 Noushin Lotfi1,2Rodolfo Thome2Nahid Rezaei3
Noushin Lotfi1,2Rodolfo Thome2Nahid Rezaei3 Guang-Xian Zhang2Abbas Rezaei1Abdolmohamad Rostami2*
Guang-Xian Zhang2Abbas Rezaei1Abdolmohamad Rostami2* Nafiseh Esmaeil1*
Nafiseh Esmaeil1*- 1Department of Immunology, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
- 2Department of Neurology, Thomas Jefferson University, Philadelphia, PA, United States
- 3Department of Immunology, School of Medicine, Lorestan University of Medical Sciences, Khorramabad, Iran
Granulocyte-macrophage colony-stimulating factor (GM-CSF) was first described as a growth factor that induces the differentiation and proliferation of myeloid progenitors in the bone marrow. GM-CSF also has an important cytokine effect in chronic inflammatory diseases by stimulating the activation and migration of myeloid cells to inflammation sites, promoting survival of target cells and stimulating the renewal of effector granulocytes and macrophages. Because of these pro-cellular effects, an imbalance in GM-CSF production/signaling may lead to harmful inflammatory conditions. In this context, GM-CSF has a pathogenic role in autoimmune diseases that are dependent on cellular immune responses such as multiple sclerosis (MS) and rheumatoid arthritis (RA). Conversely, a protective role has also been described in other autoimmune diseases where humoral responses are detrimental such as myasthenia gravis (MG), Hashimoto's thyroiditis (HT), inflammatory bowel disease (IBD), and systemic lupus erythematosus (SLE). In this review, we aimed for a comprehensive analysis of literature data on the multiple roles of GM-CSF in autoimmue diseases and possible therapeutic strategies that target GM-CSF production.
Introduction
Granulocyte-macrophage colony-stimulating factor (GM-CSF, or CSF2) was first described in the conditioned media of mouse lung tissue following LPS injection, which triggered the proliferation of bone marrow-derived macrophages and granulocytes (1). GM-CSF is produced by multiple cell types such as activated T cells, B cells, macrophages, monocytes, mast cells, vascular endothelial cells, and fibroblasts (2). GM-CSF receptor is composed of one α chain and one β chain with low and high-affinity binding to GM-CSF, respectively, and the β chain is shared with IL-3 and IL-5 receptor (3). In addition, the GM-CSF receptor (CSF2R) is found in myeloid cells and some non-hematopoietic cells, but it is not expressed by lymphoid cells such as T cells (4).
There are four main signaling pathways triggered by CSF2R (5). After binding of GM-CSF to its receptor, Janus-kinase-2 (JAK-2) is recruited to the cytoplasmic domain of the β chain, and activation of JAK-2 occurs, which subsequently induces STAT-5 phosphorylation. This signaling pathway induces migration of STAT-5 dimers to the nucleus and promotes the transcription of various genes such as pim-1 and CIS to induce cell differentiation (6). GM-CSF promotes cell survival via phosphatidylinositol-3-kinase (PI3K) and JAK/STAT-Bcl-2 signaling pathways (7). Moreover, cell differentiation and inflammation are mediated by activation of ERK1/2 and NF-kB. Accordingly, studies have shown that GM-CSF augments the LPS-induced inflammatory response by priming of TNF-alpha synthesis and also induces multipotent mesenteric mesothelial cell differentiation into macrophages through the ERK1/2 signaling pathway (8, 9).
In addition to the important role of GM-CSF as a colony-stimulating factor and its clinical application following chemo/radiotherapy to restore myeloid populations in leukemic patients, several studies suggest that GM-CSF plays a role in innate and adaptive immunity. Accumulating evidence indicates the role of this molecule in inflammatory immune response and autoimmunity (10, 11). In addition to its role in hematopoietic differentiation, GM-CSF has an effect on antigen presentation, phagocytosis, chemotaxis, and cell-adhesion as well (12, 13). Targeting GM-CSF may represent a novel approach to control undesired immune responses in autoimmune diseases and chronic inflammation (14). Interestingly, recent studies have designated GM-CSF as a player in the regulation of immune responses (15).
In this review, we discuss the role of GM-CSF in autoimmune diseases pathogenesis.
GM-CSF in Autoimmune Diseases
GM-CSF has been implicated in the inflammatory context observed in many autoimmune diseases, such as multiple sclerosis (MS) and rheumatoid arthritis (RA) (16, 17). It should be noted that GM-CSF and IL-3 are the main mediators of innate immune responses and the critical role of both GM-CSF and IL-3 is indicated in the augmentation and progression of some disorders including allergic asthma, aortic dissection, and atherosclerosis while the role of IL-3 in MS and RA pathogenesis is open to question (18).
RA pathogenesis involves the penetration of inflammatory cells into the synovial fluid, with Th1 and Th17 cells being the dominant T cell subtypes in the synovia of RA patients (19). Although IL-17 has been linked to RA pathogenesis, recent data show that GM-CSF is an important cytokine in disease development (20–22). There is some evidence that Th17 cells, innate lymphoid cells (ILCs), and stromal cells mediate inflammatory immune response in the synovia of RA patients via GM-CSF and IL-17 production. Fibroblast-like synoviocytes (FLS), which are dominant cells at the pannus-cartilage junction, produce different inflammatory mediators in RA patients, and some reports have indicated that GM-CSF production can be triggered by human chondrocytes and synovial fibroblasts (FLS) in response to IL-1 and TNFa (23, 24). In addition, Hirota et al. have shown that CD25+ IL-33Ra+ GATA-3+ ILC2s are the most common ILCs in the inflamed joints which actively secrete GM-CSF (25). Also, loss of GM-CSF production capability in FLS and other stromal cells has prevented RA progression (20).
Markis et al. have reported a higher frequency of B and T cells expressing GM-CSF in the peripheral blood of RA patients, suggesting that GM-CSF+ B cells probably contribute to autoantibody production and RA pathogenesis (26). Also, the presence of GM-CSF-producing Th cell populations is higher in synovial fluid than in peripheral blood mononuclear cells (PBMCs) in patients with juvenile idiopathic arthritis (JIA) (19).
Recently, monocyte-derived inflammatory DCs (infDCs), which are CD1c+ and share a similar transcription factor with monocyte-derived DCs (moDCs) generated in the presence of GM-CSF and IL-4, have been identified in RA synovial fluid. Reynolds et al. have indicated that CD4+ T cells are the primary source of GM-CSF in synovial and that GM-CSF production by these cells is related to Th1 cell activation and IL-15. They have also shown that CD14+ monocyte differentiation into CD1c+ infDCs is dependent on GM-CSF production by CD4+ T cells. Interestingly, the decrease in circulating MoDCs in RA patients and a higher number of these populations in rheumatoid synovial fluid and synovial tissue can be explained by the fact that these cells migrate from circulation to the synovial compartment (27). These cells are capable of producing some pro-inflammatory cytokines such as TNFa, IL-6, IL-12, and they express various activation factors that stimulate T and B cells. The stimulation of MoDCs which are induced in the presence of GM-CSF/IL-4 by TLR-2 (LTA) and TLR-4 (LPS and EDA) ligands has led to higher production of TNFa and IL-6 in RA patients compared to healthy subjects. This may indicate that the increase of different TLR ligands in the joints and serum can provoke TLRs signaling and facilitate the breakdown of tolerance in RA (28).
In addition, another study showed that the culture of ex vivo differentiated human MoDCs (CD14+CD33+) in the presence of GM-CSF is capable of class II-mediated prominent immune epitopes of two auto-antigens [type II collagen (CII) and cartilage gp39 (HCgp39)] observed in the inflamed synovial joints of patients with RA (29). Furthermore, in the presence of GM-CSF, MoDCs in the synovial fluid of RA patients have a more pro-inflammatory phenotype and are resistant to anti-inflammatory properties of IL-10 (30). In collagen-induced arthritis (CIA), a mouse model of arthritis, mice with defective GM-CSF cannot develop arthritis, and using antibodies against GM-CSF results in inhibition of disease progression and a decrease in pro-inflammatory cytokines in the joints (31). Similarly, in another mouse model of arthritis (in SKG mice), GM-CSF treatment increased the production of IL-1β or IL-6 by macrophages and promoted the differentiation and augmentation of CD4+ T cells that produce IL-17 and GM-CSF. Also, administration of anti-GM-CSF was more efficient compared to anti-IL-17 in treatment and decreased disease severity (32).
In SKG, an influx of Th17 cells, neutrophils, and GM-CSF-producing CD4+ T cells into the lungs has been observed (32). Additionally, Katano et al. have shown the effects of GM-CSF on neutrophils by MALDI-TOF/TOF MS analysis and protein database searches in RA. They cultured isolated neutrophils from healthy subjects in the presence of GM-CSF for 18 h and then extracted different parts of the cells such as cytosol, membrane/organelle, nuclei, cytoskeleton, and proteins. The digested peptides were analyzed by a MALDI-TOF mass spectrometer. They found that GM-CSF upregulates neutrophil gelatinase-associated lipocalin in neutrophils followed by transitional endoplasmic reticulum ATPase induction. They also found significantly elevated levels of neutrophil gelatinase-associated lipocalin in the synovial fluid of RA patients. They concluded that GM-CSF, through increased neutrophil gelatinase-associated lipocalin, contributes to RA pathogenesis by activation of immunologic responses and/or synoviocytes, which leads to a decrease in chondrocyte proliferation (33).
King et al. analyzed antigen-presenting cells (MHCII+ cells) in the epidermis and dermis of WT and GM-CSF−/− mice. They showed that GM-CSF is necessary for the accumulation of langerin+CD103+ CD11blo cells, which are found in the dermis and play an essential role in T cell priming (34). Additionally, elevated GM-CSF in skin lesions of psoriatic patients indicates that this cytokine promotes the function of neutrophils (35). Also, Scholz et al. have found that neutralization of GM-CSF in mice by anti-GM-CSF antibody reduced inflammation in imiquimod-induced psoriasiform dermatitis (IMQPD). However, they suggest that in the absence of GM-CSF an alternative pathway plays a role in the pathogenesis of IMQPD (36).
Overall, GM-CSF plays an important role in inflammatory responses in autoimmune disease via induction of various cells and mediators. Ongoing and complete clinical trials targeting GM-CSF and its receptor are summarized in Table 1.
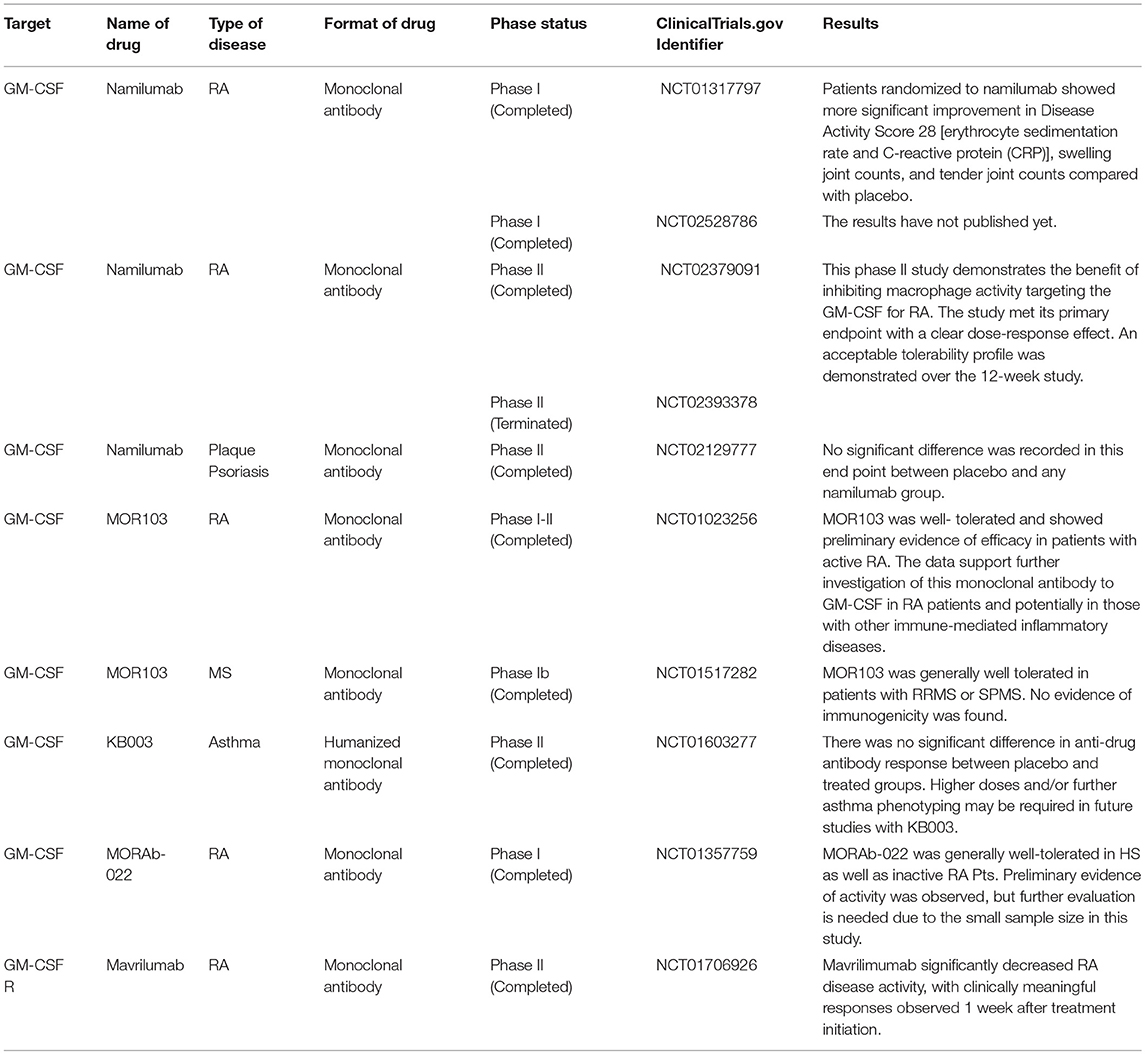
Table 1. Clinical therapeutic trials of targeting GM-CSF in autoimmune disorders.
The Inflammatory Role of GM-CSF in MS
MS is a disabling immune-mediated disease of the CNS accompanied by demyelinated plaques that result in symptoms such as vision problems, disability, depression, muscle weakness, and neurogenic bladder (37).
The Impact of GM-CSF on Blood-Brain Barrier (BBB) Permeability
Studies have demonstrated that, along with inflammatory response in the CNS, CD11b+CD62L+Ly6Chi monocytes that have formed into colonies move into the bloodstream immediately before EAE relapses in a GM-CSF dependent pathway, and trafficking of circulating Ly6Chi monocytes through the blood-brain barrier induces proinflammatory mediators and differentiation of central nervous system dendritic cells and macrophages. GM-CSF also stimulates the release of Ly6Chi precursors from bone marrow (38). High expression of both GM-CSFR subunits alpha (α) and beta (β) has been observed on microglia/macrophages and astrocytes in MS lesions (39).
In the EAE model, GM-CSF induces the proliferation and activation of microglia, which are necessary for initiation of the disease (2). Microglia secretes many mediators such as ROS, TNF-α, Interleukin-1β, Glutamate and nitrogen species (40, 41). TNF-α influences BBB permeability, which leads to further destruction via higher expression of markers such as ICAM-1, and V-CAM-1 (42). GM-CSF can also boost the differentiation of M1-like macrophages and causes the production of higher levels of inflammatory cytokines such as IL-1, IL-6, and TNF α, all of which cooperate in the destruction of the myelin sheath (43). The inflammasome processing of IL1β can be mediated by GM-CSF in myeloid cells such as monocytes and macrophages, promoting the expansion of Th17 cells and more damage to the BBB (44). Additionally, Pare et al. have recently shown an inflammatory loop between IL-1β and GM-CSF, suggesting that IL-1β plays a role in the migration of GM-CSF–activated CCR2hiLy6Chi monocytes to the CNS (17, 45). To investigate the effect of GM-CSF on BBB permeability, and to prepare a GM-CSF microenvironment, human brain microvascular endothelial cells (HBMECs) were cultured on transwell inserts as a BBB model and to mimic Alzheimer's disease (AD). Claudins and zonula occludens-1 (ZO-1), a transmembrane and cytoplasmic proteins, respectively, play an important role in maintaining tight junctions. Shang et al. have indicated that GM-CSF down-regulates the expression of ZO-1 and claudin-5 in HBMECs, which induces the disruption of tight junctions in BBB (46). Additionally, in another study, intracerebroventricular injection of GM-CSF to wild-type mice was accompanied by a decrease in ZO-1expression in comparison to the PBS group (47) (Figure 1).
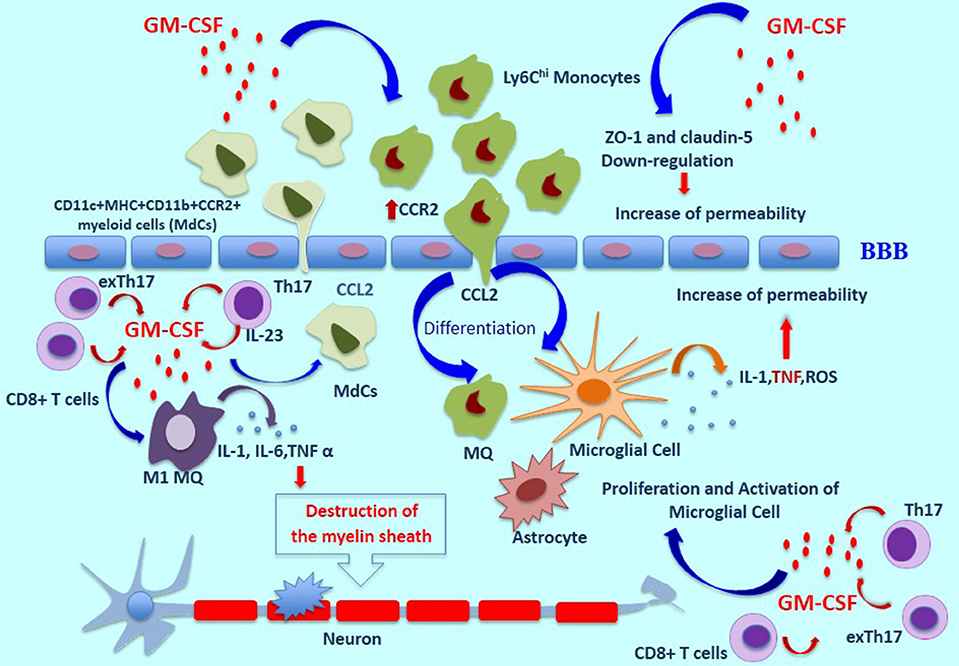
Figure 1. The role of GM-CSF in MS pathogenesis. In a GM-CSF dependent pathway, CD11b+CD62L+Ly6Chi monocytes are released and move toward the blood-brain barrier, which induces pro-inflammatory mediators and differentiation of central nervous system dendritic cells and macrophages. GM-CSF induces the expression of CCR2 on monocytes which bind to CCL-2, resulting in the migration of inflammatory cells across the BBB and into the CNS. Moreover, the proliferation and activation of microglia cells induced by GM-CSF are necessary for initiation of the disease. These cells secrete many mediators such as ROS, TNF-α, and Interleukin-1β. TNF-α influences BBB permeability, which leads to further destruction. Also, GM-CSF can boost the differentiation of M1-like macrophages and causes the production of higher levels of inflammatory cytokines such as IL-1, IL-6, and TNF α, all of which cooperate in the destruction of the myelin sheath. GM-CSF-induced expansion of CD11c+MHC+CD11b+CCR2+ myeloid cells (MdCs) population is accompanied by migration of MdCs into the CNS. GM-CSF secreted by Th17 cells is the main cytokine contributing to encephalitogenicity. IL-23 secreted by Th17 cells is necessary for the production of GM-CSF, and this cytokine causes an increase in pro-inflammatory myeloid cells. In addition, exTh17 cells produce GM-CSF, IFN-γ, and IL 17 simultaneously and play an important role in neuroinflammation. CD8+ T cells can also produce GM-CSF, and IL-17-producing CD8+ T cells (Tc17 cells) are a known source of GM-CSF. ThG cells, a subpopulation of CD4+ T cells, produce only GM-CSF and play an important role in neuroinflammation. Additionally, GM-CSF–expressing B cells play a significant role in inducing a pro-inflammatory phenotype of myeloid cells and in initiating an inflammatory response by producing GM-CSF. MS, Multiple Sclerosis Disease; BBB, Blood Brain Barrier; ROS, Reactive oxygen species.
Some evidence suggests that in EAE and MS, GM-CSF induces the expression of CCR2 on monocytes which bind to CCL2, resulting in the migration of inflammatory cells across the BBB and into the CNS (48). In agreement with this observation, another report showed, in Csf2CD4 mice, which express GM-CSF specifically in CD4+ T helper cells, a high frequency in the periphery of neutrophils and monocytes, especially CD11c+MHC+CD11b+CCR2+ myeloid cells called inflammatory monocyte-derived cells (MdCs), has been observed. GM-CSF-induced expansion of this myeloid cell population is accompanied by migration of MdCs into the CNS. Zhao et al. have suggested that, in the presence of inflammatory MdCs, endothelial cells (ECs) in the BBB are activated, enabling immune cells such as GM-CSF-overexpressing CD4+ T cells to enter the CNS (49). In addition, another study has shown that transmigration of myeloid cells across ECs of the CNS is associated with the IL-1β/IL-1R1 axis. Central nervous system ECs under the influence of IL-1β secrete GM-CSF, which induces the differentiation of monocytes into antigen-presenting cells (APCs) (17).
GM-CSF and Innate Immune Cells in MS
In an inflammatory situation, two groups of innate immune cells, dendritic cells (DCs) and macrophages, have an essential role as a link between the innate and adaptive immune responses. DCs act as antigen-presenting cells and play a significant role in presenting processed antigens to T cells. They also express co-stimulatory molecules that are essential for the interaction between DCs and T cells following T cell activation. Accumulating evidence shows that GM-CSF up-regulates MHC-II expression and the secretion of some pro-inflammatory cytokines such as TNF α, IL-6, and IL-23 (50).
DCs are recruited to MS lesions, where they mature and have an effect on the inflammatory response to myelin antigens (51). Some studies have suggested that MS patients have an increased myeloid DC population, which expresses HLA-DR, CD40, CD86, and CD80. In addition, the expression of inhibitory molecules such as PDL-1 on these cells is decreased, and they produce an elevated level of pro-inflammatory cytokines that drive Th1-Th17 immune responses, resulting in disease exacerbation (52, 53). Other studies have indicated that GM-CSF plays the main role in driving inflammatory monocytes to the CNS and its signaling in monocyte-derived DCs appears to be crucial for EAE induction (50, 54). Additionally, this cytokine promotes the differentiation of immature myeloid cells to DC in the CNS (38).
GM-CSF deficient mice (Csf2-deficient) are resistant to EAE; however, treatment with the anti-CD25 mAb PC61 induces severe and chronic EAE in these mice equivalent to that of C57BL/6 mice. Furthermore, after the induction of EAE with PC61 as a passive model of EAE, adoptive transfer of myelin-specific Csf2-deficient T cells into Csf2-deficient mice did not improve the disease course or its severity. The defective T cell response in Csf2-deficient mice is therefore likely related to an inadequate CD4+ T cell response, which is not capable of overcoming Treg cell regulatory barriers (55). King et al. have found that GM-CSF-deficient mice exhibit impairment in a particular group of migratory dermal langerin+CD103+ DCs. These DCs stimulate the expansion of naïve myelin-specific T cells, resulting in the production of IFN-γ and IL-17. Deficiency of this subset of DCs could thus inhibit the responses of these two cytokines, contributing to EAE resistance (34). In line with this theory, these cells may play an important role in the pathogenesis of autoimmune disorders via the development of CD4+ T cell differentiation.
In addition to DCs, macrophage subsets (M1 and M2) also play various roles in the immune system. M1 macrophages are associated with inflammatory response, while M2 macrophages are involved in anti-inflammatory responses and tissue repair mechanisms (12, 56). Culture of monocytes in the presence of GM-CSF and M-CSF induces M1 (CD11b+F4/80+ CD11c+ CD206−) and M2 (CD11b+F4/80+ CD11c− CD206+) macrophages, respectively, and a high M1/M2 ratio enhances the development of EAE and induces relapses but, reversing this ratio, reduces disease severity. Adoptive transfer of CD206+ M2 macrophages into EAE mice suppressed disease (56, 57). Furthermore, histological analysis of lumbar spinal cord of mice in which EAE had been induced with GM-CSF−/− T cells showed decreased CD11b+ microglia/macrophages in lesions in comparison with WT T cells. This finding suggests that the proliferation and function of residential microglia cells can be developed by GM-CSF producing T cells (2).
The Impact of GM-CSF on T Cells as an Adaptive arm of the Immune System in MS
It was initially thought that, among different subsets of CD4+ T cells, Th1 cells that can produce IFN-γ are responsible for autoimmune responses in MS. This opinion changed after it was found that IL-23 deficient mice are unable to develop EAE (58). It was later clarified that IL-23 induces the development of IL-17-producing CD4+ T cells. Furthermore, it has been found that that IL-23-driven production of IL-17 T cells is critical for pathogenicity in the CNS (59). Accordingly, myelin-specific CD4+ T cells that had been activated by APC/Ag developed EAE when transferred to naïve mice, while the transfer of myelin-specific CD4+ T activated with anti-CD3/28 did not induce EAE. A more detailed assessment showed that IL-23R signaling in APCs is critical for the generation of encephalitogenic T cells (58). A subset of Th cells was subsequently identified that could express IL-17A and IL-17F but not IFN-γ or IL-4 (60). After the discovery of Th17 cells, a study showed that these immune cells play a significant role in the induction of autoimmune disorders like MS. However, their key secreted cytokines, IL-17A and 17F, and even IL-21 and IL-22, are not essential for EAE induction (61). It was later shown that GM-CSF secreted by Th17 cells is the main cytokine contributing to encephalitogenicity (62). Interestingly, IL-23 is necessary for the production of GM-CSF, and this cytokine causes an increase in pro-inflammatory myeloid cells in the CNS, resulting in demyelination in EAE (62). Also, in EAE mice, GM-CSF is an essential factor for the secretion of IL-23 by DCs in a CCR4-dependent pathway (63). These observations suggest that there is a positive feedback loop where GM-CSF induces IL-23 production and vice versa. GM-CSF production in this situation is related to both NF-kB and RORγt transcription factors (64). However, other reports have suggested that STAT4 and Blimp-1 act as transcription factors for GM-CSF production (65, 66).
Recent studies have shown that most of the Th17 cells that infiltrate the CNS of EAE mice convert into Th1 cells, now called exTh17 cells, which are more pathogenic and promote inflammation in the CNS (67). Interestingly, Th17 cells are not as pathogenic as exTh17 cells (68). Pathogenicity of exTh17 cells correlates to their ability to produce GM-CSF, IFN-γ, and IL 17 simultaneously. Moreover, exTh17 cells express Th1-related transcription factor T-bet and Th17-related RORγt and express the chemokine receptors CXCR3 and CCR6; exTh17 cells are also called Th1/Th17 cells (69, 70). The importance of T-bet and RORγt co-expression is based on the fact that they have both been implicated in the production of GM-CSF by mouse and human CD4+ T cells (62, 64, 71) (Figure 1).
MS has also been shown to have an important T cell-dependent background as T cells are enriched in lesions and circulating T cells in the blood of MS patients and show an activated phenotype (72). As regards GM-CSF production, Hartman et al. found that MS patients have an elevated frequency of GM-CSF-producing CD4+ T cells in the blood (73). Our group showed that GM-CSF+ CD4+ T cells are also frequent in the lesions of untreated MS patients and that their numbers decrease after IFN-β treatment (74). Moreover, CD8+ T cells can produce GM-CSF, and IL-17-producing CD8+ T cells (Tc17 cells) are a known source of GM-CSF, TNF-α, IFN-γ, IL-21, and IL-22 (75–77). Our group has also shown that GM-CSF+ CD8+ T cells are present in MS lesions (74). Although the role of CD4+ T cells in CNS inflammation is well established, data in the literature on the part played by CD8+ T in demyelination and CNS inflammation are conflicting and need further elucidation (78, 79).
GM-CSF Only Producing CD4+ T Cells in MS
Recent reports have identified a subpopulation of CD4+ T cells that do not produce IFN-γ, IL-17, IL-4, IL-9, and IL-13 but produce GM-CSF in the peripheral blood of healthy individuals (71). These cells are being called ThG cells and their role in neuroinflammation is a matter of current investigation. ThG cells are increased in the peripheral blood of MS patients (71, 74, 80). These cells represent only 2% of all CD4+ T helper cells in healthy subjects and their existence in rodents has also been demonstrated (80, 81). ThG cells express low levels of T-bet, GATA3, and RORγt, which suggests that their transcriptional pathway is different from other Th cell subsets.
Human and mouse ThG cells are induced in vitro by activating naïve CD4+ T cells with agonistic anti-CD3/CD28 antibodies in the presence of IL-2 and IL-7 (58). MS patients with a polymorphism in the IL-2 receptor alpha gene have an increased frequency of ThG cells (73). Polymorphisms in the IL-7Rα chain are associated with an increased risk of developing MS (82). Given that IL-2 and IL-7 signal through a common γ chain receptor, the intracellular signaling is mediated by STAT5 and suppressed by STAT3. EAE induction in STAT5−/− mice indicates that the IL-7-STAT5 axis is needed for the development of GM-CSF/IL-3- producing T cells as STAT5−/− mice have fewer ThG cells and develop less severe EAE (80). Taken together, these observations highlight the underappreciated role of GM-CSF in the context of CNS autoimmunity.
GM-CSF Producing B Cells in MS
In addition to their role in antibody production, B cells produce large amounts of cytokines that modulate the microenvironment and inflammation (83). This “helper” function of B cells has attracted attention in the past few years, especially in MS (84, 85). In this context, it has been shown that memory B cells from MS patients produce high levels of GM-CSF, TNF-α, and IL-6 (86). In these GM-CSF–expressing memory B cells, the expression of transcription factors such as STAT5 and STAT6 is related to GM-CSF production and they suppress the formation of IL-10-producing B cells (86). Also, in vitro studies indicate that GM-CSF–expressing B cells play a significant role in inducing a pro-inflammatory phenotype of myeloid cells and in initiating an inflammatory response by producing GM-CSF (86). Interestingly, FDA-approved dimethyl fumarate (DMF) ameliorates MS and has been shown to deplete GM-CSF-producing B cells in MS patients (87, 88). As mentioned previously, GM-CSF-producing B cells promote an inflammatory phenotype of myeloid cells, and B cell depletion therapy has been accompanied by a decrease in proinflammatory myeloid cell responses. Also, anti-CD20 antibody treatment which depletes B cells has been shown to decrease Th1 and Th17 cells (86). These data indicate that the helper function of B cells plays a role in MS pathogenesis. Considering the importance of GM-CSF role in the pathogenesis of MS disease, it has recently been recognized as a therapeutic target in various studies (89).
The Role of GM-CSF in Immune Tolerance
Unlike IL-10 and TGF-β, GM-CSF has not been described as a tolerogenic or immunosuppressive cytokine. A previous study showed that regulatory T cells (Tregs) express a functional GM-CSF receptor alpha chain (CD116) and expand in response to stimulation with this cytokine independently of IL-2. Interaction of GM-CSF with CD116 on Tregs may improve immune tolerance (90). Also, GM-CSF regulates effector differentiation of invariant natural killer T (iNKT) cells, which express CD116 (91) (Figure 2). However, Ahn et al. have recently shown opposing effects for NKT cells through production of IL-4 and GM-CSF. They report that by producing GM-CSF, NKT cells contribute to the induction of inflammatory response via activation of NLRP3-dependent inflammasome (92). In any case, more detailed studies are needed for an in-depth understanding of how NKT cells regulate immune responses.
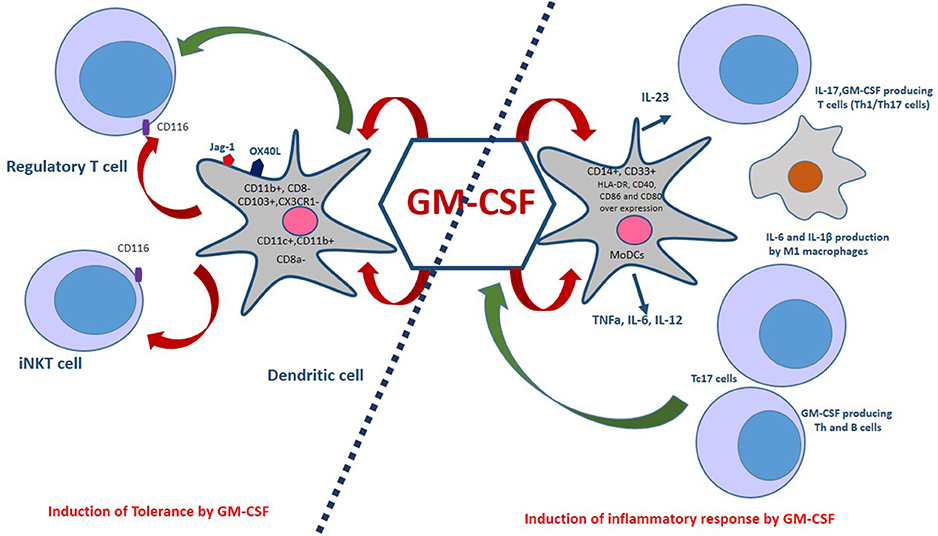
Figure 2. Dual aspects of GM-CSF immunomodulatory effects. GM-CSF-induced bone marrow-derived dendritic cells, which co-express OX40L and Jagged-1 (Jag-1), expand regulatory T cells (Tregs). Also, GM-CSF is associated with a selective expansion of CD11c+CD8a−, CD103+, CX3CR1−, and CD11c+,CD11b+ DCs. The interaction of GM-CSF with CD116 on Tregs and iNKT cells improves immune tolerance. Monocyte-derived dendritic cells (MoDCs) are generated in the presence of GM-CSF and IL-4. These cells are capable of producing pro-inflammatory cytokines such as TNFa, IL-6, and IL-12. GM-CSF induces the M1 macrophages phenotype that produces inflammatory cytokines. In addition, GM-CSF is an essential factor for the secretion of IL-23 by DCs in a CCR4- dependent pathway. Th1/Th17 cells are induced by IL-23, IL-1β in mice and IL-1 β, IL-12 in humans. Furthermore, CD8+ T cells also express GM-CSF and a subset of these cells, called Tc17, produce IL-17 cells, TNF-α, IFN-γ, IL-21, IL-22, and GM-CSF. Treg, Regulatory T cell; iNKT, Invariant natural killer T cells; Th1/Th17, T helper 1/17 cells; TNFa, Tumor necrosis factor alpha.
Additionally, high doses of GM-CSF recruit myeloid-restricted CD11b+Gr1+ precursors (MSCs) which may favor the retention of Tregs (90). Studies have shown that GM-CSF induces differentiation of bone marrow cells into bone marrow-derived dendritic cells (G-BMDCs) that co-express OX40L and Jagged-1 (Jag-1), which expand natural Tregs (Figure 2). The interaction of these surface molecules expressed in G-BMDCs with their cognate receptors (OX40, Notch3) on Treg cells triggers Treg proliferation that does not require antigen presentation or activation. In addition to the ability of G-BMDCs to expand natural Tregs, G-BMDCs secrete high levels of TGF-β, which along with TCR stimulation could convert effector T cells (Teff) into induced regulatory T cells (iTregs) (93, 94).
GM-CSF also plays an essential role in the differentiation of dendritic cells (DCs), rendering them tolerogenic and inducing T-cell-mediated tolerance (13). DCs originate from hematopoietic bone marrow progenitor cells, which play a significant role in the orchestration of immune responses. Several lines of evidence have indicated that GM-CSF broadly induces DC differentiation, which affects T cells response as an effector or regulatory cells (94, 95). Mature DCs with improved antigen-presenting capacity can induce efficient effector T cell responses while immature DCs induce anergic T cells, regulatory T cells (Tregs) and immunomodulatory cytokine-secreting T cells. The dual nature of DC immunoregulatory function mainly depends on the micromilieu during the maturation and activation of DCs. For instance, in the absence of inflammatory signals, DCs remain immature and maintain T cell tolerance in the periphery. DCs with a tolerogenic phenotype, characterized by decreased expression of co-stimulatory signal (CD80/CD86 molecules), provide a pro-tolerant environment (high IL-10, low IL-12) (96). GM-CSF expands myeloid CD11c+CD8a− and CD11c+CD11b+ DCs, two DCs subsets involved in the induction of tolerance. A study has shown that treatment with rGM-CSF for 7 days increased the percentage of myeloid DCs, making them the predominant DC population in rGM-CSF–treated C57BL/6 mice. rGM-CSF expanded only myeloid DCs, identified by their co-expression of CD11c and CD11b and their lack of expression of CD8a. These DCs showed modest increases in MHC expression and endocytotic activity compared to myeloid DCs from control mice (94, 95).
Furthermore, GM-CSF promotes the CD8a− DCs population and maintains them in a semi-mature tolerogenic status. Antigen presentation by these tolerogenic CD8a− DCs can lead to tolerance through the induction of Tregs from effector T cells (97). In another study, administration of GM-CSF before induction of experimental autoimmune myasthenia gravis (EAMG) in C57BL/6J mice suppressed disease development. The protective effect of GM-CSF was associated with a selective expansion of CD11c+CD8a− DCs. They also observed a reduction in anti-AChR Ab levels, T cell propagation and Th1 cytokine responses, and an increase in the IL-10 response. This effect was likely due to a shift in the cytokine milieu to a Th2 profile and the generation of Tregs (98). CD103+ dendritic cells have a critical role in the induction of Tregs in the gastrointestinal tract and the development of these cells from bone marrow stimulated by Flt3L and GM-CSF. It thus appears that GM-CSF also plays a vital role in the maintenance of intestinal immune tolerance (99). Accordingly, due to a predominance of either effector T cell response in autoimmune diseases, it might be feasible to use GM-CSF to modulate DC subsets in order to prevent these diseases.
The Tolerance-Regulating Role of GM-CSF in Autoimmune Diseases
Tregs are critical for the establishment and maintenance of tolerance in the periphery and play an indispensable role in the prevention of autoimmunity (100). Studies have shown that GM-CSF treatment can induce DCs with a semi-mature phenotype, and Tregs, which subsequently suppress ongoing autoimmunity in animal models (97). GM-CSF, a promoter of tolerogenic DCs, has also been reported to have a suppressive effect on autoimmune diabetes and autoimmune thyroiditis (97, 101). A correlation of lupus-like disease with a deficiency in GM-CSF has also been noted (102). GM-CSF is thought to exert its potential therapeutic effects through selective activation of DCs in non-obese diabetic (NOD) mice (97, 103).
Type 1 Diabetes (T1D)
T1D is an organ-specific autoimmune disease resulting from a breakdown of self-tolerance that leads to the destruction of T cell-mediated pancreatic beta cells. Abnormal maturation and defects in the number and function of DCs have been linked to the development of diabetes (104). There is accumulating evidence that self-tolerance can be restored and promoted by tolerogenic DCs or semi-mature DCs induced by GM-CSF. Gaudreau et al. have found that treatment of NOD mice with GM-CSF can protect them from diabetes and increase the number of splenic CD11c+CD11b+CD8a− DCs. That protection was possibly associated with the accumulation of tolerogenic immature splenic DCs and Tregs. Also, GM-CSF promotes the development of semi-mature DCs that recruit Th2 and Tr1 cells and inhibit diabetes in NOD mice as well as autoimmune thyroiditis (103). Treg cells from GM-CSF-treated mice suppressed T1D, a suppression that was dependent on IL-10 and TGF-β1 production. In addition, the transfer of GM-CSF-exposed DCs to naive mice induced Treg expansion and delayed onset of T1D. GM-CSF affects DCs primarily, causing expansion of Tregs, which are responsible for maintaining tolerance of diabetogenic T cells, and delaying the onset of T1D in NOD mice (104). Alnek et al. have found high levels of GM-CSF and other growth factors at the onset of type 1 diabetes. They have suggested that an increase in GM-CSF and IL-10 in the blood of T1D patients is likely related to their protective mechanisms (101). In another study, Surendar et al. reported an increased level of GM-CSF in patients with diagnosed type 2 diabetes, and they concluded that an activated state of myeloid DCs and plasmacytoid DCs is related to GM-CSF level (105).
Thyroiditis (EAT)
Experimental autoimmune thyroiditis (EAT) is a chronic inflammatory autoimmune disease of the thyroid that serves as a mouse model for Hashimoto's thyroiditis (HT). The condition is accompanied by infiltration of lymphocytes into the thyroid, which leads to follicular destruction. Infiltration of thyroglobulin (mTg)-specific Th cells to the thyroid are usually followed by cytokine production such as IFN-γ, which induces the expression of MHC class II on thyrocytes and eventually leads to more development. Activation of T cells and cytokine production ultimately results in apoptosis of thyrocytes and thyroid destruction (106). GM-CSF has the potential capacity not only to prevent but also to suppress EAT, and GM-CSF-induced EAT suppression in mice was accompanied by an increase in the frequency of Treg cells, which destroyed the mTg-specific T cell responses. Also, the transfer of Tregs from mTg-primed donors treated with GM-CSF into untreated recipients elicited a decrease in T cell responses against mTg (107). It has likewise been shown that mTg-immunized mice treated with GM-CSF demonstrated suppressed effector T cell response to mTg and failed to develop thyroiditis. mTg presentation by GM-CSF-exposed CD8a− DCs to T cells from mTg-primed mice induced an increase in the frequency of Tregs (108). Ganesh et al. showed that transfer of CD8a− DCs from GM-CSF-treated mice into wild-type mice prevented EAT in recipient animals following immunization with mTg (97).
Furthermore, Gangi et al. have shown that GM-CSF can induce DCs with a semi-mature phenotype that is known to have a critical role in the development and maintenance of Treg cells. They also found that IL-10 produced by Treg cells is crucial for disease suppression in GM-CSF-treated mice (107). Another study also found that adoptive transfer of G-BMDCs induces Treg expansion, increases IL-4 and IL-10 production, and suppresses EAT in recipient mice. This study showed a pivotal role for OX40L and Jag1 signaling of G-BMDC in Treg expansion (109).
Myasthenia Gravis (MG)
Myasthenia gravis (MG) is another autoimmune disease caused by autoreactive T cells and auto-antibodies against acetylcholine receptors (AChR). AChRs lose their function due to the binding of autoantibodies, which leads to a defect in neuromuscular transmission (110). Production of anti-AChR Abs is modulated by, and dependent upon, AChR-specific CD4+ T cells (111). Also, DCs are crucial in MG pathogenesis by presenting self-Ags and promoting the priming of AChR-specific T cells (112). Experimental Autoimmune Myasthenia Gravis (EAMG) is an investigational disease model for MG that provides an excellent model system for elucidating the pathogenic mechanisms, immunological nature, and novel treatment approach relevant to MG in humans (113). One study showed that immature DCs generated at a low dose of GM-CSF and pulsed in vitro with AChR could induce tolerance to EAMG (114).
Sheng et al. have reported the protective effect of GM-CSF through the expansion of CD11c+CD8a− DCs, which resulted in clinical improvement. In their study, there was a decrease in levels of circulating anti-AChR Ab as well as of T cell proliferation and Th1responses while there was an increase in IL-10. This effect of GM-CSF is related to Th2 polarization, mobilization of DCs with a tolerogenic phenotype and Treg cell induction (98). Also, Meriggioli et al. have indicated that administration of GM-CSF suppressed the development of EAMG and down-regulated anti-AChR T cell and antibody responses. These effects were linked to the activation of tolerogenic DCs, mobilization of Tregs, and enhanced production of suppressive cytokines, such as IL-10.
Furthermore, GM-CSF-treated mice had an increase in CD11c+CD8a− cells compared to the untreated group (115). Rowin et al. showed that GM-CSF treatment of a patient with a prolonged myasthenic crisis, whose disease was refractory to standard therapy, led to clinical improvement. The clinical efficacy of GM-CSF was associated with an expansion of the circulating numbers of Tregs, an enhanced intensity in Foxp3 expression levels in Tregs, and an early enhancement in Treg suppressive ability for AChR-α induced T cell proliferation (116). The function of DCs may therefore play a crucial role in the initiation and maintenance of healthy immune response in MG. Although Cao et al. have determined the phenotype of autoreactive T cells in MG by T cell library assay as the cells with high levels of IL-17, IFN-γ, and GM-CSF and a low level of IL-10, they did not discuss GM-CSF immunomodulatory effects in previous studies (117).
On the other hand, Aricha et al. have expanded Foxp3+ Treg cells ex vivo by isolating bone marrow (BM) cells. They cultured bone marrow (BM) cells in the presence of GM-CSF and induced CD11c+ MHCII+ CD45RA+ CD8− DCs (BMDCs). A co-culture of BMDCs with splenic CD4+ T cells expanded to 90% Tregs and administration of expanded Tregs to EAMG rats suppressed disease (118).
Systemic Lupus Erythematosus (SLE)
Another autoimmune disorder, juvenile systemic lupus erythematosus (JSLE), is characterized by multisystem involvement (119). Dysregulated neutrophil apoptosis may promote the development of autoimmune response. In addition, an imbalance in both pro-apoptotic and anti-apoptotic factors in both neutrophils and sera from patients with JSLE has been reported (120), and neutropenia as a consequence of accelerated apoptosis of neutrophils and their precursors can be found in patients with SLE (121). Accordingly, apoptotic bodies released by neutrophil apoptosis could be a source of auto-antigens in JSLE (122) and active disease is associated with the increased neutrophil apoptosis (120). Interestingly, neutrophil apoptosis has been ameliorated, and their function improved in the presence of GM-CSF (123). GM-CSF deficient mice have been shown to develop an SLE-like disorder associated with impaired phagocytosis of apoptotic cells (124). GM-CSF can delay neutrophil apoptosis through an increase in cellular levels of myeloid cell leukemia 1 (Mcl-1), an anti-apoptotic protein of the Bcl-2 family, and prevent caspase activation (caspase-3, caspase-7, and caspase-8) (123, 125). Hence, the therapeutic administration of GM-CSF should be considered as an alternative treatment in patients with JSLE to reduce the rate of neutrophil apoptosis.
Inflammatory Bowel Disease (IBD)
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), is characterized by chronic inflammatory disorders throughout the gastrointestinal tract (126). Impaired innate immunity (granulocytes, macrophages, and DCs) plays a critical pathogenic role in IBD (127). GM-CSF is necessary for the development of lamina propria CD103+CX3CR1− DCs that efficiently induce intestinal Tregs (128, 129). Xu et al. have indicated that administration of GM-CSF can result in clinical improvement in patients with CD. Moreover, GM-CSF-deficient mice were more susceptible to dextran sodium sulfate (DSS) induced colitis, possibly due to impaired macrophage function (130).
Similarly, Egea et al. have shown that mice deficient in GM-CSF developed more severe colitis in response to enteric exposure to DSS and that colitis was inhibited mainly by GM-CSF administration (131). In another study, Denson et al. have found that low or normal GM-CSF signaling in neutrophils is associated with a more significant number of complications in pediatric CD (132). Also, an increase in anti-GM-CSF auto-antibodies has diminished GM-CSF bioactivity, which led to an exacerbation of CD and accelerated surgical recurrence (133). Detection of GM-CSF Ab could therefore be a potential tool for monitoring disease activity and optimizing therapy. Likewise, Bernasconi et al. found a reduction in colitis severity after GM-CSF administration in DSS-treated mice and reported that GM-CSF improved accelerated ulcer healing in the colon. These effects were associated with increased CD11b+ monocytic subsets (134). Recombinant human GM-CSF (rhGM-CSF) has been used in clinical trials and is reported to have resulted in improvement and remission in patients with CD (131). Furthermore, Dieckgraefe and Korzenik have reported patients with moderate-to-severe CD who were treated with rhGM-CSF had a high rate of remission and a significant decrease in mean Crohn's disease activity index score during treatment (135). Overall, GM-CSF might be considered as an alternative to traditional immunosuppression for the treatment of Crohn's disease.
Conclusion
GM-CSF may have therapeutic value by modulating leukocyte and cytokine production. GM-CSF exerts its immunomodulatory function via the presence of other cytokines and immune cell subsets that are involved in the immune responses in different autoimmune diseases. The roles of GM-CSF in the pathogenesis of some autoimmune diseases, call our attention to the use of this cytokine or its targeting in the treatment of this type of disorder. Understanding the inflammatory and regulatory roles of GM-CSF in autoimmune disorders will therefore be useful for its application in clinical studies.
Author Contributions
NL wrote most parts of the manuscript and searched for finding data and collected information. RT helped in finding information and reviewed the article before submission not only for spelling and grammar but also for its intellectual content. NR wrote some parts of the manuscript. G-XZ and AmR were involved in the planning and organizing of manuscript. AmR was involved in the design of manuscript NE contributed to the design and implementation of the manuscript. All authors discussed the information and commented on the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to express our gratitude to Katherine Regan for assistance in the editing of the manuscript.
References
1. Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm. (2015) 2015:568543. doi: 10.1155/2015/568543
2. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:39–48. doi: 10.4049/jimmunol.178.1.39
3. Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, et al. The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell. (2008) 134:496–507. doi: 10.1016/j.cell.2008.05.053
4. van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, and Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol. (2013) 56:675–82. doi: 10.1016/j.molimm.2013.05.002
5. van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. (2012) 119:3383–93. doi: 10.1182/blood-2011-11-370130
6. Lehtonen A, Matikainen S, Miettinen M, Julkunen I. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-induced STAT5 activation and target-gene expression during human monocyte/macrophage differentiation. J Leukoc Biol. (2002) 71:511–9. doi: 10.1189/jlb.71.3.511
7. Choi JK, Kim KH, Park H, Park SR, Choi BH. Granulocyte macrophage-colony stimulating factor shows anti-apoptotic activity in neural progenitor cells via JAK/STAT5-Bcl-2 pathway. Apoptosis. (2011) 16:127–34. doi: 10.1007/s10495-010-0552-2
8. Katz S, Zsiros V, Doczi N, Kiss AL. Inflammation-induced epithelial-to-mesenchymal transition and GM-CSF treatment stimulate mesenteric mesothelial cells to transdifferentiate into macrophages. Inflammation. (2018) 2018:825. doi: 10.1007/s10753-018-0825-4
9. Yang TC, Chang PY, Kuo TL, Lu SC. Electronegative L5-LDL induces the production of G-CSF and GM-CSF in human macrophages through LOX-1 involving NF-kappaB and ERK2 activation. Atherosclerosis. (2017) 267:1–9. doi: 10.1016/j.atherosclerosis.2017.10.016
10. Xu D, Zhao M, Song Y, Song J, Huang Y, Wang J. Novel insights in preventing Gram-negative bacterial infection in cirrhotic patients: review on the effects of GM-CSF in maintaining homeostasis of the immune system. Hepatol Int. (2015) 9:28–34. doi: 10.1007/s12072-014-9588-7
11. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
12. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
13. Li BZ, Ye QL, Xu WD, Li JH, Ye DQ, Xu Y. GM-CSF alters dendritic cells in autoimmune diseases. Autoimmunity. (2013) 46:409–18. doi: 10.3109/08916934.2013.803533
14. Shiomi A, Usui T, Mimori T. GM-CSF as a therapeutic target in autoimmune diseases. Inflamm Regen. (2016) 36:8. doi: 10.1186/s41232-016-0014-5
15. Bhattacharya P, Budnick I, Singh M, Thiruppathi M, Alharshawi K, Elshabrawy H, et al. Dual role of GM-CSF as a pro-inflammatory and a regulatory cytokine: implications for immune therapy. J Interferon Cytokine Res. (2015) 35:585–99. doi: 10.1089/jir.2014.0149
16. Cook AD, Louis C, Robinson MJ, Saleh R, Sleeman MA, Hamilton JA. Granulocyte macrophage colony-stimulating factor receptor alpha expression and its targeting in antigen-induced arthritis and inflammation. Arthritis Res Ther. (2016) 18:287. doi: 10.1186/s13075-016-1185-9
17. Pare A, Mailhot B, Levesque SA, Juzwik C, Ignatius Arokia Doss PM, Lecuyer MA, et al. IL-1beta enables CNS access to CCR2(hi) monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc Natl Acad Sci USA. (2018) 115:E1194–203. doi: 10.1073/pnas.1714948115
18. Borriello F Galdiero MR, Varricchi G, Loffredo S, Spadaro G Marone G. Innate immune modulation by GM-CSF and IL-3 in health and disease. Int J Mol Sci. (2019) 20:4. doi: 10.3390/ijms20040834
19. Piper C, Pesenacker AM, Bending D, Thirugnanabalan B, Varsani H, Wedderburn LR, et al. T cell expression of granulocyte-macrophage colony-stimulating factor in juvenile arthritis is contingent upon Th17 plasticity. Arthritis Rheumatol. (2014) 66:1955–60. doi: 10.1002/art.38647
20. Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med. (2007) 204:41–7. doi: 10.1084/jem.20062259
21. Ganesan R, Rasool M. Interleukin 17 regulates SHP-2 and IL-17RA/STAT-3 dependent Cyr61, IL-23 and GM-CSF expression and RANKL mediated osteoclastogenesis by fibroblast-like synoviocytes in rheumatoid arthritis. Mol Immunol. (2017) 91:134–44. doi: 10.1016/j.molimm.2017.09.003
22. Samarpita S, Doss HM, Ganesan R, Rasool M. Interleukin 17 under hypoxia mimetic condition augments osteoclast mediated bone erosion and expression of HIF-1alpha and MMP-9. Cell Immunol. (2018) 332:39–50. doi: 10.1016/j.cellimm.2018.07.005
23. Leizer T, Cebon J, Layton JE, Hamilton JA. Cytokine regulation of colony-stimulating factor production in cultured human synovial fibroblasts: I. Induction of GM-CSF and G-CSF production by interleukin-1 and tumor necrosis factor. Blood. (1990) 76:1989–96.
24. Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol. (1991) 147:1238–46.
25. Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, et al. Autoimmune Th17 cells induced synovial stromal and innate lymphoid cell secretion of the cytokine GM-CSF to initiate and augment autoimmune arthritis. Immunity. (2018) 48:1220-1232 e1225. doi: 10.1016/j.immuni.2018.04.009
26. Makris A, Adamidi S, Koutsianas C, Tsalapaki C, Hadziyannis E, Vassilopoulos D. Increased frequency of peripheral B and T cells expressing granulocyte monocyte colony-stimulating factor in rheumatoid arthritis patients. Front Immunol. (2017) 8:1967. doi: 10.3389/fimmu.2017.01967
27. Reynolds G, Gibbon JR, Pratt AG, Wood MJ, Coady D, Raftery G, et al. Synovial CD4+ T-cell-derived GM-CSF supports the differentiation of an inflammatory dendritic cell population in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:899–907. doi: 10.1136/annrheumdis-2014-206578
28. Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, van den Hoogen FH, et al. The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. (2005) 52:2313–22. doi: 10.1002/art.21278
29. Tsark EC, Wang W, Teng YC, Arkfeld D, Dodge GR, Kovats S. Differential MHC class II-mediated presentation of rheumatoid arthritis autoantigens by human dendritic cells and macrophages. J Immunol. (2002) 169:6625–33.
30. Lebre MC, Tak PP. Dendritic cell subsets: their roles in rheumatoid arthritis. Acta Reumatol Port. (2008) 33, 35–45.
31. Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res. (2001) 3:293–8. doi: 10.1186/ar318
32. Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T. GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol. (2014) 193:849–59. doi: 10.4049/jimmunol.1303255
33. Katano M, Okamoto K, Arito M, Kawakami Y, Kurokawa MS, Suematsu N, et al. Implication of granulocyte-macrophage colony-stimulating factor induced neutrophil gelatinase-associated lipocalin in pathogenesis of rheumatoid arthritis revealed by proteome analysis. Arthritis Res Ther. (2009) 11:R3. doi: 10.1186/ar2587
34. King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. (2010) 207:953–61. doi: 10.1084/jem.20091844
35. Kelly R, Marsden RA, Bevan D. Exacerbation of psoriasis with GM-CSF therapy. Br J Dermatol. (1993) 128:468–9.
36. Scholz T, Weigert A, Brune B, Sadik CD, Bohm B, Burkhardt H. GM-CSF in murine psoriasiform dermatitis: redundant and pathogenic roles uncovered by antibody-induced neutralization and genetic deficiency. PLoS ONE. (2017) 12:e0182646. doi: 10.1371/journal.pone.0182646
37. Kister I, Bacon TE, Chamot E, Salter AR, Cutter GR, Kalina JT, et al. Natural history of multiple sclerosis symptoms. Int J MS Care. (2013) 15:146–58. doi: 10.7224/1537-2073.2012-053
38. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. (2009) 113:3190–7. doi: 10.1182/blood-2008-07-168575
39. Imitola J, Rasouli J, Watanabe F, Mahajan K, Sharan AD, Ciric B, et al. Elevated expression of granulocyte-macrophage colony-stimulating factor receptor in multiple sclerosis lesions. J Neuroimmunol. (2018) 317:45–54. doi: 10.1016/j.jneuroim.2017.12.017
40. Chuang DY, Simonyi A, Kotzbauer PT, Gu Z, Sun GY. Cytosolic phospholipase A2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J Neuroinflamm. (2015) 12:199. doi: 10.1186/s12974-015-0419-0
41. Gonzalez H, Elgueta D, Montoya A, Pacheco R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J Neuroimmunol. (2014) 274:1–13. doi: 10.1016/j.jneuroim.2014.07.012
42. Wiggins-Dohlvik K, Merriman M, Shaji CA, Alluri H, Grimsley M, Davis ML, et al. Tumor necrosis factor-alpha disruption of brain endothelial cell barrier is mediated through matrix metalloproteinase-9. Am J Surg. (2014) 208:954–60; discussion 960. doi: 10.1016/j.amjsurg.2014.08.014
43. Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, et al. GM-CSF increases LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflamm. (2012) 9:268. doi: 10.1186/1742-2094-9-268
44. Mufazalov IA, Schelmbauer C, Regen T, Kuschmann J, Wanke F, Gabriel LA, et al. IL-1 signaling is critical for expansion but not generation of autoreactive GM-CSF+ Th17 cells. EMBO J. (2017) 36:102–15. doi: 10.15252/embj.201694615
45. Pare A, Mailhot B, Levesque SA, Lacroix S. Involvement of the IL-1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: breaking the vicious cycle between IL-1beta and GM-CSF. Brain Behav Immun. (2017) 62:1–8. doi: 10.1016/j.bbi.2016.07.146
46. Shang S, Yang YM, Zhang H, Tian L, Jiang JS, Dong YB, et al. Intracerebral GM-CSF contributes to transendothelial monocyte migration in APP/PS1 Alzheimer's disease mice. J Cereb Blood Flow Metab. (2016) 36:1978–91. doi: 10.1177/0271678X16660983
47. Zhang H, Zhang S, Zhang J, Liu D, Wei J, Fang W, et al. ZO-1 expression is suppressed by GM-CSF via miR-96/ERG in brain microvascular endothelial cells. J Cereb Blood Flow Metab. (2018) 38:809–22. doi: 10.1177/0271678X17702668
48. Vogel DY, Kooij G, Heijnen PD, Breur M, Peferoen LA, van der Valk P, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol. (2015) 45:1808–19. doi: 10.1002/eji.201444960
49. Zhao J, Sun L, Li X. Commanding CNS invasion: GM-CSF. Immunity. (2017) 46:165–7. doi: 10.1016/j.immuni.2017.02.003
50. Mausberg AK, Jander S, Reichmann G. Intracerebral granulocyte-macrophage colony-stimulating factor induces functionally competent dendritic cells in the mouse brain. Glia. (2009) 57:1341–50. doi: 10.1002/glia.20853
51. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Capello E, Mancardi GL, et al. Dendritic cells in multiple sclerosis lesions: maturation stage, myelin uptake, and interaction with proliferating T cells. J Neuropathol Exp Neurol. (2006) 65:124–41. doi: 10.1097/01.jnen.0000199572.96472.1c
52. Huang G, Wang Y, Vogel P, Kanneganti TD, Otsu K, Chi H. Signaling via the kinase p38alpha programs dendritic cells to drive TH17 differentiation and autoimmune inflammation. Nat Immunol. (2012) 13:152–61. doi: 10.1038/ni.2207
53. Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D, et al. (2006). Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 177, 4196–4202
54. Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. (2015) 43:502–14. doi: 10.1016/j.immuni.2015.08.010
55. Ghosh D, Curtis, AD II, Wilkinson DS, Mannie MD. Depletion of CD4+ CD25+ regulatory T cells confers susceptibility to experimental autoimmune encephalomyelitis (EAE) in GM-CSF-deficient Csf2-/- mice. J Leukoc Biol. (2016) 100:747–60. doi: 10.1189/jlb.3A0815-359R
56. Gao J, Scheenstra MR, van Dijk A, Veldhuizen EJA, Haagsman HP. A new and efficient culture method for porcine bone marrow-derived M1- and M2-polarized macrophages. Vet Immunol Immunopathol. (2018) 200:7–15. doi: 10.1016/j.vetimm.2018.04.002
57. Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. (2011) 17:2–15. doi: 10.1177/1352458510379243
58. Sheng W, PNG CW, Reynolds JM, Zhang Y. T cell-derived GM-CSF, regulation of expression and function. Immunome Res. (2015) 11:1. doi: 10.4172/1745-7580.1000098
59. Lee PW, Smith AJ, Yang Y, Selhorst AJ, Liu Y, Racke MK, et al. (2017). IL-23R-activated STAT3/STAT4 is essential for Th1/Th17-mediated CNS autoimmunity. JCI Insight. 2:17. doi: 10.1172/jci.insight.91663
60. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. (2005) 6:1123–32. doi: 10.1038/ni1254
61. Waisman A, Hauptmann J, Regen T. The role of IL-17 in CNS diseases. Acta Neuropathologica. (2015) 129:625–37. doi: 10.1007/s00401-015-1402-7
62. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
63. Poppensieker K, Otte, DM, Schürmann B, Limmer A, Dresing P, Drews E, et al. CC chemokine receptor 4 is required for experimental autoimmune encephalomyelitis by regulating GM-CSF and IL-23 production in dendritic cells. Proc Natl Acad Sci USA. (2012) 109:3897–902. doi: 10.1073/pnas.1114153109
64. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
65. McWilliams IL, Rajbhandari R, Nozell S, Benveniste E, Harrington LE. STAT4 controls GM-CSF production by both Th1 and Th17 cells during EAE. J Neuroinflamm. (2015) 12:128. doi: 10.1186/s12974-015-0351-3
66. Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, et al. Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells. Immunity. (2016) 44:131–42. doi: 10.1016/j.immuni.2015.11.009
67. Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. (2011) 12:255–63. doi: 10.1038/ni.1993
68. Basdeo SA, Cluxton D, Sulaimani J, Moran B, Canavan M, Orr C, et al. Ex-Th17 (Nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol. (2017) 198:2249–59. doi: 10.4049/jimmunol.1600737
69. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood. (2013) 121:2402–14. doi: 10.1182/blood-2012-09-378653
70. Duhen T, Campbell DJ. IL-1beta promotes the differentiation of polyfunctional human CCR6+CXCR3+ Th1/17 cells that are specific for pathogenic and commensal microbes. J Immunol. (2014) 193:120–9. doi: 10.4049/jimmunol.1302734
71. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. (2014) 6:241ra280. doi: 10.1126/scitranslmed.3008706
72. Kaskow BJ, Baecher-Allan C. Effector T cells in multiple sclerosis. Cold Spring Harb Perspect Med. (2018) 8:4. doi: 10.1101/cshperspect.a029025
73. Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. (2014) 5:5056. doi: 10.1038/ncomms6056
74. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol. (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
75. Ciric B, El-behi M, Cabrera R, Zhang GX, Rostami A. IL-23 drives pathogenic IL-17-producing CD8+ T cells. J Immunol. (2009) 182:5296–305. doi: 10.4049/jimmunol.0900036
76. Salehi Z, Doosti R, Beheshti M, Janzamin E, Sahraian MA, Izad M. Differential frequency of CD8+ T cell subsets in multiple sclerosis patients with various clinical patterns. PLoS ONE. (2016) 11:e0159565. doi: 10.1371/journal.pone.0159565
77. Srenathan U, Steel K, Taams LS. IL-17+ CD8+ T cells: differentiation, phenotype and role in inflammatory disease. Immunol Lett. (2016) 178:20–6. doi: 10.1016/j.imlet.2016.05.001
78. York NR, Mendoza JP, Ortega SB, Benagh A, Tyler AF, Firan M, et al. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J Autoimmun. (2010) 35:33–44. doi: 10.1016/j.jaut.2010.01.003
79. Ortega SB, Kashi VP, Tyler AF, Cunnusamy K, Mendoza JP, Karandikar NJ. The disease-ameliorating function of autoregulatory CD8 T cells is mediated by targeting of encephalitogenic CD4 T cells in experimental autoimmune encephalomyelitis. J Immunol. (2013) 191:117–26. doi: 10.4049/jimmunol.1300452
80. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. (2014) 24:1387–402. doi: 10.1038/cr.2014.154
81. Stojic-Vukanic Z, Nacka-Aleksic M, Pilipovic I, Vujnovic I, Blagojevic V, Kosec D, et al. Aging diminishes the resistance of AO rats to EAE: putative role of enhanced generation of GM-CSF Expressing CD4+ T cells in aged rats. Immun Ageing. (2015) 12:16. doi: 10.1186/s12979-015-0044-x
82. Tavakolpour S. Interleukin 7 receptor polymorphisms and the risk of multiple sclerosis: a meta-analysis. Mult Scler Relat Disord. (2016) 8:66–73. doi: 10.1016/j.msard.2016.05.001
83. Fillatreau S. B cells and their cytokine activities implications in human diseases. Clin Immunol. (2018) 186:26–31. doi: 10.1016/j.clim.2017.07.020
84. Touil H, Kobert A, Lebeurrier N, Rieger A, Saikali P, Lambert C, et al. Human central nervous system astrocytes support survival and activation of B cells: implications for MS pathogenesis. J Neuroinflamm. (2018) 15:114. doi: 10.1186/s12974-018-1136-2
85. Fraussen J, Claes N, Van Wijmeersch B, van Horssen J, Stinissen P, Hupperts R, et al. B cells of multiple sclerosis patients induce autoreactive proinflammatory T cell responses. Clin Immunol. (2016) 173:124–32. doi: 10.1016/j.clim.2016.10.001
86. Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. (2015). Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 7, 310ra166. doi: 10.1126/scitranslmed.aab4176
87. Li R, Rezk A, Ghadiri M, Luessi F, Zipp F, Li H, et al. Dimethyl fumarate treatment mediates an anti-inflammatory shift in B cell subsets of patients with multiple sclerosis. J Immunol. (2017) 198:691–8. doi: 10.4049/jimmunol.1601649
88. Smith MD, Martin KA, Calabresi PA, Bhargava P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann Clin Transl Neurol. (2017) 4:351–5. doi: 10.1002/acn3.411
89. Aram J, Francis A, Tanasescu R, Constantinescu CS. Granulocyte-macrophage colony-stimulating factor as a therapeutic target in multiple sclerosis. Neurol Ther. (2018) 1:120. doi: 10.1007/s40120-018-0120-1
90. Kared H, Leforban B, Montandon R, Renand A, Layseca Espinosa E, Chatenoud L, et al. Role of GM-CSF in tolerance induction by mobilized hematopoietic progenitors. Blood. (2008) 112:2575–8. doi: 10.1182/blood-2008-02-140681
91. Bezbradica JS, Gordy LE, Stanic AK, Dragovic S, Hill T, Hawiger J, et al. Granulocyte-macrophage colony-stimulating factor regulates effector differentiation of invariant natural killer T cells during thymic ontogeny. Immunity. (2006) 25:487–97. doi: 10.1016/j.immuni.2006.06.017
92. Ahn S, Jeong D, Oh SJ, Ahn J, Lee SH, Chung DH. GM-CSF and IL-4 produced by NKT cells inversely regulate IL-1beta production by macrophages. Immunol Lett. (2017) 182:50–6. doi: 10.1016/j.imlet.2017.01.003
93. Bhattacharya P, Gopisetty A, Ganesh BB, Sheng JR, Prabhakar BS. GM-CSF-induced, bone-marrow-derived dendritic cells can expand natural Tregs and induce adaptive Tregs by different mechanisms. J Leukoc Biol. (2011) 89:235–49. doi: 10.1189/jlb.0310154
94. Schutt CR, Gendelman HE, Mosley RL. Tolerogenic bone marrow-derived dendritic cells induce neuroprotective regulatory T cells in a model of Parkinson's disease. Mol Neurodegener. (2018) 13:26. doi: 10.1186/s13024-018-0255-7
95. Kenkel JA, Tseng WW, Davidson MG, Tolentino LL, Choi O, Bhattacharya N, et al. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res. (2017) 77:4158–70. doi: 10.1158/0008-5472.CAN-16-2212
96. Adorini L. Tolerogenic dendritic cells induced by vitamin D receptor ligands enhance regulatory T cells inhibiting autoimmune diabetes. Ann N Y Acad Sci. (2003) 987:258–61. doi: 10.1111/j.1749-6632.2003.tb06057.x
97. Ganesh BB, Cheatem DM, Sheng JR, Vasu C, Prabhakar BS. GM-CSF-induced CD11c+CD8a–dendritic cells facilitate Foxp3+ and IL-10+ regulatory T cell expansion resulting in suppression of autoimmune thyroiditis. Int Immunol. (2009) 21:269–82. doi: 10.1093/intimm/dxn147
98. Sheng JR, Li L, Ganesh BB, Vasu C, Prabhakar BS, Meriggioli MN. Suppression of experimental autoimmune myasthenia gravis by granulocyte-macrophage colony-stimulating factor is associated with an expansion of FoxP3+ regulatory T cells. J Immunol. (2006) 177:5296–306. doi: 10.4049/jimmunol.177.8.5296
99. Park JH, Choi AJ, Kim SJ, Jeong SY. 3,3'-Diindolylmethane inhibits Flt3L/GM-CSF-induced-bone marrow-derived CD103(+) dendritic cell differentiation regulating phosphorylation of STAT3 and STAT5. Immune Netw. (2015) 15:278–90. doi: 10.4110/in.2015.15.6.278
100. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
101. Alnek K, Kisand K, Heilman K, Peet A, Varik K, Uibo R. Increased blood levels of growth factors, proinflammatory cytokines, and Th17 cytokines in patients with newly diagnosed type 1 diabetes. PLoS ONE. (2015) 10:e0142976. doi: 10.1371/journal.pone.0142976
102. Willeke P, Schluter B, Schotte H, Erren M, Mickholz E, Domschke W, et al. Increased frequency of GM-CSF secreting PBMC in patients with active systemic lupus erythematosus can be reduced by immunoadsorption. Lupus. (2004) 13:257–62. doi: 10.1191/0961203304lu1009oa
103. Gaudreau S, Guindi C, Menard M, Besin G, Dupuis G, Amrani A. Granulocyte-macrophage colony-stimulating factor prevents diabetes development in NOD mice by inducing tolerogenic dendritic cells that sustain the suppressive function of CD4+CD25+ regulatory T cells. J Immunol. (2007) 179:3638–47. doi: 10.4049/jimmunol.179.6.3638
104. Cheatem D, Ganesh BB, Gangi E, Vasu C, Prabhakar BS. Modulation of dendritic cells using granulocyte-macrophage colony-stimulating factor (GM-CSF) delays type 1 diabetes by enhancing CD4+CD25+ regulatory T cell function. Clin Immunol. (2009) 131:260–70. doi: 10.1016/j.clim.2008.12.001
105. Surendar J, Mohan V, Pavankumar N, Babu S, Aravindhan V. Increased levels of serum granulocyte-macrophage colony-stimulating factor is associated with activated peripheral dendritic cells in type 2 diabetes subjects (CURES-99). Diabetes Technol Ther. (2012) 14:344–9. doi: 10.1089/dia.2011.0182
106. Ramos-Levi AM, Marazuela M. Pathogenesis of thyroid autoimmune disease: the role of cellular mechanisms. Endocrinol Nutr. (2016) 63:421–9. doi: 10.1016/j.endonu.2016.04.003
107. Gangi E, Vasu C, Cheatem D, Prabhakar BS. IL-10-producing CD4+CD25+ regulatory T cells play a critical role in granulocyte-macrophage colony-stimulating factor-induced suppression of experimental autoimmune thyroiditis. J Immunol. (2005) 174:7006–13. doi: 10.4049/jimmunol.174.11.7006
108. Vasu C, Dogan RN, Holterman MJ, Prabhakar BS. Selective induction of dendritic cells using granulocyte macrophage-colony stimulating factor, but not fms-like tyrosine kinase receptor 3-ligand, activates thyroglobulin-specific CD4+/CD25+ T cells and suppresses experimental autoimmune thyroiditis. J Immunol. (2003) 170:5511–22. doi: 10.4049/jimmunol.170.11.5511
109. Gopisetty A, Bhattacharya P, Haddad C, Bruno JCJr, Vasu C, Miele L, et al. OX40L/Jagged1 cosignaling by GM-CSF-induced bone marrow-derived dendritic cells is required for the expansion of functional regulatory T cells. J Immunol. (2013) 190:5516–25. doi: 10.4049/jimmunol.1202298
110. Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet. (2001) 357:2122–8. doi: 10.1016/S0140-6736(00)05186-2
111. Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol. (2002) 2:797–804. doi: 10.1038/nri916
112. Nagane Y, Utsugisawa K, Obara D, Yamagata M, Tohgi H. Dendritic cells in hyperplastic thymuses from patients with myasthenia gravis. Muscle Nerve. (2003) 27:582–9. doi: 10.1002/mus.10362
113. Fuchs S, Aricha R, Reuveni D, Souroujon MC. Experimental Autoimmune Myasthenia Gravis (EAMG): from immunochemical characterization to therapeutic approaches. J Autoimmun. (2014) 54:51–9. doi: 10.1016/j.jaut.2014.06.003
114. Li L, Sun S, Cao X, Wang Y, Chang L, Yin X. Experimental study on induction of tolerance to experimental autoimmune myasthenia gravis by immature dendritic cells. J Huazhong Univ Sci Technolog Med Sci. (2005) 25:215–8.
115. Meriggioli MN, Sheng JR, Li L, Prabhakar BS. Strategies for treating autoimmunity: novel insights from experimental myasthenia gravis. Ann N Y Acad Sci. (2008) 1132:276–82. doi: 10.1196/annals.1405.023
116. Rowin J, Thiruppathi M, Arhebamen E, Sheng J, Prabhakar BS, Meriggioli MN. Granulocyte macrophage colony-stimulating factor treatment of a patient in myasthenic crisis: effects on regulatory T cells. Muscle Nerve. (2012) 46:449–53. doi: 10.1002/mus.23488
117. Cao Y, Amezquita RA, Kleinstein SH, Stathopoulos P, Nowak RJ, O'Connor KC. Autoreactive T cells from patients with myasthenia gravis are characterized by elevated IL-17, IFN-gamma, and GM-CSF and diminished IL-10 production. J Immunol. (2016) 196:2075–84. doi: 10.4049/jimmunol.1501339
118. Aricha R, Reuveni D, Fuchs S, Souroujon MC. Suppression of experimental autoimmune myasthenia gravis by autologous T regulatory cells. J Autoimmun. (2016) 67:57–64. doi: 10.1016/j.jaut.2015.09.005
119. Fernandes H, Brito I. Juvenile systemic lupus erythematosus: neuropsychiatric manifestations. Acta Reumatol Port. (2012) 37:117–25.
120. Midgley A, McLaren Z, Moots RJ, Edwards SW, Beresford MW. The role of neutrophil apoptosis in juvenile-onset systemic lupus erythematosus. Arthritis Rheum. (2009) 60:2390–401. doi: 10.1002/art.24634
121. Matsuyama W, Yamamoto M, Higashimoto I, Oonakahara K, Watanabe M, Machida K, et al. TNF-related apoptosis-inducing ligand is involved in neutropenia of systemic lupus erythematosus. Blood. (2004) 104:184–91. doi: 10.1182/blood-2003-12-4274
122. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. (1994) 179:1317–30.
123. Chiewchengchol D, Midgley A, Sodsai P, Deekajorndech T, Hirankarn N, Beresford MW, et al. The protective effect of GM-CSF on serum-induced neutrophil apoptosis in juvenile systemic lupus erythematosus patients. Clin Rheumatol. (2015) 34:85–91. doi: 10.1007/s10067-014-2800-2
124. Enzler T, Gillessen S, Manis JP, Ferguson D, Fleming J, Alt FW, et al. Deficiencies of GM-CSF and interferon gamma link inflammation and cancer. J Exp Med. (2003) 197:1213–9. doi: 10.1084/jem.20021258
125. Derouet M, Thomas L, Cross A, Moots RJ, Edwards SW. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J Biol Chem. (2004) 279:26915–21. doi: 10.1074/jbc.M313875200
126. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. (2015) 12:205–17. doi: 10.1038/nrgastro.2015.34
127. Marks DJ, Rahman FZ, Sewell GW, Segal AW. Crohn's disease: an immune deficiency state. Clin Rev Allergy Immunol. (2010) 38:20–31. doi: 10.1007/s12016-009-8133-2
128. Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. (2007) 204:1757–64. doi: 10.1084/jem.20070590
129. Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, et al. Origin of the lamina propria dendritic cell network. Immunity. (2009) 31:513–25. doi: 10.1016/j.immuni.2009.08.010
130. Xu Y, Hunt NH, Bao S. The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res. (2008) 18:1220–9. doi: 10.1038/cr.2008.310
131. Egea L, Hirata Y, Kagnoff MF. GM-CSF: a role in immune and inflammatory reactions in the intestine. Expert Rev Gastroenterol Hepatol. (2010) 4:723–31. doi: 10.1586/egh.10.73
132. Denson LA, Jurickova I, Karns R, Shaw KA, Cutler DJ, Okou D, et al. Genetic and transcriptomic variation linked to neutrophil granulocyte-macrophage colony-stimulating factor signaling in pediatric crohn's disease. Inflamm Bowel Dis. (2018) 2018:265. doi: 10.1093/ibd/izy265
133. Gathungu G, Zhang Y, Tian X, Bonkowski E, Rowehl L, Krumsiek J, et al. Impaired granulocyte-macrophage colony-stimulating factor bioactivity accelerates surgical recurrence in ileal Crohn's disease. World J Gastroenterol. (2018) 24:623–30. doi: 10.3748/wjg.v24.i5.623
134. Bernasconi E, Favre L, Maillard MH, Bachmann D, Pythoud C, Bouzourene H, et al. Granulocyte-macrophage colony-stimulating factor elicits bone marrow-derived cells that promote efficient colonic mucosal healing. Inflamm Bowel Dis. (2010) 16:428–41. doi: 10.1002/ibd.21072
Keywords: GM-CSF, inflammation, tolerance, modulation, autoimmune diseases
Citation: Lotfi N, Thome R, Rezaei N, Zhang G-X, Rezaei A, Rostami A and Esmaeil N (2019) Roles of GM-CSF in the Pathogenesis of Autoimmune Diseases: An Update. Front. Immunol. 10:1265. doi: 10.3389/fimmu.2019.01265
Received: 12 February 2019; Accepted: 17 May 2019;
Published: 04 June 2019.
Edited by:
Silvano Sozzani, University of Brescia, ItalyReviewed by:
Tiziana Musso, University of Turin, ItalyVanessa Pinho, Federal University of Minas Gerais, Brazil
Copyright © 2019 Lotfi, Thome, Rezaei, Zhang, Rezaei, Rostami and Esmaeil. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abdolmohamad Rostami, YS5tLnJvc3RhbWlAamVmZmVyc29uLmVkdQ==; Nafiseh Esmaeil, bl9lc21hZWlsQG1lZC5tdWkuYWM=