 Thaiz Rivera Vargas
Thaiz Rivera Vargas Lionel Apetoh
Lionel Apetoh- 1INSERM, U1231, Dijon, France
- 2Faculté de Médecine, Université de Bourgogne Franche Comté, Dijon, France
The unprecedented clinical activity of checkpoint blockade in several types of cancers has formally demonstrated that anti-tumor immune responses are crucial in cancer therapy. Durable responses seen in patients treated with immune checkpoint inhibitors (ICI) show that they can trigger the establishment of long-lasting immunologic memory. This beneficial outcome is however achieved for a limited number of patients. In addition, late relapses are emerging suggesting the development of acquired resistances that compromise the anticancer efficacy of ICI. How can this be prevented through combination therapies? We here review the functions of immune checkpoints, the successes of ICI in treating cancer and their therapeutic limits. We discuss how conventional cancer therapies can be properly selected to set up combinatorial approaches with ICI leading to treatment improvement. We finally summarize clinical data showing the ongoing progress in cancer treatment involving ICI and chemotherapy combination strategies.
Introduction
Immune cells prevent the outgrowth of cancer cells before they form a clinically detectable tumor (1). Thus, while chronic inflammation supports cancer cell growth, defective immune responses also contribute to tumor formation and development. The complex relationships between immune cells and tumor cells are underscored by the paradoxical role of the immune system during the course of tumor progression. During the early stages of tumor development, immune cells eliminate cancer cells. However, resistant clones eventually develop in the tumor microenvironment and tumors can not only become resistant to the immune driven cytotoxicity but immune responses are also subverted to further promote tumor growth (2). This concept of “cancer immunoediting” pioneered by Robert Schrieber has been elegantly reviewed (2).
Downregulation of effector T cell function is one of the mechanisms used by cancer cells to escape recognition and elimination by the immune system. This results in the tumor microenvironment in the emergence of dysfunctional T cells, with a progressively compromised ability to secrete effector cytokines and kill cancer cells (3–8). Dysfunctional T cells can stem from functional effector cells, which gradually lost their effector function due to chronic activation through the T cell receptor (TCR) (3, 9). This is for instance illustrated in mice with persistent chronic viral infections, where virus-specific CD8 T cells undergo dysfunction, which can however be reversible during the early stages of infection (10, 11). The induction of T cell dysfunction is also a physiological process that prevents the development of autoimmunity. This was exemplified in mouse studies where interfering with T cell dysfunction led to severe CD8 T cell-driven immunopathologies (12, 13). Thus, tumors subvert physiological immune responses that normally prevent excessive immune activation to further promote their growth.
Dysfunctional T cells can be characterized by the concomitant expression of several inhibitory receptors (IR), a reduced secretion of effector cytokines, such as IL-2, TNFα, and IFNγ as well as altered cell metabolism and a markedly different transcriptional profile (5, 6, 8, 14–18). Accordingly, compounds blocking IR engagement and signaling, i.e., immune checkpoint inhibitors (ICI), were developed and led to durable disease control in advanced cancers (19, 20). These successes ultimately translated into the clinic. Indeed, antibodies directed the immune checkpoints CTLA-4 (cytotoxic T lymphocyte-associated protein 4) and PD-1 (programmed cell death protein-1) were shown to prolong the survival of cancer patients suffering from metastatic cancers. While these achievements were initially noted in patients with metastatic melanoma (21, 22), they therapies are now approved to treat multiple types of cancers (23–26).
Despite the tremendous success of ICI, some tumors are resistant to these therapies while others gain resistance during the course of the treatments, thereby compromising their efficacy (27). Generally, immunogenic tumors with limited size and strong T cell-infiltration respond to checkpoint blockade. Therefore, increasing tumor immunogenicity and T cell presence in the tumor bed while reducing tumor burden are key factors to improve disease outcome. Because it has been proposed that some chemotherapies could trigger immune activation (28, 29), these drugs may act valuable partners with checkpoint blockade and combinatorial approaches are warranted. We first discuss the consequences of the use of ICI on immune and cancer cells. We then review key chemotherapy properties that are able to improve patient's response to checkpoint blockade and discuss how this knowledge could be exploited to translate immunotherapy-based approaches into more successful therapeutic combinations.
The Success Story of Checkpoint Blockade-Based Cancer Therapies
Cancer Treatment: A Tightrope Walker on the Fine Line Between T Cell Activation and T Cell Dysfunction
A deep knowledge of the mechanisms that drive T cell activation is required to design improved therapeutics unleashing T cell anticancer properties without triggering T cell dysfunction. Given the recent clinical success of ICI therapies, this area of research has attracted not only academic scientists but also pharmaceutical industries. T cell activation relies on TCR-driven signals, cytokines as well as co-stimulatory signals that will ultimately drive the outcome following T cell stimulation (30–32). Co-signaling receptors present on T cells can either convey stimulatory or inhibitory signals. The expression of these co-signaling receptors is dependent on the environment surrounding the T cells. Thus, neighboring cells harboring ligands of these receptors will profoundly affect T cell activation. This is thoroughly documented for antigen presenting cells (APCs), which profoundly shape T cell activation (33–35). The outcome of T cell activation thus results from the integration of activating and inhibitory signals.
From a therapeutic perspective, strategies altering T cell costimulation have been successfully exploited to restore T cell activation and anticancer immune responses. The initial characterization of CTLA-4 as molecule negatively regulating T cell activation has indeed prompted the development of antibody-mediated therapies that disrupt T cell dysfunction, reinvigorate effector T cells and dampen the activity of regulatory T cells, resulting in anticancer immunity (22, 36, 37). Manipulation of the PD-1 signaling pathway has similarly led to remarkable activation of anticancer T cells [reviewed in (38)]. These strategies have been successfully implemented into the clinic and the administration of antibodies preventing the engagement of PD-1 in cancer patients has resulted in response rates ranging from 20 to 90% in different cancer types (21, 23–26, 38–42). These remarkable results led to the routine use of these therapies to treat patients suffering from different cancer types, including metastatic melanoma and non-small-cell lung cancer (NSCLC). ICI treatments thus represent a major advance in cancer immunotherapy, which was further underscored by the scientific community that awarded of the Nobel Prize in Physiology or Medicine to the two researchers who identified CTLA-4 and PD-1 in 2018. However, as discussed below, other cancers, such as microsatellite stable (MSS) colon cancers respond poorly to ICI therapies, highlighting the need to pursue efforts to decipher the mechanisms explaining the resistance of some cancers to ICI therapies.
Mechanisms of Resistance to Immune Checkpoint Inhibitors
Patients with melanoma and Hodgkin lymphoma exhibit the best response rates following treatment with ICI therapies (38). About 20% of melanoma patients who received anti-CTLA-4 (ipilimumab) still exhibit a complete response 10 years after treatment initiation (36). Likewise, melanoma patients administered anti-PD-1 (pembrolizumab) featured a 3-year response rate of 33% (39). The added value of combination therapies of ICI has been tested in metastatic melanoma patients. Upon combined treatment with anti-CTLA-4 and anti-PD-1, a high response rate of 58% was achieved but was associated with severe toxicity (40, 41). Patient treatment with ICI therapies can lead to various outcomes. Unfortunately, even in cancers susceptible to ICI therapies, a substantial fraction of patients will not respond to treatment. Conversely, other patients exhibit a desirable response that will be long-lasting and lead to a complete response. However, some patients, who initially benefit from the therapy, eventually become resistant to treatment, resulting in cancer outgrowth (27, 43–45).
The observation that about two-third of patients do not respond to the administration of ICI given as a monotherapy prompted scientists and physicians to identify factors able to predict treatment efficacy (26, 40, 46–50). It now established that one of the important factors in determining the clinical response to ICI is the number of tumor mutations (51–57). This is illustrated for instance by the better response to anti-PD-1 therapy of colorectal cancers with microsatellite instability compared to MSS colorectal cancers. An additional parameter that affects ICI clinical efficacy is the presence and level of expression of the ligand of the targeted checkpoint by tumor cells and immune cells infiltrating the tumor bed (26, 58, 59). Finally, patients featuring an increased frequency of proliferating CD8 T cells following treatment with ICI was associated with a beneficial response (60). Thus, the defective generation of effector and memory CD8 T cell responses following ICI treatments can compromise their efficacy (27, 45, 61, 62). The quality of T cell responses will critically depend on the availability of tumor-derived neoantigens that will trigger the development of tumor-specific T cells (45). In addition, overcoming the immunosuppression present in the tumor microenvironment is essential for effective and long-lasting T cell responses to be maintained following treatment (43, 63, 64). Combination therapies should thus be designed with the aim to reinforce tumor immunogenicity, alleviate immunosuppression, and enhance T cell trafficking and memory.
Chemotherapy to Improve Checkpoint Blockade-Based Cancer Therapies
Most chemotherapeutic agents are involved in disrupting DNA replication leading to apoptosis of dividing cells (65). Mechanisms of direct tumor killing by chemotherapy involve DNA damage, inhibition of DNA replication, and prevention of mitosis (66). The cytotoxic properties of chemotherapy against dividing cells, which include immune cells, explain their well-known ability to trigger immunosuppression. This knowledge has led to implement treatments like high-dose cyclophosphamide to alleviate the course of severe autoimmune diseases (67). However, preclinical findings reported in the last fifty years suggested that chemotherapies could favor anti-tumor immunity [(68) and reviewed in (28, 29, 69)]. These beneficial effects on anticancer immunity were not only attributable to chemotherapy directly affecting tumor cells, thereby enhancing tumor cell immunogenicity, but also through the direct killing of immunosuppressive cells (70–73). All these properties provide a solid rationale for strategies combining checkpoint inhibitors and chemotherapy agents (Figure 1).
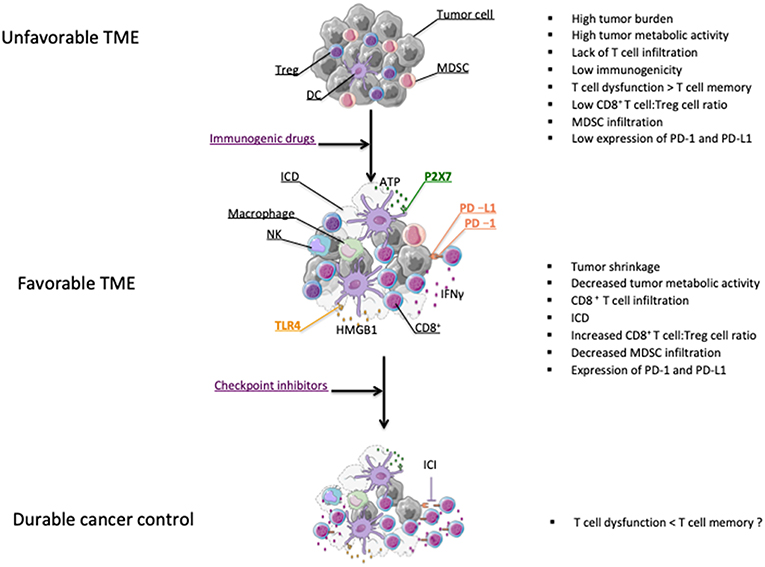
Figure 1. Effect of chemotherapy on the modification of the tumor microenvironment (TME). Chemotherapies leading to tumor shrinkage can shift the tumor microenvironment from tumor promoting to tumor suppressive (38). Chemotherapeutic drugs can induce ICD leading to the release of danger signal molecules that will contribute to the induction of anticancer immunity (74). Chemotherapy can also enhance the CD8+ T cell:Treg cell ratio and eliminate myeloid-derivative suppressive cells (MDSCs) (75–77). Some chemotherapy treatments promote the activation of CD8 T cells as well as PD-L1 expression on tumor cells (75). All these effects may partly explain why chemotherapies and ICI therapies can possibly synergize for durable cancer control (75, 78).
Chemotherapy Enhances the Development of T Cell-Dependent Anticancer Responses
Some chemotherapeutics like doxorubicin, cyclophosphamide and oxaliplatin can induce an immunogenic form of tumor cell death that will contribute to the development of T cell-dependent anticancer responses. Immunogenic cell death (ICD) is characterized by a series of molecular events responsible for the induction of anticancer immunity. The first feature of ICD is the cell surface expression of calreticulin, which is responsible for the phagocytosis of tumor cells undergoing ICD by dendritic cells (DC) (79). Another key event dictating the immunostimulatory activity of ICD is the release of the High mobility group box 1 (HMGB1) protein that is essential for an efficient cross-presentation of tumor-derived antigens by DC (80). In addition, HMGB1, together with ATP, which is liberated following tumor cell insult, will drive the secretion of IL-1β from DC, thereby enhancing CD8 T cell activation (81). We have investigated the importance of this immunoadjuvant pathway in humans by studying a retrospective cohort of breast cancer patients with lymph node involvement and treated with anthracyclines and radiotherapy. We found that patients with polymorphisms affecting the signaling of TLR4 and the high affinity ATP receptor, P2X7, were associated with a faster time to progression, thereby suggesting that anticancer immune responses driven through ICD are relevant in humans (80–82) (Figure 1).
In the context of an unfavorable TME two major questions are: 1) How to restore T cell infiltration and anticancer immunity? Is this process able to sensitize tumors to ICI therapy? To address this, Pfirschke et al. used a conditional lung adenocarcinoma mouse model with Kras and Trp53 mutations (KP model) (78). This model features molecular characteristics that mimic the development of human lung cancer. An important feature in this model is that T cells weakly infiltrate the TME. Accordingly, in this context anti-PD-1 treatment given alone failed to delay tumor progression. The authors then introduced neoantigens to sensitize the tumors to ICI therapy. For this, they generated KP-OVA mice, whose tumors expressed ovalbumin-derived peptides. However, even in the presence of these exogenous antigens, tumors from KP-OVA mice were resistant to combined treatment with anti-PD-1 and anti-CTLA-4 (78). The authors then screened different chemotherapies to test their anticancer efficacy in this model. Among several NSCLC chemotherapeutics (paclitaxel, docetaxel, carboplatin, mitoxantrone), they found that the combination of oxaliplatin and cyclophosphamide significantly increased nuclear HMGB1 staining in tumor nodules. While the combination paclitaxel-carboplatin failed to suppress cancer progression, oxaliplatin-cyclophosphamide (OX-CTX) combination controlled tumor growth. Accordingly, anticancer immune responses were enhanced in the lungs of the treated mice, as illustrated by increased presence of CD8 T cells along with reduced Treg frequency. Importantly, proliferating effector T cells were noted in the TME following the combined treatment (78). This chemotherapy-driven activation of adaptive immune responses was critical for the beneficial effect of the treatment as illustrated by the inability of Rag2−/− KP mice, which lack T and B cells, to control cancer growth upon OX-CTX treatment. The authors also tested the involvement of TLR4 in the ability of KP mice to respond to combined chemotherapeutic treatment. They found that mice deficient for TLR4 featured a decreased ability to resist KP tumor progression. Thus, TLR4 is required for the effective induction of T-cell dependent anticancer immunity in this genetic lung cancer tumor model following treatment with chemotherapy. These results are in line with our pioneering studies obtained using transplantable tumor models indicating that TLR4 dictates the immunogenicity of chemotherapy (80).
In addition to noting tumor-specific CD8 T cell infiltration in the lungs of KP-OVA mice upon combined treatment with OX-CTX, the authors found that following this treatment these tumor-specific cells expressed PD-1. Moreover, cells of the neighboring TME expressed PD-L1. This suggested that in that context the efficacy of the treatment could possibly be dampened by the engagement of the PD-1/PD-L1 pathway in tumor-specific CD8 T cells. To test this, KP-OVA mice were treated with anti-CTLA-4 and anti-PD-1, OX-CTX or the combined chemo-immunotherapeutic treatment. They found that the latter treatment featured a superior ability to control tumor growth compared to the monotherapies. Collectively, these results suggest that chemotherapy can restore T cell infiltration and activation even in the non-immunogenic KP lung cancer model and sensitize tumors to subsequent and/or concomitant treatment with ICI (78) (Figure 1).
In line with Pfirschke's work, we recently showed that 5-Fluorouracil plus Oxaliplatin (Folfox), in contrast to monotherapies, drove complete tumor cures in two mouse colorectal cancer models when combined to anti-PD-1 treatment, thereby suggesting that Folfox administration renders colorectal tumors sensitive to PD-1 blockade (75). Interestingly, the ability of Folfox to synergize with anti-PD-1 therapy was directly associated with the Folfox-driven induction of IFNγ-producing CD8 T cells as well as of enhanced PD-L1 expression on tumor cells in vivo. In line with this, we found in the CT26 colon adenocarcinoma model that Mitomycin C, which does not promote tumor PD-L1 expression on tumor cells in vivo, failed to synergize with anti-PD-1 treatment. Thus, the therapeutic success of the Folfox/Anti-PD-1 combination therapy is linked to the Folfox-driven induction of an anticancer immune response that drives tumor adaptive immune resistance through the PD-1/PD-L1 pathway (75).
We have further studied the molecular events leading to the development of anticancer immunity following Folfox administration. We first identified that Folfox drove the induction of TLR4-dependent immunity, resulting in CD8 T cell activation. In addition, this combined treatment not only induced the depletion of Myeloid derived suppressor cells (MDSCs) in vivo but also induced CD8 T cells expressing higher levels of the transcription factor T-bet compared to mice treated with monotherapies. We have investigated the relevance of this observation using mice lacking conditionally the expression of T-bet in CD4 and CD8 T cells. We noted that in these mice the therapeutic effect of Folfox against MC38 colon carcinomas was lost, indicating that T-bet expression in this context was required for the induction of T cell-dependent anticancer immune responses. We also unraveled the signaling pathway driving PD-L1 expression on tumor cells in vivo following Folfox administration. Using either T cell-deficient nude mice, mice depleted of CD8 T cells as well as mice receiving IFNγ neutralizing antibodies, we identified IFNγ-secreting CD8 T cells as a major driver of PD-L1 tumor expression in vivo following Folfox treatment. While we were unable to rule out a contribution of other IFNγ-producing cells in our observations, it is notable that we identified a strong correlation between the ability of different chemotherapies to induce CD8 T cell infiltration in the tumor in vivo and the induction of PD-L1 tumor expression. Overall, Folfox triggers a CD8 T cell-dependent anticancer immune response that in turn drives tumor PD-L1 expression, which thus acts as an adaptive resistance mechanism to the combined therapy. This resistance is successfully overcome by the addition of ICI therapy and our results therefore prompt for the combination of immunogenic drugs with ICI (75) (Figure 1).
Successful chemo-immunotherapy combinations involving the use of ICI are not restricted to antibodies targeting CTLA-4 or PD-1. Indeed, De Mingo Pulido et al. have just reported in mouse models of breast cancer that anti-Tim-3 treatment could improve the anticancer effect of paclitaxel (PTX) while anti-PD-1 therapy could not do so (83). Tim-3 was initially characterized as an immunoglobulin expressed on highly polarized Th1 cells (84). We and others subsequently showed that Tim-3 was also present on dysfunctional CD8 T cells in mouse and human tumors (17, 18). These findings were relevant as blockade of Tim-3 and PD-L1 in vivo could prevent tumor outgrowth (17). Interestingly, while Tim-3 was weakly expressed on CD8 T cells from mouse MMTV-PyMT tumors, the combined therapy induced CD8 T cell anticancer immunity (83). In fact, myeloid cells from both mouse and human tumors expressed Tim-3 and combined therapy with PTX and anti-Tim-3 triggered CXCL9 expression on DCs, possibly enhancing DC/T cell interactions and resulting in anticancer immunity. Accordingly, in human breast cancer patients, CXCL9 expression correlates with response to neoadjuvant chemotherapy (83). Thus, Tim-3 represents a molecular target, which can be exploited in the setting of combinatorial treatments relying on chemotherapy.
During ICD certain chemotherapies can also induce the release of various danger signals. For instance, DNA leakage into the cytosol can lead to the engagement of cytosolic DNA sensors, which will trigger the secretion of type I interferon from tumor cells, thereby leading to the induction of anticancer immune responses (74, 85). Chemotherapy also favors the generation of mutations in cancer cells, thereby increasing their antigenicity and rendering them more sensitive to ICI therapy (54, 86). Some chemotherapies will enhance tumor expression of MHC molecules, which enhances their ability to present tumor antigens and thus immunogenicity (85, 87, 88). Drugs like CTX can also drive lymphopenia, which can be exploited therapeutically in the context of combination therapies to drive immune activation and anticancer immunity (89–92). Thus, chemotherapy can be an attractive partner of ICI that can overcome ICI resistance due to insufficient anti-tumor T cell generation.
Chemotherapy Resets the TME to Favor T-cell Effector Function
Immunosuppressive cells present in the TME compromise the anticancer efficacy of ICI. Mouse studies have documented that myeloid cells, including tumor-associated macrophages (TAMs) and MDSCs, as well as Tregs and Th2 lymphocytes can contribute to the repression of anticancer T cell responses following ICI administration (27, 43, 45). Accordingly, preventing the accumulation of these immunosuppressive cells in the TME enhances the efficacy of ICI therapy (93, 94). The mechanisms accounting for these observations are progressively being unraveled. It was for instance shown that TAMs featured the ability to capture anti-PD-1 antibodies, which are thus no longer able to target CD8 T cells (95). MDSC are likewise able to suppress immune responses because of their immunosuppressive enzymes like indolamine-2,3-dioxygenase and arginase 1 that will dampen DC and T cell effector functions (96, 97).
Chemotherapy has the ability to eliminate immunosuppressive cells from the TME. CTX was shown to have a remarkable ability to eliminate Treg cells (98). These results have been extended to the human setting. Treg frequency in cancer patients treated with repeated, lose-dose cyclophosphamide was indeed reduced, underscoring the relevance of these observations initially made in preclinical studies (99, 100). We and others have also demonstrated that MDSC could be selectively targeted by some chemotherapies (101). Initially, gemcitabine was identified in mouse studies as a drug capable of eliminating MDSC in tumor-bearing mice in vivo (102). MDSC elimination using gemcitabine improved CD8 T cell functions and favored tumor control (102). Upon screening different chemotherapies to test their ability to eliminate MDSC in vivo, we not only could confirm the results of Li et al. but also noted that 5-Fluorouracil (5-FU) efficiently killed MDSC (76). In CT26 tumor-bearing mice, 5-FU was indeed able to eliminate MDSC without strongly affecting the frequency of T, B, or NK cells (76). We found that the high sensitivity of MDSC to 5-FU could be attributed to their low expression of Thymidylate synthase. In the setting of breast cancer, doxorubicin was likewise shown to target MDSC, leading to an improved efficacy of T cell adoptive transfer (103). Overall, chemotherapy has the ability to profoundly affect the TME and drive immune activation through the direct elimination of immunosuppressive cells, thereby enhancing the activity of ICI therapies.
Does Chemotherapy Influence the Formation of T-cell Memory?
Most if not all tumor-infiltrating lymphocytes feature memory T cell phenotypes (14, 104–106). CD8 T cells infiltrating the TME lack the expression of naïve T cell markers and instead express surface marker proteins found on activated and dysfunctional T cells (14). Thus, TILs harvested from cancer patients, which were shown to induce potent anticancer responses after in vitro expansion and reinfusion (107), are actually stemming from memory T cells (108–111). These observations are significant as an explanation for the ability of ICI therapies to induce anticancer responses is that these treatments actually reactivate preexisting immune responses. In addition, the most striking successes of ICI therapies lie in their abilities to induce long-lasting anticancer immune responses, resulting in long-term complete responses. This evidence altogether suggests that ICI therapies could affect the generation of anticancer memory T cells. This would actually be in line with previous preclinical and clinical observations showing CD8 T cell expansion following interference with the PD-1/PD-L1 pathway (58). Finally, this would also be congruent with recent preclinical findings obtained on isolated CD8+ TILs following combined treatment with ICI (112). Indeed, combined therapies induced profound transcriptional changes in PD-1 negative TILs that exhibited a memory precursor-like phenotype as well as effector functions in various cancer models (112). However, the induction or restoration of memory T cell responses in patients with high tumor loads is challenging (60), underscoring the need to design alternative strategies to improve therapeutic outcome. In this regard, the knowledge that chemotherapy-induced ICD shapes the quality of T cell responses (113, 114), will be key to combine selected chemotherapeutic agents with immunomodulation to generate potent memory T cell responses.
Clinical Activity of Therapeutic Strategies Combining ICI and Chemotherapy
The discoveries relating to molecular rationales for combinatorial approaches involving ICI and chemotherapy led to several clinical studies. We here discuss a few examples underscoring the relevance of combining ICI with chemotherapy. In NSCLC, the efficacy of the combination of anti-PD-1 (pembrolizumab) with chemotherapy was evaluated in the KEYNOTE-021 phase II clinical trial. Results revealed that the combination of anti-PD-1 treatment with chemotherapy (carboplatin and pemetrexed) led to significantly higher response rate in comparison to chemotherapy alone (Table 1) (116). The addition of immunomodulation together with chemotherapy was not associated with enhanced toxicity (116, 127). An updated analysis confirmed a beneficial effect both in terms of improved response rate and progression free survival (127). This led the FDA to grant accelerated approval for this combination therapy to treat metastatic NSCLC patients who have not been previously received chemotherapy and without targetable mutations. Interestingly, higher tumor PD-L1 expression (≥50%) was associated with an enhanced response rate of 80% but given the relatively low numbers of patients studied and observations that patients with <1% PD-L1 expression and with ≥1% PD-L1 expression featured comparable response rate of 54 and 57%, respectively, this study did not conclude on any link between combined treatment efficacy and tumor PD-L1 expression (116). Nevertheless, these observations were of clinical relevance because they provided a therapeutic option to NSCLC patients harboring below 50% tumor PD-L1 expression. Indeed, the efficacy of anti-PD-1 (pembrolizumab) treatment given alone was initially confirmed in patients featuring above 50% PD-L1 expression (24, 128). The KEYNOTE-189 phase III trial subsequently tested the benefit of the addition of anti-PD-1 therapy to chemotherapy in nonsquamous NSCLC (118). Results revealed, independently of the tumor PD-L1 status, that the addition of anti-PD-1 antibody to chemotherapy led to improved overall survival and progression free survival compared to chemotherapy alone. Full approval was then granted by the FDA to anti-PD-1 treatment (pembrolizumab) in combination with chemotherapy for the treatment of NSCLC patients. Whether this combination can also benefit patients with squamous NSCLC is currently being tested in a phase 3 trial (KEYNOTE-407). Overall, the addition of ICI to chemotherapy has changed the standard of care of metastatic lung cancer patients.

Table 1. Examples of completed and ongoing clinical trials evaluating immunomodulation using anti-PD-1, anti-PD-L1, or/and anti-CTLA-4 in combination with chemotherapy.
Therapeutic successes have been achieved when combining other ICI with chemotherapy. In the phase 1 CHECKMATE-012 trial, NSCLC patients received first-line therapy with a combination of anti-PD-1 (Nivolumab) and chemotherapy associations (120). While enhanced toxicity was observed, a beneficial activity of the combination was noted especially for the paclitaxel-carboplatin association. Other combinations of ICI with chemotherapy agents are being evaluated in clinical trials in other cancer types (Table 1). Whether pembrolizumab efficacy in combination with chemotherapy can be extended to other cancer types in addition to lung cancer will be investigated in a phase I/II study (NCT02331251) that will include different chemotherapies and solid tumors, including sarcoma, breast and ovarian cancer. Likewise, the ability of Pembrolizumab to bring additional benefit to chemotherapy will also be searched in the KEYNOTE-062 study in gastric cancer patients.
Importantly, interfering with the PD-L1/PD-1 pathway in combination with chemotherapies using PD-L1 inhibitors has recently led to important clinical achievements. The Impower 150 study underscored the superior efficacy of the addition of atezolizumab to bevacizumab plus chemotherapy in metastatic nonsquamous NSCLC (122). These results were independent of the PD-L1 status of the tumor. The success of this study has led to the approval of this combined therapy in late 2018 for the treatment of metastatic non-squamous NSCLC with atezolizumab with chemotherapy and bevacizumab as first-line treatment. Other phase III studies currently assess the benefit of atezolizumab with different chemotherapy combinations in lung cancer (Table 1). Likewise, the therapeutic benefit of combining other anti-PD-L1 antibodies to chemotherapy in various malignancies is currently being evaluated [Table 1 and reviewed in (129)]. For instance, based on our initial preclinical findings suggesting the ability of chemotherapy to synergize with the blockade of the PD-1/PD-L1 pathway in colon cancer [(75) and discussed above], the MEDITREME trial was initiated at the anticancer center Georges François in Dijon, France. The aim will be to determine the safety and efficacy of the anti-PD-L1/anti-CTLA-4 treatment given in combination with chemotherapy (FOLFOX) (Table 1).
While these different examples illustrate the successes and the possibilities of combining chemotherapy with ICI, even leading in some situations in changing the routine clinical practice in non-squamous NSCLC, it should be noted that some combination therapies have yielded disappointing results. It is indeed notable that in lung cancer the targeting of CTLA-4 combined to chemotherapy has brought no additional therapeutic benefit over chemotherapy treatment given alone in phase 3 clinical trials (Table 1). Thus, while some exciting novel treatment options have been provided by combination therapies using ICI and chemotherapy, the identification of additional successful combinations is warranted as discussed below.
Conclusion
Some chemotherapies have been coined with the term “immunogenic,” i.e., the ability to induce an immune response. The latter can stem from the elimination of suppressive cells, the induction of ICD and/or of effector T cell functions, and possibly lead to tumor eradication (Figure 1). It should be re-emphasized that the ability of chemotherapies to induce ICD is only one of the mechanisms by which they can promote immunity. For instance the drug cisplatin, despite its weak ability to induce ICD, was shown to be immunogenic. Indeed, cisplatin enhances antitumor adaptive immunity by increasing tumor cell killing by CD8 T cells (130, 131). Because high dose chemotherapy can promote immunosuppression as discussed above, a careful selection of the optimal chemotherapy dose to be administered is required to harness the immunogenic properties of chemotherapies while minimizing their immunosuppressive effects (130). Can immunogenic chemotherapies then be used to relieve cancer cell resistance to immune checkpoint inhibitors? While the answer in preclinical models clearly seems to be yes [(75, 78) and reviewed in (129)], the translation of these findings to the clinic has been challenging. While combined treatments with chemotherapy and ICI targeting PD-1/PD-L1 signaling have been effective in human non-squamous NSCLC, disappointing results were obtained when administering chemotherapy and anti-CTLA-4 (Table 1). The lack of relevant biomarkers indicative of anti-CTLA-4 treatment efficacy may explain the difficulties to harness the full potential of this strategy (132). Despite these hurdles, the recent proof-of-principle study showing the ability to successfully combine other ICI such as Tim-3 with chemotherapy opens up a myriad of novel possibilities to be subsequently tested in a clinical setting (83). Identifying and validating additional biomarkers indicative of the efficacy of ICI will help to optimize the most effective ICI—chemotherapy combinations to be given to cancer patients.
Author Contributions
TRV drafted the review and prepared the figure. LA revised and edited the review.
Funding
Our work was supported by grants from the Fondation de France, the Conseil Régional de Bourgogne, the FEDER, the Agence Nationale de la Recherche [ANR-11-LABX-0021], the Acteria Foundation and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 677251).
Conflict of Interest Statement
LA has performed consultancy work for Roche, Merck, and Bristol-Myers Squibb. LA is a recipient of a research grant from Sanofi.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Burnet M. Cancer; a biological approach. I. The processes of control. Br Med J. (1957) 1:779–86. doi: 10.1136/bmj.1.5022.779
2. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
3. Apetoh L, Smyth MJ, Drake CG, Abastado JP, Apte RN, Ayyoub M, et al. Consensus nomenclature for CD8(+) T cell phenotypes in cancer. Oncoimmunology. (2015) 4:e998538. doi: 10.1080/2162402X.2014.998538
4. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
5. Fourcade J, Kudela P, Sun Z, Shen H, Land SR, Lenzner D, et al. PD-1 is a regulator of NY-ESO-1-specific CD8+ T cell expansion in melanoma patients. J Immunol. (2009) 182:5240–9. doi: 10.4049/jimmunol.0803245
6. Gassner FJ, Zaborsky N, Neureiter D, Huemer M, Melchardt T, Egle A, et al. Chemotherapy-induced augmentation of T cells expressing inhibitory receptors is reversed by treatment with lenalidomide in chronic lymphocytic leukemia. Haematologica. (2014) 99:67–9. doi: 10.3324/haematol.2013.098459
7. Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med. (1999) 5:677–85. doi: 10.1038/9525
8. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
9. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
10. Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. (2012) 86:8161–70. doi: 10.1128/JVI.00889-12
11. Brooks DG, McGavern DB, Oldstone MB. Reprogramming of antiviral T cells prevents inactivation and restores T cell activity during persistent viral infection. J Clin Invest. (2006) 116:1675–85. doi: 10.1172/JCI26856
12. Frebel H, Nindl V, Schuepbach RA, Braunschweiler T, Richter K, Vogel J, et al. Programmed death 1 protects from fatal circulatory failure during systemic virus infection of mice. J Exp Med. (2012) 209:2485–99. doi: 10.1084/jem.20121015
13. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. (1999) 11:141–51. doi: 10.1016/S1074-7613(00)80089-8
14. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
15. Radoja S, Saio M, Schaer D, Koneru M, Vukmanovic S, Frey AB. CD8(+) tumor-infiltrating T cells are deficient in perforin-mediated cytolytic activity due to defective microtubule-organizing center mobilization and lytic granule exocytosis. J Immunol. (2001) 167:5042–51. doi: 10.4049/jimmunol.167.9.5042
17. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
18. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
19. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
20. Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. (2013) 14:1212–8. doi: 10.1038/ni.2762
21. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
22. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
23. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
24. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. (2015) 372:2018–28. doi: 10.1056/NEJMoa1501824
25. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
26. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
27. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
28. Lake RA, Robinson BW. Immunotherapy and chemotherapy–a practical partnership. Nat Rev Cancer. (2005) 5:397–405. doi: 10.1038/nrc1613
29. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol. (2008) 8:59–73. doi: 10.1038/nri2216
30. June CH, Ledbetter JA, Gillespie MM, Lindsten T, Thompson CB. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol Cell Biol. (1987) 7:4472–81. doi: 10.1128/MCB.7.12.4472
31. Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. (1970) 169:1042–9. doi: 10.1126/science.169.3950.1042
32. Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. (1989) 7:445–80. doi: 10.1146/annurev.iy.07.040189.002305
33. Liechtenstein T, Dufait I, Lanna A, Breckpot K, Escors D. Modulating co-stimulation during antigen presentation to enhance cancer immunotherapy. Immunol Endocr Metab Agents Med Chem. (2012) 12:224–35. doi: 10.2174/187152212802001875
34. Zhu Y, Yao S, Chen L. Cell surface signaling molecules in the control of immune responses: a tide model. Immunity. (2011) 34:466–78. doi: 10.1016/j.immuni.2011.04.008
35. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
36. Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from Phase II and Phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. (2015) 33:1889–94. doi: 10.1200/JCO.2014.56.2736
37. Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. (2018) 28:416–32. doi: 10.1038/s41422-018-0011-0
38. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. (2018) 33:581–98. doi: 10.1016/j.ccell.2018.03.005
39. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. (2016) 315:1600–9. doi: 10.1001/jama.2016.4059
40. Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:1270–1. doi: 10.1056/NEJMc1509660
41. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
42. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. (2016) 7:550. doi: 10.3389/fimmu.2016.00550
43. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity. (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
44. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. (2016) 16:121–6. doi: 10.1038/nrc.2016.2
45. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
46. Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. (2014) 32:1020–30. doi: 10.1200/JCO.2013.53.0105
47. Rexer H, Bedke J. [First-line therapy in advanced renal cell carcinoma: a randomized, open-label phase III study evaluating the efficacy and safety of pembrolizumab (MK-3475) in combination with axitinib compared to sunitinib monotherapy as first-line treatment for locally advanced or metastatic renal cell carcinoma (mRCC) (Keynote-426) - AN 39/16 of the AUO]. Urologe A. (2017) 56:385–6. doi: 10.1007/s00120-017-0335-2
48. Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S, et al. FDA approval summary: pembrolizumab for the treatment of recurrent or metastatic head and neck squamous cell carcinoma with disease progression on or after platinum-containing chemotherapy. Oncologist. (2017) 22:873–8. doi: 10.1634/theoncologist.2016-0496
49. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
50. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
51. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
52. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577–81. doi: 10.1038/nature13988
53. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
54. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) 348:124–8. doi: 10.1126/science.aaa1348
55. Chan TA, Wolchok JD, Snyder A. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. (2015) 373:1984. doi: 10.1056/NEJMc1508163
56. Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. (2017) 18:1009–21. doi: 10.1016/S1470-2045(17)30516-8
57. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. (2013) 31:e439–42. doi: 10.1200/JCO.2012.47.7521
58. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. (2014) 515:563–7. doi: 10.1038/nature14011
59. Wang Q, Liu F, Liu L. Prognostic significance of PD-L1 in solid tumor: an updated meta-analysis. Medicine. (2017) 96:e6369. doi: 10.1097/MD.0000000000006369
60. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. (2017) 545:60–5. doi: 10.1038/nature22079
61. Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. (2000) 74:181–273. doi: 10.1016/S0065-2776(08)60911-6
62. Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. (2005) 201:1257–68. doi: 10.1084/jem.20042028
63. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. (2016) 354:1160–5. doi: 10.1126/science.aaf2807
64. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science. (2016) 354:1165–9. doi: 10.1126/science.aae0491
65. Puyo S, Montaudon D, Pourquier P. From old alkylating agents to new minor groove binders. Crit Rev Oncol Hematol. (2014) 89:43–61. doi: 10.1016/j.critrevonc.2013.07.006
66. DeVita VT Jr., Chu E. A history of cancer chemotherapy. Cancer Res. (2008) 68:8643–53. doi: 10.1158/0008-5472.CAN-07-6611
67. Dezern AE, Styler MJ, Drachman DB, Hummers LK, Jones RJ, Brodsky RA. Repeated treatment with high dose cyclophosphamide for severe autoimmune diseases. Am J Blood Res. (2013) 3:84–90.
68. Schwartz HS, Grindey GB. Adriamycin and daunorubicin: a comparison of antitumor activities and tissue uptake in mice following immunosuppression. Cancer Res. (1973) 33:1837–44.
69. Apetoh L, Mignot G, Panaretakis T, Kroemer G, Zitvogel L. Immunogenicity of anthracyclines: moving towards more personalized medicine. Trends Mol Med. (2008) 14:141–51. doi: 10.1016/j.molmed.2008.02.002
70. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
71. Apetoh L, Ladoire S, Coukos G, Ghiringhelli F. Combining immunotherapy and anticancer agents: the right path to achieve cancer cure? Ann Oncol. (2015) 26:1813–23. doi: 10.1093/annonc/mdv209
72. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. (2013) 39:74–88. doi: 10.1016/j.immuni.2013.06.014
73. Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer. (2017) 17:286–301. doi: 10.1038/nrc.2017.17
74. Rivera Vargas T, Apetoh L. Danger signals: chemotherapy enhancers? Immunol Rev. (2017) 280:175–93. doi: 10.1111/imr.12581
75. Dosset M, Vargas TR, Lagrange A, Boidot R, Vegran F, Roussey A, et al. PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. (2018) 7:e1433981. doi: 10.1080/2162402X.2018.1433981
76. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
77. Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, et al. Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res. (2008) 14:2413–20. doi: 10.1158/1078-0432.CCR-07-4491
78. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. (2016) 44:343–54. doi: 10.1016/j.immuni.2015.11.024
79. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. (2007) 13:54–61. doi: 10.1038/nm1523
80. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. (2007) 13:1050–9. doi: 10.1038/nm1622
81. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. (2009) 15:1170–8. doi: 10.1038/nm.2028
82. Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. (2011) 8:151–60. doi: 10.1038/nrclinonc.2010.223
83. de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 regulates CD103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. (2018) 33:60–74 e6. doi: 10.1016/j.ccell.2017.11.019
84. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. (2002) 415:536–41. doi: 10.1038/415536a
85. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. (2014) 20:1301–9. doi: 10.1038/nm.3708
86. Bezu L, Gomes-da-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Corrigendum: “combinatorial strategies for the induction of immunogenic cell death”. Front Immunol. (2015) 6:275. doi: 10.3389/fimmu.2015.00187
87. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-treatment with chemotherapy can enhance the antigenicity and immunogenicity of tumours by promoting adaptive immune responses. Br J Cancer. (2010) 102:115–23. doi: 10.1038/sj.bjc.6605465
88. Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. (2006) 6:715–27. doi: 10.1038/nri1936
89. Sanchez-Perez L, Suryadevara CM, Choi BD, Reap EA, Sampson JH. Leveraging chemotherapy-induced lymphopenia to potentiate cancer immunotherapy. Oncoimmunology. (2014) 3:e944054. doi: 10.4161/21624011.2014.944054
90. Penel N, Adenis A, Bocci G. Cyclophosphamide-based metronomic chemotherapy: after 10 years of experience, where do we stand and where are we going? Crit Rev Oncol Hematol. (2012) 82:40–50. doi: 10.1016/j.critrevonc.2011.04.009
91. Moschella F, Valentini M, Arico E, Macchia I, Sestili P, D'Urso MT, et al. Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer Res. (2011) 71:3528–39. doi: 10.1158/0008-5472.CAN-10-4523
92. Moschella F, Torelli GF, Valentini M, Urbani F, Buccione C, Petrucci MT, et al. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients' blood cells: implications for cancer chemoimmunotherapy. Clin Cancer Res. (2013) 19:4249–61. doi: 10.1158/1078-0432.CCR-12-3666
93. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med. (2014) 6:237ra67. doi: 10.1126/scitranslmed.3007974
94. Ngiow SF, Young A, Jacquelot N, Yamazaki T, Enot D, Zitvogel L, et al. A threshold level of intratumor CD8+ T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res. (2015) 75:3800–11. doi: 10.1158/0008-5472.CAN-15-1082
95. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. (2017) 9:eaal3604. doi: 10.1126/scitranslmed.aal3604
96. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. (2014) 5:673. doi: 10.3389/fimmu.2014.00673
97. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. (2017) 46:233–44. doi: 10.1016/j.immuni.2017.01.005
98. Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. (2004) 34:336–44. doi: 10.1002/eji.200324181
99. Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. (2007) 56:641–8. doi: 10.1007/s00262-006-0225-8
100. Alizadeh D, Larmonier N. Chemotherapeutic targeting of cancer-induced immunosuppressive cells. Cancer Res. (2014) 74:2663–8. doi: 10.1158/0008-5472.CAN-14-0301
101. Apetoh L, Vegran F, Ladoire S, Ghiringhelli F. Restoration of antitumor immunity through selective inhibition of myeloid derived suppressor cells by anticancer therapies. Curr Mol Med. (2011) 11:365–72. doi: 10.2174/156652411795976574
102. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. (2009) 9:900–9. doi: 10.1016/j.intimp.2009.03.015
103. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. (2014) 74:104–18. doi: 10.1158/0008-5472.CAN-13-1545
104. Fang L, Lowther DE, Meizlish ML, Anderson RC, Bruce JN, Devine L, et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro Oncol. (2013) 15:1479–90. doi: 10.1093/neuonc/not110
105. Zhou J, Nagarkatti P, Zhong Y, Nagarkatti M. Characterization of T-cell memory phenotype after in vitro expansion of tumor-infiltrating lymphocytes from melanoma patients. Anticancer Res. (2011) 31:4099–109.
106. Wang QJ, Hanada K, Robbins PF, Li YF, Yang JC. Distinctive features of the differentiated phenotype and infiltration of tumor-reactive lymphocytes in clear cell renal cell carcinoma. Cancer Res. (2012) 72:6119–29. doi: 10.1158/0008-5472.CAN-12-0588
107. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. (2015) 348:62–8. doi: 10.1126/science.aaa4967
108. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. (2002) 298:850–4. doi: 10.1126/science.1076514
109. Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. (2008) 26:5233–9. doi: 10.1200/JCO.2008.16.5449
110. Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Hershkovitz L, et al. Clinical responses in a phase II study using adoptive transfer of short-term cultured tumor infiltration lymphocytes in metastatic melanoma patients. Clin Cancer Res. (2010) 16:2646–55. doi: 10.1158/1078-0432.CCR-10-0041
111. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. (2011) 17:4550–7. doi: 10.1158/1078-0432.CCR-11-0116
112. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, et al. Checkpoint blockade immunotherapy induces dynamic changes in PD-1(-)CD8(+) tumor-infiltrating T cells. Immunity. (2019) 50:181–94 e6. doi: 10.1016/j.immuni.2018.11.014
113. Inoue H, Tani K. Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ. (2014) 21:39–49. doi: 10.1038/cdd.2013.84
114. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, et al. RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science. (2015) 350:328–34. doi: 10.1126/science.aad0395
115. Gadgeel SM, Stevenson JP, Langer CJ, Gandhi L, Borghaei H, Patnaik A, et al. Pembrolizumab and platinum-based chemotherapy as first-line therapy for advanced non-small-cell lung cancer: Phase 1 cohorts from the KEYNOTE-021 study. Lung Cancer. (2018) 125:273–81. doi: 10.1016/j.lungcan.2018.08.019
116. Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. (2016) 17:1497–508. doi: 10.1016/S1470-2045(16)30498-3
117. Borghaei H, Langer CJ, Gadgeel S, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. Updated results from KEYNOTE-021 Cohort G: a randomized, phase 2 study of pemetrexed and carboplatin (PC) with or without pembrolizumab (pembro) as first-line therapy for advanced nonsquamous NSCLC. Ann Oncol. (2017) 28. doi: 10.1093/annonc/mdx440.052
118. Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
119. Weiss GJ, Waypa J, Blaydorn L, Coats J, McGahey K, Sangal A, et al. A phase Ib study of pembrolizumab plus chemotherapy in patients with advanced cancer (PembroPlus). Br J Cancer. (2017) 117:33–40. doi: 10.1038/bjc.2017.145
120. Rizvi NA, Hellmann MD, Brahmer JR, Juergens RA, Borghaei H, Gettinger S, et al. Nivolumab in combination with platinum-based doublet chemotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. (2016) 34:2969–79. doi: 10.1200/JCO.2016.66.9861
121. Liu SV, Camidge DR, Gettinger SN, Giaccone G, Heist RS, Hodi FS, et al. Long-term survival follow-up of atezolizumab in combination with platinum-based doublet chemotherapy in patients with advanced non-small-cell lung cancer. Eur J Cancer. (2018) 101:114–22. doi: 10.1016/j.ejca.2018.06.033
122. Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. (2018) 378:2288–301. doi: 10.1056/NEJMoa1716948
123. Govindan R, Szczesna A, Ahn MJ, Schneider CP, Gonzalez Mella PF, Barlesi F, et al. Phase III trial of ipilimumab combined with paclitaxel and carboplatin in advanced squamous non-small-cell lung cancer. J Clin Oncol. (2017) 35:3449–57. doi: 10.1200/JCO.2016.71.7629
124. Patel SP, Kim DW, Bassett RL, Cain S, Washington E, Hwu WJ, et al. A phase II study of ipilimumab plus temozolomide in patients with metastatic melanoma. Cancer Immunol Immunother. (2017) 66:1359–66. doi: 10.1007/s00262-017-2030-y
125. Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W, et al. Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol. (2016) 34:3740–8. doi: 10.1200/JCO.2016.67.6601
126. Yamazaki N, Uhara H, Fukushima S, Uchi H, Shibagaki N, Kiyohara Y, et al. Phase II study of the immune-checkpoint inhibitor ipilimumab plus dacarbazine in Japanese patients with previously untreated, unresectable or metastatic melanoma. Cancer Chemother Pharmacol. (2015) 76:969–75. doi: 10.1007/s00280-015-2870-0
127. Borghaei H, Langer CJ, Gadgeel S, Papadimitrakopoulou VA, Patnaik A, Powell SF, et al. 24-month overall survival from KEYNOTE-021 Cohort G: pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous non-small cell lung cancer. J Thorac Oncol. (2018) 14:124–9. doi: 10.1016/j.jtho.2018.08.004
128. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
129. Yan Y, Kumar AB, Finnes H, Markovic SN, Park S, Dronca RS, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front Immunol. (2018) 9:1739. doi: 10.3389/fimmu.2018.01739
130. Tran L, Allen CT, Xiao R, Moore E, Davis R, Park SJ, et al. Cisplatin alters antitumor immunity and synergizes with PD-1/PD-L1 inhibition in head and neck squamous cell carcinoma. Cancer Immunol Res. (2017) 5:1141–51. doi: 10.1158/2326-6066.CIR-17-0235
131. Merritt RE, Mahtabifard A, Yamada RE, Crystal RG, Korst RJ. Cisplatin augments cytotoxic T-lymphocyte-mediated antitumor immunity in poorly immunogenic murine lung cancer. J Thorac Cardiovasc Surg. (2003) 126:1609–17. doi: 10.1016/S0022-5223(03)00707-4
Keywords: chemotherapy, checkpoint, immunomodulation, cancer, T cells
Citation: Rivera Vargas T and Apetoh L (2019) Can Immunogenic Chemotherapies Relieve Cancer Cell Resistance to Immune Checkpoint Inhibitors? Front. Immunol. 10:1181. doi: 10.3389/fimmu.2019.01181
Received: 21 February 2019; Accepted: 09 May 2019;
Published: 29 May 2019.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelCopyright © 2019 Rivera Vargas and Apetoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lionel Apetoh, bGlvbmVsLmFwZXRvaEBpbnNlcm0uZnI=