 Anne M. Stevens1
Anne M. Stevens1 Kathryn S. Torok2
Kathryn S. Torok2 Suzanne C. Li3
Suzanne C. Li3 Sarah F. Taber4,5
Sarah F. Taber4,5 Theresa T. Lu4,6,7
Theresa T. Lu4,6,7 Francesco Zulian8*
Francesco Zulian8*- 1Division of Rheumatology, Department of Pediatrics, University of Washington, Seattle, WA, United States
- 2Division of Pediatric Rheumatology, Department of Pediatrics, Children's Hospital of Pittsburgh, University of Pittsburgh, Pittsburgh, PA, United States
- 3Division of Pediatric Rheumatology, Department of Pediatrics, Hackensack University Medical Center, Hackensack Meridian School of Medicine at Seton Hall University, Hackensack, NJ, United States
- 4Division of Pediatric Rheumatology, Department of Rheumatology, Hospital for Special Surgery, New York, NY, United States
- 5Department of Pediatrics, Weill Cornell Medicine, New York, NY, United States
- 6HSS Research Institute, Hospital for Special Surgery, New York, NY, United States
- 7Department of Microbiology and Immunology, Weill Cornell Medicine, New York, NY, United States
- 8Rheumatology Unit, Department of Woman's and Child's Health, University of Padua, Padua, Italy
Juvenile-onset systemic sclerosis (jSSc) is a rare and severe autoimmune disease with associated life-threatening organ inflammation and evidence of fibrosis. The organ manifestations of jSSc resemble adult SSc, but with better outcomes and survival. The etiology of jSSc appears to reflect adult-onset SSc, with similar inflammatory mediators and autoantibodies, but with a significant population of children with uncharacterized anti-nuclear antibodies. The genetics of patients with jSSc differ from women with SSc, resembling instead the genes of adult males with SSc, with additional HLA genes uniquely associated with childhood-onset disease. Current treatments are aimed at inhibiting the inflammatory aspect of disease, but important mechanisms of fibrosis regulated by dermal white adipose tissue dendritic cells may provide an avenue for targeting and potentially reversing the fibrotic stage.
Clinical Aspects
General Demographics and Clinical Characteristics
Juvenile systemic sclerosis (jSSc) is a multisystem connective tissue disease characterized by skin induration and widespread fibrosis of internal organs. The incidence has been reported at 0.27–0.50 per million children per year in the UK and Finland (1, 2), with a prevalence of 3 per million (3–5). Less than 5% of SSc has an onset in childhood (6–8). As in adults, there are three main subtypes of jSSc: diffuse cutaneous, limited cutaneous, and overlap (6, 9–12).
The onset of jSSc is usually insidious. Isolated Raynaud phenomenon is often the presenting symptom along with positive antinuclear antibodies (ANA) and nailfold capillary changes. Months or years later, tightening of the skin, especially of fingers and face, cutaneous telangiectasias and symptoms related to internal organ involvement gradually develop [Table 1, (6, 9–13)].
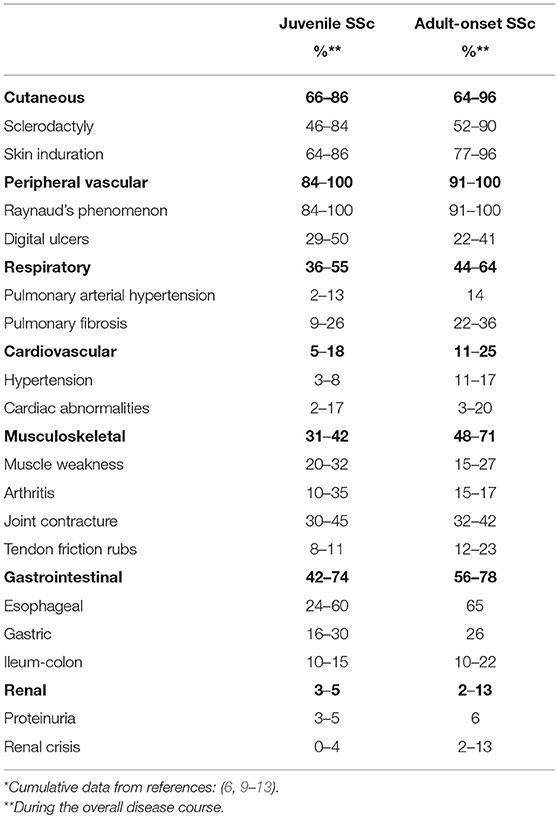
Table 1. Clinical features of juvenile systemic sclerosis in comparison with adult-onset systemic sclerosis*.
Cutaneous changes characteristically evolve in a sequence beginning with oedema, followed by induration and eventually atrophy. Nailfold capillary abnormalities are common, and include capillary dropout, tortuous dilated loops, and occasionally distorted capillary architecture (14, 15). Musculoskeletal symptoms are common in jSSc and characteristically occur at or near onset of the disease in one-third of children (6, 13).
Gastrointestinal involvement affects 42–74% of patients, and has been associated with interstitial lung disease, malnutrition, and poor quality of life (6, 11, 13, 16). Although the esophagus is often involved quite early, many patients are asymptomatic and rarely present heartburn or dysphagia. Malabsorptive diarrhea and delayed colonic transit, when present, reflect long-standing disease.
Cardiopulmonary involvement is the leading cause of morbidity and mortality in jSSc (17, 18). Cardiac inflammation and fibrosis may lead to conduction defects, arrhythmias, and impaired ventricular function. In the largest published study on jSSc, cardiac involvement was found in 8.4% of cases at onset, and in 24% during the overall course of disease (13). Cardiac involvement, especially pericarditis, is a major prognostic factor in jSSc, with a cardiac cause of death in 50% of deceased patients in a large international case series (odds ratio 41.3) (17, 18).
Pulmonary artery hypertension occurs in a minority of jSSc patients and, as in adults, may be either an isolated vascular complication or a consequence of pulmonary fibrosis (6, 13). Interstitial pulmonary fibrosis is reported in approximately one of four children (11, 13). The kidney is rarely affected (1).
Morbidity and Survival
As compared with adults, children with jSSc have overall better outcomes, related to a lower frequency of major visceral organ involvement and lower mortality (6, 13, 17). During follow-up, interstitial lung involvement, gastroesophageal dysmotility, and renal involvement are significantly more common in adults, while arthritis and muscle inflammation are more common in children, because of the higher prevalence of the overlap form of jSSc (3, 4, 6).
Morbidity is a major issue for jSSc, with most patients having multi-organ manifestations. In a recent North American study (n = 64 jSSc), 38% were found to have >4 organ systems involved, likely associating with the findings that 36% of the jSSc patients were found to have impaired function (ACR functional scores >1), and 64% reported having pain in the prior week (11). Poorer QOL scores were found to be associated with gastrointestinal symptoms, arthritis and pulmonary diseases (19).
Data from the jSSc Pediatric Rheumatology European Society registry reported a 5, 10, 15, and 20 years survival of 89, 87.4, 87.4, and 82.5%, respectively—significantly better than in adult-onset disease (17). Death in jSSc is usually related to the involvement of cardiac, renal, and pulmonary systems (9, 17, 18). A small subset of jSSc patients can have a rapidly progressive disease course that leads to early death as 60% of deaths are within 5 years (13). A raised creatinine level, pericarditis, and signs of fibrosis on chest X ray at diagnosis are potential risk factors for early mortality, similar to adult-onset SSc (17). In contrast to adult disease, however, malignancy is not an additional risk factor in jSSc for mortality as it has not been associated with jSSc (9, 13, 20, 21).
Autoantibodies and Clinical Associations
Autoimmunity plays an important role in juvenile and adult SSc, with a high prevalence of positive ANA and scleroderma-related autoantibodies, which can further assist clinical characterization and organ risk assessment as an adjunct to skin thickening distribution assignment of limited or diffuse cutaneous (6, 11, 13, 22–24). For example, Anti-topoisomerase antibodies (ATA; Scl-70) would be expected in a patient with diffuse cutaneous SSc, and would be worrisome for rapid skin progression and development of interstitial lung disease (ILD), prompting more aggressive pulmonary monitoring (6, 13, 23).
ANA positivity is reported in 78–97% of patients in the jSSc cohorts (6, 9–13), with scleroderma-associated antibodies reflecting the majority of extractable nuclear antigens causing ANA reactivity, such as ATA (Scl-70), centromere, and U1-RNP as reported in adult SSc, but in divergent frequencies, mirroring the slightly different clinical phenotype in children with SSc (Table 2). Another observation is that a significant proportion of jSSc patients, up to 23%, have a positive ANA without a specific extractable nuclear antigen identified, which is rarely observed in adults (6, 11, 13).
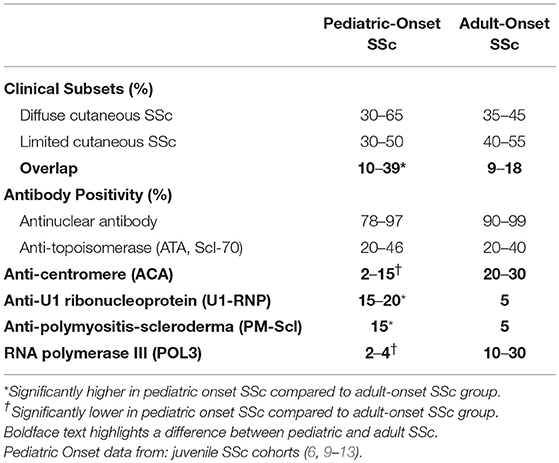
Table 2. Comparison between pediatric and adult-onset systemic sclerosis (SSc) subtype and autoantibody distribution.
Table 2 shows the general distribution of SSc clinical subtypes and the SSc-associated auto-antibodies in pediatric-onset compared to adult SSc. The diffuse cutaneous SSc subtype and associated ATA frequencies are similar between pediatric and adult SSc. A higher proportion of overlap patients and associated antibodies U1-RNP and PM-Scl was observed across several jSSc cohorts (6, 9–12) and accounts for the relatively common disease presentation in children of the combination of arthritis, myositis, Raynaud phenomenon and digital ulcers (6). The jSSc patients with overlap syndrome and associated antibodies (U1-RNP, Pm-Scl, U3-RNP) that have myositis may be of the most concern, as myopathy of the peripheral skeletal muscle is likely related to cardiac skeletal myopathy, and these patients have been demonstrated to have more conduction defects and other cardiac manifestations, a major contributor to mortality (6, 12, 13, 18).
Another main difference from adults with SSc is the lack of anti-centromere antibody–positive patients with jSSc (leq5% in most cohorts) (6, 9–13, 25), especially with pre-pubertal patients (<10 years at onset) (10). This is true despite ~30% to 50% of patients with jSSc being clinically classified as limited cutaneous SSc (Table 2). The relatively low rate of centromere antibodies found in jSSc does not seem to correlate with pulmonary arterial hypertension (PAH), as PAH is found with a similar frequency in juvenile and adult SSc (Table 1) (6, 9–13, 23). Specifically, in the Scalapino jSSc cohort manuscript (n = 111, 4 with intrinsic PAH) it was noted there was no association of ACA positivity with manifestation of PAH (6).
There are several other SSc-related autoantibodies that associate with different organ manifestations in adult SSc; however, they are much less common in childhood onset SSc. One of these is RNA polymerase III (POL3) antibody, which relates to severe renal disease in the form of scleroderma renal crisis in adult SSc. POL3 is rarely observed in jSSc (<5% in all cohorts; 4 or fewer patients per cohort), but can be found in up to 30% of adult onset SSc patients and reflects the clinically significant higher proportion of renal disease in adult SSc (6, 23). POL3 antibodies have also been associated with malignancy in SSc, a clinical outcome not reported in jSSc (21). Some of this data in jSSc is limited by the incomplete serological testing of scleroderma-associated antibodies in children. A more comprehensive evaluation for less common autoantibodies, such as POL3 and Th/To, to complete the full scleroderma-associated auto-antibody profile in jSSc cohorts may allow for better disease characterization and allow for more significant clinical associations.
Genetic Basis of Juvenile Systemic Sclerosis
Evidence for a Genetic Contribution to jSSc
Although the etiology of SSc is undoubtedly multifactorial, association studies have revealed immunoregulatory genes that most likely contribute to the pathogenesis of disease. The genetic risk factors appear to be lower for adult-onset SSc than for other autoimmune diseases [reviewed recently in (26)]. Only one study has examined history of autoimmunity in family members of SSc patients, and found a rate of 11% for a first or second degree relative of jSSc patients, suggesting that the disease is not monogenic (13).
The concordance rate for adult-onset SSc in twins is low (5.6% for dizygotic (DZ) twins and 4.2% for monozygotic (MZ) twins) (27). However, the genetic predisposition for loss of immune self-tolerance is high in families with SSc, with greatly increased concordance for ANA in MZ twins (90%) compared with DZ twins (40%) (27). Some of the discordance in disease penetrance may be attributed to differential methylation of X-encoded genes (28). A genetic contribution has also been suggested by reports of familial clustering in three cohorts in which first degree relatives of patients with SSc carried a 10–16-fold relative risk for disease and siblings a 10–27-fold risk (29).
In families with more than one case of SSc, affected individuals shared cutaneous subsets of disease severity and SSc-associated autoantibodies, with similar ages of onset (30). Finally, gene expression studies in cultured dermal fibroblasts suggest a heritable profibrotic program. In this study, healthy adult-onset MZ twins shared fibroblast gene expression patterns with their twins with SSc, whereas gene expression in unaffected adult-onset DZ twins resembled that of healthy controls (31). Moreover, serum from SSc patients or MZ twins could induce a SSc gene expression pattern in fibroblasts from healthy controls (31).
Peculiar Genetic Aspects of Juvenile SSc
Although numerous studies have investigated HLA associations in adult SSc, age of onset has not generally been considered. In pediatric studies, some HLA alleles found associated with adult SSc have also been associated with jSSc (32). An initial study in the UK examined HLA alleles in 27 patients with jSSc-dermatomyositis overlap, demonstrated an increase of DQA1*05 and DRB1*03 and a trend toward decreased DRB1*15. This finding was confirmed in a larger study of 76 Caucasian patients with a mean onset of SSc onset at 10 years of age in the US (25). Children with SSc did not have an increased frequency of DRB1*01, which has been associated with adult limited cutaneous SSc and ACA or HLA-DRB1*11 alleles, the group most consistently described in association with SSc in Caucasian adults.
One novel HLA association was discovered in jSSc patients: DRB1*10. DRB1*10 appeared to be a risk factor specific for jSSc, present in 10.5% of jSSc compared to 1.5% of controls, similar to one report of adult Han Chinese, but not otherwise described in adult SSc (33).
Protective HLA alleles in adult-onset SSc (DRB1*07, DQB1*02:02) were not found to be protective for jSSc. Instead, DQB1*06 was protective for jSSc (25).
Age and Sex Matter
The frequency of the adult limited cutaneous SSc-associated allele DRB1*01 differed in children with age of disease onset and autoantibody status (25). DRB1*01 was less prevalent in patients who were <6 years of age compared to patients 11–16 years at disease onset. DRB1*01 was under-represented among anti-topoisomerase 1 (ATA) -positive jSSc patients, consistent with a similar observation reported in adult SSc (34). In this study, just as the ATA-associated alleles decreased with age, the prevalence of ATA significantly decreased with increasing age (25).
Thus, children with SSc genetically resemble adult males with SSc, rather than the more prevalent women with SSc, with an increase frequency of DQA1*05 but not DRB1*11. This phenomenon could be explained by pregnancy immunopathology. It has been hypothesized that fetal microchimerism could trigger or perpetuate a graft-vs. -host type chronic inflammatory response (35). Women with SSc more often have been pregnant, and carry high levels of fetal microchimerism, whereas males and children do not. About 1 in 10 children with SSc carry an HLA allele not found in adults with SSc (DRB1*10), suggesting a unique childhood antigen trigger.
Pathology of Systemic Sclerosis
Histology of the Skin in SSc
The histology of the skin reflects the main three pathways affected in SSc, endothelial/vascular, immune/inflammatory, and fibrosis. The epidermis is relatively spared, though may be thinned, and rete ridges reduced. The dermis is thickened from the fibrosis and the connective tissue appears homogenous from collagen deposition. There is a loss of adnexal structures such as the pilosebaceous and eccrine glands (36). There are also vascular changes characterized by capillary rarefaction in the papillary dermis, thickened arteriolar walls and intimal thickening (37, 38). There can be inflammatory infiltrates of lymphocytes and macrophages, similar to that found in localized scleroderma (37), though may limited to early disease in SSc.
The Role of the Adipocytes
The dermal alterations that are most often discussed are the features that are apparent on a full thickness skin biopsy. However, intrinsic to the dermis but sitting mainly under the connective tissue-rich portions of the dermis and immediately adjacent to subcutaneous adipose tissue in humans is the dermal white adipose tissue (DWAT) (39). Fleishmajer et al. performed excisional biopsies in SSc patients, sampling skin down to the fascia, and thereby were able to characterize the DWAT (36). They reported that DWAT was replaced by connective tissue comprised of immature disorganized collagen fibrils and massive ground substance. Interestingly, this loss of DWAT is also observed in multiple murine models of scleroderma skin fibrosis (40–42), and reflects at least in part transdifferentiation of adipocytes into myofibroblasts (40, 41). Furthermore, this DWAT loss can further compromise skin function, as adipocytes can express anti-fibrotic molecules such as adiponectin and are involved in hair regeneration. DWAT is a niche for regenerative and reparative adipose-derived stromal cells (ADSCs) that participate in wound healing (41, 43, 44).
Upon loss of DWAT in murine skin fibrosis models, there is an 80% loss of ADSCs, likely due to cell death, which could potentially contribute to the inability to reverse and heal the fibrosis (42). Interestingly, dendritic cells helped to maintain the survival of the remaining ADSCs in fibrotic skin, and stimulating with the dendritic cell-derived signal lymphotoxin can enhance the survival and therapeutic effectiveness of intradermally injected ADSCs in a murine skin fibrosis model. ADSCs or a related type of mesenchymal stromal cells (MSCs) given either locally or systemically are being tested in human SSc (45); if clinical trials show efficacy of these approaches, it may be helpful to investigate whether adding lymphotoxin beta receptor stimulation can enhance these approaches (Figure 1).
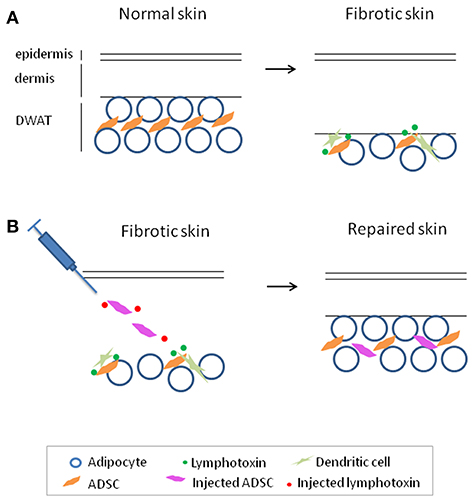
Figure 1. Adipose-derived stromal cells (ADSC) maintenance by dendritic cell-derived lymphotoxin in fibrotic skin and enhancing ADSC therapy with lymphotoxin stimulation to treat fibrosis. (A) Reparative and regenerative ADSCs are lost with skin fibrosis and are maintained in fibrotic skin by dendritic cell-derived lymphotoxin. (B) Adding lymphotoxin stimulation to therapeutic ADSC injection enhances survival and effectiveness of the injected ADSCs in skin repair in a model of skin fibrosis. This model may be helpful to investigate its potential application for the treatment of fibrosis.
Cytokine and Cellular Signatures
There have been multiple studies in adult SSc evaluating the cellular and cytokine profiles of tissue and peripheral blood, with only a few studies in jSSc owing to the rarity of disease in children. Skin lesions in jSSc show lymphocytes as the predominant cell type within the dermal and subcutaneous infiltrate (46, 47).
The lymphocytes have been further characterized, with T lymphocytes (CD4 and CD8) as the predominant cell type in the dermis of SSc patients (48, 49). T cell activation and associated cytokine release are thought to play a pivotal role in SSc pathogenesis through stimulating fibroblasts to promote fibrosis (50–52). CD4+ T-helper (TH) cell lineage and its effector cytokines have been identified in biopsies of skin lesions (48, 53, 54), peripheral circulation (adult and pediatric) (55–58) and culture in supernatants of peripheral blood mononuclear cells (PBMC) of SSc patients (59).
TH cells consist of three main effector cell types including TH1, TH2, and TH17; each are differentiation states characterized by the predominant cytokines they produce, namely interferon-γ (IFN-γ), interleukin-4 (IL-4)/IL-13, and IL-17, respectively. Many autoimmune diseases, including scleroderma, are thought to be propagated by an imbalance of TH cell subsets and their associated cytokines (48, 53–57, 59–61).
There is an extensive amount of literature (mainly adult studies) supporting a TH2 cellular and associated cytokine predominance in SSc, with peripheral blood and tissue derived IL-4 and IL-13, and more recently identified TH2 associated cytokines, IL-33, IL-34, and IL-35 (62, 63), correlating with the degree of skin and lung fibrosis and disease burden (50, 51, 57, 64–67). Both IL-4 and IL-13 are effector cytokines of the TH2 lineage characterized as pro-fibrotic and anti-inflammatory due to their respective actions as initiators of extracellular matrix production and inhibitors of TH1 function (68).
Therefore, it is suspected that the TH2 cytokine signature in SSc supports the fibrotic component of the disease, with peripheral cytokines serving as prognostic markers in SSc patients (50, 51). A recent study analyzing 14 pediatric SSc subjects compared to 24 healthy pediatric controls found a significantly elevated proportion of circulating CD4+IL-4+ (TH2) cells in pediatric SSc, and notably 10 of the 14 subjects had later stage disease (>2 years from onset) (58). Conversely, although higher than pediatric LS, the TH17 (CD4+IL-17+) profile of these pediatric SSc patients was significantly lower than pediatric healthy controls, perhaps reflecting their later stage of disease (58).
Cytokines associated with a TH1 profile (IL-1, TNF-α, and IFN-γ) have been identified as elevated in the peripheral blood of adult SSc patients compared to healthy controls, and their levels decrease overtime signifying their elevation during the earlier, active phase of the disease (69, 70). Specifically, the IFN-γ associated chemokines, IFN-inducible protein 10 (IP-10/CXCL10) and monocyte chemoattractant protein 1 (MCP-1/CCL2), have been demonstrated in more early, inflammatory adult SSc (71) as well as preliminary data in jSSc (72). The sera levels of jSSc are even more elevated than adult SSc and reflect those of juvenile LS (73), which in general, is considered the more inflammatory scleroderma subset. The “hallmark” effector cytokine of TH17 cells, IL-17a, has been demonstrated in significant amounts in the skin, lungs, and sera of adult SSc patients during the early, more active, stages of the disease (55). These findings suggest that TH17 and TH1 cells may contribute to cellular inflammation in SSc via production of associated pro-inflammatory cytokines (51) in the earlier stage of disease, as hypothesized in the pediatric SSc findings (58, 72).
It is thought that these T cell–associated chemokines and cytokines then stimulate fibroblasts and endothelial cells to produce TGF-β and connective tissue growth factor (CTGF) to stimulate tissue fibrosis (via increased collagen production) and endothelial cell damage (via influence of adhesion molecules ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 (vascular cell adhesion molecule-1), with elevation of these adhesion molecules demonstrated in the peripheral blood of SSc patients (74–78).
The counterbalance to the inflammatory TH cell phenotype, the regulatory T cell (Treg) immunophenotype, has been recently reported by Torok and Reiff in jSSc patients (58, 79), and collectively demonstrate decrease in functional Tregs compared to healthy controls, as well as association to more severe clinical phenotype and longer disease duration. This could, in part, be due to the differentiation of Tregs in the blood and skin to proinflammatory (TH17) and fibrotic (TH2) cells, therefore decreasing their frequency and effectiveness, as reported in adult SSc (80, 81).
Although the focus has been more on lymphocytes, innate cells such as macrophages and plasmacytoid dendritic cells are becoming increasingly recognized as potential contributors to systemic sclerosis, especially during early disease (42, 82–85). In addition to directly stimulating fibroblasts, innate cells can contribute to activating endothelial cells, which then further recruits circulating cells into the tissue, fueling the inflammation and further injury of the endothelium (86–88). While most patient data is gathered from adult SSc patients, a recent gene expression and expression quantitative trait loci analysis of monocyte-derived macrophages generated from pediatric and adult SSc patients suggested that changes in macrophage gene expression is an important contributor to disease and that upregulation of GSMDA, a regulator of pyroptosis in macrophages but not other cell types, contributes to disease risk (89). Further delineation of innate cell phenotype and function in pediatric SSc awaits.
Figure 2 summarizes, in a schematic way, the main immunologic/genetic pathways involved in the pathogenesis of the disease, in its different stages.
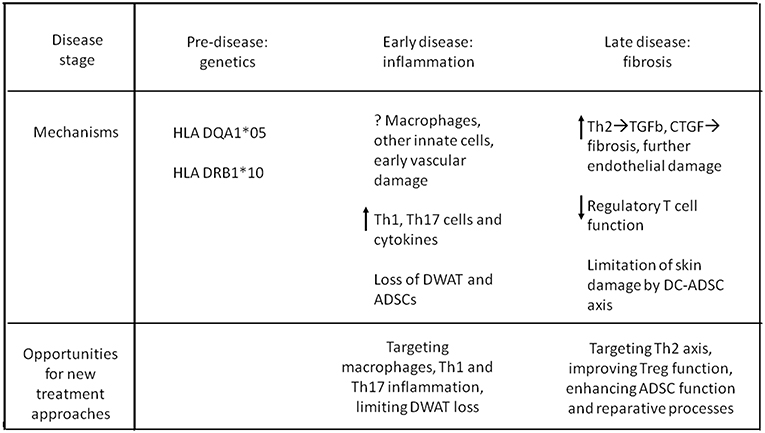
Figure 2. Schematic representation of proposed disease pathogenesis of pediatric SSc.
Future Directions For Research
In summary, early-onset SSc seems to be a distinct entity, whereas genetically-programmed manifestations in older children resemble adult disease. Disease manifestations and histopathology of jSSc resemble adult SSc, albeit with less organ involvement and superior outcomes.
Although genome-wide association studies cannot be performed on a disease as rare as jSSc, a genetic basis is likely in early-onset disease, and HLA class II associations with jSSc resemble adult males rather than adult females with SSc.
A pediatric-specific association of DRB1*10 suggests a unique trigger leading to younger onset disease. Younger children with SSc tended to carry genes associated with ATA, diffuse cutaneous SSc type, and not DRB1*01. More extensive genetic studies in families of children with early-onset (<6 years old) disease, with special focus on those with rapidly progressive organ fibrosis (90) are needed.
Novel therapeutic approaches could target specific HLA molecules, inflammatory chemokine, and cytokine genes over-expressed in jSSc. Recent reports of promising results of hematopoietic stem cell transplantation in adult-onset SSc encourage optimism for this therapeutic approach in children, who may have less organ damage when disease is recognized (91) Anti-fibrotic cellular therapies with adipose-derived stromal cells are being investigated, and recent findings in the laboratory suggest the potential power of stromal cell survival factors to enhance cellular therapy of the future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by NIH R01AI079178, Scleroderma Foundation, St. Giles Foundation, the Lupus Research Alliance (all TL), and A Lasting Mark Foundation (TL and ST).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res. (2010) 62:213–8. doi: 10.1002/acr.20070
2. Pelkonen PM, Jalanko HJ, Lantto RK, Mäkelä AL, Pietikäinen MA, Savolainen HA, et al. Incidence of systemic connective tissue diseases in children: a nationwide prospective study in Finland. J Rheumatol. (1994) 21:2143–6.
3. Royle JG, Lanyon PC, Grainge MJ, Abhishek A, Pearce FA. Correction to: the incidence, prevalence, and survival of systemic sclerosis in the UK Clinical Practice Research Datalink. Clin Rheumatol. (2018) 37:2601. doi: 10.1007/s10067-018-4219-7
4. Royle JG, Lanyon PC, Grainge MJ, Abhishek A, Pearce FA. The incidence, prevalence, and survival of systemic sclerosis in the UK Clinical Practice Research Datalink. Clin Rheumatol. (2018) 37:2103–11. doi: 10.1007/s10067-018-4182-3
5. Beukelman T, Xie F, Foeldvari I. Assessing the prevalence of juvenile systemic sclerosis in childhood using administrative claims data from the United States. J Scler Relat Disord. (2018) 3:189–90. doi: 10.1177/2397198318763701
6. Scalapino K, Arkachaisri T, Lucas M, Fertig N, Helfrich DJ, Londino AV, et al. Childhood onset systemic sclerosis: classification, clinical and serologic features, and survival in comparison with adult onset disease. J Rheumatol. (2006) 33:1004–13.
7. Misra R, Singh G, Aggarwal P, Aggarwal A. Juvenile onset systemic sclerosis: a single center experience of 23 cases from Asia. Clin Rheumatol. (2007) 2007:1259–62. doi: 10.1007/s10067-006-0483-z
8. Li SC. Scleroderma in children and adolescents: localized scleroderma and systemic sclerosis. Pediatr Clin North Am. (2018) 65:757–81. doi: 10.1016/j.pcl.2018.04.002
9. Foeldvari I, Nihtyanova SI, Wierk A, Denton CP. Characteristics of patients with juvenile onset systemic sclerosis in an adult single-center cohort. J Rheumatol. (2010) 37:2422–6. doi: 10.3899/jrheum.100001
10. Foeldvari I, Tyndall A, Zulian F, Müller-Ladner U, Czirjak L, Denton C, et al. Juvenile and young adult-onset systemic sclerosis share the same organ involvement in adulthood: data from the EUSTAR database. Rheumatology. (2012) 51:1832–7. doi: 10.1093/rheumatology/kes144
11. Stevens BE, Torok KS, Li SC, Hershey N, Curran M, Higgins GC, et al. Clinical characteristics and factors associated with disability and impaired quality of life in children with juvenile systemic sclerosis. Arthritis Care Res. (2018) 70:1806–13. doi: 10.1002/acr.23547
12. Foeldvari I, Klotsche J, Torok KS, et al. Are diffuse and limited juvenile systemic sclerosis different in clinical presentation? Clinical characteristics of a juvenile systemic sclerosis cohort. J Scler Relat Disord. (2018) 4:49–61. doi: 10.1177/2397198318790494
13. Martini G, Foeldvari I, Russo R, Cuttica R, Eberhard A, Ravelli A, et al. Systemic sclerosis in childhood: clinical and immunologic features of 153 patients in an international database. Arthritis Rheum. (2006) 54:3971–8. doi: 10.1002/art.22207
14. Ingegnoli F, Boracchi P, Gualtierotti R, Smith V, Cutolo M, Foeldvari I, et al. A comparison between nailfold capillaroscopy patterns in adulthood in juvenile and adult-onset systemic sclerosis: a EUSTAR exploratory study. Microvasc Res. (2015) 102:19–24. doi: 10.1016/j.mvr.2015.07.007
15. Dolezalova P, Young SP, Bacon PA, Southwood TR. Nailfold capillary microscopy in healthy children and in childhood rheumatic diseases: a prospective single blind observational study. Ann Rheum Dis. (2003) 62:444–9. doi: 10.1136/ard.62.5.444
16. Ambartsumyan L, Zheng HB, Iyer RS, Soares J, Henstorf G, Stevens AM. Relationship between esophageal abnormalities on fluoroscopic esophagram and pulmonary function testing in juvenile systemic sclerosis. Arthritis Care Res. (2018). doi: 10.1002/acr.23778. [Epub ahead of print].
17. Martini G, Vittadello F, Kasapçopur O, Magni Manzoni S, Corona F, Duarte-Salazar C, et al. Factors affecting survival in juvenile systemic sclerosis. Rheumatology. (2009) 48:119–22. doi: 10.1093/rheumatology/ken388
18. Quartier P, Bonnet D, Fournet JC, Bodemer C, Acar P, Ouachée-Chardin M, et al. Severe cardiac involvement in children with systemic sclerosis and myositis. J Rheumatol. (2002) 29:1767–73.
19. Baildam EM, Ennis H, Foster HE, Shaw L, Chieng AS, Kelly J, et al. Influence of childhood scleroderma on physical function and quality of life. J Rheumatol. (2011) 38:167–73. doi: 10.3899/jrheum.100447
20. Hatta Y, Hasegawa M, Matsushita T, Hamaguchi Y, Fujimoto M, Takehara K. The clinical characteristics of juvenile-onset systemic sclerosis in Japanese patients. Mod Rheumatol. (2013). doi: 10.1007/s10165-013-0884-1
21. Maria ATJ, Partouche L, Goulabchand R, Rivière S, Rozier P, Bourgier C, et al. Intriguing relationships between cancer and systemic sclerosis: role of the immune system and other contributors. Front Immunol. (2018) 9:3112. doi: 10.3389/fimmu.2018.03112
22. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. (1988) 15:202–5.
23. Foeldvari I, Zhavania M, Birdi N, Cuttica RJ, de Oliveira SH, Dent PB, et al. Favourable outcome in 135 children with juvenile systemic sclerosis: results of a multi-national survey. Rheumatology. (2000) 39:556–9. doi: 10.1093/rheumatology/39.5.556
24. Steen VD. The many faces of scleroderma. Rheum Dis Clin North Am. (2008) 34:1–15; v. doi: 10.1016/j.rdc.2007.12.001
25. Stevens AM, Kanaan SB, Torok KS, Medsger TA, Mayes MD, Reveille JD, et al. Brief report: HLA-DRB1, DQA1, and DQB1 in juvenile-onset systemic sclerosis. Arthritis Rheumatol. (2016) 68:2772–7. doi: 10.1002/art.39765
26. Angiolilli C, Marut W, van der Kroef M, Chouri E, Reedquist KA, Radstake TRDJ. New insights into the genetics and epigenetics of systemic sclerosis. Nat Rev Rheumatol. (2018) 14:657–73. doi: 10.1038/s41584-018-0099-0
27. Feghali-Bostwick C, Medsger TA Jr., Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. (2003) 48:1956–63. doi: 10.1002/art.11173
28. Selmi C, Feghali-Bostwick CA, Lleo A, Lombardi SA, De Santis M, Cavaciocchi F, et al. X chromosome gene methylation in peripheral lymphocytes from monozygotic twins discordant for scleroderma. Clin Exp Immunol. (2012) 169:253–62. doi: 10.1111/j.1365-2249.2012.04621.x
29. Arnett FC, Cho M, Chatterjee S, Aguilar MB, Reveille JD, Mayes MD. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. (2001) 44:1359–62. doi: 10.1002/1529-0131(200106)44:6<1359::AID-ART228>3.0.CO;2-S
30. Stephens CO, Briggs DC, Whyte J, Artlett CM, Scherbakov AB, Olsen N, et al. Familial scleroderma–evidence for environmental versus genetic trigger. Br J Rheumatol. (1994) 33:1131–5. doi: 10.1093/rheumatology/33.12.1131
31. Zhou X, Tan FK, Xiong M, Arnett FC, Feghali-Bostwick CA. Monozygotic twins clinically discordant for scleroderma show concordance for fibroblast gene expression profiles. Arthritis Rheum. (2005) 52:3305–14. doi: 10.1002/art.21355
32. Wedderburn LR, McHugh NJ, Chinoy H, Cooper RG, Salway F, Ollier WE, et al. HLA class II haplotype and autoantibody associations in children with juvenile dermatomyositis and juvenile dermatomyositis-scleroderma overlap. Rheumatology (Oxford). (2007) 46:1786–91. doi: 10.1093/rheumatology/kem265
33. He D, Wang J, Yi L, Guo X, Guo S, Guo G, et al. Association of the HLA-DRB1 with scleroderma in Chinese population. PLoS ONE. (2014) 9:e106939. doi: 10.1371/journal.pone.0106939
34. Beretta L, Rueda B, Marchini M, Santaniello A, Simeón CP, Fonollosa V, et al. Analysis of class II human leucocyte antigens in Italian and Spanish systemic sclerosis. Rheumatology. (2012) 51:52–9. doi: 10.1093/rheumatology/ker335
35. Nelson JL. The otherness of self: microchimerism in health and disease. Trends Immunol. (2012) 33:421–7. doi: 10.1016/j.it.2012.03.002
36. Fleischmajer R, Damiano V, Nedwich A. Scleroderma and the subcutaneous tissue. Science. (1971) 171:1019–21. doi: 10.1126/science.171.3975.1019
37. Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheum. (1977) 20:975–84. doi: 10.1002/art.1780200410
38. Fleming JN, Nash RA, McLeod DO, Fiorentino DF, Shulman HM, Connolly MK, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS ONE. (2008) 3:e1452. doi: 10.1371/annotation/6b021f46-17bd-4ffe-a378-a1b8d24a1398
39. Driskell RR, Jahoda CA, Chuong CM, Watt FM, Horsley V. Defining dermal adipose tissue. Exp Dermatol. (2014) 23:629–31. doi: 10.1111/exd.12450
40. Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. (2015) 67:1062–73. doi: 10.1002/art.38990
41. Marangoni RG, Lu TT. The roles of dermal white adipose tissue loss in scleroderma skin fibrosis. Curr Opin Rheumatol. (2017) 29:585–90. doi: 10.1097/BOR.0000000000000437
42. Chia JJ, Zhu T, Chyou S, Dasoveanu DC, Carballo C, Tian S, et al. Dendritic cells maintain dermal adipose-derived stromal cells in skin fibrosis. J Clin Invest. (2016) 126:4331–45. doi: 10.1172/JCI85740
43. Festa E, Fretz J, Berry R, Schmidt B, Rodeheffer M, Horowitz M, et al. Adipocyte lineage cells contribute to the skin stem cell niche to drive hair cycling. Cell. (2011) 146:761–71. doi: 10.1016/j.cell.2011.07.019
44. Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. (2013) 504:277–81. doi: 10.1038/nature12783
45. Peltzer J, Aletti M, Frescaline N, Busson E, Lataillade JJ, Martinaud C. Mesenchymal stromal cells based therapy in systemic sclerosis: rational and challenges. Front Immunol. (2018) 9:2013. doi: 10.3389/fimmu.2018.02013
46. Succaria F, Kurban M, Kibbi AG, Abbas O. Clinicopathological study of 81 cases of localized and systemic scleroderma. J Eur Acad Dermatol Venereol. (2013) 27:e191–6. doi: 10.1111/j.1468-3083.2012.04581.x
47. Torres JE, Sánchez JL. Histopathologic differentiation between localized and systemic scleroderma. Am J Dermatopathol. (1998) 20:242–5. doi: 10.1097/00000372-199806000-00003
48. Roumm AD, Whiteside TL, Medsger TA Jr., Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum. (1984) 27:645–53. doi: 10.1002/art.1780270607
49. Kalogerou A, Gelou E, Mountantonakis S, Settas L, Zafiriou E, Sakkas L. Early T cell activation in the skin from patients with systemic sclerosis. Ann Rheum Dis. (2005) 64:1233–5. doi: 10.1136/ard.2004.027094
50. Baraut J, Farge D, Jean-Louis F, Kesmandt H, Durant C, Verrecchia F, et al. [Cytokines in systemic sclerosis]. Pathol Biol. (2012) 60:127–39. doi: 10.1016/j.patbio.2009.11.003
51. Hasegawa M, Takehara K. Potential immunologic targets for treating fibrosis in systemic sclerosis: a review focused on leukocytes and cytokines. Semin Arthritis Rheum. (2012) 42:281–96. doi: 10.1016/j.semarthrit.2012.03.014
52. Fuschiotti P. T cells and cytokines in systemic sclerosis. Curr Opin Rheumatol. (2018) 30:594–9. doi: 10.1097/BOR.0000000000000553
53. Keystone EC, Lau C, Gladman DD, Wilkinson S, Lee P, Shore A. Immunoregulatory T cell subpopulations in patients with scleroderma using monoclonal antibodies. Clin Exp Immunol. (1982) 48:443–8.
54. Inoshita T, Whiteside TL, Rodnan GP, Taylor FH. Abnormalities of T lymphocyte subsets in patients with progressive systemic sclerosis (PSS, scleroderma). J Lab Clin Med. (1981) 97:264–77.
55. Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, et al. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. (2000) 43:2455–63. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K
56. Matsushita T, Hasegawa M, Hamaguchi Y, Takehara K, Sato S. Longitudinal analysis of serum cytokine concentrations in systemic sclerosis: association of interleukin 12 elevation with spontaneous regression of skin sclerosis. J Rheumatol. (2006) 33:275–84.
57. Needleman BW, Wigley FM, Stair RW. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. (1992) 35:67–72. doi: 10.1002/art.1780350111
58. Mirizio E, Marathi A, Hershey N, Ross C, Schollaert K, Salgado C, et al. Identifying the signature immune phenotypes present in pediatric localized scleroderma. J Invest Dermatol. (2018) 139:715–8. doi: 10.1016/j.jid.2018.09.025
59. Valentini G, Baroni A, Esposito K, Naclerio C, Buommino E, Farzati A, et al. Peripheral blood T lymphocytes from systemic sclerosis patients show both Th1 and Th2 activation. J Clin Immunol. (2001) 21:210–7. doi: 10.1023/A:1011024313525
60. Ihn H, Sato S, Fujimoto M, Kikuchi K, Takehara K. Demonstration of interleukin-2, interleukin-4 and interleukin-6 in sera from patients with localized scleroderma. Arch Dermatol Res. (1995) 287:193–7. doi: 10.1007/BF01262331
61. Hasegawa M, Sato S, Nagaoka T, Fujimoto M, Takehara K. Serum levels of tumor necrosis factor and interleukin-13 are elevated in patients with localized scleroderma. Dermatology. (2003) 207:141–7. doi: 10.1159/000071783
62. Zhang YJ, Zhang Q, Yang GJ, Tao JH, Wu GC, Huang XL, et al. Elevated serum levels of interleukin-1beta and interleukin-33 in patients with systemic sclerosis in Chinese population. Z Rheumatol. (2018) 77:151–9. doi: 10.1007/s00393-016-0202-3
63. Kuzumi A, Yoshizaki A, Toyama S, Fukasawa T, Ebata S, Nakamura K, et al. Serum interleukin-34 levels in patients with systemic sclerosis: Clinical association with interstitial lung disease. J Dermatol. (2018) 45:1216–20. doi: 10.1111/1346-8138.14538
64. Atamas SP, Yurovsky VV, Wise R, Wigley FM, Goter Robinson CJ, Henry P, et al. Production of type 2 cytokines by CD8+ lung cells is associated with greater decline in pulmonary function in patients with systemic sclerosis. Arthritis Rheum. (1999) 42:1168–78. doi: 10.1002/1529-0131(199906)42:6<1168::AID-ANR13>3.0.CO;2-L
65. Parel Y, Aurrand-Lions M, Scheja A, Dayer JM, Roosnek E, Chizzolini C. Presence of CD4+CD8+ double-positive T cells with very high interleukin-4 production potential in lesional skin of patients with systemic sclerosis. Arthritis Rheum. (2007) 56:3459–67. doi: 10.1002/art.22927
66. Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol. (1997) 24:328–32.
67. Fuschiotti P, Medsger TA Jr., Morel PA. Effector CD8+ T cells in systemic sclerosis patients produce abnormally high levels of interleukin-13 associated with increased skin fibrosis. Arthritis Rheum. (2009) 60:1119–28. doi: 10.1002/art.24432
68. Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. (2004) 4:583–94. doi: 10.1038/nri1412
69. Gourh P, Arnett FC, Assassi S, Tan FK, Huang M, Diekman L, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. (2009) 11:R147. doi: 10.1186/ar2821
70. Dantas AT, Almeida AR, Sampaio MCPD, Cordeiro MF, Oliveira PSS, Mariz HA, et al. Different profile of cytokine production in patients with systemic sclerosis and association with clinical manifestations. Immunol Lett. (2018) 198:12–6. doi: 10.1016/j.imlet.2018.03.011
71. Liu X, Mayes MD, Tan FK, Wu M, Reveille JD, Harper BE, et al. Correlation of interferon-inducible chemokine plasma levels with disease severity in systemic sclerosis. Arthritis Rheum. (2013) 65:226–35. doi: 10.1002/art.37742
72. Kurzinski KKC, Arkachaisri T, Feghali-Bostwick C, Torok KS. Circulating IP-10 and MCP-1 levels in active localized scleroderma. Arthritis Rheum. (2011) 63:S115.
73. Magee KE, Kelsey CE, Kurzinski KL, Ho J, Mlakar LR, Feghali-Bostwick CA, et al. Interferon-gamma inducible protein-10 as a potential biomarker in localized scleroderma. Arthritis Res Ther. (2013) 15:R188. doi: 10.1186/ar4378
74. Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. (1987) 247:597–604. doi: 10.1042/bj2470597
75. Gore-Hyer E, Pannu J, Smith EA, Grotendorst G, Trojanowska M. Selective stimulation of collagen synthesis in the presence of costimulatory insulin signaling by connective tissue growth factor in scleroderma fibroblasts. Arthritis Rheum. (2003) 48:798–806. doi: 10.1002/art.10953
76. Uziel Y, Feldman BM, Krafchik BR, Laxer RM, Yeung RS. Increased serum levels of TGFbeta1 in children with localized scleroderma. Pediatr Rheumatol Online J. (2007) 5:22. doi: 10.1186/1546-0096-5-22
77. Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C. Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud's phenomenon. Arthritis Rheum. (1994) 37:278–88. doi: 10.1002/art.1780370218
78. Thakkar V, Patterson KA, Stevens W, Wilson M, Roddy J, Sahhar J, et al. Increased serum levels of adhesion molecules ICAM-1 and VCAM-1 in systemic sclerosis are not specific for pulmonary manifestations. Clin Rheumatol. (2018) 37:1563–71. doi: 10.1007/s10067-018-4081-7
79. Reiff A, Weinberg KI, Triche T, Masinsin B, Mahadeo KM, Lin CH, et al. T lymphocyte abnormalities in juvenile systemic sclerosis patients. Clin Immunol. (2013) 149:146–55. doi: 10.1016/j.clim.2013.07.005
80. Slobodin G, Rimar D. Regulatory T cells in systemic sclerosis: a comprehensive review. Clin Rev Allergy Immunol. (2017) 52:194–201. doi: 10.1007/s12016-016-8563-6
81. MacDonald KG, Dawson NA, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. (2015) 135:946–.e9. doi: 10.1016/j.jaci.2014.12.1932
82. Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med. (2018) 10:eaam8458. doi: 10.1126/scitranslmed.aam8458
83. Raker V, Haub J, Stojanovic A, Cerwenka A, Schuppan D, Steinbrink K. Early inflammatory players in cutanous fibrosis. J Dermatol Sci. (2017) 87:228–35. doi: 10.1016/j.jdermsci.2017.06.009
84. Stifano G, Sornasse T, Rice LM, Na L, Chen-Harris H, Khanna D, et al. Skin gene expression is prognostic for the trajectory of skin disease in patients with diffuse cutaneous systemic sclerosis. Arthritis Rheumatol. (2018) 70:912–9. doi: 10.1002/art.40455
86. Volkmann ER, Varga J. Emerging targets of disease-modifying therapy for systemic sclerosis. Nat Rev Rheumatol. (2019) 15:208–24. doi: 10.1038/s41584-019-0184-z
87. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheumatism. (2013) 65:1953–62. doi: 10.1002/art.37988
88. Sierra-Sepúlveda A, Esquinca-González A, Benavides-Suárez SA, Sordo-Lima DE, Caballero-Islas AE, Cabral-Castañeda AR, et al. Systemic sclerosis pathogenesis and emerging therapies, beyond the fibroblast. BioMed Res Int. (2019) 2019:4569826. doi: 10.1155/2019/4569826
89. Moreno-Moral A, Bagnati M, Koturan S, Ko JH, Fonseca C, Harmston N, et al. Changes in macrophage transcriptome associate with systemic sclerosis and mediate GSDMA contribution to disease risk. Ann Rheum Dis. (2018) 77:596–601. doi: 10.1136/annrheumdis-2017-212454
90. Stock CJW, Renzoni EA. Genetic predictors of systemic sclerosis-associated interstitial lung disease: a review of recent literature. Eur J Hum Genet. (2018) 26:765–77. doi: 10.1038/s41431-018-0104-8
91. Sullivan KM, Majhail NS, Bredeson C, Carpenter PA, Chatterjee S, Crofford LJ, et al. Systemic sclerosis as an indication for autologous hematopoietic cell transplantation: position statement from the American society for blood and marrow transplantation. Biol Blood Marrow Transplant. (2018) 24:1961–4. doi: 10.1016/j.bbmt.2018.06.025
Keywords: systemic sclerosis, Pediatric Rheumatology, genetics, disease etiology, autoimmune disease, skin, fibrosis
Citation: Stevens AM, Torok KS, Li SC, Taber SF, Lu TT and Zulian F (2019) Immunopathogenesis of Juvenile Systemic Sclerosis. Front. Immunol. 10:1352. doi: 10.3389/fimmu.2019.01352
Received: 13 December 2018; Accepted: 28 May 2019;
Published: 25 June 2019.
Edited by:
David Bending, University of Birmingham, United KingdomReviewed by:
Giacomo Emmi, University of Florence, ItalyKazuhiro Komura, Municipal Tsuruga Hospital, Japan
Maureen Davidica Mayes, University of Texas Health Science Center at Houston, United States
Copyright © 2019 Stevens, Torok, Li, Taber, Lu and Zulian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Zulian, ZnJhbmNlc2NvenVsaWFuNThAZ21haWwuY29t