 Ágata C. Cevey
Ágata C. Cevey Federico N. Penas
Federico N. Penas Catalina D. Alba Soto
Catalina D. Alba Soto Gerardo A. Mirkin
Gerardo A. Mirkin Nora B. Goren
Nora B. Goren- 1Facultad de Medicina, Instituto de Investigaciones Biomédicas en Retrovirus y SIDA (INBIRS), CONICET—Universidad de Buenos Aires, Buenos Aires, Argentina
- 2Facultad de Medicina, Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM), CONICET—Universidad de Buenos Aires, Buenos Aires, Argentina
Anti-parasitic treatment for Chagas disease mainly relies on benznidazole, which is virtually the only drug available in the market. Besides its anti-parasitic effects, benznidazole has anti-inflammatory properties. In this work we studied the mechanisms involved in the latter, demonstrating the participation of the IL-10/STAT3/SOCS3 pathway. To achieve this goal, the anti-inflammatory properties of benznidazole were studied using an in vitro model of cardiomyocyte primary culture stimulated with LPS. LPS increased both SOCS3 expression and STAT3 phosphorylation. The addition of benznidazole increased their expression even further. Specific inhibition of STAT3 precluded this effect, suggesting a role for STAT3 in the increase of SOCS3 expression induced by benznidazole. To assess the participation of SOCS3 in the anti-inflammatory effect of benznidazole, we accomplished specific knockdown of SOCS3 with siRNA. Silencing of SOCS3 in cardiomyocytes precluded the inhibitory effects of benznidazole on TNF-α, IL-6, iNOS expression and NO release. Moreover, in the absence of SOCS3, benznidazole could neither prevent IKK phosphorylation nor IκBα degradation, supporting the notion that SOCS3 is required for the benznidazole-mediated inhibition of the NF-κB pathway. Previously, we demonstrated that IL-10 increases the expression of SOCS3 in cultured cardiomyocytes. Here, we found that benznidazole shows a trend to increased IL-10 expression. To evaluate whether benznidazole increased SOCS3 in an IL-10-dependent manner, cardiomyocytes from IL-10 knockout mice were pre-treated with benznidazole and stimulated with LPS. Benznidazole neither inhibited NO release nor avoid IKK phosphorylation or IκBα degradation, showing that IL-10 is required for benznidazole-mediated inhibition of NF-κB. Moreover, exogenous addition of IL-10 to IL-10 knockout cardiomyocytes restored the inhibitory effect of benznidazole on NO release. The results reported herein show, for the first time, that the IL-10/STAT3/SOCS3 axis is involved in the anti-inflammatory effects of benznidazole. These findings may add up to new therapeutic strategies for chronic Chagas disease given its inflammatory nature.
Introduction
American trypanosomiasis is an endemic parasitic disease caused by infection with the obligate intracellular protozoan parasite Trypanosoma cruzi (T. cruzi). It spans throughout Central and South America and represents a major public health concern due to its high morbidity and mortality rates (1). Benznidazole (N-benzyl-2-(2-nitroimidazol-1-yl)acetamide) is one of the drugs of choice for its treatment (2) despite its toxicity and limited efficacy, particularly during the chronic phase of the disease. Although benznidazole has long been used in clinical settings, its mechanisms of action have not been fully elucidated yet. Indeed, there is a general premise that the etiological treatment contributes to a reduction of the parasite load and to fit the host immune response, leading to a balanced inflammatory response which is crucial to control Chagas disease morbidity (3, 4). In a previous work, Campi-Azevedo et al. highlighted that treatment with benznidazole in the indeterminate phase of the disease not only leads to parasite clearance but also would contribute to a general immunomodulation. Induction of a broad change in the immune response was observed in patients in the cardiac phase, shaping an intricate phenotypic/functional network compatible with beneficial and protective immunological events (5). In this regard, we have recently shown in an experimental murine model of acute Chagas disease, using a highly virulent benznidazole-susceptible T. cruzi strain, that the optimal effects of benznidazole, in terms of parasite clearance from blood and heart tissue, as well as the reduction of inflammatory reaction, can be achieved at doses significantly lower than those usually used for the treatment (6).
In addition to its antiparasitic activity, benznidazole exerts immunomodulatory effects on macrophages stimulated with lipopolysaccharide (LPS) and treated with a high concentration of benznidazole (1 mM), by inhibiting the NF-κB pathway (7). These in vitro effects have also been described in LPS-challenged mice pre-treated with high doses of benznidazole (200 mg/Kg/day) (8). Like LPS, certain T. cruzi components, such as glycoinositolphospholipids, are recognized by TLR4 and induce pro-inflammatory cytokines. Thus, in order to distinguish the anti-inflammatory effects from the anti-parasitic effects of benznidazole, we used LPS in experimental settings aimed at exploring its anti-inflammatory mechanism of action (9, 10). Interestingly, anti-inflammatory as well as anti-parasitic effects were achieved using 15 μM of benznidazole, a concentration rendering parasite DNA almost undetectable by qPCR (6).
Interleukin-10 (IL-10), a well-known anti-inflammatory cytokine, is produced by a range of cells such as T cells, B cells, macrophages, and dendritic cells. Its expression is regulated by multiple signaling molecules, including p38 MAPK and ERK1/2 (11). One of the main biological function of IL-10 is to counter the production of inflammatory mediators, especially in response to TLR signaling (12–16). The binding of IL-10 to the IL-10R results in the activation of JAK1 which induces STAT3 phosphorylation. It has been demonstrated that STAT3 is a key effector molecule of IL-10 action. Its activation is necessary for the IL-10-regulated anti-inflammatory effects (17–20). Although better studied in macrophages (17, 21–26), the IL-10/STAT3 anti-inflammatory pathway has long been known to extend to other cells of the immune system (19, 27–30), and non-immune cells (31–35).
Both IL-10 and Interleukin-6 (IL-6) induce the activation of STAT3, yet generate different cellular responses. While IL-6 stimulation promotes a pro-inflammatory response, IL-10 signaling induces a strong anti-inflammatory one (36). Activated STATs not only drive transcription of many genes related to cell proliferation, function, and survival, but also induce the transcription of SOCS genes (37). SOCS3, one of the better studied members of the SOCS family, controls critical cellular processes such as cell growth, apoptosis, and transcription of inflammatory genes (38). It also regulates the kinetics of STAT3 activation, determining the pattern of responsive genes in the case of IL-6 and IL-10 (36).
Induction of SOCS3 by STAT3 results in a negative feedback loop, via binding to the gp130 subunit of the IL-6 receptor, which results in transient STAT3 activation with a rapid decline in phosphorylation and nuclear localization. In contrast, SOCS3 does not block IL-10 activation of STAT3, thereby inducing sustained STAT3 activation (39, 40). This confers a particular timing to the initiation and perpetuation of the inflammatory response.
We have described that 15 μM of benznidazole has the ability to inhibit the NF-κB pathway, demonstrating that the anti-inflammatory effects of this drug may be attained at a concentration lower than that reported elsewhere (6).
The aim of this work is to deepen into the mechanisms involved in the anti-inflammatory effects of benznidazole, as they have not yet been fully elucidated. Our results show that benznidazole leads to STAT3 activation and up-regulation of SOCS3. Moreover, we demonstrate for the first time that IL-10 is required for the benznidazole-mediated inhibition of NF-κB, through IL-10/STAT3/SOCS3 axis.
Materials and Methods
Ethics Statement
To carry out this work, CF1, BALB/c, and BALB/c-background IL-10 knockout mice, homozygous for the targeted mutation Il10tm1Cgn (Stock Number 004333; The Jackson Laboratory, USA) (41) were used. All the animals were bred and maintained in the animal facility at the Instituto de Investigaciones en Microbiología y Parasitología Médica, Universidad de Buenos Aires—CONICET. All the procedures were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, Facultad de Medicina de la Universidad de Buenos Aires, CD N° 2271/2014), in line with guidelines of the Argentinean National Administration of Medicines, Food, and Medical Devices (ANMAT), Argentinean National Service of Agri-Food Health and Quality (SENASA) and with the Guide for the Care and Use of Laboratory Animals (NIH, USA).
In vitro Model: Neonatal Mouse Primary Cardiomyocytes Culture and Stimulation
For each experiment, 20–30 neonatal mice (One- to three-day old, male, and female, 2–3 g weight) were euthanized by decapitation after CO2 exposure, and cardiomyocytes were obtained as described previously (6). Briefly, the hearts were removed aseptically, pooled and maintained in Hank's Buffer Saline Solution (HBSS). The hearts were mechanically disaggregated, and the cells were isolated after several digestions with porcine pancreas trypsin (0.25% w/v in PBS). The cells were plated in culture plates in order to generate five replicates per experimental group, with complete medium (10% FBS-DMEM-M199-PenStrep®) at 37°C in a 5% CO2 atmosphere, up to 80% confluence. Then, the cells were maintained with 1% FBS-DMEM-M199 -PenStrep®, at least for a day before the experiments were performed. For SOCS3 silencing experiments using SOCS3 siRNA and oligofectamine®, cells were cultured up to 30% confluence with complete medium, and maintained with DMEM-M199 medium (without FBS and PenStrep®), at least for a day before stimulation.
In vitro Treatments
Benznidazole (Abarax®, ELEA, Argentina. PubChem Compound Database CID = 31,593, Figure 1A) was suspended in PBS. According to the experiment, stimulated cells were pre-treated with benznidazole 15 μM for 30 min before stimulation with LPS in DMEM.
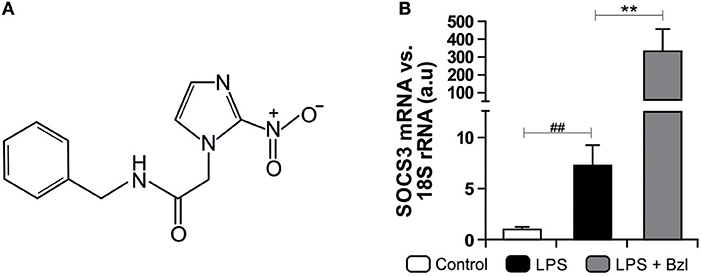
Figure 1. The chemical structure of benznidazole (Bzl) used in this study is shown (A). Cardiomyocytes were treated with 15 μM of Bzl for 30 min and then with 10 mg/L LPS for 24 h. SOCS3 mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA (B). Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS stimulated cells. Gray bar: LPS stimulated and Bzl treated cells **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.
Recombinant Mouse Interleukin-10 (rm IL-10, Immunotools Cat. #12340102) was suspended in PBS. Cardiomyocytes were treated with 50 ng/mL of rmIL-10 for 30 min before benznidazole treatment and LPS stimulation.
In vitro Stimulation
The cardiomyocytes were stimulated with LPS (10 μg/ml) from Escherichia coli O26:B6 (Sigma-Aldrich Co, USA), for the indicated period of time, in polystyrene culture plates. Each independent experiment was carried out three times, with five replicates per experimental group each one.
STAT3 Inhibition
Cardiomyocytes were treated with 10 μM of the specific STAT3 Inhibitor V, Stattic (Santa Cruz Biotechnology, USA), 30 min before benznidazole treatment and LPS stimulation. Stattic inhibits the activation of the STAT3 transcription factor by blocking phosphorylation and dimerization events (31, 42). Briefly, Static was re-suspended in DMSO to generate a 50 mM stock solution. Working solution (500 μM) was generated diluting the stock solution in PBS (Final concentration of DMSO: 1%). PBS/DMSO was added to each untreated well in order to perform vehicle controls (Final concentration of DMSO, 1% in PBS).
SOCS3 Knockdown With Small Interfering RNA (siRNA)
Cardiomyocytes were cultured up to 30–50% of confluence in DMEM: M-199 medium containing 5% FCS without antibiotics for 24 h. Thereafter, cells were transfected with 20 μM of SOCS3-BLOCKiT™siRNA that targets SOCS3 mRNA (siRNA duplex: 5′-GUAUGAUGCUCCACUUUAATT-3′, 5′-UUAAAGUGGAGCAUCAUACTT-3′), following the manufacturer's instructions (Life Technologies Inc., USA). Transfections were performed with Oligofectamine (Life Technologies, Inc., USA) as specified by the manufacturer. Assays for gene activity were performed at 72 h post-transfection. The impact of SOCS3-siRNA interference on SOCS3 mRNA was evaluated by RT-qPCR.
RNA Purification
Total RNA was obtained from cardiomyocytes using Quickzol reagent (Kalium Technologies, Argentina), treated with RQ1 RNase-Free DNase (Promega Co., USA). Total RNA was reverse-transcribed using M-MLV Reverse Transcriptase (Promega Co., USA), according to manufacturer's instructions.
Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
mRNA expression was determined using 5X HOT FIREPOL EVAGREEN qPCR (Solis BioDyne, Estonia) in a StepOnePlus Real-Time PCR System. Parameters were: 52°C for 2 min, 95°C for 15 min, and 40 cycles at 95°C for 15 s, specific Tm °C for 30 s and 72°C for 1 min. Normalization was carried out using 18S rRNA. Quantification was performed using the comparative threshold cycle (Ct) method, as all the primer pairs (target gene/reference gene) were amplified using comparable efficiencies (relative quantity, 2−ΔΔCt) (43, 44).
Primer Sequences

Cytokine ELISA
IL-6, TNF-α, and IL-10 concentrations were measured in culture supernatants using cytokine-specific OptEIA™ ELISA kits according to the manufacturer's instructions (BD Biosciences, USA). The reaction was detected using peroxidase-conjugated Streptavidin, followed by incubation with hydrogen peroxide as a substrate, and with ABTS (Sigma Aldrich Co., USA) as a chromogen. Cytokine concentrations in samples were interpolated from recombinant IL-6, TNF-α, and IL-10 standard curves. Absorbance readings were made at 405 nm.
Protein Extraction and Western Blot Analysis
Total protein extracts were prepared as described previously by our group (6). Protein concentration was determined by the Bradford method using a commercial protein assay (Bio-Rad, USA) and bovine serum albumin (BSA, Sigma-Aldrich Co, USA.) as a standard (45).
Fifty microgram of protein extracts separated in 8–15% SDS-PAGE gels were blotted onto a Hybond-P membrane (GE Health-care, Spain) to detect SOCS3 (Elabscience®, Cat# E-AB-10603, USA), p-STAT3 (Tyr705) (BioLegend Antibody Cat# 690402, RRID:AB_2629819, USA), and α-actin (Santa Cruz Biotechnology Cat# sc-1615, RRID:AB_630835, USA), using specific antibodies, in a 1:500 dilution in PBS. Blots were revealed by enhanced chemiluminescence in a BioSpectrum® Imaging Systems (UVP, Analytik Jena Company, USA). Band intensity was analyzed using the NIH Image J software (ImageJ, RRID:SCR_003070).
NO Measurement
To determine the amount of NO released into the culture medium, nitrate was reduced to nitrite and measured spectrophotometrically by the Griess reaction (46, 47). The amount of NO in the culture supernatants was calculated by interpolation of the samples absorbances at 540 nm using a standard curve of NaNO2.
Immunofluorescence
Myocardial cells grown on round glass coverslips were fixed with methanol and blocked with 3% BSA in PBS. The expression of p-IKK and IκBα was determined by immunofluorescence, as described previously (6). For this purpose, rabbit polyclonal IgG anti- IκBα (Santa Cruz Biotechnology Cat# sc-1643, RRID:AB_627772) or rabbit polyclonal IgG anti- p-IKK (Ser176) (Santa Cruz Biotechnology Cat# sc-21661, RRID:AB_669364) were used as primary antibodies at a 1:50 dilution, and Goat anti-rabbit IgG Alexa Fluor 488 nm (Jackson ImmunoResearch Labs Cat# 111-545-003, RRID:AB_2338046), or Goat anti-Rabbit IgG Alexa Fluor 594 nm (Jackson ImmunoResearch Labs Cat# 111-585-003, RRID:AB_2338059) were used at a 1:500 dilution as secondary antibodies. Cells nuclei were stained with DAPI (300 nM). At least 30 random microscopic fields (400X) and 1,000 cells per culture were acquired using a Spot RT digital camera attached to an Eclipse 600 fluorescence microscope (Nikon Inc., USA). Mean fluorescence intensity (MFI) were quantified using Fiji Image J software (Fiji, RRID:SCR_002285) (48).
Statistical Analysis
Data are expressed as the mean of three independent experiments ± SEM (n = 3) for each experimental group (Five culture replicates/group). One-way ANOVA was used to analyze the statistical significance of the differences observed between infected, treated or untreated groups. The Tukey post-hoc test was performed to compare every mean with every other mean. Differences were considered statistically significant when P < 0.05. All analyses were performed using the Prism 7.0 Software (GraphPad Prism, RRID:SCR_002798).
Results
LPS and Benznidazole Promote STAT3 Activation and SOCS3 Expression
In a previous work we found that T. cruzi and IL-10 promote STAT3 phosphorylation and up-regulate the expression of SOCS3 thereby preventing NF-κB nuclear translocation and ERK1/2 phosphorylation (31). Besides, we determined that the treatment with benznidazole 15 μM was the optimal concentration to clear parasite load in T. cruzi-infected primary cultured cardiomyocytes, as measured by qPCR (6). These findings led us to study its anti-inflammatory properties given the optimal parasiticidal concentration found. We determined that benznidazole 15 μM reduced inflammatory mediators via inhibition of the NF-κB pathway (6). In this work, we sought to determine whether SOCS3 was involved in the anti-inflammatory effects of benznidazole. To this aim, we set up a model to dissociate the anti-parasitic effect of benznidazole from its anti-inflammatory properties using LPS instead of live parasites. Cultured cardiomyocytes were pre-treated with 15 μM benznidazole and stimulated with 10 mg/L of LPS. Interestingly, an increase in SOCS3 mRNA level was observed 24 h after LPS stimulation. Moreover, pre-treatment with benznidazole further increased SOCS3 expression significantly, as shown in Figure 1B.
Moreover, both LPS and benznidazole are able to induce phosphorylation of STAT3. As expected, this effect was inhibited by 10 μM of Stattic, a STAT3 inhibitor (Figure 2A). Interestingly, SOCS3 increase was precluded by Stattic, suggesting that benznidazole increased SOCS3 mRNA expression in a STAT3-dependent manner (Figure 2B). As expected, Western blot showed an increase in SOCS3 protein expression after LPS stimulation and even more in LPS-stimulated and benznidazole-treated cardiomyocytes. Furthermore, lower SOCS3 expression upon treatment with Stattic of LPS-stimulated and benznidazole-treated cardiomyocytes was observed (Figure 2B).
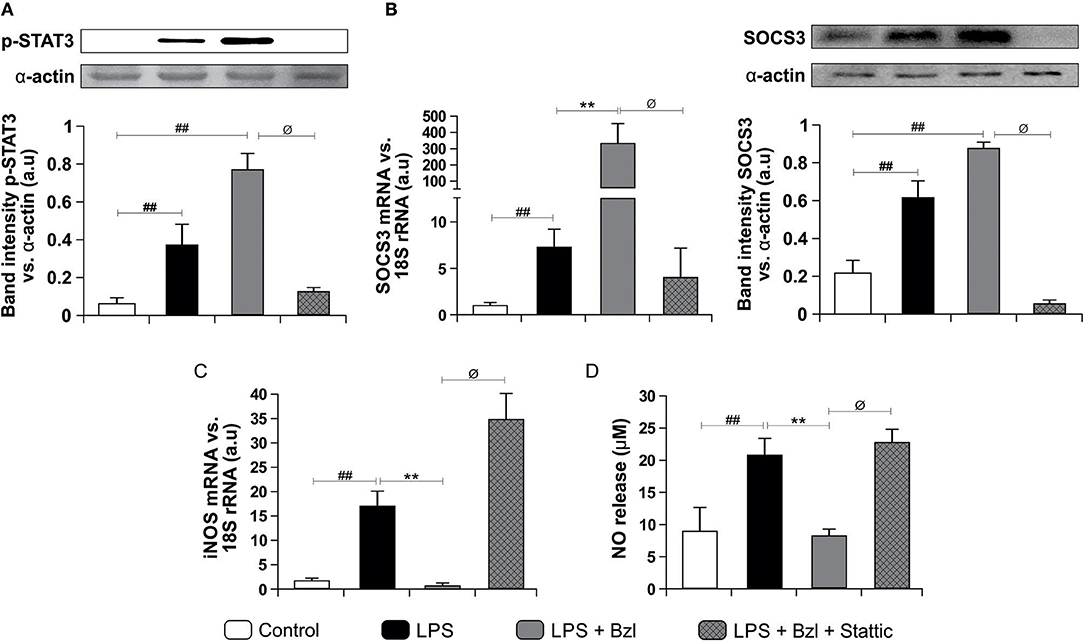
Figure 2. Cardiomyocytes were treated with 10 μM Stattic, a specific inhibitor of STAT3, 30 min before the addition of 15 μM benznidazole (Bzl). Cells were stimulated with 10 mg/L LPS 30 min after Bzl treatment. After 15 min, p-STAT expression was determined by Western blot with a specific antibody and normalized against α-actin (A). After 24 h, SOCS3 mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA. After 48 h SOCS3 protein expression was determined by Western blot with a specific antibody and normalized against α-actin (B). After 48 h, iNOS mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA (C), and NO levels were quantified by the Griess reaction in culture supernatants (D). Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS stimulated cells. Gray bar: LPS stimulated and Bzl treated cells. Hatched gray bar: Stattic pre-treated, LPS stimulated and Bzl treated cells **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.øP < 0.05 vs. Bzl-treated and LPS-stimulated cells.
Earlier, we reported that while LPS increases the expression of iNOS and the release of NO by cardiomyocytes, benznidazole inhibits these responses (6). In this work we assessed the participation of STAT3 in the inhibitory effect of benznidazole on these inflammatory mediators. When STAT3 is inhibited, benznidazole neither prevents iNOS expression (Figure 2C) nor NO release (Figure 2D), pointing out that STAT3 is involved in the anti-inflammatory effect of benznidazole.
SOCS3 Is Involved in the Anti-inflammatory Effects Exerted by Benznidazole
In order to determine whether SOCS3 participates in the inhibition of inflammatory mediators, we accomplished specific knockdown of SOCS3 with siRNA in cultured cardiomyocytes. Then, cells were pre-treated with benznidazole and stimulated with LPS. IL-6 and TNF-α expression and their concentrations in culture supernatants were evaluated. Figure 3A shows that benznidazole inhibited IL-6 and TNF-α mRNA expression. Conversely, when SOCS3 was silenced, benznidazole was unable to modulate the expression of these pro-inflammatory cytokines. In agreement with this benznidazole could not inhibit the release of IL-6 and TNF-α to culture supernatants upon SOCS3 silencing (Figure 3B). Also, under silencing conditions of SOCS3, benznidazole neither inhibited iNOS mRNA expression nor NO production in LPS-stimulated cells (Figure 3C).
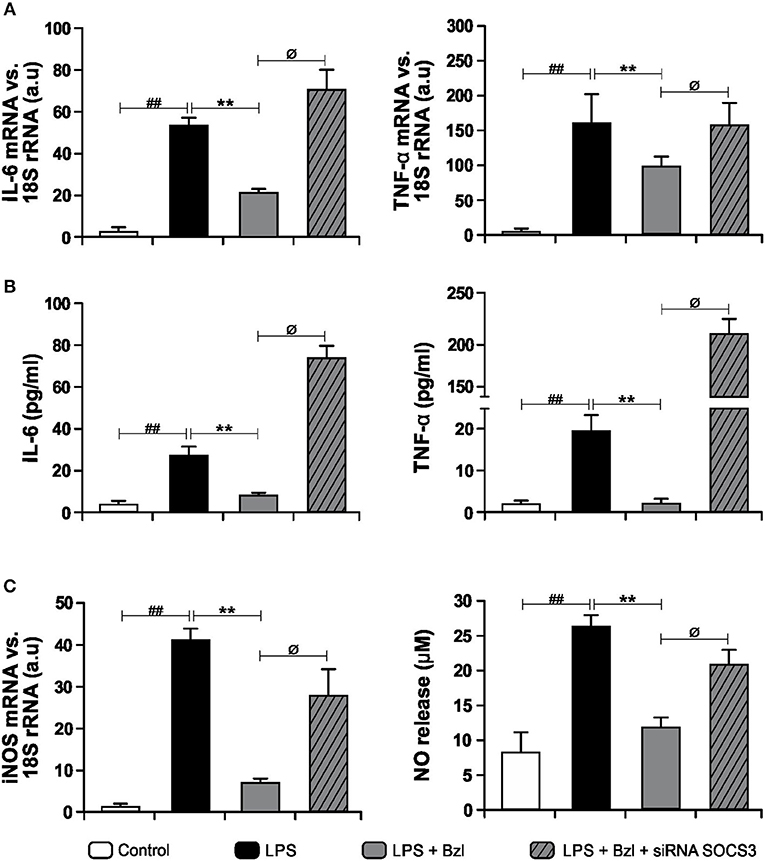
Figure 3. Cardiomyocytes were transfected with SOCS3 siRNA. After 72 h, cells were treated with 15 μM benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. After 4 h, IL-6 and TNF-α mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA (A). Concentration of cytokines in culture supernatants was measured by ELISA (B). After 48 h, iNOS mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA and NO levels were quantified by the Griess reaction in culture supernatants (C). Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells. Hatched gray bar: SOCS3 silenced, LPS-stimulated and Bzl-treated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.øP < 0.05 vs. Bzl-treated and LPS-stimulated cells.
Benznidazole Inhibits IKK/NF-κB Activation Through SOCS3
The transcription factor NF-κB is known to participate in the induction of pro-inflammatory genes. We previously showed that a low dose of benznidazole reduces the expression of inflammatory mediators by inhibiting NF-κB pathway, although the mechanism involved is poorly understood (6). Therefore, in this work we designed a set of experiments to evaluate whether SOCS3 participates in the effects of benznidazole on the NF-κB pathway. Figure 4A shows IKK phosphorylation visualized using a fluorescence microscopy upon challenge for 15 min with LPS. When benznidazole was added, IKK phosphorylation was inhibited. However, when SOCS3 was silenced, benznidazole could not inhibit IKK phosphorylation. Similar results were obtained when we evaluated cytosolic IκBα degradation. The NF-κB inhibitor, IκBα, underwent degradation upon stimulation with LPS and this effect was prevented in the presence of benznidazole. The participation of SOCS3 was confirmed, since benznidazole did not inhibit the degradation of IκBα promoted by LPS stimulation in the cells in which SOCS3 was silenced (Figure 4B).
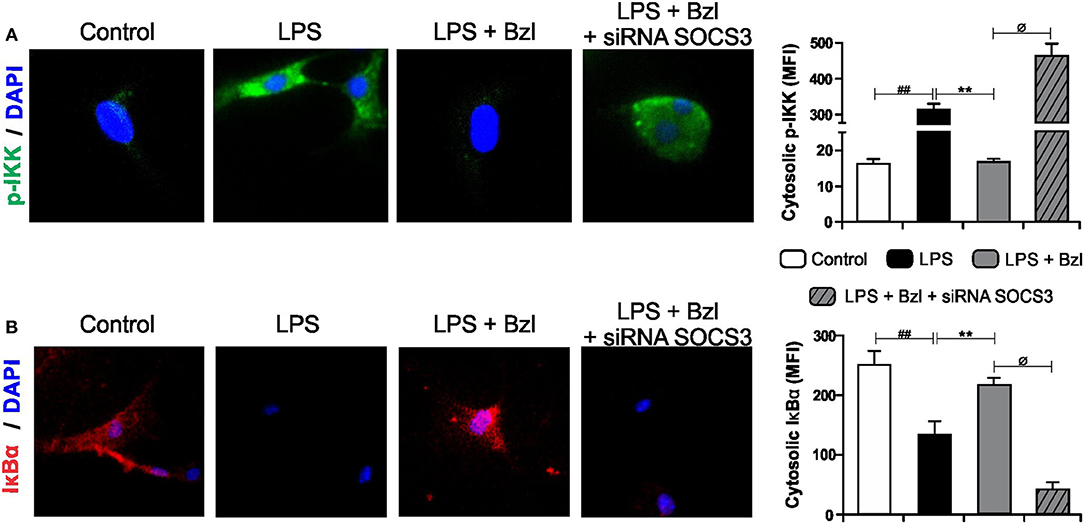
Figure 4. Cardiomyocytes were transfected with SOCS3 siRNA. After 72 h, cells were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. p-IKK (15 min) (A), and IκBα (30 min) (B) expression was detected by immunofluorescence with a rabbit polyclonal IgG anti- p-IKK or anti- IκBα (primary antibodies), and Goat anti-rabbit IgG Alexa Fluor 488 or Alexa Fluor 594 (secondary antibodies), respectively. Cell nuclei were stained with 300 nM DAPI. Representative microphotographs (400x) are shown. Mean fluorescence intensity (MFI) was quantified using Fiji Software, and the results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells. Hatched gray bar: SOCS3- silenced, LPS-stimulated and Bzl-treated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.øP < 0.05 vs. Bzl-treated and LPS-stimulated cells.
IL-10 Participates in Benznidazole Anti-inflammatory Effects
IL-10 is a key inhibitor of many aspects of the inflammatory response. In previous reports, we highlighted the relevance of IL-10 in the modulation of the pro-inflammatory response of cardiomyocytes in Chagas disease. We showed that recombinant mouse IL-10 acts as a negative regulator of pro-inflammatory signaling by up-regulation of SOCS3 in cardiomyocytes infected with T. cruzi (31). Moreover, it has been reported that peripheral blood mononuclear cells isolated from patients in the indeterminate phase of Chagas disease produce higher levels of IL-10 than those isolated from patients with chronic cardiomyopathy (49, 50).
In order to establish whether IL-10 could be involved in the effects of benznidazole, we evaluated the expression of IL-10 and its receptor in LPS-stimulated and benznidazole pre-treated cardiomyocytes as well as the release of IL-10 to culture supernatants. Figure 5 shows a trend to increase the expression of IL-10 and its release to culture medium, as well as the expression of IL-10R in cardiomyocytes stimulated with LPS, and that pre-treatment with benznidazole further increases this trend.
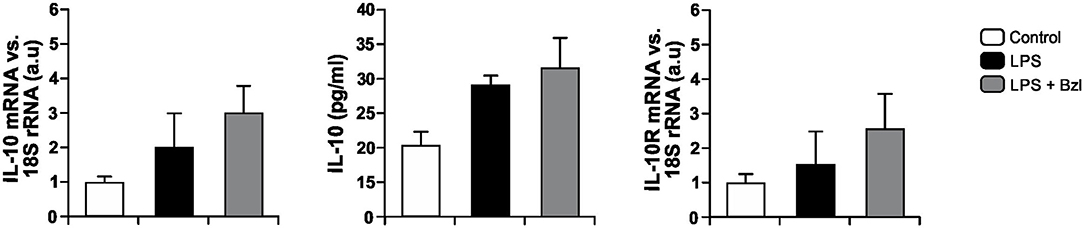
Figure 5. Cardiomyocytes were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. After 24 h, IL-10 and IL-10 receptor (IL-10R) mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA. Concentration of IL-10 in culture supernatants was measured by ELISA. Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells.
Moreover, to determine whether benznidazole requires IL-10 for the increase of SOCS3, we evaluated the expression of SOCS3 in neonatal cardiomyocytes isolated from IL-10-knockout mice (41). Figure 6 shows that LPS increases SOCS3 mRNA expression in IL-10-knockout cardiomyocytes. However, when these cells were pre-incubated with benznidazole, its expression did not increase, strongly suggesting that benznidazole induction of SOCS3 depends on IL-10 signaling.
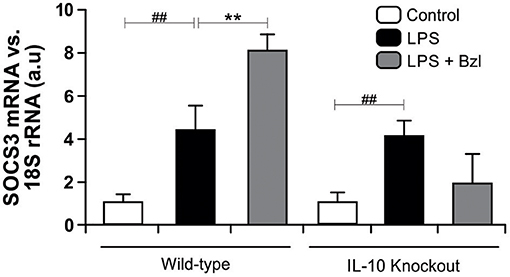
Figure 6. Cardiomyocytes from BALB/c and IL-10-knockout (IL-10-KO) mice of BALB/c background were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS for 24 h. SOCS3 mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA. Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.
In the same direction we observed that benznidazole fails to modulate the expression of IL-6, TNF-α, and iNOS in cardiomyocytes lacking IL-10 (Figure 7). In addition, when we evaluated NO release to culture supernatant, the secretion induced by LPS cannot be inhibited by benznidazole in IL-10 knockout cardiomyocytes (Figure 8A), and this was restored in the presence of recombinant IL-10 (Figure 8B).
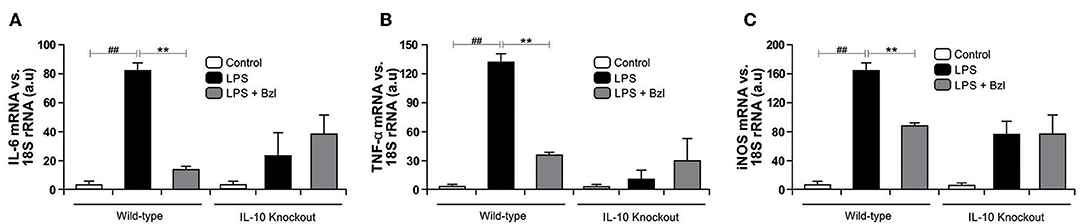
Figure 7. Cardiomyocytes from BALB/c and IL-10-knockout (IL-10-KO) mice of BALB/c background were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. After 4 h, IL-6 (A) and TNF-α (B) mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA. After 48 h, iNOS mRNA levels were analyzed by RT-qPCR and normalized against 18S rRNA (C). Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.
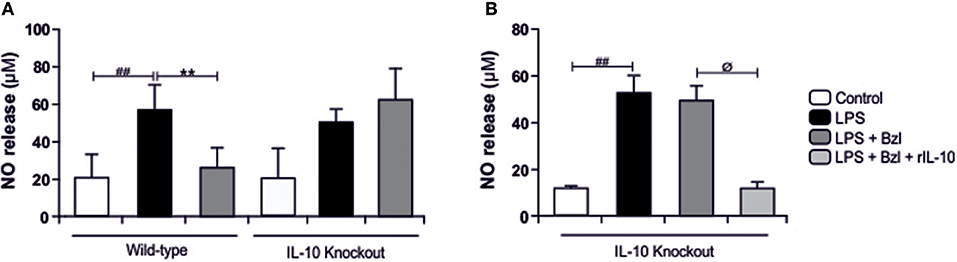
Figure 8. Cardiomyocytes from BALB/c and IL-10-knockout (IL-10-KO) mice of BALB/c background were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. After 48 h NO levels were quantified by the Griess reaction in culture supernatants (A). Cardiomyocytes from IL-10-KO mice were treated with 50 ng/ml of recombinant mouse IL-10 (rIL-10) for 30 min before Bzl treatment and LPS stimulation. After 48 h NO levels were quantified by the Griess reaction in culture supernatants (B). Results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl treated cells. Light-Gray bar: rIL-10- and Bzl-treated, LPS stimulated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.øP < 0.05 vs. Bzl treated-LPS-stimulated cells.
Furthermore, benznidazole could not avoid the phosphorylation of IKK (Figure 9A) and degradation of IκBα (Figure 9B) in IL-10-knockout mice, as shown by immunofluorescence microscopy. This confirms that IL-10 is required for the benznidazole-mediated inhibition of NF-κB.
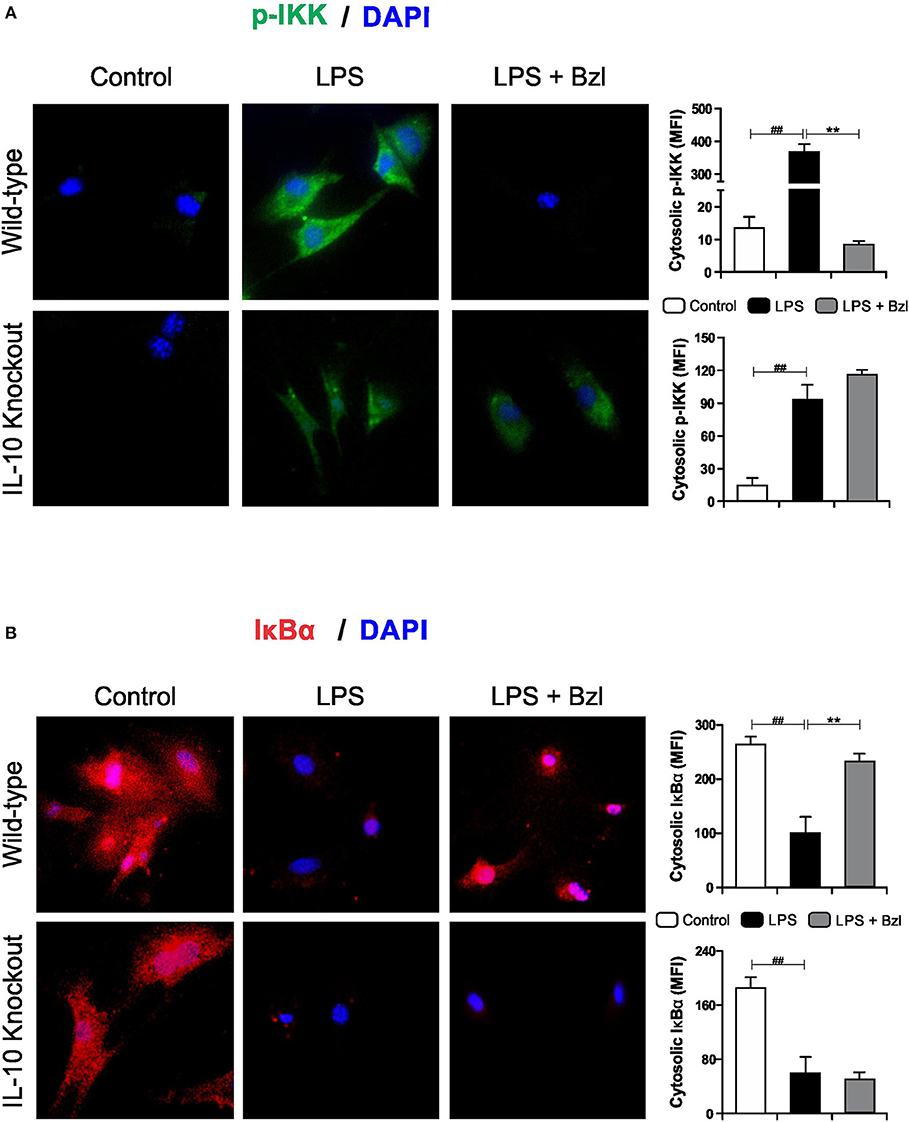
Figure 9. Cardiomyocytes from BALB/c and IL-10-knockout (IL-10-KO) mice of BALB/c background were treated with 15 μM of benznidazole (Bzl) for 30 min and then with 10 mg/L LPS. p-IKK (15 min) (A) and IκBα (30 min) (B) expression was detected by immunofluorescence with a rabbit polyclonal IgG anti- p-IKK or anti- IκBα (primary antibodies), and Goat anti-rabbit IgG Alexa Fluor 488 or Alexa Fluor 594 (secondary antibodies), respectively. Cell nuclei were stained with 300 nM DAPI. Representative microphotographs (400x) are shown. Mean fluorescence intensity (MFI) was quantified using Fiji Software, and the results are expressed as the mean of three independent experiments (n = 3, 5 replicates/treatment) ± SEM. White bar: Unstimulated control cells. Black bar: LPS-stimulated cells. Gray bar: LPS-stimulated and Bzl-treated cells. **P < 0.001, vs. LPS-stimulated cells; ##P < 0.001 vs. control cells.
Discussion
Benznidazole is one of the anti-parasitic drugs currently used in the treatment of Chagas disease. This drug has also anti-inflammatory effects, the mechanisms of which have not been fully elucidated yet. During the acute infection, the inflammatory response must be critically balanced. Although necessary for the control of parasite proliferation, inflammation results in tissue damage, leading to progressive myocardial fibrosis, and cardiac remodeling, ultimately leading to heart failure. The evolution of this disease causes major disabilities with high economic and social impact (51). For these reasons, besides controlling the parasite load, control of the inflammatory response both in the early and chronic stages of the infection is especially important.
Although benznidazole is one of the main drugs used in the treatment of acute and early chronic Chagas disease, there are serious limitations, such as the side effects exhibited by a significant proportion of patients (52). These include organic manifestations indicative of systemic toxicity. Adverse effects symptoms are evident in the first days of treatment and represent a major threat of benznidazole in clinical use with frequent therapy discontinuation (52–56). In a previous work we described, for the first time, that benznidazole has parasiticidal properties, in vivo as well as in vitro, at doses or concentrations lower than those previously reported for a highly virulent T. cruzi strain. In addition, treatment with a low dose of benznidazole was also able to inhibit the expression of inflammatory mediators and tissue inflammation in an experimental model of Chagas disease (6). In vitro studies using primary culture cardiomyocytes infected with the virulent RA strain of T. cruzi showed that 15 μM benznidazole was the optimal concentration to reduce the parasite load as well as the inflammatory mediators in the infected cells (6). This finding led us to search for the mechanism of action involved in the anti-inflammatory effects, knowing the optimal parasiticidal concentration and considering that this is the only antiparasitic treatment currently available in many countries.
In a previous work, we found that T. cruzi and IL-10 promotes STAT3 phosphorylation and up-regulates the expression of SOCS3 thereby preventing NF-κB nuclear translocation and ERK1/2 phosphorylation, two of the pathways known to promote the expression of pro-inflammatory genes (31).
In this work we focused on the pathways through which benznidazole exerts anti-inflammatory effects, demonstrating the IL-10/STAT3/SOCS3 axis is involved. To this aim we proceeded to dissociate the antiparasitic effects and anti-inflammatory properties of benznidazole using an in vitro model of primary culture cardiomyocytes pre-treated with benznidazole and stimulated with LPS. We report, for the first time, that benznidazole increases SOCS3 expression.
It has been described that phosphorylated STAT3 is dimerized and induces transcription of the SOCS3 gene (57). Also, it has been reported that the induction of SOCS3 expression in macrophages infected with Mycobacterium tuberculosis leads to the inhibition of NF-κB activation as a mechanism of immune evasion (58). Moreover, Carow et al. demonstrated that SOCS3, both in myeloid and T cells, is essential for resistance against M. tuberculosis via discrete mechanisms (59).
In the 1960s, it was postulated that the production of reactive oxygen species (ROS) would be one of the mechanisms capable of killing pathogens during the respiratory outbreak by phagocytes (60). In the same way, the trypanocidal activity of benznidazole, for instance, was credited to its capability to cause oxidative damage to the parasite (61). Paradoxically, it is also suggested that oxidative environments would stimulate parasite growth (62). However, it must be borne in mind that excessive ROS production and secretion to the tissue milieu can be detrimental to the host in the context of persistent inflammation.
Taking into consideration that T. cruzi can activate STAT3, and also increase SOCS3 expression (31), we wondered whether benznidazole was also able to promote STAT3 activation. In the current work we show that LPS also increases SOCS3. Moreover, the addition of benznidazole increases SOCS3 more than 50-fold with respect to LPS. Interestingly, both IL-10 and IL-6 induce the activation of STAT3, yet generate different cellular responses. While IL-6 stimulation promotes a pro-inflammatory response, IL-10 signaling induces a strong anti-inflammatory one (36). The increase in SOCS3 in LPS-stimulated cardiomyocytes might be dependent on the IL-6 pathway of STAT3 activation, since we observed a significant amount of IL-6 in the cell supernatants at 48 h. We hypothesized that up-regulation of SOCS3 in benznidazole-treated cardiomyocytes was mediated through IL-10. This was confirmed in our model using IL-10KO cardiomyocytes, in which upregulation of SOCS3 is not observed. Furthermore, the increase of SOCS3 was precluded by Stattic, suggesting that benznidazole increases SOCS3 expression in a STAT3-dependent manner. Previously, we reported that benznidazole inhibits iNOS and consequently NO release in LPS stimulated cardiomyocytes (6). In this work, we demonstrate the participation of STAT3 in the inhibitory effect of benznidazole since when STAT3 was inhibited by Stattic, benznidazole could neither inhibit the expression of iNOS nor the release of NO. Furthermore, when SOCS3 was silenced in cardiomyocytes stimulated with LPS, using SOCS3-specific siRNA, benznidazole neither inhibited the expression nor the release of inflammatory cytokines. Similar effects were observed for the expression of iNOS and release of NO to the culture medium.
It has been described that SOCS3 exerts its anti-inflammatory effects by inhibition of the activation of the NF-κB pathway (63). In this regard, it has been demonstrated that the PPE18 protein of M. tuberculosis up-regulates the expression as well as tyrosine phosphorylation of SOCS3. Phosphorylated SOCS3 physically interacts with the IκBα-NFκB/rel complex, inhibiting phosphorylation of IκBα and thus inactivates the NF-κB pathway (58). Moreover, we have demonstrated that knockdown of SOCS3 by specific siRNA, impedes the IL-10-mediated inhibition of NFκB and ERK1/2 activation in cardiomyocytes infected with T. cruzi (31). Furthermore, we previously showed that a low dose of benznidazole reduces the expression of inflammatory mediators by inhibiting the NFκB pathway (6). In this work we demonstrate the participation of SOCS3 in the effects of benznidazole, because when this cytokine suppressor was silenced in cardiomyocytes stimulated with LPS, the treatment with benznidazole could neither inhibit the IKK phosphorylation nor prevent IκBα degradation. Therefore, under conditions that preclude SOCS3 signaling, the NF-κB pathway cannot be inhibited.
Previously, we have demonstrated that the treatment with mouse recombinant IL-10, acts as a negative regulator of pro-inflammatory signaling by up-regulation of SOCS3 in cardiomyocytes infected with T. cruzi (31). Then, we sought to determine whether IL-10 was involved in the anti-inflammatory effects mediated by benznidazole. Our results showed, first of all, that LPS and LPS plus benznidazole increase the release of IL-10 and the expression of its receptor. Interestingly, the assays show that the increase in the expression of SOCS3 depends on the presence of IL0, since benznidazole was unable to increase the expression of SOCS3 in cardiomyocytes from IL-10-knockout mice. Secondly, benznidazole failed to inhibit the expression of inflammatory mediators in IL-10-knockout cells. There are evidences about the participation of SOCS3 in the mechanism by which IL-10 modulates the inflammatory response. In this regard, it has been reported that SOCS3 is the key mediator of the inhibitory effects of IL-10 in macrophages stimulated with LPS (64–66), and in an in vivo model of collagen induced arthritis (67). Moreover, our previous results highlight the relevance of IL-10 in the modulation of pro-inflammatory response in cardiomyocytes infected with T. cruzi, as we demonstrated that the IL-10/STAT3/SOCS3 axis negatively regulates NF-κB and ERK/MAPK–dependent gene expression of pro-inflammatory mediators (31). Furthermore, the relevance of IL-10 was evident as we demonstrated that benznidazole can neither inhibit IKK phosphorylation nor prevent the degradation of IκBα in cardiac cells from IL-10-knockout mice. Thus, IL-10 is required for the inhibitory effects of benznidazole on the NF-κB pathway to take place. Lastly, it has been widely described that the modulatory effects of IL-10 on the inflammatory response are associated with the inhibition of the NF-κB pathway (68–70), making IL-10 a reasonable mediator of benznidazole anti-inflammatory mechanisms.
In summary, we show for the first time, that the IL-10/STAT3/SOCS3 axis is involved in the anti-inflammatory effects of benznidazole (Figure 10). These findings may add up to new therapeutic strategies for the treatment of chronic Chagas disease given both its anti-parasitic and inflammatory properties.
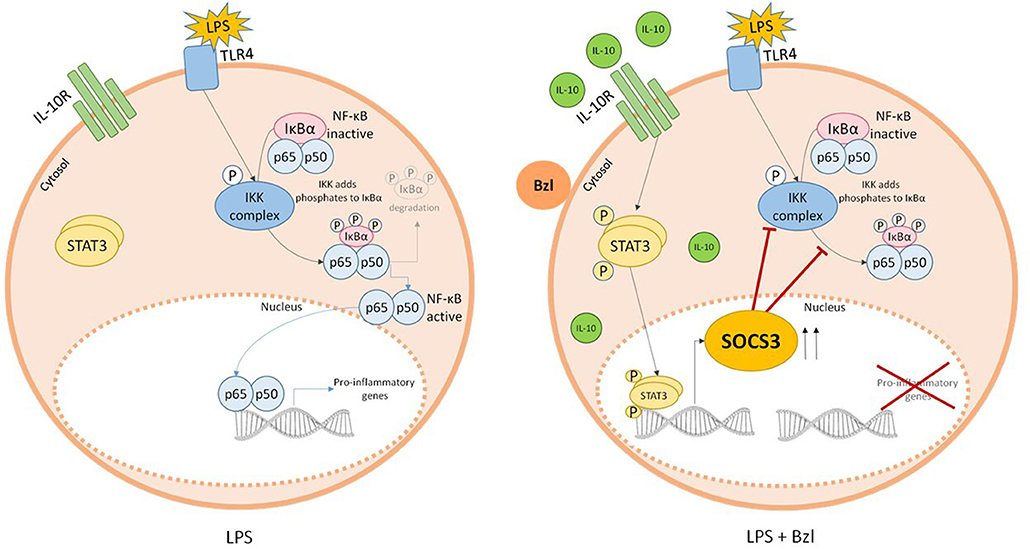
Figure 10. Parasiticidal concentrations of benznidazole (Bzl) exert anti-inflammatory effects through IL-10/STAT3/SOCS3 axis. When cardiomyocytes are stimulated with LPS and pre-treated with 15 μM of Bzl, IL-10 expression tends to increase. This induces the phosphorylation of STAT3 resulting in an increase in SOCS3 expression, thereby preventing NF-κB activation.
Ethics Statement
To carry out this work, CF1, BALB/c, and BALB/c-background IL-10 knockout mice, homozygous for the targeted mutation Il10tm1Cgn (Stock Number 004333; The Jackson Laboratory, USA) (41) were used. All the animals were bred and maintained in the animal facility at the Instituto de Investigaciones en Microbiología y Parasitología Médica, Universidad de Buenos Aires—CONICET. All the procedures were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUAL, Facultad de Medicina de la Universidad de Buenos Aires, CD N° 2271/2014), in line with guidelines of the Argentinean National Administration of Medicines, Food, and Medical Devices (ANMAT), Argentinean National Service of Agri-Food Health and Quality (SENASA) and with the Guide for the Care and Use of Laboratory Animals (NIH, USA).
Author Contributions
NG and ÁC designed experiments and analyzed data. ÁC, NG, and GM contributed to the writing of the manuscript. ÁC and FP did experiments. CA provided the IL-10 knockout mice. NG, GM, FP, CA, and ÁC contributed to final approval of the version to be published.
Funding
This work was supported by Universidad de Buenos Aires [Grant number 20020170100562BA] and Agencia Nacional de Promoción Científica y Tecnológica [Grant numbers PICT 2014-1049, PICT 2016-0629].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Carla Pascuale for their excellent technical assistance performing fluorescence microscopy analysis. We would also like to thank Mr. Sergio Mazzini for his assistance in English grammar and spelling corrections.
References
1. WHO–World Health Organization. WHO Chagas disease (American trypanosomiasis). World Health Organization (2017). Available online at: http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed April 17, 2019).
2. WHO Expert Committee. Control of Chagas disease. World Health Organization Technical Report Series (2002) 905:i–vi, 1–109, back cover.
3. Garcia S, Ramos CO, Senra JFV, Vilas-Boas F, Rodrigues MM, Campos-de-Carvalho AC, et al. Treatment with benznidazole during the chronic phase of experimental chagas' disease decreases cardiac alterations. Antimicrob Agents Chemother. (2005) 49:1521–8. doi: 10.1128/AAC.49.4.1521-1528.2005
4. Viotti R, Vigliano C, Lococo B, Bertocchi G, Petti M, Alvarez MG, et al. Long-term cardiac outcomes of treating chronic chagas disease with benznidazole versus no treatment: a nonrandomized trial. Ann Intern Med. (2006) 144:724–34. doi: 10.7326/0003-4819-144-10-200605160-00006
5. Campi-Azevedo AC, Gomes JAS, Teixeira-Carvalho A, Silveira-Lemos D, Vitelli-Avelar DM, Sathler-Avelar R, et al. Etiological treatment of chagas disease patients with benznidazole lead to a sustained pro-inflammatory profile counterbalanced by modulatory events. Immunobiology. (2015) 220:564–74. doi: 10.1016/j.imbio.2014.12.006
6. Cevey ÁC, Mirkin GA., Penas FN, Goren NB. Low-dose benznidazole treatment results in parasite clearance and attenuates heart inflammatory reaction in an experimental model of infection with a highly virulent trypanosoma cruzi strain. Int J Parasitol Drugs Drug Resist. (2016) 6:12–22. doi: 10.1016/j.ijpddr.2015.12.001
7. Piaggio E, Sancéau J, Revelli S, Bottasso O, Wietzerbin J, Serra E. Trypanocidal drug benznidazole impairs lipopolysaccharide induction of macrophage nitric oxide synthase gene transcription through inhibition of nf-kappaB activation. J Immunol. (2001) 167:3422–6. doi: 10.4049/jimmunol.167.6.3422
8. Pascutti MF, Pitashny M, Nocito AL, Guermonprez P, Amigorena S, Wietzerbin J, et al. Benznidazole, a drug used in chagas' disease, ameliorates LPS-induced inflammatory response in mice. Life Sci. (2004) 76:685–97. doi: 10.1016/j.lfs.2004.09.013
9. Dos-Santos ALA, Carvalho-Kelly LF, Dick CF, Meyer-Fernandes JR. Innate immunomodulation to trypanosomatid parasite infections. Exp Parasitol. (2016) 167:67–75. doi: 10.1016/j.exppara.2016.05.005
10. Rosadini CV, Kagan JC. Early innate immune responses to bacterial LPS. Curr Opin Immunol. (2017) 14–9. doi: 10.1016/j.coi.2016.10.005
11. Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. (2010) 10:170–81. doi: 10.1038/nri2711
12. Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. (2006) 6:379–86. doi: 10.1016/j.coph.2006.01.010
13. Hamidullah, Changkija B, Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat. (2012) 133:11–21. doi: 10.1007/s10549-011-1855-x
14. Redford PS, Murray PJ, O'Garra A. The role of IL-10 in immune regulation during M. tuberculosis infection. Mucosal Immunol. (2011) 4:261–70. doi: 10.1038/mi.2011.7
15. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. (2011) 51:170–82. doi: 10.1007/s12026-011-8262-6
16. Wilson EB, Brooks DG. The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol. (2011) 350:39–65. doi: 10.1007/82_2010_96
17. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. (2002) 169:2253–63. doi: 10.4049/jimmunol.169.5.2253
18. Williams L, Bradley L, Smith A, Foxwell B. Signal Transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. (2004) 172:567–76. doi: 10.4049/jimmunol.172.1.567
19. Hutchins AP, Diez D, Miranda-Saavedra D. The IL-10/STAT3-mediated anti-inflammatory response: recent developments and future challenges. Brief Funct Genomics. (2013) 12:489–98. doi: 10.1093/bfgp/elt028
20. Rottenberg ME, Carow B. SOCS3 and STAT3, major controllers of the outcome of infection with Mycobacterium tuberculosis. Semin Immunol. (2014) 26:518–32. doi: 10.1016/j.smim.2014.10.004
21. Seo Y-J, Jeong M, Lee K-T, Jang DS, Choi J-H. Isocyperol, isolated from the rhizomes of cyperus rotundus, inhibits LPS-induced inflammatory responses via suppression of the NF-κB and STAT3 pathways and ROS stress in LPS-stimulated RAW 264.7 cells. Int Immunopharmacol. (2016) 38:61–9. doi: 10.1016/j.intimp.2016.05.017
22. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5′-monophosphate–activated protein kinase regulates IL-10–mediated anti-inflammatory signaling pathways in macrophages. J Immunol. (2015) 194:584–94. doi: 10.4049/jimmunol.1401024
23. Shirakawa K, Endo J, Kataoka M, Katsumata Y, Yoshida N, Yamamoto T, et al. IL-10-STAT3-galectin-3 axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation. (2018) 138:2021–35. doi: 10.1161/CIRCULATIONAHA.118.035047
24. Yu Q, Zeng K, Ma X, Song F, Jiang Y, Tu P, et al. Resokaempferol-mediated anti-inflammatory effects on activated macrophages via the inhibition of JAK2/STAT3, NF-κB and JNK/p38 MAPK signaling pathways. Int Immunopharmacol. (2016) 38:104–14. doi: 10.1016/j.intimp.2016.05.010
25. Gemelli C, Zanocco Marani T, Bicciato S, Mazza EMC, Boraschi D, Salsi V, et al. MafB is a downstream target of the IL-10/STAT3 signaling pathway, involved in the regulation of macrophage de-activation. Biochim Biophys Acta Mol Cell Res. (2014) 1843:955–64. doi: 10.1016/j.bbamcr.2014.01.021
26. Barrett JP, Henry RJ, Villapol S, Stoica BA, Kumar A, Burns MP, et al. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflammation. (2017) 14:1–17. doi: 10.1186/s12974-017-0843-4
27. Akdis CA, Blaser K. Mechanisms of interleukin-10-mediated immune suppression. Immunology. (2001) 103:131–6. doi: 10.1046/j.1365-2567.2001.01235.x
28. Schmetterer KG, Pickl WF. The IL−10/STAT3 axis: contributions to immune tolerance by thymus and peripherally derived regulatory t-cells. Eur J Immunol. (2017) 47:1256–65. doi: 10.1002/eji.201646710
29. Hutchins AP, Takahashi Y, Miranda-Saavedra D. Genomic analysis of LPS-stimulated myeloid cells identifies a common pro-inflammatory response but divergent IL-10 anti-inflammatory responses. Sci Rep. (2015) 5:1–12. doi: 10.1038/srep09100
30. Gao Y, Basile JI, Classon C, Gavier-Widen D, Yoshimura A, Carow B, et al. STAT3 expression by myeloid cells is detrimental for the T-cell-mediated control of infection with Mycobacterium tuberculosis. PLoS Pathog. (2018) 14:e1006809. doi: 10.1371/journal.ppat.1006809
31. Hovsepian E, Penas F, Siffo S, Mirkin GA, Goren NB. IL-10 inhibits the NF-κB and ERK/MAPK-mediated production of pro-inflammatory mediators by up-regulation of SOCS-3 in trypanosoma cruzi-infected cardiomyocytes. PLoS ONE. (2013) 8:e79445. doi: 10.1371/journal.pone.0079445
32. Frangogiannis NGN. Regulation of the inflammatory response in cardiac repair. Circ Res. (2012) 110:159–73. doi: 10.1161/CIRCRESAHA.111.243162
33. Barry JC, Shakibakho S, Durrer C, Simtchouk S, Jawanda KK, Cheung ST, et al. Hyporesponsiveness to the anti-inflammatory action of interleukin-10 in type 2 diabetes. Sci Rep. (2016) 6:1–9. doi: 10.1038/srep21244
34. Cianciulli A, Dragone T, Calvello R, Porro C, Trotta T, Lofrumento DD, et al. IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int Immunopharmacol. (2015) 24:369–76. doi: 10.1016/j.intimp.2014.12.035
35. Deng G, Li K, Chen S, Chen P, Zheng H, Yu B, et al. Interleukin 10 promotes proliferation and migration, and inhibits tendon differentiation via the JAK/Stat3 pathway in tendon derived stem cells in vitro. Mol Med Rep. (2018) 18:5044–52. doi: 10.3892/mmr.2018.9547
36. Braun DA, Fribourg M, Sealfon SC. Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem. (2013) 288:2986–93. doi: 10.1074/jbc.M112.386573
37. Palmer DC, Restifo NP. Suppressors of cytokine signaling (SOCS) in T cell differentiation, maturation, and function. Trends Immunol. (2009) 30:592–602. doi: 10.1016/j.it.2009.09.009
38. Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. (2014) 5:58. doi: 10.3389/fimmu.2014.00058
39. Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. (2003) 4:551–6. doi: 10.1038/ni938
40. Camporeale A, Poli V. IL-6, IL-17 and STAT3: a holy trinity in auto-immunity? Front Biosci. (2012) 17:2306. doi: 10.2741/4054
41. Berg DJ, Davidson N, Kühn R, Müller W, Menon S, Holland G, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+Th1-like responses. J Clin Invest. (1996) 98:1010–20. doi: 10.1172/JCI118861
42. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. (2006) 13:1235–42. doi: 10.1016/j.chembiol.2006.09.018
43. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. (2008) 3:1101–8. doi: 10.1038/nprot.2008.73
44. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. (2009) 55:611–22. doi: 10.1373/clinchem.2008.112797
45. Kruger NJ. The bradford method for protein quantitation. Methods Mol Biol. (1994) 32:9–15. doi: 10.1385/0-89603-268-X:9
46. Bryan NS, Grisham MB. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic Biol Med. (2007) 43:645–57. doi: 10.1016/j.freeradbiomed.2007.04.026
47. Díaz-Guerra MJ, Velasco M, Martín-Sanz P, Boscá L. Evidence for common mechanisms in the transcriptional control of type II nitric oxide synthase in isolated hepatocytes. requirement of NF-kappaB activation after stimulation with bacterial cell wall products and phorbol esters. J Biol Chem. (1996) 271:30114–20.
48. Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. (2012) 9:676–82. doi: 10.1038/nmeth.2019
49. Gomes JAS, Bahia-Oliveira LMG, Rocha MOC, Martins-Filho OA, Gazzinelli G, Correa-Oliveira R. Evidence that development of severe cardiomyopathy in human chagas' disease is due to a Th1-specific immune response. Infect Immun. (2003) 71:1185–93. doi: 10.1128/iai.71.3.1185-1193.2003
50. Souza PEA, Rocha MOC, Rocha-Vieira E, Menezes CAS, Chaves ACL, Gollob KJ, et al. Monocytes from patients with indeterminate and cardiac forms of chagas' disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun. (2004) 72:5283–91. doi: 10.1128/IAI.72.9.5283-5291.2004
51. Benziger CP, do Carmo GAL, Ribeiro ALP. Chagas cardiomyopathy. Cardiol Clin. (2017) 35:31–47. doi: 10.1016/j.ccl.2016.08.013
52. Castro JA, de Mecca MM, Bartel LC. Toxic side effects of drugs used to treat chagas' disease (American trypanosomiasis). Hum Exp Toxicol. (2006) 25:471–9. doi: 10.1191/0960327106het653oa
53. Rodriques Coura J, de Castro SL. A critical review on chagas disease chemotherapy. Mem Inst Oswaldo Cruz. (2002) 97:3–24. doi: 10.1590/S0074-02762002000100001
54. Viotti R, Vigliano C, Lococo B, Alvarez MG, Petti M, Bertocchi G, et al. Side effects of benznidazole as treatment in chronic chagas disease: fears and realities. Expert Rev Anti Infect Ther. (2009) 157–63. doi: 10.1586/14787210.7.2.157
55. Pinazo M-J, Guerrero L, Posada E, Rodríguez E, Soy D, Gascon J. Benznidazole-related adverse drug reactions and their relationship to serum drug concentrations in patients with chronic chagas disease. Antimicrob Agents Chemother. (2013) 57:390–5. doi: 10.1128/AAC.01401-12
56. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas heart disease: pathophysiologic mechanisms, prognostic factors and risk stratification. Mem Inst Oswaldo Cruz. (2009) 104(Suppl):152–8. doi: 10.1590/S0074-02762009000900021
57. He B, You L, Uematsu K, Matsangou M, Xu Z, He M, et al. Cloning and characterization of a functional promoter of the human SOCS-3 gene. Biochem Biophys Res Commun. (2003) 301:386–91. doi: 10.1016/S0006-291X(02)03071-1
58. Nair S, Pandey AD, Mukhopadhyay S. The PPE18 protein of Mycobacterium tuberculosis inhibits NF-κB/rel-mediated proinflammatory cytokine production by upregulating and phosphorylating suppressor of cytokine signaling 3 protein. J Immunol. (2011) 186:5413–24. doi: 10.4049/jimmunol.1000773
59. Carow B, Reuschl AK, Gavier-Widén D, Jenkins BJ, Ernst M, Yoshimura A, et al. Critical and independent role for SOCS3 in either myeloid or T cells in resistance to Mycobacterium tuberculosis. PLoS Pathog. (2013) 9: e1003442. doi: 10.1371/journal.ppat.1003442
60. Baehner RL, Karnovsky ML. Deficiency of reduced nicotinamide-adenine dinucleotide oxidase in chronic granulomatous disease. Science. (1968) 162:1277–9. doi: 10.1126/science.162.3859.1277
61. Pedrosa RC, de Bem AF, Locatelli C, Pedrosa RC, Geremias R, Filho DW. Time-dependent oxidative stress caused by benznidazole. Redox Rep. (2001) 6:265–70. doi: 10.1179/135100001101536328
62. Paiva CN, Medei E, Bozza MT. ROS and trypanosoma cruzi: fuel to infection, poison to the heart. PLoS Pathog. (2018) 14:e1006928. doi: 10.1371/journal.ppat.1006928
63. Mahony R, Ahmed S, Diskin C, Stevenson NJ. SOCS3 revisited: a broad regulator of disease, now ready for therapeutic use? Cell Mol Life Sci. (2016) 73:3323–36. doi: 10.1007/s00018-016-2234-x
64. Berlato C, Cassatella MA, Kinjyo I, Gatto L, Yoshimura A, Bazzoni F. Involvement of suppressor of cytokine signaling-3 as a mediator of the inhibitory effects of IL-10 on lipopolysaccharide-induced macrophage activation. J Immunol. (2002) 168:6404–11. doi: 10.4049/jimmunol.168.12.6404
65. Qasimi P, Ming-Lum A, Ghanipour A, Ong CJ, Cox ME, Ihle J, et al. Divergent mechanisms utilized by SOCS3 to mediate interleukin-10 inhibition of tumor necrosis factor α and nitric oxide production by macrophages. J Biol Chem. (2006) 281:6316-6324. doi: 10.1074/jbc.M508608200
66. Huang N, Rizshsky L, Hauck CC, Nikolau BJ, Murphy PA, Birt DF. The inhibition of lipopolysaccharide-induced macrophage inflammation by 4 compounds in hypericum perforatum extract is partially dependent on the activation of SOCS3. Phytochemistry. (2012) 76:106–16. doi: 10.1016/j.phytochem.2011.12.001
67. Veenbergen S, Bennink MB, Affandi AJ, Bessis N, Biton J, Arntz OJ, et al. A pivotal role for antigen-presenting cells overexpressing SOCS3 in controlling invariant NKT cell responses during collagen-induced arthritis. Ann Rheum Dis. (2011) 70:2167–75. doi: 10.1136/ard.2011.154815
68. Wang P, Wu P, Siegel MI, Egan RW, Billah MM. Interleukin (IL)-10 inhibits nuclear factor κB (NFκB) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. (1995) 270:9558–63. doi: 10.1074/jbc.270.16.9558
69. Shames BD, Selzman CH, Meldrum DR, Pulido EJ, Barton HA, Meng XZ, et al. Interleukin-10 stabilizes inhibitory kappa b-alpha in human monocytes. Shock. (1998) 10:389–94. doi: 10.1097/00024382-199812000-00002
Keywords: benznidazole, anti-inflammatory effects, cardiomyocytes, mechanism of action, inflammatory mediators
Citation: Cevey ÁC, Penas FN, Alba Soto CD, Mirkin GA and Goren NB (2019) IL-10/STAT3/SOCS3 Axis Is Involved in the Anti-inflammatory Effect of Benznidazole. Front. Immunol. 10:1267. doi: 10.3389/fimmu.2019.01267
Received: 30 January 2019; Accepted: 17 May 2019;
Published: 04 June 2019.
Edited by:
Heiko Mühl, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Sanjiv Dhingra, University of Manitoba, CanadaGeorge W. Booz, University of Mississippi Medical Center School of Dentistry, United States
Copyright © 2019 Cevey, Penas, Alba Soto, Mirkin and Goren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nora B. Goren, bmdvcmVuQGZtZWQudWJhLmFy