining a precipitation reaction with desoxyribonucleic acid]. Compt Rend Acad Sci. (1957) 245:243–5.
10. Miescher F. Ueber die chemische Zusammensetzung der Eiterzellen. Med. Chem. Untersuchungen. (1871) 4:441–60.
11. Dahm R. Friedrich Miescher and the discovery of DNA. Dev Biol. (2005) 278:274–88. doi: 10.1016/j.ydbio.2004.11.028
12. Levene PA. On the chemistry of the chromatin substance of the nerve cell. J Med Res. (1903) 10:204–11.
13. Chargaff E, Lipshitz R, Green C. Composition of the desoxypentose nucleic acids of four genera of sea-urchin. J Biol Chem. (1952) 195:155–60.
14. Elson D, Chargaff E. On the desoxyribonucleic acid content of sea urchin gametes. Experientia. (1952) 8:143–5. doi: 10.1007/BF02170221
16. Franklin RE, Gosling RG. Evidence for 2-chain helix in crystalline structure of sodium deoxyribonucleate. Nature. (1953) 172:156–7. doi: 10.1038/172156a0
19. Pulleyblank DE, Haniford DB, Morgan AR. A structural basis for S1 nuclease sensitivity of double-stranded DNA. Cell. (1985) 42:271–80. doi: 10.1016/S0092-8674(85)80122-7
20. Stollar BD, Papalian M. Secondary structure in denatured DNA is responsible for its reaction with antinative DNA antibodies of systemic lupus erythematosus sera. J Clin Invest. (1980) 66:210–9. doi: 10.1172/JCI109846
21. Menzel AEO, Heidelberger M. Cell protein fractions of bovine and avian tubercle bacillus strains and of the timothy-grass bacillus. J Biol Chem. (1938) 124:301–7.
22. Sevag MG, Lackman DB, Smolen J. The isolation of the components of streptococcal nucleoproteins in serologically active form. J Biol Chem. (1938) 124:425–36.
23. Winkenwerder WL, Buell MV, Howard JE. The sensitizing properties of the nucleic acids and their derivatives. Science. (1939) 90:356. doi: 10.1126/science.90.2337.356
24. Darrah E, Andrade F. NETs: the missing link between cell death and systemic autoimmune diseases? Front Immunol. (2012) 3:428. doi: 10.3389/fimmu.2012.00428
25. Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. (2016) 12:402–13. doi: 10.1038/nrneph.2016.71
26. Dwivedi N, Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. (2014) 73:483–91. doi: 10.1136/annrheumdis-2013-203844
27. Munoz LE, Leppkes M, Fuchs TA, Hoffmann M, Herrmann M. Missing in action-the meaning of cell death in tissue damage and inflammation. Immunol Rev. (2017) 280:26–40. doi: 10.1111/imr.12569
28. Munoz LE, Herrmann M. When autologous chromatin becomes a foe. Autoimmunity. (2012) 45:565–7. doi: 10.3109/08916934.2012.719949
29. Pieterse E, van der Vlag j Breaking immunological tolerance in systemic lupus erythematosus. Front Immunol. (2014) 5:164. doi: 10.3389/fimmu.2014.00164
30. Gaipl US, Kuhn A, Sheriff A, Munoz LE, Franz S, Voll RE, et al. Clearance of apoptotic cells in human SLE. Curr Dir Autoimmun. (2006) 9:173–87. doi: 10.1159/000090781
31. Gaipl US, Sheriff A, Franz S, Munoz LE, Voll RE, Kalden JR, et al. Inefficient clearance of dying cells and autoreactivity. Curr Top Microbiol Immunol. (2006) 305:161–76. doi: 10.1007/3-540-29714-6_8
32. Berden JH. Lupus nephritis: consequence of disturbed removal of apoptotic cells? Neth J Med. (2003) 61:233–8.
33. Berden JH, Grootscholten C, Jurgen WC, van der Vlag J. Lupus nephritis: a nucleosome waste disposal defect? J Nephrol. (2002) 15(Suppl. 6):S1–10.
34. Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. (2009) 60:1733–42. doi: 10.1002/art.24535
35. Dwivedi N, Neeli I, Schall N, Wan H, Desiderio DM, Csernok E, et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. (2014) 28:2840–51. doi: 10.1096/fj.13-247254
36. Beyer C, Pisetsky DS. The role of microparticles in the pathogenesis of rheumatic diseases. Nat Rev Rheumatol. (2010) 6:21–9. doi: 10.1038/nrrheum.2009.229
37. Pisetsky DS. The complex role of DNA, histones and HMGB1 in the pathogenesis of SLE. Autoimmunity. (2014) 47:487–93. doi: 10.3109/08916934.2014.921811
38. Mjelle JE, Kalaaji M, Rekvig OP. Exposure of chromatin and not high affinity for dsDNA determines the nephritogenic impact of anti-dsDNA antibodies in (NZBxNZW)F1 mice. Autoimmunity. (2009) 42:104–11. doi: 10.1080/08916930802375729
39. Kruse K, Janko C, Urbonaviciute V, Mierke CT, Winkler TH, Voll RE, et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis. (2010) 15:1098–113. doi: 10.1007/s10495-010-0478-8
40. Goilav B, Putterman C. The role of Anti-DNA antibodies in the development of lupus nephritis: a complementary, or alternative, viewpoint? Semin Nephrol. (2015) 35:439–43. doi: 10.1016/j.semnephrol.2015.08.005
41. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
42. Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, et al. Cutting edge: role of Toll-like receptor 9 in CpG DNA-induced activation of human cells. J Immunol. (2001) 167:3555–8. doi: 10.4049/jimmunol.167.7.3555
43. Doyle SL, Jefferies CA, Feighery C, O'Neill LA. Signaling by toll-like receptors 8 and 9 requires Bruton's tyrosine kinase. J Biol Chem. (2007) 282:36953–60. doi: 10.1074/jbc.M707682200
44. Kono DH, Haraldsson MK, Lawson BR, Pollard KM, Koh YT, Du X, et al. Endosomal TLR signaling is required for anti-nucleic acid and rheumatoid factor autoantibodies in lupus. Proc Natl Acad Sci USA. (2009) 106:12061–6. doi: 10.1073/pnas.0905441106
45. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. (2002) 416:603–7. doi: 10.1038/416603a
46. Cerutti ML, Zarebski LM, de Prat GG, Goldbaum FA. A viral DNA-binding domain elicits anti-DNA antibodies of different specificities. Mol Immunol. (2005) 42:327–33. doi: 10.1016/j.molimm.2004.09.003
47. Moens U, Seternes OM, Hey AW, Silsand Y, Traavik T, Johansen B, et al. In vivo expression of a single viral DNA-binding protein generates systemic lupus erythematosus-related autoimmunity to double-stranded DNA and histones. Proc Natl Acad Sci USA. (1995) 92:12393–7. doi: 10.1073/pnas.92.26.12393
48. Rekvig OP, Moens U, Sundsfjord A, Bredholt G, Osei A, Haaheim H, et al. Experimental expression in mice and spontaneous expression in human SLE of polyomavirus T-antigen. A molecular basis for induction of antibodies to DNA and eukaryotic transcription factors. J Clin Invest. (1997) 99:2045–54. doi: 10.1172/JCI119373
49. Pisetsky DS, Vrabie IA. Antibodies to DNA: infection or genetics? Lupus. (2009) 18:1176–80. doi: 10.1177/0961203309106492
50. Sundar K, Jacques S, Gottlieb P, Villars R, Benito ME, Taylor DK, et al. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J Autoimmun. (2004) 23:127–40. doi: 10.1016/j.jaut.2004.06.001
51. Pisetsky DS. Anti-DNA antibodies - quintessential biomarkers of SLE. Nat Rev Rheumatol. (2016) 12:102–10. doi: 10.1038/nrrheum.2015.151
52. Stollar BD. Immunochemical analyses of nucleic acids. Prog Nucleic Acid Res Mol Biol. (1992) 42:39–77. doi: 10.1016/S0079-6603(08)60573-5
53. Stollar BD. An overview of the anti-DNA antibody workshop: expansion of molecular structural analysis. Lupus. (1997) 6:346–8. doi: 10.1177/096120339700600333
54. Polymenis M, Brigido MM, Sanford DG, Stollar BD. The targets and genes for antibodies to Z-DNA. Biotechnol Appl Biochem. (1993) 18(Pt 2):175–83.
55. Rahman A, Hiepe F. Anti-DNA antibodies–overview of assays and clinical correlations. Lupus. (2002) 11:770–3. doi: 10.1191/0961203302lu313rp
56. Lahita RG, Tsokos G, Buyon BP, Koike T. Systemic Lupus Erythematosus. 5 ed. New York, NY: Academic Press (2010).
57. Tsokos GC, Lo MS, Costa RP, Sullivan KE. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol. (2016) 12:716–30. doi: 10.1038/nrrheum.2016.186
58. Doaty S, Agrawal H, Bauer E, Furst DE. Infection and lupus: which causes which? Curr Rheumatol Rep. (2016) 18:13. doi: 10.1007/s11926-016-0561-4
59. Weinberg I, Vasiliev L, Gotsman I. Anti-dsDNA antibodies in sarcoidosis. Semin Arthritis Rheum. (2000) 29:328–31. doi: 10.1016/S0049-0172(00)80019-0
60. Noble PW, Bernatsky S, Clarke AE, Isenberg DA, Ramsey-Goldman R, Hansen JE. DNA-damaging autoantibodies and cancer: the lupus butterfly theory. Nat Rev Rheumatol. (2016) 12:429–34. doi: 10.1038/nrrheum.2016.23
61. Attar SM, Koshak EA. Medical conditions associated with a positive anti-double-stranded deoxyribonucleic acid. Saudi Med J. (2010) 31:781–7.
62. Sthoeger ZM, Wakai M, Tse DB, Vinciguerra VP, Allen SL, Budman DR, et al. Production of autoantibodies by CD5-expressing B lymphocytes from patients with chronic lymphocytic leukemia. J Exp Med. (1989) 169:255–68. doi: 10.1084/jem.169.1.255
63. Maheshwari A, Pandey M, Rath B, Chandra J, Singh S, Sharma S. Clinical and laboratory observation systemic lupus erythematosus and acute lymphocytic leukemia: an unusual case. Indian J Med Paediatr Oncol. (2011) 32:154–6. doi: 10.4103/0971-5851.92816
64. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. (2010) 107:13075–80. doi: 10.1073/pnas.1004594107
65. Aluoch AO, Farbman M, Gladue H. An unusual mimicker of systemic lupus erythematosus: a case report. Open Rheumatol J. (2015) 9:27–9. doi: 10.2174/18743129014090100027
66. Rosman A, Atsumi T, Khamashta MA, Ames PR, Hughes GR. Development of systemic lupus erythematosus after chemotherapy and radiotherapy for malignant thymoma. Br J Rheumatol. (1995) 34:1175–6. doi: 10.1093/rheumatology/34.12.1175
67. Lv S, Zhang J, Wu J, Zheng X, Chu Y, Xiong S. Origin and anti-tumor effects of anti-dsDNA autoantibodies in cancer patients and tumor-bearing mice. Immunol Lett. (2005) 99:217–27. doi: 10.1016/j.imlet.2005.03.019
68. Cao Q, Xu W, Wen Z, Xu L, Li K, Chu Y, et al. An anti-double-stranded DNA monoclonal antibody induced by tumor cell-derived DNA inhibits the growth of tumor in vitro and in vivo via triggering apoptosis. DNA Cell Biol. (2008) 27:91–100. doi: 10.1089/dna.2007.0633
69. Blaes F, Klotz M, Huwer H, Straub U, Kalweit G, Schimrigk K, et al. Antineural and antinuclear autoantibodies are of prognostic relevance in non-small cell lung cancer. Ann Thorac Surg. (2000) 69:254–8. doi: 10.1016/S0003-4975(99)01198-4
70. Syrigos KN, Charalambopoulos A, Pliarchopoulou K, Varsamidakis N, Machairas A, Mandrekas D. The prognostic significance of autoantibodies against dsDNA in patients with colorectal adenocarcinoma. Anticancer Res. (2000) 20:4351–3.
71. Madrid FF, Maroun MC, Olivero OA, Long M, Stark A, Grossman LI, et al. Autoantibodies in breast cancer sera are not epiphenomena and may participate in carcinogenesis. BMC Cancer. (2015) 15:407. doi: 10.1186/s12885-015-1385-8
72. Madrid FF, Maroun MC. Serologic laboratory findings in malignancy. Rheum Dis Clin North Am. (2011) 37:507–25. doi: 10.1016/j.rdc.2011.09.006
73. Desai DD, Krishnan MR, Swindle JT, Marion TN. Antigen-specific induction of antibodies against native mammalian DNA in nonautoimmune mice. J Immunol. (1993) 151:1614–26.
74. Dong X, Hamilton KJ, Satoh M, Wang J, Reeves WH. Initiation of autoimmunity to the p53 tumor suppressor protein by complexes of p53 and SV40 large T antigen. J Exp Med. (1994) 179:1243–52. doi: 10.1084/jem.179.4.1243
75. Reeves WH, Dong X, Wang J, Hamilton K. Initiation of autoimmunity to self-proteins complexed with viral antigens. Ann N Y Acad Sci. (1997) 815:139–54. doi: 10.1111/j.1749-6632.1997.tb52056.x
76. Zur Hausen HH. Viruses in human cancers. Science. (1991) 254:1167–73. doi: 10.1126/science.1659743
77. Hirsch HH, Steiger J. Polyomavirus BK. Lancet Infect Dis. (2003) 3:611–23. doi: 10.1016/S1473-3099(03)00770-9
78. Bernatsky S, Ramsey-Goldman R, Labrecque J, Joseph L, Boivin JF, Petri M, et al. Cancer risk in systemic lupus: an updated international multi-centre cohort study. J Autoimmun. (2013) 42:130–5. doi: 10.1016/j.jaut.2012.12.009
79. Pedersen HL, Horvei KD, Thiyagarajan D, Seredkina N, Rekvig OP. Murine and human lupus nephritis: pathogenic mechanisms and theoretical strategies for therapy. Semin Nephrol. (2015) 35:427–38. doi: 10.1016/j.semnephrol.2015.08.004
80. Seredkina N, van d V, Berden J, Mortensen E, Rekvig OP. Lupus nephritis: enigmas, conflicting models and an emerging concept. Mol Med. (2013) 19:161–9. doi: 10.2119/molmed.2013.00010
81. van der Vlag V, Berden JH. Lupus nephritis: role of antinucleosome autoantibodies. Semin Nephrol. (2011) 31:376–89. doi: 10.1016/j.semnephrol.2011.06.009
82. Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti- DNA antibodies. Annu Rev Immunol. (1994) 12:487–520. doi: 10.1146/annurev.iy.12.040194.002415
83. Heidelberger M, Scherp HW. Protein fractions of a strain of Group “A” hemolytic streptococci. J Immunol. (1939) 37:563–70.
84. Fredriksen K, Skogsholm A, Flaegstad T, Traavik T, Rekvig OP. Antibodies to dsDNA are produced during primary BK virus infection in man, indicating that anti-dsDNA antibodies may be related to virus replication in vivo. Scand J Immunol. (1993) 38:401–6. doi: 10.1111/j.1365-3083.1993.tb01744.x
85. Rekvig OP, Bendiksen S, Moens U. Immunity and autoimmunity induced by polyomaviruses: clinical, experimental and theoretical aspects. Adv Exp Med Biol. (2006) 577:117–47. doi: 10.1007/0-387-32957-9_9
86. Thiyagarajan D, Fismen S, Seredkina N, Jacobsen S, Elung-Jensen T, Kamper AL, et al. Silencing of renal DNaseI in murine lupus nephritis imposes exposure of large chromatin fragments and activation of toll like receptors and the Clec4e. PLoS ONE. (2012) 7:e34080. doi: 10.1371/journal.pone.0034080
87. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
88. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. (2011) 18:581–8. doi: 10.1038/cdd.2011.1
89. Yousefi S, Simon HU. NETosis - Does it really represent nature's “suicide bomber”? Front Immunol. (2016) 7:328. doi: 10.3389/fimmu.2016.00328
90. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights into neutrophil extracellular traps: mechanisms of formation and role in inflammation. Front Immunol. (2016) 7:302. doi: 10.3389/fimmu.2016.00302
91. Dieker J, Berden JH, Bakker M, Briand JP, Muller S, Voll R, et al. Autoantibodies against modified histone peptides in SLE patients are associated with disease activity and lupus nephritis. PLoS ONE. (2016) 11:e0165373. doi: 10.1371/journal.pone.0165373
92. Muller S, Radic M. Oxidation and mitochondrial origin of NET DNA in the pathogenesis of lupus. Nat Med. (2016) 22:126–7. doi: 10.1038/nm.4044
93. Dwived N, Marko R. Burning controversies in NETs and autoimmunity: the mysteries of cell death and autoimmune disease. Autoimmunity. (2018) 51:267–80. doi: 10.1080/08916934.2018.1523395
94. Dieker JW, Fransen JH, van Bavel CC, Briand JP, Jacobs CW, Muller S, et al. Apoptosis-induced acetylation of histones is pathogenic in systemic lupus erythematosus. Arthritis Rheum. (2007) 56:1921–33. doi: 10.1002/art.22646
95. Muller S, Isabey A, Couppez M, Plaue S, Sommermeyer G, Van Regenmortel MH. Specificity of antibodies raised against triacetylated histone H4. Mol Immunol. (1987) 24:779–89. doi: 10.1016/0161-5890(87)90062-9
96. Gordon RA, Herter JM, Rosetti F, Campbell AM, Nishi H, Kashgarian M, et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight. (2017) 2:92926. doi: 10.1172/jci.insight.92926
97. Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. (2012) 4:157ra141. doi: 10.1126/scitranslmed.3004801
98. Kienhofer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight. (2017) 2:92920. doi: 10.1172/jci.insight.92920
99. Rother N, Pieterse E, Lubbers J, Hilbrands L, van der Vlag J. Acetylated histones in apoptotic microparticles drive the formation of neutrophil extracellular traps in active lupus nephritis. Front Immunol. (2017) 8:1136. doi: 10.3389/fimmu.2017.01136
100. van Bavel CC, Dieker JW, Kroeze Y, Tamboer WP, Voll R, Muller S, et al. Apoptosis-induced histone H3 methylation is targeted by autoantibodies in systemic lupus erythematosus. Ann Rheum Dis. (2011) 70:201–7. doi: 10.1136/ard.2010.129320
101. Tillack K, Breiden P, Martin R, Sospedra M. T lymphocyte priming by neutrophil extracellular traps links innate and adaptive immune responses. J Immunol. (2012) 188:3150–9. doi: 10.4049/jimmunol.1103414
102. Peng SL, Craft J. T cells in murine lupus: propagation and regulation of disease. Mol Biol Rep. (1996) 23:247–51. doi: 10.1007/BF00351176
103. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
104. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
105. Dwivedi N, Hedberg A, Zheng YY, Neeli I, Satoh M, Morel L, et al. B Cell tolerance to deiminated histones in BALB/c, C57BL/6, and autoimmune-prone mouse strains. Front Immunol. (2017) 8:362. doi: 10.3389/fimmu.2017.00362
106. Biermann MHC, Boeltz S, Pieterse E, Knopf J, Rech J, Bilyy R, et al. autoantibodies recognizing secondary NEcrotic cells promote neutrophilic phagocytosis and identify patients with systemic lupus erythematosus. Front Immunol. (2018) 9:989. doi: 10.3389/fimmu.2018.00989
107. Mjelle JE, Rekvig OP, Fenton KA. Nucleosomes possess a high affinity for glomerular laminin and collagen IV and bind nephritogenic antibodies in murine lupus-like nephritis. Ann Rheum Dis. (2007) 66:1661–8. doi: 10.1136/ard.2007.070482
108. Fismen S, Hedberg A, Fenton K, Jacobsen S, Krarup E, Kamper A, et al. Circulating chromatin-anti-chromatin antibody complexes bind with high affinity to dermo-epidermal structures in murine and human lupus nephritis. Lupus. (2009) 18:597–607. doi: 10.1177/0961203308100512
109. Van Bruggen MC, Kramers C, Berden JH. Autoimmunity against nucleosomes and lupus nephritis. Ann Med Interne. (1996) 147:485–9.
110. Kramers C, Hylkema MN, Van Bruggen MC, van de LR, Dijkman HB, Assmann KJ, et al. Anti-nucleosome antibodies complexed to nucleosomal antigens show anti-DNA reactivity and bind to rat glomerular basement membrane in vivo. J Clin Invest. (1994) 94:568–77. doi: 10.1172/JCI117371
111. Van Bruggen MC, Kramers C, Hylkema MN, Smeenk RJ, Berden JH. Pathophysiology of lupus nephritis: the role of nucleosomes. Neth J Med. (1994) 45:273–9.
112. van Bavel CC, Fenton KA, Rekvig OP, van der Vlag J, Berden JH. Glomerular targets of nephritogenic autoantibodies in systemic lupus erythematosus. Arthritis Rheum. (2008) 58:1892–9. doi: 10.1002/art.23626
113. Deocharan B, Qing X, Lichauco J, Putterman C. Alpha-actinin is a cross-reactive renal target for pathogenic anti-DNA antibodies. J Immunol. (2002) 168:3072–8. doi: 10.4049/jimmunol.168.6.3072
114. Krishnan MR, Wang C, Marion TN. Anti-DNA autoantibodies initiate experimental lupus nephritis by binding directly to the glomerular basement membrane in mice. Kidney Int. (2012) 82:184–92. doi: 10.1038/ki.2011.484
115. Qureshi F, Yang Y, Jaques SM, Johnson MP, Naparstek Y, Ulmansky R, et al. Anti-DNA antibodies cross-reacting with laminin inhibit trophoblast attachment and migration: implications for recurrent pregnancy loss in SLE patients. Am J Reprod Immunol. (2000) 44:136–42. doi: 10.1111/j.8755-8920.2000.440302.x
116. Mostoslavsky G, Fischel R, Yachimovich N, Yarkoni Y, Rosenmann E, Monestier M, et al. Lupus anti-DNA autoantibodies cross-react with a glomerular structural protein: a case for tissue injury by molecular mimicry. Eur J Immunol. (2001) 31:1221–7. doi: 10.1002/1521-4141(200104)31:4<1221::AID-IMMU1221>3.0.CO;2-P
117. Ben-Yehuda A, Rasooly L, Bar-Tana R, Breuer G, Tadmor B, Ulmansky R, et al. The urine of SLE patients contains antibodies that bind to the laminin component of the extracellular matrix. J Autoimmun. (1995) 8:279–91. doi: 10.1006/jaut.1995.0021
118. Franchin G, Son M, Kim SJ, Ben-Zvi I, Zhang J, Diamond B. Anti-DNA antibodies cross-react with C1q. J Autoimmun. (2013) 44:34–9. doi: 10.1016/j.jaut.2013.06.002
119. Sabbaga J, Line SR, Potocnjak P, Madaio MP. A murine nephritogenic monoclonal anti-DNA autoantibody binds directly to mouse laminin, the major non-collagenous protein component of the glomerular basement membrane. Eur J Immunol. (1989) 19:137–43. doi: 10.1002/eji.1830190122
120. Termaat RM, Assmann KJ, van Son JP, Dijkman HB, Koene RA, Berden JH. Antigen-specificity of antibodies bound to glomeruli of mice with systemic lupus erythematosus-like syndromes. Lab Invest. (1993) 68:164–73.
121. Zhang W, Dang S, Wang J, Nardi MA, Zan H, Casali P, et al. Specific cross-reaction of anti-dsDNA antibody with platelet integrin GPIIIa49-66. Autoimmunity. (2010) 43:682–9. doi: 10.3109/08916934.2010.506207
122. Zhang H, Fu R, Guo C, Huang Y, Wang H, Wang S, et al. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J Transl Med. (2016) 14:156. doi: 10.1186/s12967-016-0911-z
123. DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. (2001) 7:1189–93. doi: 10.1038/nm1101-1189
124. Raz E, Ben Bassat H, Davidi T, Shlomai Z, Eilat D. Cross-reactions of anti-DNA autoantibodies with cell surface proteins. Eur J Immunol. (1993) 23:383–90. doi: 10.1002/eji.1830230213
125. Takeda I, Rayno K, Wolfson-Reichlin M, Reichlin M. Heterogeneity of anti-dsDNA antibodies in their cross-reaction with ribosomal P protein. J Autoimmun. (1999) 13:423–8. doi: 10.1006/jaut.1999.0330
126. Mageed RA, Zack DJ. Cross-reactivity and pathogenicity of anti-DNA autoantibodies in systemic lupus erythematosus. Lupus. (2002) 11:783–6. doi: 10.1191/0961203302lu317oa
127. Putterman C, Limpanasithikul W, Edelman M, Diamond B. The double edged sword of the immune response: mutational analysis of a murine anti-pneumococcal, anti-DNA antibody. J Clin Invest. (1996) 97:2251–9. doi: 10.1172/JCI118666
128. Yadav P, Carr MT, Yu R, Mumbey-Wafula A, Spatz LA. Mapping an epitope in EBNA-1 that is recognized by monoclonal antibodies to EBNA-1 that cross-react with dsDNA. Immun Inflamm Dis. (2016) 4:362–75. doi: 10.1002/iid3.119
129. Saxena R, Bygren P, Butkowski R, Wieslander J. Entactin: a possible auto-antigen in the pathogenesis of non-Goodpasture anti-GBM nephritis. Kidney Int. (1990) 38:263–72. doi: 10.1038/ki.1990.195
130. Rekvig OP, Thiyagarajan D, Pedersen HL, Horvei KD, Seredkina N. Future perspectives on pathogenesis of lupus nephritis: facts, problems, and potential causal therapy modalities. Am J Pathol. (2016) 186:2772–82. doi: 10.1016/j.ajpath.2016.06.026
131. Ehrenstein MR, Katz DR, Griffiths MH, Papadaki L, Winkler TH, Kalden JR, et al. Human IgG anti-DNA antibodies deposit in kidneys and induce proteinuria in SCID mice. Kidney Int. (1995) 48:705–11. doi: 10.1038/ki.1995.341
132. Fenton KA, Tommeras B, Marion TN, Rekvig OP. Pure anti-dsDNA mAbs need chromatin structures to promote glomerular mesangial deposits in BALB/c mice. Autoimmunity. (2010) 43:179–88. doi: 10.3109/08916930903305633
133. Adu D, Dobson J, Williams DG. DNA-anti-DNA circulating complexes in the nephritis of systemic lupus erythematosus. Clin Exp Immunol. (1981) 43:605–14.
134. Eilat D. Cross-reactions of anti-DNA antibodies and the central dogma of lupus nephritis. Immunol Today. (1985) 6:123–7. doi: 10.1016/0167-5699(85)90077-5
135. Fenton K, Fismen S, Hedberg A, Seredkina N, Fenton C, Mortensen ES, et al. Anti-dsDNA antibodies promote initiation, and acquired loss of renal Dnase1 promotes progression of lupus nephritis in autoimmune (NZBxNZW)F1 mice. PLoS ONE. (2009) 4:e8474. doi: 10.1371/journal.pone.0008474
136. Fu SM, Dai C, Zhao Z, Gaskin F. Anti-dsDNA antibodies are one of the many autoantibodies in systemic lupus erythematosus. F1000Res. (2015) 4:939. doi: 10.12688/f1000research.6875.1
137. Rekvig OP. Anti-dsDNA antibodies as a classification criterion and a diagnostic marker for systemic lupus erythematosus: critical remarks. Clin Exp Immunol. (2015) 179:5–10. doi: 10.1111/cei.12296
138. Marion TN, Postlethwaite AE. Chance, genetics, and the heterogeneity of disease and pathogenesis in systemic lupus erythematosus. Semin Immunopathol. (2014) 36:495–517. doi: 10.1007/s00281-014-0440-x
139. Lackman D, Mudd S, Sevag MG, Smolens J, Wiener M. The serological reactivity of nucleic acid. J Immunol. (1941) 40:11–20.
140. Seligmann M. [Demonstration in the blood of patients with disseminated lupus erythematosus a substance determining a precipitation reaction with desoxyribonucleic acid.]. C R Hebd Seances Acad Sci. (1957) 245:243–5.
141. Izui S, Lambert PH, Fournie GJ, Turler H, Miescher PA. Features of systemic lupus erythematosus in mice injected with bacterial lipopolysaccharides: identification of circulating DNA and renal localization of DNA-anti-DNA complexes. J Exp Med. (1977) 145:1115–30. doi: 10.1084/jem.145.5.1115
142. Winfield JB, Faiferman I, Koffler D. Avidity of anti-DNA antibodies in serum and IgG glomerular eluates from patients with systemic lupus erythematosus. Association of high avidity antinative DNA antibody with glomerulonephritis. J Clin Invest. (1977) 59:90–6. doi: 10.1172/JCI108626
143. Xie C, Liang Z, Chang S, Mohan C. Use of a novel elution regimen reveals the dominance of polyreactive antinuclear autoantibodies in lupus kidneys. Arthritis Rheum. (2003) 48:2343–52. doi: 10.1002/art.11092
144. Van Bruggen MC, Kramers C, Hylkema MN, Smeenk RJ, Berden JH. Significance of anti-nuclear and anti-extracellular matrix autoantibodies for albuminuria in murine lupus nephritis; a longitudinal study on plasma and glomerular eluates in MRL/l mice. Clin Exp Immunol. (1996) 105:132–9. doi: 10.1046/j.1365-2249.1996.d01-731.x
145. van Bavel CC, van der Vlag J, Berden JH. Glomerular binding of anti-dsDNA autoantibodies: the dispute resolved? Kidney Int. (2007) 71:600–1. doi: 10.1038/sj.ki.5002126
146. Zhao Z, Weinstein E, Tuzova M, Davidson A, Mundel P, Marambio P, et al. Cross-reactivity of human lupus anti-DNA antibodies with alpha-actinin and nephritogenic potential. Arthritis Rheum. (2005) 52:522–30. doi: 10.1002/art.20862
147. Jang YJ, Stollar BD. Anti-DNA antibodies: aspects of structure and pathogenicity. Cell Mol Life Sci. (2003) 60:309–20. doi: 10.1007/s000180300026
148. Jang YJ, Sanford D, Chung HY, Baek SY, Stollar BD. The structural basis for DNA binding by an anti-DNA autoantibody. Mol Immunol. (1998) 35:1207–17. doi: 10.1016/S0161-5890(98)00095-9
149. Spatz L, Saenko V, Iliev A, Jones L, Geskin L, Diamond B. Light chain usage in anti-double-stranded DNA B cell subsets: role in cell fate determination. J Exp Med. (1997) 185:1317–26. doi: 10.1084/jem.185.7.1317
150. Amital H, Heilweil M, Ulmansky R, Szafer F, Bar-Tana R, Morel L, et al. Treatment with a laminin-derived peptide suppresses lupus nephritis. J Immunol. (2005) 175:5516–23. doi: 10.4049/jimmunol.175.8.5516
151. Koga T, Ichinose K, Tsokos GC. T cells and IL-17 in lupus nephritis. Clin Immunol. (2017) 185:95–9. doi: 10.1016/j.clim.2016.04.010
152. Bagavant H, Deshmukh US, Wang H, Ly T, Fu SM. Role for nephritogenic T cells in lupus glomerulonephritis: progression to renal failure is accompanied by T cell activation and expansion in regional lymph nodes. J Immunol. (2006) 177:8258–65. doi: 10.4049/jimmunol.177.11.8258
153. Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. (2013) 24:1357–66. doi: 10.1681/ASN.2013010026
154. Lorenz G, Desai J, Anders HJ. Lupus nephritis: update on mechanisms of systemic autoimmunity and kidney immunopathology. Curr Opin Nephrol Hypertens. (2014) 23:211–7. doi: 10.1097/01.mnh.0000444816.57378.21
155. Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. (1999) 189:1639–48. doi: 10.1084/jem.189.10.1639
156. Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. (1998) 160:51–9.
157. Wellmann U, Letz M, Herrmann M, Angermuller S, Kalden JR, Winkler TH. The evolution of human anti-double-stranded DNA autoantibodies. Proc Natl Acad Sci USA. (2005) 102:9258–63. doi: 10.1073/pnas.0500132102
158. Yaniv G, Twig G, Shor DB, Furer A, Sherer Y, Mozes O, et al. A volcanic explosion of autoantibodies in systemic lupus erythematosus: a diversity of 180 different antibodies found in SLE patients. Autoimmun Rev. (2015) 14:75–9. doi: 10.1016/j.autrev.2014.10.003
159. Kalaaji M, Fenton KA, Mortensen ES, Olsen R, Sturfelt G, Alm P, et al. Glomerular apoptotic nucleosomes are central target structures for nephritogenic antibodies in human SLE nephritis. Kidney Int. (2007) 71:664–72. doi: 10.1038/sj.ki.5002133
160. Kalaaji M, Sturfelt G, Mjelle JE, Nossent H, Rekvig OP. Critical comparative analyses of anti-alpha-actinin and glomerulus-bound antibodies in human and murine lupus nephritis. Arthritis Rheum. (2006) 54:914–26. doi: 10.1002/art.21622
161. Van Bruggen MC, Kramers C, Walgreen B, Elema JD, Kallenberg CG, van den Born J, et al. Nucleosomes and histones are present in glomerular deposits in human lupus nephritis. Nephrol Dial Transplant. (1997) 12:57–66. doi: 10.1093/ndt/12.1.57
162. Seredkina S, Rekvig OP. Acquired loss of renal nuclease activity is restricted to DNaseI and is an organ-selective feature in murine lupus nephritis. Am J Pathol. (2011) 179:1120–8. doi: 10.1016/j.ajpath.2011.05.011
Keywords: chromatin, dsDNA, autoimmunity, lupus nephritis, enigma, controversies
Citation: Rekvig OP (2019) The dsDNA, Anti-dsDNA Antibody, and Lupus Nephritis: What We Agree on, What Must Be Done, and What the Best Strategy Forward Could Be. Front. Immunol. 10:1104. doi: 10.3389/fimmu.2019.01104
Received: 09 November 2018; Accepted: 30 April 2019;
Published: 15 May 2019.
Edited by:
Reinhard Edmund Voll, Freiburg University Medical Center, GermanyReviewed by:
Umesh S. Deshmukh, Oklahoma Medical Research Foundation, United StatesRaju P. Khubchandani, Jaslok Hospital, India
Copyright © 2019 Rekvig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ole Petter Rekvig, b3ByMDAwQHBvc3QudWl0Lm5v