 Henrike J. Fischer
Henrike J. Fischer Tobias L. K. Finck
Tobias L. K. Finck Hannah L. Pellkofer3‡
Hannah L. Pellkofer3‡ Holger M. Reichardt
Holger M. Reichardt Fred Lühder
Fred Lühder- 1Institute for Neuroimmunology and Multiple Sclerosis Research, University Medical Center Goettingen, Göttingen, Germany
- 2Institute for Cellular and Molecular Immunology, University Medical Center Goettingen, Göttingen, Germany
- 3Department of Neurology, University Medical Center Goettingen, Göttingen, Germany
Multiple Sclerosis (MS) is an autoimmune disease of the central nervous system (CNS), characterized by the infiltration of mononuclear cells into the CNS and a subsequent inflammation of the brain. Monocytes are implicated in disease pathogenesis not only in their function as potential antigen-presenting cells involved in the local reactivation of encephalitogenic T cells but also by independent effector functions contributing to structural damage and disease progression. However, monocytes also have beneficial effects as they can exert anti-inflammatory activity and promote tissue repair. Glucocorticoids (GCs) are widely used to treat acute relapses in MS patients. They act on a variety of cell types but their exact mechanisms of action including their modulation of monocyte function are not fully understood. Here we investigated effects of the therapeutically relevant GC methylprednisolone (MP) on monocytes from healthy individuals and MS patients in vitro and in vivo. The monocyte composition in the blood was different in MS patients compared to healthy individuals, but it was only marginally affected by MP treatment. In contrast, application of MP caused a marked shift toward an anti-inflammatory monocyte phenotype in vitro and in vivo as revealed by an altered gene expression profile. Chemotaxis of monocytes toward CCL2, CCL5, and CX3CL1 was increased in MS patients compared to healthy individuals and further enhanced by MP pulse therapy. Both of these migration-promoting effects were more pronounced in MS patients with an acute relapse than in those with a progressive disease. Interestingly, the pro-migratory GC effect was independent of chemokine receptor levels as exemplified by results obtained for CCR2. Collectively, our findings suggest that GCs polarize monocytes toward an anti-inflammatory phenotype and enhance their migration into the inflamed CNS, endowing them with the capacity to suppress the pathogenic immune response.
Introduction
Multiple Sclerosis (MS) is an inflammatory autoimmune disease of the central nervous system (CNS) involving different types of immune cells including T cells, B cells, and monocytes. The most common disease course is characterized by acute relapses with complete or incomplete remission. This relapsing-remitting phenotype (RRMS) is observed in the majority of the MS patients, with young adults being most affected. RRMS can convert into a secondary-progressive form (SPMS) later in life, which is characterized by progressive worsening of the disease with or without additional relapses. The hallmark of the third form of MS, termed primary-progressive (PPMS), is a continuous worsening of the symptoms without intermittent improvements (1). Although numerous new drugs have been developed within the last decade, the most widely used treatment of acute relapses is still high-dose methylprednisolone (MP) pulse therapy to which most patients respond well, resulting in an amelioration of symptoms within a few days (2). Patients suffering from SPMS and PPMS are also treated with MP pulse therapy in the case that the disease is not stable. Mechanistically, various activities of glucocorticoids (GCs) affecting immune cells but also non-hematopoietic cell types are discussed (3, 4). Furthermore, the GC response was recently reported to be highly cell type-specific, both in magnitude and even direction of transcriptional regulation (5). In the context of MS therapy it is believed that patients profit most from direct or indirect dampening effects on T cells. It has been reported that GCs down-regulate expression levels of pro-inflammatory cytokines and adhesion molecules required to pass the blood-brain barrier (BBB). They also promote apoptosis induction in immune cells, inhibit T cell activation, and additionally exert inhibitory effects on inflammatory mediators such as nitric oxide (NO) (6). Our own preclinical studies using the animal model experimental autoimmune encephalomyelitis (EAE) further revealed that T cells are the major target cells of free administered GCs (7, 8). However, effects on myeloid cells were also shown to be crucial if GC were encapsulated in liposomes (9) or nanoparticles (10). In addition, we found that altered T cell migration along chemokine gradients was a mechanism accounting for the therapeutic activity of GCs in the treatment of neuroinflammation, whereas apoptosis induction in T cells unexpectedly turned out to be of minor importance (11).
T cells are the target of most current immunotherapies for MS patients, highlighting the importance of this cell population for the pathogenesis of MS. Nevertheless, myeloid cells including monocytes play important roles for innate immune responses and indirectly also influence adaptive immune responses by serving as antigen-presenting cells, and with both functions they also play a crucial role in MS and EAE (12). They are found in CNS lesions in EAE and MS and often outnumber infiltrating T cells. In animal models it has been shown that monocytic infiltration contributes to disease progression (13), and monocyte-derived macrophages are key players in the reactivation of infiltrating T cells (14). Consequently, elimination of macrophages (15–17) or selective depletion of CCR2+ Ly-6Chi monocytes (18) reduced CNS inflammation. Alterations in the composition of monocyte subpopulations in the peripheral blood and cerebrospinal fluid (CSF) of MS patients have been reported as well, thus further highlighting their substantial role in human neuroinflammation (19).
In humans, monocytes are a heterogeneous cell population, constituting ~10% of total leukocytes in the blood. They have a short life span and evolve in three different subsets: the most prevalent being CD14++CD16− classical (or inflammatory) monocytes, CD14++CD16+ intermediate state monocytes, and CD14+CD16++ non-classical monocytes (20). They can give rise to macrophages that encompass a dynamic spectrum of phenotypes with classical or M1 macrophages (producing IL-12, IL-1β, NO and reactive oxygen species, and acting in a pro-inflammatory fashion) and alternatively activated or M2 macrophages (expressing CD163, CD206, Arg1, and acting in an anti-inflammatory fashion) being the extreme ends of this spectrum (21). M2 myeloid cells were found to contribute to an improvement of autoimmune diseases such as MS and EAE (22–25). Similarly, skewed proportions of the different monocyte subsets have been reported for many human inflammatory and autoimmune diseases. For instance, in rheumatoid arthritis, systemic lupus erythematodes, sepsis, uveitis and sarcoidosis, intermediate-state monocytes were expanded (26–31). In contrast, data for MS patients concerning monocyte subsets are less consistent (19, 32, 33). Classical pro-inflammatory CD14++CD16− monocytes are recruited to the CNS in response to CCL2. Non-classical CD14+CD16++ monocytes, however, are not necessarily beneficial in the context of MS. Namely, it has been shown that the latter cells can adhere to the endothelium and help T cells to extravasate at the site of inflammation and thereby contribute to MS pathology. Accordingly, they are found in active and demyelinating lesions and the CSF (33). Beyond their disease-promoting activity, however, monocytes are also able to dampen inflammation depending on their subtype and status.
The influence of different drugs used in the long-term treatment of MS such as glatiramer acetate (23), dimethyl fumarate (34), or fingolimod (35) on myeloid cell function has been intensively investigated. In contrast, effects of GCs on myeloid cells in the context of MS are less clear. Treatment of monocytes with GCs in vitro induces a stable anti-inflammatory gene expression profile (36). Consequently, such monocytes interfere with T-cell-mediated inflammation in vivo, where they were shown to directly suppress the secretion of IL-17 and IFNγ without inducing a direct Th2 shift. Additionally, treatment with GCs enables monocytes to induce regulatory T cells (Treg) at the site of inflammation (37, 38).
Here we investigate the influence of MP—it being the most widely applied GC in MS therapy—on human monocytes from healthy individuals and MS patients in vitro and ex vivo. We found evidence that monocyte polarization becomes skewed toward the M2 phenotype by MP treatment and that the migration of monocytes along chemokine gradients is increased without any significant changes in the level of their respective receptors. These findings suggest that GCs also exert their beneficial effects on MS bouts by tuning monocyte function and not necessarily solely by suppressing T cells.
Materials and Methods
Patients
Thirty patients with established diagnosis of MS according to the McDonald Criteria revised in 2017 were included in the current study (14 RRMS, 8 SPMS, 8 PPMS). All patients received high-dose MP (1,000 mg) intravenously on three consecutive days according to medical indication (due either to MS relapse or progressive worsening of neurologic symptoms in patients with progressive MS). Peripheral blood was drawn in Li-Heparin monovettes (Sarstedt, Nürnbrecht, Germany) before and 24 h after the first injection of MP. Due to the small volume of blood that could be obtained from each patient and due to the sometimes limited recovery of blood after MP therapy, not all types of analyses were performed for every patient. The number of patients included in each experiment is therefore indicated in the figure legends.
Information about MS patients included in this study (disease subtype, age, gender, severity of clinical symptoms as assessed by the Expanded Disability Score Scale (EDSS), acute relapse, disease duration, treatment) are summarized in Table 1. SPMS patients that were treated with MP due to an acute relapse (Table 1) were combined with the RRMS group and collectively referred to as “MS patients with acute relapse.” In contrast, SPMS patients without an acute relapse (Table 1) were combined with the PPMS group and referred to as “MS patients with progressive disease.” In addition, 24 healthy donors (age and gender summarized in Table 1) were included. The investigations were conducted according to the Declaration of Helsinki and national and international guidelines. The study was approved by the local ethics committee of the University Medical Center Göttingen. Informed written consent was obtained from each subject prior to the collection of blood.
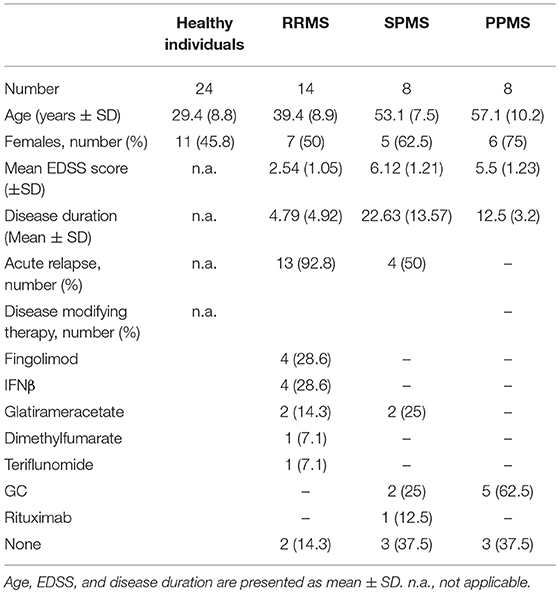
Table 1. Summary of the characteristics of patients and healthy individuals included in the study.
Purification and Short-Term Culture of Human Monocytes
Peripheral blood lymphocytes were enriched using a lymphoprep gradient (Axis Shield, Oslo, Norway) as described (11), and monocytes were purified with magnetic beads (Stemcell Technologies, Köln, Germany). Purity was assessed on the basis of CD14/CD16 staining by flow cytometry using a FACSCanto II device (BD Biosciences, Heidelberg, Germany), and routinely >95% (Figure 1). Monocytes were analyzed directly or cultured for 3 h in RPMI 1640 medium supplemented with 0.5% fatty acid-free BSA under serum-starved conditions in the presence or absence of 10−6 M MP. One portion of the cells was used for RNA isolation and surface marker analyses and the other portion served to assess the migratory capacity.
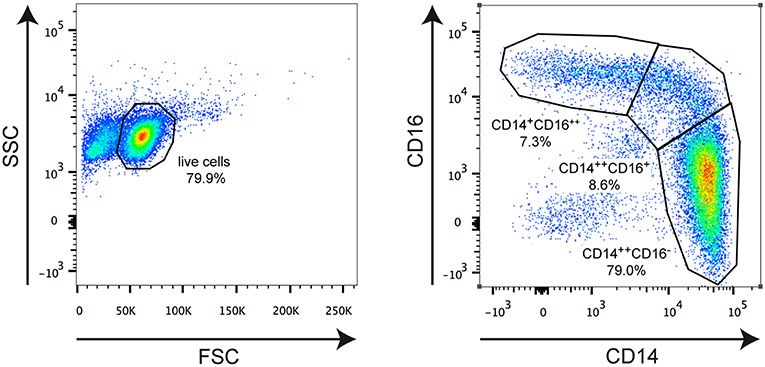
Figure 1. Representative FACS analysis illustrating the applied gating strategy. Monocytes were isolated from an MS patient and stained for CD14 and CD16 surface expression using fluorochrome-conjugated monoclonal antibodies. The left plot depicts the gating for living cells based on forward scatter (FSC) and side scatter (SSC). The right plot shows gating for classical CD14++CD16− monocytes, intermediate state CD14++CD16+ monocytes, and non-classical CD14+CD16++ monocytes. The borders of the gates and the percentages of cells therein are indicated in each plot.
Flow Cytometry
Flow cytometric analysis of monocytes was performed as previously described (11). To this end, cells were stained with the following monoclonal antibodies (BioLegend, Uithoorn, The Netherlands) in PBS supplemented with 0.1% BSA and 0.01% NaN3: anti-human CD14-PE/Cy7 (clone: HCD14), anti-human CD16-APC/Cy7 (clone: 3G8), anti-human CD163-PE (clone GHI/61), anti-human CD192 (CCR2)-PerCP/Cy5.5 (clone: K036C2), and anti-human CX3CR1-FITC (clone: 2A9-1). Data were acquired on a FACS Canto II device (BD Bioscience) and analyzed using FlowJo® software (Tree Star, Ashland, OR).
Boyden Camber Assay
After 3 h in vitro cultivation with or without MP (see above), 5 × 105 monocytes per well were subjected to a transwell assay using a pore size of 5 μm (Corning Life Sciences, NY, USA) as previously described (11). Cells were allowed to migrate along a gradient of 10 ng/ml CCL2, 10 ng/ml CCL5, or 1 ng/ml CX3CL1 (ImmunoTools, Friesoythe, Germany) for 1 h. The medium in the lower chamber was harvested and the transmigrated monocytes attached to the plate were incubated with 2 mM EDTA in PBS for 20 min at 37°C. Detached cells were scratched off the well bottom and pooled with the harvested medium for analysis. Finally, cells were quantified by flow cytometric analysis using Calibrite Beads (BD Bioscience).
Quantitative RT-PCR
Quantitative RT-PCR was performed as previously described (11). To this end, total RNA was isolated using the Quick-RNA MiniPrep Kit (Zymo, Irvine, CA) and cDNA was prepared with the iScript Kit (Bio-Rad, Munich, Germany). Quantitative RT-PCR was performed on an ABI 7500 instrument (Applied Biosystems, Darmstadt, Germany) using the SYBR mastermix from the same company. Results were normalized to the mRNA expression of HPRT and evaluated using the ΔΔCt method. Primer sequences are depicted in Table 2.
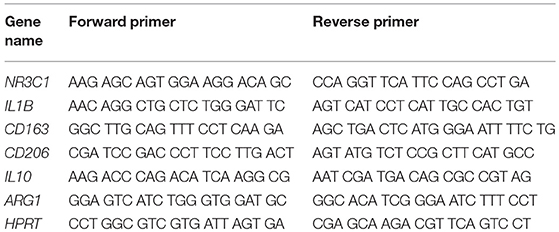
Table 2. Primer sequences used for quantitative RT-PCR analysis.
Statistical Analysis
Data sets were initially subjected to the Shapiro-Wilk normality test to analyze Gaussian distribution. Depending on the results, either a parametric or a non-parametric test was employed, and in the case of matched data, a paired test was used. Accordingly, the experimental groups were compared with a t-test, Mann Whitney test, Wilcox matched-pairs signed rank test, or a One-way ANOVA followed by Newman-Keuls Multiple Comparison test as outlined in the figure legends. Analyses were performed with GraphPad Prism software (San Diego, CA). Data are depicted as box-and-whiskers plots showing the minimum, maximum and median, or as the mean ± SEM in all other types of graphs. Levels of significance are as follows: n.s. p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
Results
The Abundance of Classical CD14++CD16− Monocytes Is Increased in MS Patients Independently of Disease Activity
Monocytes were purified from the peripheral blood of healthy individuals and MS patients and analyzed for the distribution of cellular subsets by flow cytometry (Figure 1). Classical CD14++CD16− monocytes were significantly more abundant in MS patients than in healthy control subjects whereas non-classical CD14+CD16++ monocytes were less frequent in MS patients (Figure 2). In contrast, the percentage of intermediate state CD14++CD16+ monocytes was unaltered. Noteworthy, these findings are in line with previous reports (19). Furthermore, we did not observe any differences concerning the abundance of monocyte subtypes between MS patients with progressive disease and those undergoing an acute relapse (Figure 2).
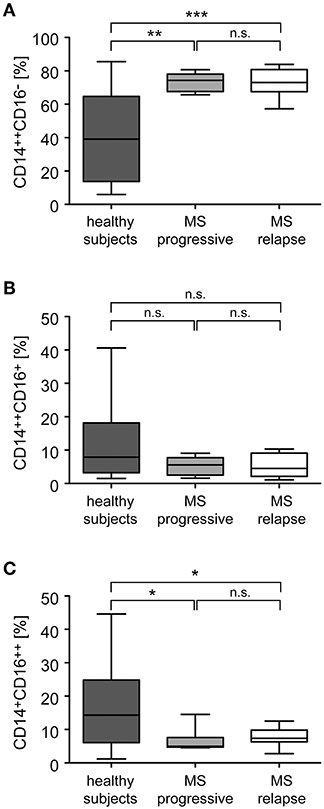
Figure 2. Distribution of monocyte subsets in the peripheral blood of healthy subjects and MS patients with progressive disease or an acute relapse. Monocytes were isolated from the peripheral blood and the percentages of CD14++CD16− inflammatory monocytes (A), CD14++CD16+ intermediate state monocytes (B), and CD14+CD16++ non-classical monocytes (C) were determined by flow cytometry. MS patients were divided into two groups according to their disease activity (progressive, relapse). Data are presented as box-and-whiskers plots showing the minimum, maximum and median; n = 20 (healthy subjects), n = 8 (MS progressive), n = 12 (MS relapse). Statistical analysis was performed using a One-way ANOVA and Newman-Keuls Multiple Comparison test. Levels of significance: n.s. p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
GCs Have Only a Minor Impact on Monocyte Subset Distribution
Monocytes were isolated from MS patients with progressive disease or an acute relapse before they received a bolus injection of MP. To study short term effects of GCs, the ex vivo retrieved cells were incubated for 3 h in vitro in the absence (control) or presence of 10−6 M MP. In addition, monocytes were isolated from the same MS patients again 24 h after MP pulse therapy to determine long term effects of GC treatment in vivo. In the case of patients with an acute relapse, monocyte subset distribution remained unaltered by MP treatment with regard to both short and long term effects (Figure 3). In contrast, we observed an increased frequency of classical CD14++CD16− monocytes in patients with progressive disease after long term MP pulse therapy, and a concomitant but non-significant reduction of non-classical CD14+CD16++ monocytes (Figure 3).
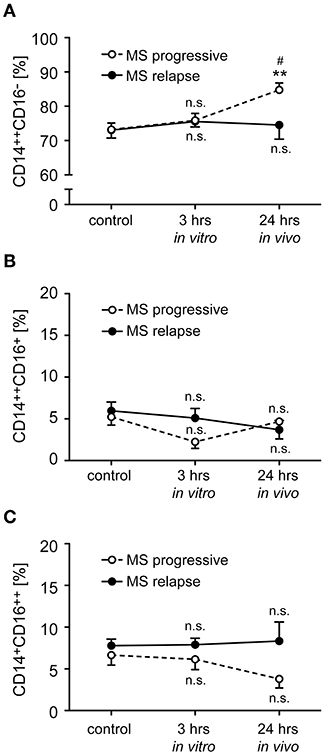
Figure 3. Impact of GC treatment on monocyte subset distribution in MS patients with progressive disease or an acute relapse. Monocytes were isolated from MS patients before MP pulse therapy and incubated for 3 h without (control) or with 10−6 M MP in vitro. A second blood sample was obtained from the same MS patients 24 h after MP pulse therapy in vivo. The percentages of CD14++CD16− inflammatory monocytes (A), CD14++CD16+ intermediate state monocytes (B), and CD14+CD16++ non-classical monocytes (C) were determined by flow cytometry. MS patients were divided into two groups according to their disease activity (progressive, relapse). Data are presented as the mean ± SEM; n = 8 (MS progressive), n = 12 (MS relapse). Statistical analysis was performed using a One-way ANOVA and Newman-Keuls Multiple Comparison test. Levels of significance: n.s. p ≥ 0.05; **p < 0.01 (control vs. 24 h); #p < 0.05 (3 vs. 24 h).
GCs Induce Monocyte Polarization Toward an Anti-inflammatory M2 Phenotype
In addition to the classification of monocytes on the basis of cell surface receptors, their phenotype can be characterized by determining their gene expression profile. To this end, we performed an mRNA expression analysis of genes that have been linked to either an M1 or M2 polarization. Monocytes were isolated from healthy subjects and MS patients, incubated with or without 10−6 M MP in vitro for 3 h and analyzed by quantitative RT-qPCR. In addition, long term GC effects were investigated 24 h after MP pulse therapy in vivo (only patients). Initially, we analyzed mRNA levels of NR3C1, the gene encoding the GC receptor (GR). NR3C1 expression did not significantly differ between healthy subjects and MS patients and was reduced by MP treatment as expected (39, 40). However, the latter effect reached statistical significance only in the case of the in vivo therapy (Figure 4A). Expression analysis further revealed that mRNA levels of IL1B, a pro-inflammatory cytokine that is typical for an M1 polarization of monocytes, were reduced by MP treatment in healthy subjects and MS patients both in vitro and in vivo (Figure 4B). Concomitantly, the M2 marker genes ARG1, CD163, and CD206 as well as the gene encoding the anti-inflammatory cytokine IL10 were all increased in monocytes of healthy subjects and MS patients following MP treatment, although the differences were not always statistically significant (Figures 4C–F). It is noteworthy that in general, GC effects were more pronounced after high-dose MP pulse therapy than following in vitro culture (Figure 4). Gene expression levels for CD163 and IL10 were found to be elevated in MS patients compared to healthy individuals in the steady state (Figure 4). To confirm our results at the protein level, we analyzed surface expression of CD163 as an example by flow cytometry. There were no differences in CD163 levels after short term MP treatment in vitro, either for healthy subjects or MS patients (Figure 5A and data not shown). However, 24 h after MP pulse therapy in vivo, CD163 surface levels were strongly elevated in a subgroup of MS patients (Figure 5). Interestingly, 6 out of 7 patients in whom CD163 surface expression on monocytes was upregulated were suffering from an acute relapse. Collectively, MP induces a shift toward the anti-inflammatory M2 monocyte phenotype, which is most evident in MS patients receiving high-dose MP pulse therapy.
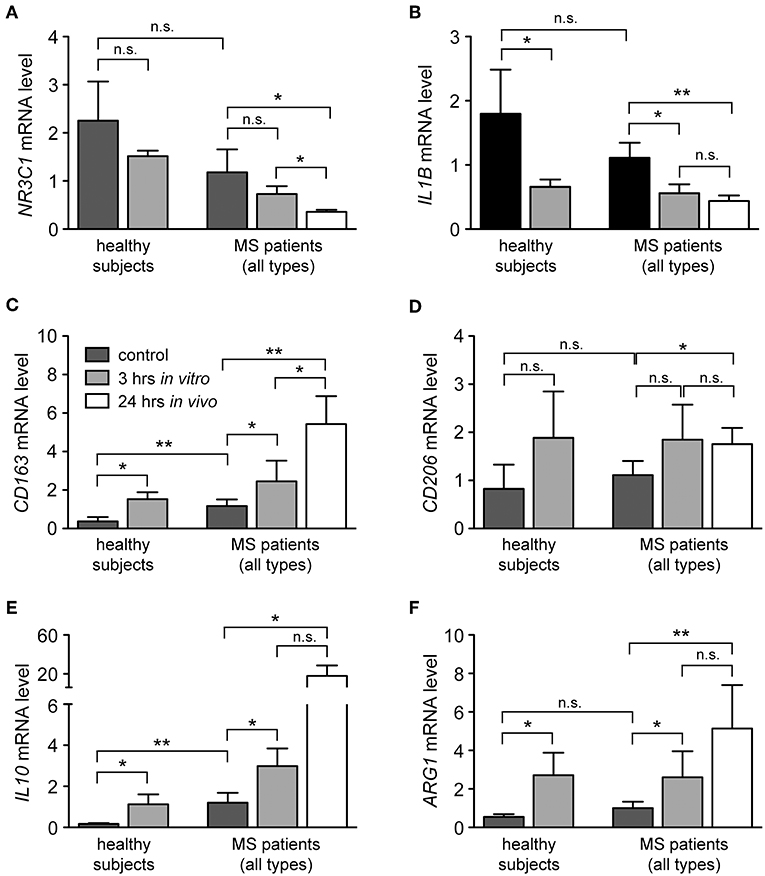
Figure 4. Modulation of the phenotype of monocytes from healthy subjects and MS patients by GCs. Monocytes were isolated from the peripheral blood and cultured without (control) or with 10−6 M MP for 3 h in vitro. A second blood sample was obtained from the same MS patients 24 h after MP pulse therapy in vivo. Thereafter, RNA was prepared and analyzed by quantitative RT-PCR for mRNA levels of NR3C1 (A), IL1B (B), CD163 (C), CD206 (D), IL10 (E), and ARG1 (F). Gene expression was evaluated using the ΔΔCt method and normalized to HPRT. Data are presented as the mean ± SEM; n = 6 (healthy individuals), n = 9 (MS patients). Statistical analysis was performed using a paired t-test (IL1B, CD206) or a Wilcox matched-pairs signed rank test (NR3C1, CD163, IL10, ARG1). Levels of significance: n.s. p ≥ 0.05; *p < 0.05; **p < 0.01.
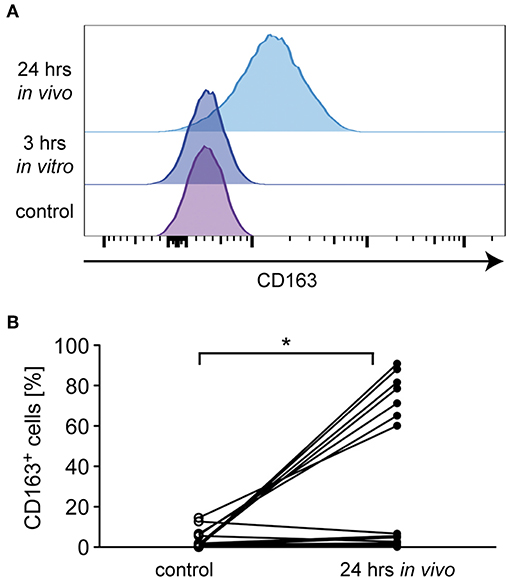
Figure 5. Analysis of monocyte CD163 surface levels in MS patients. Monocytes were isolated from MS patients before MP pulse therapy and cultured without (control) or with 10−6 M MP for 3 h in vitro. A second blood sample was obtained from the same MS patients 24 h after MP pulse therapy in vivo. CD163 surface expression was analyzed by flow cytometry on all cells independently of the CD14/CD16 status. (A) Representative stacked histograms are depicted for an MS patient in which CD163 surface levels were upregulated after MP pulse therapy. (B) Percentages of CD163+ monocytes before (control) and 24 h after MP pulse therapy in vivo. The corresponding values for each patient are connected by a line. n = 15. Statistical analysis was performed using a Mann Whitney test. Levels of significance: *p < 0.05.
GCs Enhance the Migratory Capacity of Monocytes Along Chemokine Gradients
Transmigration of monocytes across the BBB and infiltration into the meninges and parenchyma is a hallmark of MS and guided by a set of pro-inflammatory chemokines (41). It is against this background that we determined the migratory capacity of monocytes from healthy subjects and MS patients after GC treatment in vitro and in vivo. The spontaneous basal migration rate of monocytes in the absence of a chemokine gradient was low and independent of disease status and MP treatment (Figure 6A). Expectedly, monocytes migrated toward the chemokines CCL2, CCL5, and CX3CL1, with a higher migratory activity observed for monocytes from MS patients compared to healthy individuals (Figures 6B–D). Short term in vitro culture slightly increased the migratory capacity of monocytes retrieved from healthy subjects and MS patients, although significance was missed in most cases (Figures 6B–D). In contrast, in vivo MP pulse therapy of MS patients strongly and significantly enhanced the migratory capacity of monocytes in response to all three chemokines (Figures 6B–D). In addition, we further dissected the migratory capacity of monocytes toward CCL2, the chemokine that caused the largest effects, for MS patients according to their individual disease activity. It turned out that the basal migration was the same in both groups, whereas MS patients with progressive disease had a lower CCL2-directed migration than MS patients with an acute relapse (Figure 7). Importantly, the results for monocyte migration toward CCL2 were comparable for both groups with regard to short and long term MP effects (Figure 7B). Furthermore, the same tendency was observed for patients from different MS subtypes (RRMS, SPMS, PPMS), although statistical significance was not reached here due to limited numbers of patients (data not shown). In summary, our data indicate that high-dose MP pulse therapy of MS patients enhances monocyte chemotaxis.
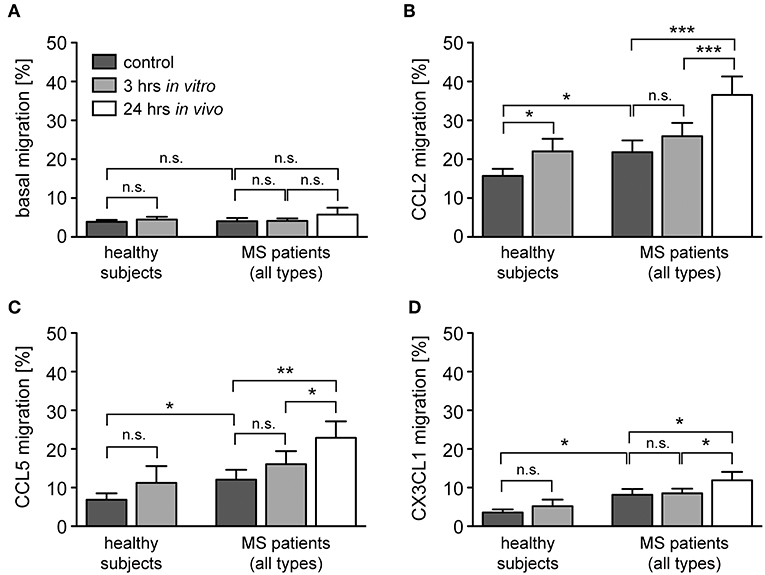
Figure 6. Monocyte migration along chemokine gradients in healthy subjects and MS patients under the influence of GCs. Monocytes were isolated from healthy subjects and MS patients before and after (24 h in vivo) MP pulse therapy. Cells were cultured in the absence (control) or presence of 10−6 M MP for 3 h in vitro and then transferred into the upper part of a Boyden chamber. Basal monocyte migration without a chemokine gradient (A) and migration toward a gradient of CCL2 (B), CCL5 (C), or CX3CL1 (D) into the lower part of the Boyden chamber were analyzed by flow cytometry and results are depicted as the percentage of transmigrated cells (mean ± SEM). n = 19/19/11/9 (healthy subjects), n = 13/15/12/17 (MS patients). For statistical analysis, untreated samples were compared to each other using a t-test, comparison of untreated vs. MP-treated samples from healthy subjects was performed using a paired t-test, and comparison of samples from MS patients to each other was performed using a One-way ANOVA and Newman-Keuls Multiple Comparison test. Levels of significance: n.s. p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
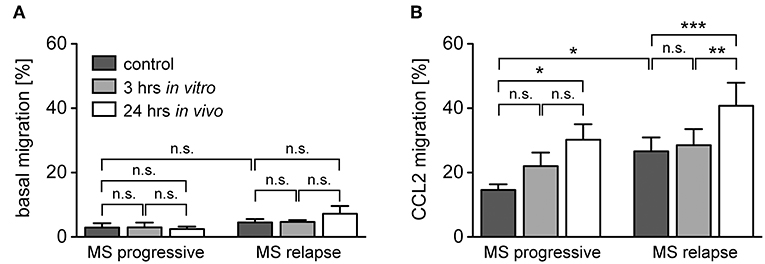
Figure 7. Monocyte migration along a CCL2-gradient in MS patients with progressive disease or an acute relapse under the influence of GCs. The data are the same as in the experiment presented in Figure 4, but the MS patients are now divided into two groups according to their disease activity. Basal monocyte migration without a chemokine gradient (A) and migration toward a gradient of CCL2 (B) into the lower part of the Boyden chamber were analyzed by flow cytometry and are depicted as the percentage of transmigrated cells (mean ± SEM); n = 4/6 (progressive), n = 9/9 (relapse). For statistical analysis, untreated samples were compared using a t-test and comparison of samples from MS patients to each other was performed using a One-way ANOVA and Newman-Keuls Multiple Comparison test. Levels of significance: n.s. p ≥ 0.05; *p < 0.05; **p < 0.01; ***p < 0.001.
The Frequency of CCR2+ Monocytes and Their CCR2 Surface Expression Levels Are Unaffected by GC Treatment
Monocyte migration along chemokine gradients depends on the surface expression of the respective receptors as well as intracellular signaling pathways and cytoskeletal rearrangements. To distinguish between these mechanisms, we tested alterations in chemokine receptor expression levels exemplified for CCR2, the receptor of CCL2 which is the chemokine that induced the most robust migration and alteration by GC treatment (Figures 6, 7). The percentage of CCR2+ monocytes in MS patients was significantly higher than in healthy subjects (Figure 8A), which is in agreement with their higher percentage of classical inflammatory CD14++CD16− monocytes (Figure 2). In contrast, the surface density of this receptor was not significantly changed (Figure 8B). Importantly, MP pulse therapy of MS patients neither altered the abundance of CCR2+ monocytes nor the surface expression levels of the receptor (Figures 8A,B), indicating that the increased migration of monocytes toward CCL2 after MP treatment was unrelated to GC effects on the chemokine receptor itself. Notably, CX3CR1+ monocytes in MS patients were less abundant than in healthy subjects and unaffected by MP pulse therapy (data not shown), which is also in line with the lower abundance of non-classical CD14+CD16++ monocytes in MS patients regardless of their treatment (Figure 2).
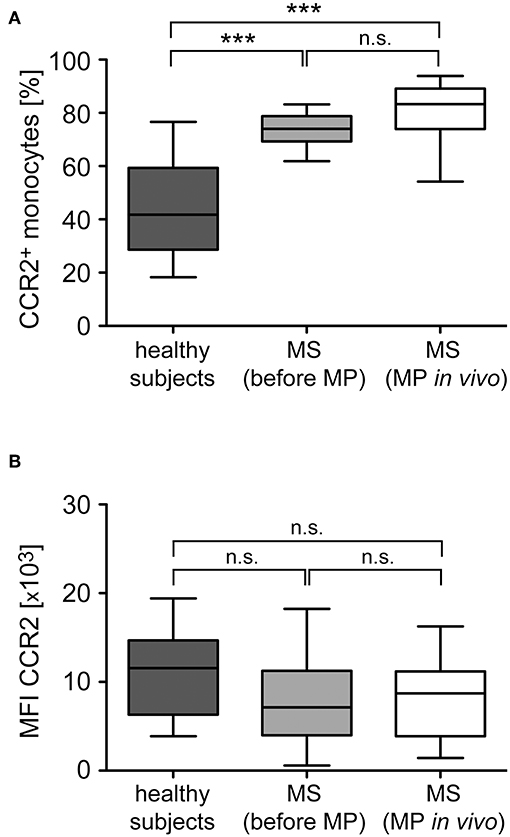
Figure 8. Analysis of CCR2 surface expression levels in monocytes from healthy subjects and MS patients before and after GC treatment in vivo. Monocytes were isolated from healthy subjects as well as MS patients before and 24 h after MP pulse therapy in vivo. CCR2 surface expression was analyzed by flow cytometry and subsequently the percentage of CCR2+ monocytes (A) and the surface level of CCR2 based on the mean fluorescence intensity (MFI) were determined (B). Data are presented as box-and-whiskers plots showing the minimum, maximum and median; n = 13/22/17. Statistical analysis was performed using a One-way ANOVA and Newman-Keuls Multiple Comparison test. Levels of significance: n.s. p ≥ 0.05; ***p < 0.001.
Discussion
MS is a complex disease involving multiple interactions between different immune cell populations. Although T cells undoubtedly play a very important role in the pathogenesis of MS, monocytes are implicated in disease pathogenesis too and therefore represent potential therapeutic targets. They can contribute to inflammatory processes by influencing Th17 cell differentiation (30) and impact T-cell activation and differentiation, e.g., by down-regulation of cytokine production or induction of Treg cells at the site of inflammation (37). Although progress has been made in the understanding of these processes (42, 43), the role of different monocyte subsets in autoimmune diseases such as MS remains incompletely understood. Monocytes are rapidly mobilized in large numbers to inflamed sites and also possess T cell-independent effector functions such as phagocytic activity and the secretion of pro-inflammatory cytokines and chemokines. Interestingly, these cells can even be found in the healthy human brain. Especially intermediate state CD14++CD16+ monocytes are present in the CSF of healthy subjects, where they account for >50% of all monocytes (19), which highlights their importance for the immune surveillance of the CNS. In pathological conditions such as MS, monocytes are found in active and early demyelinating lesions and thus may contribute to the breakdown of the BBB. Nevertheless, depletion of this cell type is not advisable as they can also have beneficial effects in the resolution phase of inflammation and repair processes. For instance, when myeloid cells transduced with the innate immune receptor TREM2 were applied in EAE mice, they created an anti-inflammatory milieu in the CNS resulting in the amelioration of clinical symptoms and reduced structural damage (44). It is further noteworthy that a removal of monocytes would be difficult to achieve because they only have a short half-life of a few days. Selectively employing the anti-inflammatory capacity of monocytes while avoiding a general immune suppression might, however, be favorable for the treatment of autoimmune diseases like MS. In this respect it is relevant that GCs induce a stable anti-inflammatory phenotype in mouse monocytes following their treatment in vitro (36). After transfer into recipient mice, such monocytes were found to maintain their polarization and were able to repress T-cell-mediated inflammation even when it was already established (37). Astonishingly, relatively little is known about the mechanisms by which GCs impact this immune cell population during MS pulse therapy, albeit myeloid cells are known to contribute to the pathogenesis of MS and EAE. Expression of the GC receptor in myeloid cells was found to be dispensable for the treatment of EAE with free dexamethasone (7). However, when the GCs were targeted to myeloid cells by encapsulation into liposomes (9) or nanoparticles (10), their effects on monocytes/macrophages turned out to be crucial for their therapeutic efficacy in the same EAE model. In this case, GC treatment resulted in a strong M2 polarization of myeloid cells, which was essential for an amelioration of the disease symptoms (9, 10).
In this study, we report that GCs also have pronounced effects on human monocytes, especially those from MS patients. In vitro culture of monocytes from MS patients with MP resulted in an increased gene expression of the M2 markers ARG1, CD163, and CD206 and the anti-inflammatory cytokine IL10, and the concomitant down-regulation of the mRNA level of the pro-inflammatory cytokine IL1B. This effect was even more pronounced after MS patients were subjected to 24 h of MP pulse therapy. Hence, GCs induce an anti-inflammatory M2 monocyte phenotype in MS patients. Surprisingly, we found that the migration of monocytes toward several pro-inflammatory chemokines was enhanced after the exposure to MP in vitro and in vivo. Presumably, this effect is mostly independent of a modulation of the expression levels of the respective chemokine receptors. Neither the frequency of CCR2+ monocytes nor the surface level of this receptor were significantly increased after MS patients underwent MP pulse therapy. Hence it is likely that GCs influence processes other than chemokine receptor levels leading to an enhanced chemotaxis of monocytes. It is noteworthy that we previously described a similar phenomenon for the impact of GCs on the migratory behavior of T cells toward CCL19 and CXCL12, which turned out to be independent of the levels of the respective chemokine receptor as well (11). Therefore, it appears likely that downstream signaling pathways are responsible for the altered migration of monocytes after GC treatment. It has been reported that phospholipase C and phosphokinase C are involved in CCR2 signaling (45), leading to an activation of focal adhesion kinase (FAK) (46). The observation that FAK is phosphorylated in response to GCs in T cells (11) provides a possible explanation for synergistic effects of GCs and CCL2 on monocyte migration. Further support for this notion comes from a report that paxillin, a downstream signaling molecule of FAK, is induced by GCs in human mesenchymal stem cells and thereby enhances their migration (47), and that GCs influence the cytoskeleton of T cells via phospholipase C (48), an effect which could also impact the migratory behavior of monocytes.
Interestingly, frequencies of inflammatory and non-classical monocytes were not substantially influenced by MP treatment although our gene expression analysis revealed a shift toward an anti-inflammatory phenotype under these conditions. Apparently, the cell populations defined by either surface expression of CD14 and CD16 or gene expression of anti-inflammatory molecules are different. It has been shown that human M2 polarized macrophages display a more motile phenotype and migrated more directed and over longer distances toward CCL2 as compared to M1 or M0 macrophages (49). Additionally, it has been hypothesized that the inflammatory chemokine CCL2 might have a beneficial role in MS because its levels are higher in the remission phase than during relapses, although no clear explanation for this phenomenon could be provided (50). This suggests that in phases of disease remission M2 polarized macrophages and monocytes are recruited to the site of inflammation by CCL2, where they promote repair and remyelination. We postulate that the application of GCs enhances this natural repair mechanism by affecting two different aspects of this process. First, GC application promotes M2 polarization of monocytes and second, it enhances the migration of these M2 polarized monocytes toward different chemokines. It is tempting to speculate that under these circumstances anti-inflammatory monocytes already reach the CNS at time points when natural repair mechanisms have not yet been initiated, thereby accelerating and optimizing the repair process and facilitating the remission of the disease.
Although T cells are still widely considered to be the major target cells of MP pulse therapy, resulting in changes in cytokine expression, adhesion molecule expression, migration and apoptosis (51), some effects of GCs on myeloid cells have already been described in the past. For instance, the phagocytic potential of human monocytes was enhanced by the incubation with dexamethasone in vitro (52). Interestingly, enhanced phagocytosis of macrophages in vitro is associated with an M2 polarization (53, 54). Furthermore, high-dose MP pulse therapy resulted in a decreased frequency of monocytes producing IL-8, which is typical for the inflammatory CD14++CD16− subset (55). While we did not observe a change in the frequency of inflammatory or non-classical monocytes 24 h after MP pulse therapy, it is noteworthy that the aforementioned decrease of IL-8-producing monocytes was described 5 days after treatment, suggesting that such a change might be evident only at a later time point. In addition, GC treatment of EAE in mice resulted in a reduced expression of beta-arrestin-1 and enhanced mRNA levels of A1AR (56), which is thought to regulate cytokine expression and release and NO production in myeloid cells (57). In fact, mRNA levels of cytokine genes were at least partially affected by GCs in our study: we observed a reduced expression of the pro-inflammatory cytokine IL1B and an increased expression of the anti-inflammatory cytokine IL10. Interestingly, previous reports indicated that IL-6 levels were not changed by GCs in human monocytes (36), which is in contrast to mouse monocytes (58). The reason for this species difference, however, is unclear. Furthermore, analysis of human monocyte-derived dendritic cells showed that GCs induced IL-10 secretion in vitro (59), and analysis of human monocyte-derived macrophages revealed that GCs repressed IL-6 and TNFα responses induced by LPS stimulation in vitro (60). Collectively, these findings are in line with our finding that GC treatment modulates cytokine expression by human monocytes.
Our data suggest that CCL2 is the chemokine that controls monocyte migration into the CNS to a higher degree compared to the other chemokines tested in this study. The migration rate of untreated monocytes from healthy subjects and MS patients toward CCL2 was higher compared to CCL5 and CX3CL1, which confirms previous data also showing higher migration rates of human monocytes toward CCL2 in comparison to CX3CL1 (19). Hence, it does not come as a surprise that CCL2-directed migration is also the predominant target of GCs in the context of chemotaxis. Still, it is somewhat contradictory at first sight that chemotaxis toward an inflammatory chemokine is increased by GCs rather than decreased. We believe that this observation needs to be interpreted in the light of the concurrent phenotypic changes that lead to an anti-inflammatory polarization of monocytes. It appears that GCs promote the infiltration of those monocytes into the CNS that are able to terminate inflammation and initiate repair processes, thus contributing to an amelioration of disease symptoms after MP pulse therapy of MS patients. Of note, the occurrence of anti-inflammatory activity of myeloid cells in inflammatory CNS diseases has been reported previously (61, 62) but the exact mechanisms remained incompletely understood.
In summary, GCs exert marked effects on monocytes from MS patients, which could in part explain the therapeutic efficacy of MP pulse therapy. Apparently, these effects are achieved by a combination of M2 polarization and enhanced chemotaxis and certainly play an important role in addition to the well-described impact of GCs on T-cell function. Therefore, GCs should not only be considered as T-cell suppressors but also as modulators of myeloid cells in MS therapy.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
This study was carried out in accordance with the recommendations of the local ethics committee of the University Medical Center Göttingen with written informed consent from all subjects, in accordance with the Declaration of Helsinki.
Author Contributions
HF performed and analyzed most of the experiments, and wrote the manuscript. TF performed and analyzed experiments. HP provided blood samples of MS patients. HR designed the project, analyzed experiments, and wrote the manuscript. FL designed the project, analyzed experiments, and wrote the manuscript.
Funding
This work was supported by grants from Deutsche Forschungsgemeinschaft (Lu 634/8-1, Lu 634/9-1, Re 1631/15-1).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Amina Bassibas for expert technical assistance, Cathy Ludwig for language editing, and Alexander Flügel for critically reading the manuscript.
References
1. Sospedra M, Martin R. Immunology of multiple sclerosis. Semin Neurol. (2016) 36:115–27. doi: 10.1055/s-0036-1579739
2. Milligan NM, Newcombe R, Compston DAS. A double-blind controlled trial of high-dose methylprednisolone in patients with multiple-sclerosis.1. Clinical effects. J Neurol Neurosurg Psychiatry. (1987) 50:511–6. doi: 10.1136/jnnp.50.5.511
3. Buttgereit F, Straub RH, Wehling M, Burmester GR. Glucocorticoids in the treatment of rheumatic diseases - an update on the mechanisms of action. Arthritis Rheum. (2004) 50:3408–17. doi: 10.1002/art.20583
4. Zen M, Canova M, Campana C, Bettio S, Nalotto L, Rampudda M, et al. The kaleidoscope of glucorticoid effects on immune system. Autoimmun Rev. (2011) 10:305–10. doi: 10.1016/j.autrev.2010.11.009
5. Franco LM, Gadkari M, Howe KN, Sun J, Kardava L, Kumar P, et al. Immune regulation by glucocorticoids can be linked to cell type-dependent transcriptional responses. J Exp Med. (2019) 216:384–406. doi: 10.1084/jem.20180595
6. Sloka JS, Stefanelli M. The mechanism of action of methylprednisolone in the treatment of multiple sclerosis. Mult Scler. (2005) 11:425–32. doi: 10.1191/1352458505ms1190oa
7. Wüst S, van den BJ, Tischner D, Kleiman A, Tuckermann JP, Gold R, Lühder F, Reichardt HM. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. Immunol J. (2008) 180:8434–43. doi: 10.4049/jimmunol.180.12.8434
8. Wüst S, Tischner D, John M, Tuckermann JP, Menzfeld C, Hanisch UK, et al. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS ONE. (2009) 4:e8202. doi: 10.1371/journal.pone.0008202
9. Schweingruber N, Haine A, Tiede K, Karabinskaya A, van den Brandt J, Wüst S, et al. Liposomal encapsulation of glucocorticoids alters their mode of action in the treatment of experimental autoimmune encephalomyelitis. Immunol J. (2011) 187:4310–8. doi: 10.4049/jimmunol.1101604
10. Montes-Cobos E, Ring S, Fischer HJ, Heck J, Strauss J, Schwaninger M, et al. Targeted delivery of glucocorticoids to macrophages in a mouse model of multiple sclerosis using inorganic-organic hybrid nanoparticles. Control Release J. (2016) 245:157–69. doi: 10.1016/j.jconrel.2016.12.003
11. Schweingruber N, Fischer HJ, Fischer L, van den Brandt J, Karabinskaya A, Labi V, et al. Chemokine-mediated redirection of T cells constitutes a critical mechanism of glucocorticoid therapy in autoimmune CNS responses. Acta Neuropathol. (2014) 127:713–29. doi: 10.1007/s00401-014-1248-4
12. Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. (2011) 1812:265–74. doi: 10.1016/j.bbadis.2010.07.008
13. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci. (2011) 14:1142–9. doi: 10.1038/nn.2887
14. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. (2014) 211:1533–49. doi: 10.1084/jem.20132477
15. Brosnan CF, Bornstein MB, Bloom BR. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. Immunol J. (1981) 126:614–20.
16. Huitinga I, van RN, de Groot CJ, Uitdehaag BM, Dijkstra CD. Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med. (1990) 172:1025–33. doi: 10.1084/jem.172.4.1025
17. Tran EH, Hoekstra K, van RN, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. Immunol J. (1998) 161:3767–75.
18. Mildner A, Mack M, Schmidt H, Brück W, Djukic M, Zabel MD, et al. CCR2+Ly-6Chi monocytes are crucial for the effector phase of autoimmunity in the central nervous system. Brain. (2009) 132:2487–500. doi: 10.1093/brain/awp144
19. Waschbisch A, Schröder S, Schraudner D, Sammet L, Weksler B, Melms A, et al. Pivotal role for CD16+ monocytes in immune surveillance of the central nervous system. Immunol J. (2016) 196:1558–67. doi: 10.4049/jimmunol.1501960
20. Ziegler-Heitbrock L, Hofer TP. Toward a refined definition of monocyte subsets. Front Immunol. (2013) 4:23. doi: 10.3389/fimmu.2013.00023
21. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
22. Kim HJ, Ifergan I, Antel JP, Seguin R, Duddy M, Lapierre Y, et al. Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol. (2004) 172:7144–53. doi: 10.4049/jimmunol.172.11.7144
23. Weber MS, Prod'homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med. (2007) 13:935–43. doi: 10.1038/nm1620
24. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur Immunol J. (2012) 42:1804–14. doi: 10.1002/eji.201141947
25. Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. (2011) 17:2–15. doi: 10.1177/1352458510379243
26. Fingerle G, Pforte A, Passlick B, Blumenstein M, Strobel M, Ziegler-Heitbrock HW. The novel subset of CD14+/CD16+ blood monocytes is expanded in sepsis patients. Blood. (1993) 82:3170–6.
27. Hepburn AL, Mason JC, Davies KA. Expression of Fcgamma and complement receptors on peripheral blood monocytes in systemic lupus erythematosus and rheumatoid arthritis. Rheumatology. (2004) 43:547–54. doi: 10.1093/rheumatology/keh112
28. Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, et al. CD14+,CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. (2002) 46:2578–86. doi: 10.1002/art.10545
29. Okamoto H, Mizuno K, Horio T. Circulating CD14+ CD16+ monocytes are expanded in sarcoidosis patients. Dermatol J. (2003) 30:503–9. doi: 10.1111/j.1346-8138.2003.tb00424.x
30. Rossol M, Kraus S, Pierer M, Baerwald C, Wagner U. The CD14(bright) CD16+ monocyte subset is expanded in rheumatoid arthritis and promotes expansion of the Th17 cell population. Arthritis Rheum. (2012) 64:671–7. doi: 10.1002/art.33418
31. Walscheid K, Weinhage T, Foell D, Heinz C, Kasper M, Heiligenhaus A. Phenotypic changes of peripheral blood mononuclear cells upon corticosteroid treatment in idiopathic intermediate uveitis. Clin Immunol. (2016). 173:27–31 doi: 10.1016/j.clim.2016.10.013
32. Kouwenhoven M, Teleshova N, Ozenci V, Press R, Link H. Monocytes in multiple sclerosis: phenotype and cytokine profile. Neuroimmunol J. (2001) 112:197–205. doi: 10.1016/S0165-5728(00)00396-9
33. Chuluundorj D, Harding SA, Abernethy D, La Flamme AC. Expansion and preferential activation of the CD14(+)CD16(+) monocyte subset during multiple sclerosis. Immunol Cell Biol. (2014) 92:509–17. doi: 10.1038/icb.2014.15
34. Michell-Robinson MA, Moore CS, Healy LM, Osso LA, Zorko N, Grouza V, et al. Effects of fumarates on circulating and CNS myeloid cells in multiple sclerosis. Ann Clin Transl Neurol. (2016) 3:27–41. doi: 10.1002/acn3.270
35. Di DM, Colombo E, Govi C, De FD, Messina MJ, Romeo M, et al. Myeloid cells as target of fingolimod action in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e157. doi: 10.1212/NXI.0000000000000157
36. Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. (2007) 109:1265–74. doi: 10.1182/blood-2006-02-001115
37. Varga G, Ehrchen J, Brockhausen A, Weinhage T, Nippe N, Belz M, et al. Immune suppression via glucocorticoid-stimulated monocytes: a novel mechanism to cope with inflammation. Immunol J. (2014) 193:1090–9. doi: 10.4049/jimmunol.1300891
38. Liu B, Dhanda A, Hirani S, Williams EL, Sen HN, Martinez EF, et al. CD14++CD16+ monocytes are enriched by glucocorticoid treatment and are functionally attenuated in driving effector T cell responses. Immunol J. (2015) 194:5150–60. doi: 10.4049/jimmunol.1402409
39. Vedeckis WV, Ali M, Allen HR. Regulation of glucocorticoid receptor protein and mRNA levels. Cancer Res. (1989) 49:2295s–302s
40. Burnstein KL, Bellingham DL, Jewell CM, Powell-Oliver FE, Cidlowski JA. Autoregulation of glucocorticoid receptor gene expression. Steroids. (1991) 56:52–8. doi: 10.1016/0039-128X(91)90124-E
41. Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta. (2011) 1812:220–30. doi: 10.1016/j.bbadis.2010.07.019
42. Katsiari CG, Liossis SN, Sfikakis PP. The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. (2010) 39:491–503. doi: 10.1016/j.semarthrit.2008.11.002
43. Shemer A, Jung S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin Immunopathol. (2015) 37:613–23. doi: 10.1007/s00281-015-0519-z
44. Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. (2007) 4:e124. doi: 10.1371/journal.pmed.0040124
45. Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoattractant protein-1 signalling in disease. Exp Opn Ther Targets. (2003) 7:35–48. doi: 10.1517/14728222.7.1.35
46. O'Hayre M, Salanga CL, Handel TM, Allen SJ. Chemokines and cancer: migration, intracellular signalling and intracellular communication in the microenvironment. Biochem J. (2008) 409:635–49. doi: 10.1042/BJ20071493
47. Yun SP, Ryu JM, Han HJ. Involvement of ß1-integrin via pip complex and FAK/paxillin in dexamethason-induced human mesenchymal stem cell migration. J Cell Physiol. (2011) 226:683–92. doi: 10.1002/jcp.22383
48. Müller N, Fischer HJ, Tischner D, van den Brandt J, Reichardt HM. Glucocorticoids induce effector T cell depolarization via ERM proteins, thereby impeding migration and APC conjugation. Immunol J. (2013) 190:4360–70. doi: 10.4049/jimmunol.1201520
49. Vogel DY, Heijnen PD, Breur M, de Vries HE, Tool AT, Amor S, et al. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. Neuroinflammation J. (2014) 11:23. doi: 10.1186/1742-2094-11-23
50. Iarlori C, Reale M, De LG, Di IA, Feliciani C, Tulli A, et al. Interferon beta-1b modulates MCP-1 expression and production in relapsing-remitting multiple sclerosis. Neuroimmunol J. (2002) 123:170–9. doi: 10.1016/S0165-5728(01)00487-8
51. Lühder F, Reichardt HM. Traditional concepts and future avenues of glucocorticoid action in experimental autoimmune encephalomyelitis and multiple sclerosis therapy. Crit Rev Immunol. (2009) 29:255–73. doi: 10.1615/CritRevImmunol.v29.i3.50
52. van der Goes A, Hoekstra K, van den Berg TK, Dijkstra CD. Dexamethasone promotes phagocytosis and bacterial killing by human monocytes/macrophages in vitro. J Leukoc Biol. (2000) 67:801–7. doi: 10.1002/jlb.67.6.801
53. Leidi M, Gotti E, Bologna L, Miranda E, Rimoldi M, Sica A, et al. M2 macrophages phagocytose rituximab-opsonized leukemic targets more efficiently than m1 cells in vitro. Immunol J. (2009) 182:4415–22. doi: 10.4049/jimmunol.0713732
54. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. (2011) 108:985–95. doi: 10.1161/CIRCRESAHA.110.233775
55. Mirowska-Guzel DM, Kurowska K, Skierski J, Koronkiewicz M, Wicha W, Kruszewska J, et al. High dose of intravenously given glucocorticosteroids decrease IL-8 production by monocytes in multiple sclerosis patients treated during relapse. Neuroimmunol J. (2006) 176:134–40. doi: 10.1016/j.jneuroim.2006.03.024
56. Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SSG, Power C. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. Faseb J. (2008) 22:786–96. doi: 10.1096/fj.07-9002com
57. Hasko G, Szabo C, Nemeth ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. Immunol J. (1996) 157:4634–40.
58. Varga G, Ehrchen J, Tsianakas A, Tenbrock K, Rattenholl A, Seeliger S, et al. Glucocorticoids induce an activated, anti-inflammatory monocyte subset in mice that resembles myeloid-derived suppressor cells. J Leukoc Biol. (2008) 84:644–50. doi: 10.1189/jlb.1107768
59. Segerer SE, Müller N, van den Brandt J, Kapp M, Dietl J, Reichardt HM, et al. The glycoprotein-hormones activin A and inhibin A interfere with dendritic cell maturation. Reprod Biol Endocrinol. (2008) 6:17. doi: 10.1186/1477-7827-6-17
60. Wang C, Nanni L, Novakovic B, Megchelenbrink W, Kuznetsova T, Stunnenberg HG, et al. Extensive epigenomic integration of the glucocorticoid response in primary human monocytes and in vitro derived macrophages. Sci Rep. (2019) 9:2772. doi: 10.1038/s41598-019-39395-9
61. Wlodarczyk A, Cedile O, Jensen KN, Jasson A, Mony JT, Khorooshi R, et al. Pathologic and protective roles for microglial subsets and bone marrow- and blood-derived myeloid cells in central nervous system inflammation. Front Immunol. (2015) 6:463. doi: 10.3389/fimmu.2015.00463
Keywords: multiple sclerosis, methylprednisolone therapy, monocytes, M2 polarization, chemokines
Citation: Fischer HJ, Finck TLK, Pellkofer HL, Reichardt HM and Lühder F (2019) Glucocorticoid Therapy of Multiple Sclerosis Patients Induces Anti-inflammatory Polarization and Increased Chemotaxis of Monocytes. Front. Immunol. 10:1200. doi: 10.3389/fimmu.2019.01200
Received: 12 February 2019; Accepted: 13 May 2019;
Published: 29 May 2019.
Edited by:
Alexandra K. Kiemer, Saarland University, GermanyReviewed by:
Mieke Gouwy, KU Leuven, BelgiumKatarzyna Barczyk-Kahlert, Universitätsklinikum Münster, Germany
Copyright © 2019 Fischer, Finck, Pellkofer, Reichardt and Lühder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Holger M. Reichardt, aHJlaWNoYXJkdEBtZWQudW5pLWdvZXR0aW5nZW4uZGU=; Fred Lühder, ZnJlZC5sdWVoZGVyQG1lZC51bmktZ29ldHRpbmdlbi5kZQ==
†These authors have contributed equally to this work
‡Present Address: Henrike J. Fischer, Institute for Immunology, Medical Faculty, RWTH Aachen University, Aachen, Germany
Hannah L. Pellkofer, Institute of Clinical Neuroimmunology, Ludwig Maximilians University, Munich, Germany