 Deborah Cross
Deborah Cross Ruth Drury
Ruth Drury Jennifer Hill
Jennifer Hill Andrew J. Pollard
Andrew J. Pollard- Oxford Vaccine Group, Department of Paediatrics, NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom
Sepsis has a complex pathophysiology in which both excessive and refractory inflammatory responses are hallmark features. Pro-inflammatory cytokine responses during the early stages are responsible for significant endothelial dysfunction, loss of endothelial integrity, and organ failure. In addition, it is now well-established that a substantial number of sepsis survivors experience ongoing immunological derangement and immunosuppression following a septic episode. The underpinning mechanisms of these phenomena are incompletely understood yet they contribute to a significant proportion of sepsis-associated mortality. Epigenetic mechanisms including DNA methylation, histone modifications, and non-coding RNAs, have an increasingly clear role in modulating inflammatory and other immunological processes. Recent evidence suggests epigenetic mechanisms are extensively perturbed as sepsis progresses, and particularly play a role in endothelial dysfunction and immunosuppression. Whilst therapeutic modulation of the epigenome is still in its infancy, there is substantial evidence from animal models that this approach could reap benefits. In this review, we summarize research elucidating the role of these mechanisms in several aspects of sepsis pathophysiology including tissue injury and immunosuppression. We also evaluate pre-clinical evidence for the use of “epi-therapies” in the treatment of poly-microbial sepsis.
Introduction
An Overview of Sepsis Pathophysiology
Sepsis is a syndrome with a broad clinical manifestation, defined by The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) as “a life-threatening organ dysfunction caused by dysregulated host responses to infection” (1). Due to the numerous possible presentations, sepsis can be a difficult clinical condition to recognize, especially during the early stages if patients exhibit non-specific symptoms of being unwell (1–4) or if archetypal signs of infection are absent, e.g., in young infants, the elderly, and the immunocompromised (5–8). Signs which are highly suggestive of sepsis include (but are not limited to) acute confusion, hypotension, tachycardia, and tachypnoea, hypoxia, reduced urine production, a high blood lactate level and a non-blanching rash. Only one of these signs may be present, and none are unique to sepsis (9).
Screening tools have been developed to aid identification of patients who are seriously ill with suspected sepsis (10–12). An example of one such tool, the Sequential Organ Failure Assessment (SOFA) score, codifies the progression of sepsis-related organ failure (13). However, despite these efforts to improve diagnostics, sepsis still can be missed, leading to delays in treatment which can dramatically worsen outcomes. Rapid administration of antibiotics is critical; for every hour of delayed treatment, mortality risk increases by 7.6% (14). The development of tests that accurately predict the onset of sepsis before organ failure occurs would be useful for improving outcomes. The broad manifestation and rapid onset of sepsis make this very challenging, but it nevertheless continues to be an active area of research (15–22).
Infection-driven inflammation causes substantial tissue injury and organ dysfunction during acute sepsis and represents a major cause of mortality (22, 23). The binding of commonly expressed, conserved pathogen antigens (pathogen-associated molecular patterns, PAMPs) to pattern recognition receptors activates NF-κB signaling and promotes transcription of a wide range of pro-inflammatory factors (24–26). The endothelium becomes activated, increasing its permeability as well as the adherence and migration of leucocytes (27). Loss of endothelial integrity drives intravascular leak, hypotension, and widespread oedema (27, 28). The production of damage-associated molecular patterns (DAMPs) from host cells feeds the inflammatory response, resulting in more tissue injury, and thereby, creating a vicious circle. Concurrent to this, cytokines with anti-inflammatory properties are produced in efforts to promote resolution of inflammation and tissue repair; antigen presenting cells become less responsive to lipopolysaccharide (LPS) and other PAMPs, widespread apoptosis of leucocytes is observed, and myeloid-derived suppressor cells (MDSCs) are substantially increased (9, 29). It was once thought that acute hyperinflammatory responses preceded an immunosuppressive phase, however, it is now believed that there two phases can exist simultaneously (30).
Individuals who clear infection can still exhibit protracted, deranged immune responses following a septic episode. Persistent inflammation, immunosuppression, and catabolism syndrome (PICS), whilst not a universal phenomenon in sepsis, describes a clinical syndrome that patients with longer ICU stays can exhibit (31, 32). One characteristic of PICS is increased susceptibility to opportunistic infections and reactivation of latent viruses, which contributes to morbidity and mortality after the initial infective insult has resolved (33, 34). The factors which contribute to PICS are multi-factorial but the significant risk of rehospitalization with infection may suggest an ongoing perturbation of the immune response (35). Indeed, a study by Arens et al. demonstrated persistent immunoparalysis weeks to years after sepsis (36). There is a paucity of studies which investigate the persistence of PICS after hospital discharge, however, it is notable that sepsis survivors have a significantly reduced survival rate over the years following acute infection vs. age matched individuals, occurring independently of health status preceding the septic episode (37–39). The increased death rate may result from persisting sequalae of sepsis; e.g., increased frailty, irreversible impairment in organ function and/or from sepsis-associated, sustained changes in immune function e.g., immunosuppression. Work is underway to elucidate the mechanisms behind these modifications, with some studies suggesting they arise from changes to the epigenome of leucocytes.
Epigenetics: Definition and Mechanisms
Epigenetics refers to the regulation of gene expression not caused by underlying changes in DNA sequence (40). In eukaryotes, DNA forms a stable structure with octomers of histone proteins; this stable structure is known as chromatin [Figure 1, (41)]. The “openness” of chromatin structure affects the accessibility of DNA to transcription factors and RNA polymerase II, and is therefore a key factor in determining the rate of mRNA expression (42). Three major epigenetic mechanisms are described, two of which exert their effect by influencing chromatin compaction (see Figure 1). DNA methylation is a modification of cytosine residues mainly in the context of cytosine-guanine (CpG) motifs. The majority of the mammalian genome is CpG poor, with enriched regions occurring at transcriptional regulatory loci such as promotors and enhancers (termed CpG islands). Around 60–70% of promotors contain CpG islands (43). The second mechanism is histone modification; post-translational modifications of the amino acids in the tail region of histone proteins which include acetylation, phosphorylation, ubiquitylation, and methylation. Modifications of amino acids at specific locations in the protruding tails either strengthen or weaken the interaction between DNA and histones. The final mechanism involves non-coding RNAs (ncRNAs), which can modulate gene expression by binding to either sites in the genome to prevent gene transcription or mRNA transcripts to prevent translation (44, 45).

Figure 1. (A) Histone modifications: The negative charge of DNA allows it to bind tightly to positively charged histone proteins. DNA wraps around octomers of histone proteins and forms discrete units known as nucleosomes, the basis of chromatin. The overall structure and openness of chromatin is dictated by chemical modifications of the N terminal amino acid tails of the histone proteins. Chemical modifications include acetylation, methylation, phosphorylation, SUMOylation, citrullination, and ADP-ribosylation. (B) CpG methylation: The majority of cytosines found in cytosine-guanine dinucleotides (gray circles) are methylated. CpG-rich sections of the genome (CpG islands) occurs in areas requiring transcriptional control e.g., retrotransposons and gene promotors. Here, methylation status is more dynamic, with some hypomethylated CpGs (white circles) facilitating promotor accessibility and gene transcription. (C) Small non-coding RNAs interact with complementary sequences in DNA and on mRNA to interfere with gene transcription and translation respectively. A well-known species of small RNAs are microRNAs. Mature single stranded microRNAs molecules (21–24 nt long) are incorporated into the RNA induced silencing complex (RISC) and then bind to a complementary sequence in the 3'UTRs of mRNA molecules. This binding inhibits mRNA translation and results in either mRNA degradation or storage.
There is growing evidence that modifications of the epigenome impacts the phenotype of immune cells in such a way as to affect responses to infection, and are involved in propagating inflammatory disorders (46, 47). Whilst most epigenetic marks are generally stable over time, those at certain loci show high plasticity in response to environmental factors such as smoking, diet, and disease, making them of interest in the context of various pathologies (48–50). The rewritable nature of epigenetic modifications and the responsiveness of epigenetic enzymes to inhibitor therapy creates great potential for this avenue of treatment in patients both with chronic and acute inflammatory diseases such as sepsis.
In this review, the epigenetic modifications associated with various stages of sepsis will be discussed. Specifically, we cover mechanisms involved in endothelial dysfunction during the hyperinflammatory response and those underpinning aspects of immunosuppression in PICS. The pre-clinical evidence for use of epi-therapies will also be described.
Search Strategy
References were identified through Ovid using search terms (“sepsis” OR “septic shock” OR “endotoxin tolerance (ET)”) AND (“epigenomics” OR “epigenetic” OR “DNA methylation” OR “Histone modifications” OR “histone” OR “non-coding RNA” OR “micro RNA”). Bibliographies of papers of interest were searched by hand to identify additional studies. Relevant papers identified in the database were included.
Epigenetic Changes Associated With Sepsis Pathophysiology
Histone Acetyltransferases (HATs) and Histone Deacetylases (HDACs) as Regulators of Inflammation
Histone acetylation is a key process involved in regulating inflammatory response genes (51, 52). Addition or removal of acetyl groups is mediated by two families of antagonistic enzymes, histone acetyltransferases (HATs), and histone deacetylases (HDACs). There are numerous studies that associate levels of histone acetylation with expression of pro-inflammatory cytokines and other anti-microbial products (52). Therefore, understanding the relative activities of these two enzymatic groups has great relevance to sepsis. To date, five families of HATs enzymes have been discovered. Using acetyl-CoA as a substrate, these enzymes target primarily lysine residues on histones 3 and 4 (53). In humans, 18 HDACs have been discovered, grouped into four classes based on sequence homology with their yeast counterparts. Classes I, II, and IV represent the “classical” HDACs and are the most extensively studied. Class III HDACs, otherwise known as sirtuins, utilize a distinct mechanism for lysine deacetylation requiring NAD+ as a substrate, in contrast with classical HDACs which are Zn2+-dependent metalloproteases (54).
Whilst acetylation is generally considered a pro-transcriptional modification, increasing evidence suggests this is an over-simplistic view and that the effect of acetylation on chromatin structure is in fact site-specific (55, 56). Therefore, the roles of these enzymes in transcriptional regulation is likely to be highly complex and requires detailed elucidation as they are pursued as targets of therapeutics. In addition to histones, HATs, and HDACs have multiple non-histone targets that are critical for a range of cellular processes including metabolism and cell cycle (54).
Epigenetic Changes Associated With Endothelial Dysfunction, Tissue Injury, and Organ Failure in Sepsis
Endothelial damage, as a result of an excessive cytokine response, is one of the initiating steps that ultimately leads to sepsis-associated organ dysfunction. Other than provision of fluids and use of inotropic drugs, there are no interventions available to restore loss in arterial partial pressure and organ perfusion (57). During sepsis, the endothelium is activated and adhesion molecules including ICAM, VCAM, and E-selectin are upregulated (58, 59). These adhesion molecules are critical for leucocyte infiltration into tissues. Entry of neutrophils into the endothelium in particular has been paradoxically associated with both containment of infection and exacerbation of tissue injury (60). Besides adhesion molecule upregulation, endothelial cell junctions become “loose,” leading to an increase in permeability and a loss of fluid from the vascular system into the surrounding tissues. Whilst neither of these processes are pathological in themselves, the extent to which they occur in sepsis is a major driver of organ failure. Therefore, stabilizing endothelial disruption could be an effective avenue of therapeutic intervention in sepsis.
Loss of histone acetylation during acute lung injury may partially drive the over-expression of adhesion molecules and regulate endothelial permeability. Acetylation loss at the promotors of Angp1, Tek, and Kdr–genes with critical roles in both Tie2/Angiopoietin and vascular endothelial growth factor (VEGF/VEGFR) signaling cascades–was observed in lung and extra-pulmonary organs in a mouse sepsis model (61). Loss of acetylation was suggested to be responsible for a significant reduction in gene expression 6 h post-induction of sepsis and for increased albumin leak. Despite the limitations of this study [as discussed in detail by Bataille et al. (62)], these findings highlight a potential mechanism by which inflammatory factors can influence epigenetic regulation and drive maladaptive changes in endothelium. Indeed, ICAM-1, and E-selectin expression are markedly reduced in the lungs of mice with poly-microbial sepsis if they are pretreated with histone deacetylase inhibitors (HDACi) (63, 64). Neutrophil infiltration and albumin leak were both minimized and associated with improved survival.
Other epigenetic mechanisms are also implicated in mediating tissue injury [extensively reviewed in (44)]. Perturbation of several micro RNAs (miRNAs) during sepsis has been described in plasma and the endothelium (65). MiR-181b expression in endothelial cells minimizes leucocyte invasion of tissues by reducing expression of adhesion molecules, mediated by suppression of NF-κB signaling (66). Injection of miR-181b mimics in a mouse model of endotoxemia downregulated VCAM-1 in the lung and reduced leucocyte adhesion and lung histopathology scores (66). Interestingly, 24 intensive care patients with sepsis have lower circulating levels of miR-181b than those with other inflammatory conditions, suggesting this mechanism is particularly pertinent in driving sepsis-associated overexpression of adhesion molecules (66). Suppression of NF-κB signaling by miRNAs may also confer protective effects in other organs. Animal models of sepsis-associated cardiac dysfunction have shown that miR-146a expression can attenuate NF-κB activation and inflammatory responses in both the myocardium and peripheral blood, changes associated with improved survival (67–69). Further mechanistic studies would be helpful to fully elucidate the functions of these miRNAs, and beyond this should explore whether their therapeutic modulation would be of benefit in sepsis.
Persistent Inflammation, Immunosuppression, and Catabolism Syndrome (PICS)
Immunosuppression in critically ill patients was first noted in 1970's when it was discovered that these patients did not develop delayed hypersensitivity responses to common antigens (70). It is now recognized that ongoing immunological disturbance following sepsis occurs in a subset of patients, keeping them in intensive care with a milieu of symptoms despite clearance of initiating infection. These symptoms are collectively referred to as PICS. Individuals may exhibit inappropriately elevated protein catabolism (leading to loss of lean body mass and thus increased frailty), poor wound healing, an increased susceptibility to infection, and prolonged immunosuppression. Current prognosis is poor with many requiring extensive stays in intensive care and high rates of mortality (71). Understanding of the mechanistic features that drive immunosuppression is essential and likely to involve epigenetic elements.
Expansion of Myeloid-Derived Suppressor Cells (MDSCs)
Several studies have highlighted the extensive apoptosis of immune cells during acute sepsis as a prominent driver of subsequent immune dysfunction. Besides a depletion in sheer numbers of cells, several studies have noted functional abnormalities in the remaining subsets of the immune system. The proportional number of regulatory T-cells (Tregs) and other immunosuppressor subsets is significantly increased in patients with PICS. MDSCs are a subset of immature myeloid cells with highly immunosuppressive properties [reviewed extensively by Schrijver et al. (72)]. Specifically, their production of arginase-1, reactive oxygen species, TGF-β, and IL-10 critically suppress T-cell and NK cell function (73). These cells are largely absent in healthy individuals but form a major component of tumor micro-environments in cancer and are detectable in blood following sepsis (74). MDSCs associate with deleterious outcomes and are highly elevated in patients with PICS. Their considerable expansion following sepsis and function as immunosuppressors make understanding of their development an important area of research.
Epigenetic modulation of myeloid progenitors may explain the disproportional expansion in MDSCs. Transcriptional regulators in these cells are precisely controlled by numerous epigenetic mechanisms. Regulation of nuclear factor 1A (NFI-A) has been shown to be critical for myeloid cell differentiation (75). MiR-181b and miR-21, through a synergistic mechanism, negatively modulate NFI-A expression in mice subjected to cecal ligation and puncture (76). In this study, miR-181b and miR-21 were both upregulated in bone marrow, and blockade of these miRNAs substantially impeded MDSC expansion, improved the capacity of these mice to clear peritoneal infection, and increased survival. Other transcription factors, including C/EBP-β and Runx1, are also epigenetically regulated and drive MDSC expansion. HDAC11 is recruited to the C/EBP- β promoter and negatively controls its expression; in knockout models, loss of HDAC11 significantly increases MDSC populations (77). Whether HDAC11 is upregulated during sepsis is unclear. MDSC differentiation and suppressive capacity can also be altered by regulation of miR-9 which in turn exerts its effect by regulation of Runx1 (78). Elevation of miR-9 expression during sepsis is not confirmed, however it should be noted that it is inducible by LPS and several pro-inflammatory cytokines (79), making a strong case for activity during sepsis. Whether targeting the mechanisms that drive MDSC development could be of therapeutic benefit in sepsis is an unanswered question. The miRNAs and acetylation enzymes highlighted here have multiple targets and their inhibition may have other undesirable effects. Furthermore, specific targeting of regulatory mechanisms in these cells alone may prove a challenge. However, further exploration of this area is undoubtedly warranted.
Endotoxin Tolerance as a Mechanism Mediating Immunosuppression
The second notable aspect of immunosuppression is the hypo-responsiveness, particularly of innate immune cells, to subsequent challenge. ET is a well-described clinical phenomenon whereby pro-inflammatory responses to LPS are repressed during secondary encounter. Not all refractory responses are necessarily harmful–acute modulation of pro-inflammatory responses may in fact be beneficial during early sepsis. However, protracted suppression has detrimental consequences, potentially making patients more vulnerable to Gram-negative infections. Several epigenetic mechanisms have been linked to the persistence of ET (80, 81). Elevated miR-221 and miR-222 following prolonged LPS exposure were recently found to have a role in regulating Brahma-related gene 1, which in turn mediated transcriptional silencing of several pro-inflammatory products (80). In addition, failure to induce pro-transcriptional histone modifications–namely, acetylation and tri-methylation of histone 3– was shown by Foster et al. to repress pro-inflammatory gene expression on secondary LPS encounter, whilst leaving anti-microbial, and metabolic gene expression intact (81). Other studies have also demonstrated alterations in promotor histone profiles during sepsis–including loss of marks of active transcription–downregulating genes involved in pro-inflammatory responses and antigen presentation (82).
Some of the epigenetic machinery responsible for modifying histones has been demonstrated to be directly regulated by LPS [reviewed in (9)], potentially providing a mechanistic link between epigenetics and immune regulation. Expression of histone demethylase enzyme, JMJD3, was shown to be induced by LPS stimulation via NF-κB signaling in macrophages (83, 84). Furthermore, the activity of HAT and HDAC enzymes can also be modulated by LPS, although the extent to which their activity contributes to ET remains unclear. CREB-binding protein (CBP), a transcriptional co-activator with HAT activity, is critically involved in NF-κB signaling and regulation of the inflammatory responses (85). LPS exposure increases CBP stability by stimulating the removal of ubiquitin and blocking proteosomal degradation (86). This in turn correlates with increased histone acetylation and cytokine release. Stabilization of histone acetyltransferase HBO1 via a similar mechanism has also been reported (87). Conversely, sirtuins (class III HDAC) have demonstrable suppressive roles in cytokine regulation. Sirtuin 1 (SIRT1) rapidly accumulates at the proximal promotors of TNFα and IL1B following LPS stimulation and induces facultative heterochromatin formation thus silencing gene expression (88). In the same study, SIRT1 was additionally shown to deacetylate (and deactivate) the transcription factor NF-κB p65, a critical inducer of inflammatory signaling, preventing further transcription of pro-inflammatory genes. Another prominent sirtuin family member, SIRT6, can also act as an inflammatory repressor by deacetylating histone 3 at lysine 9 (H3K9) and inducing heterochromatin formation at NF-κB target gene promoters (89). In addition to acetylation changes, methylation and the enzymes which regulate methylation state are observed to negatively regulate expression of some of pro-inflammatory gene loci such as TNFα during sepsis immunosuppression (90).
Cytokines, TNF-α and type I interferons, have also been shown to modulate monocyte responsiveness to LPS through changes in the epigenome. Pre-treatment of monocytes in vitro with TNF-α prior to LPS stimulation was shown to block accumulation of euchromatin-associated H4ac and H3K4me3 at promotor regions of NF- κB target genes (90). When stimulated with LPS pre-treated monocytes had significantly lower pro-inflammatory mRNA expression than those without prior TNF-α exposure. Conversely, type I interferons propagated LPS responses by priming chromatin to respond, heightening sensitivity to weak upstream signaling. The ability of the immune response to self-modulate may represent a beneficial protective mechanism in the short-term. It is the timing and extent of the immunosuppression, specifically an inappropriate continuation once infection has cleared, which generates harm. It is notable that in most of these studies a very small selection of cytokines have been investigated (typically TNF-α and IL-6). Furthermore, no studies have characterized the persistence of epigenetic modifications, for example, in re-hospitalized patients after sepsis. Therefore, the ability of these described changes to potentiate long term suppression is difficult to assess. Repression of pro-inflammatory cytokines represents only one element of immunosuppression. Therefore, the contribution of epigenetic modulation of immune function to overall patient outcome remains to be fully elucidated.
Additional Epigenetic States of Potential Relevance to Sepsis
Other epigenetic states have been associated with modulation of immune function and may be pertinent to inflammatory disorders such as sepsis. Trained immunity in innate cells was reported in 2011 by Netea et al. (91), defined as a heightened immune response to secondary challenge following sub-lethal exposure to an initial stimulus. This phenomenon was subsequently linked to deposition of permissive histone modifications, H3K4me3, at promotors of tnfα, il6, and tlr4 in monocytes following antigen exposure (92). Primed monocytes were found to mount a stronger pro-inflammatory cytokine response during secondary challenge. Priming with other antigens such as fungal β-glucan has also been shown to increase H3K4me3 occupancy at pro-inflammatory gene promotors and correlate with increased cytokine release (93).
The induction of ET or trained immunity appears to be dependent on the microbial stimulus itself and antigen concentration (94). Stimulation of monocytes via Nod Like Receptor (NLR) or Toll Like Receptor (TLR) pathways resulted in unique effector functions, epigenetic and metabolic profiles (95, 96). Whilst TLR stimulation via LPS induced strongly immunosuppressive effects, NLR engagement had the opposite effect, enhancing effector function in a dose-dependent manner. Interestingly, tolerized monocytes regain responsiveness when stimulated with β-glucan (97). These findings underline the complexity of proposed innate immunological “memory.” A plethora of factors including the host cytokine milieu, the antigen in question and antigen concentration all influence the development of either refractory or enhanced effector function. In a complex immune response such as during sepsis, it is likely that a combination of these features occurs simultaneously. Which factors, epigenetic or otherwise, contribute to persistence of ET still require complete elucidation. That immune states such as trained immunity have been shown to propagate via progenitor cells suggests that alteration of host epigenetic regulation can persist extensively (98, 99). Therefore, characterization of histone alterations in sepsis survivors over a prolonged period of time would provide useful information on the longevity of sepsis-induced changes.
Epigenetic Therapeutics: Potential and Limitations in Treatment of Sepsis-Associated Tissue Injury
Histone Deacetylase Inhibitors (HDACi)
A significant amount of research has examined the effect of modulating epigenetic enzymes upon sepsis-associated organ dysfunction and outcome. Numerous histone deacetylase inhibitor studies in pre-clinical models of sepsis have been conducted (summarized in Table 1), discussed in detail in this section. Characterizing the effect of HDACi at the tissue level is difficult in humans. Animal models circumvent this limitation and have brought valuable insights.
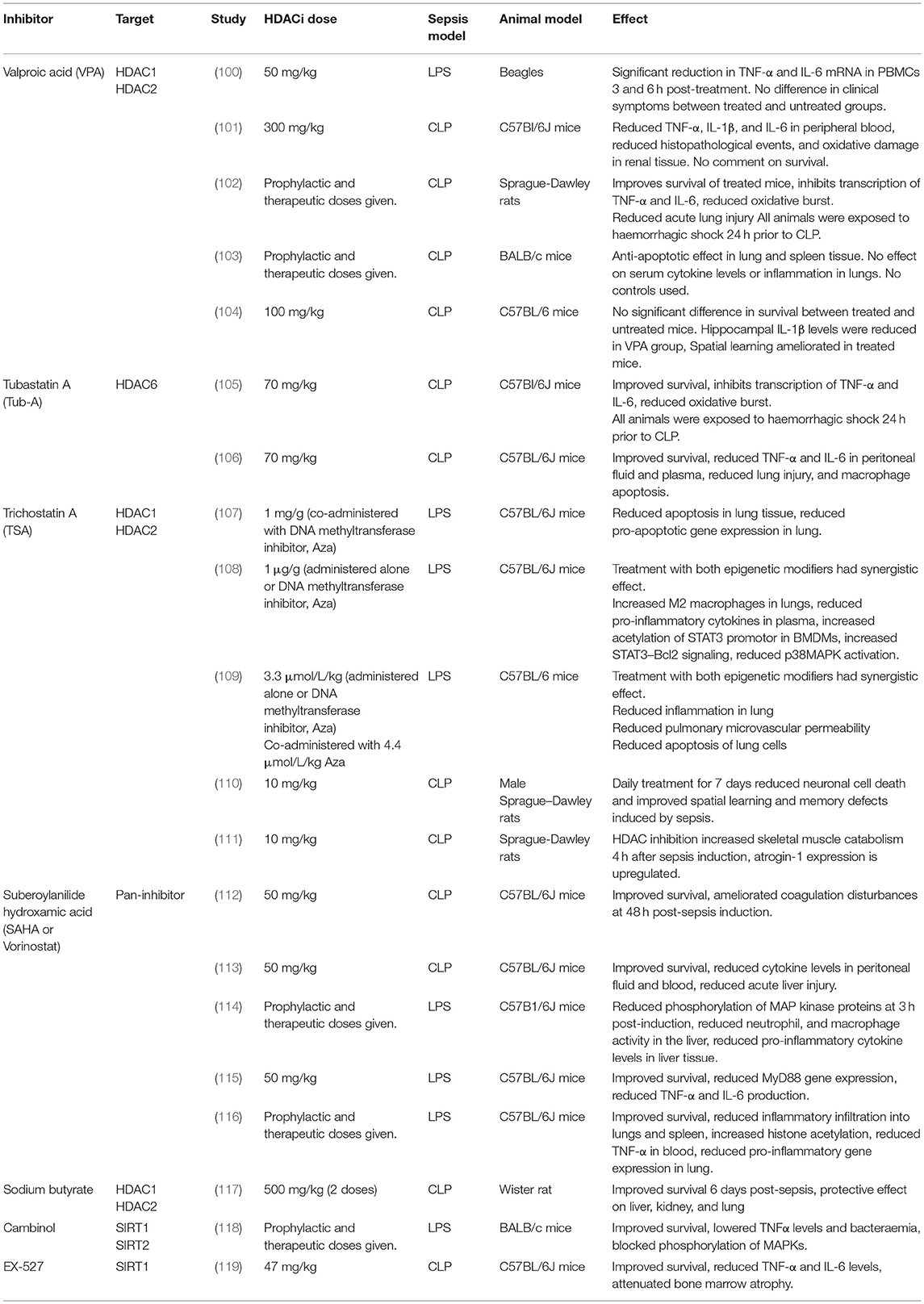
Table 1. Summary of pre-clinical studies investigating the therapeutic potential of various HDACi inhibitors.
Histone deacetylases inhibitors targeting classical HDACs are currently used in a number of clinical contexts including cancer. HDACi administration has been shown to attenuate tumor growth and cause apoptosis in tumorigenic cells though the exact mechanism of action is unknown. Synergistic beneficial effects of combinational HDACi use have been demonstrated, although how synergy is achieved is unclear. Vorinostat (or suberoylanilide hydroxamic acid, SAHA) was licensed in 2006 for the treatment of relapsed and/or refractory cutaneous T-cell lymphoma (120). Two other pan-HDACi, for peripheral T-cell lymphoma and multiple myeloma, are also in use (121, 122). Outside of oncology, valproic acid (VPA) is used as an anticonvulsant which acts on class I and II HDACs, and trichostatin A (TSA) is an antifungal which also acts on class I and II HDACs. These examples demonstrate that HDACi treatment, in principle, has an acceptable safety profile, therefore, their use in sepsis is a realistic option should they prove effective.
Pre-clinical Evidence of HDACi Efficacy in Sepsis
In addition to the positive effects of HDACi on the endothelium during sepsis in mice (discussed above), HDACi treatment has been shown to curb other pro-inflammatory and innate immune responses in pre-clinical models of sepsis. Leoni et al. were the first to report anti-inflammatory properties of Vorinostat both in vitro and in vivo (123). Prophylactic administration of Vorinostat in mice reduced pro-inflammatory cytokine production upon challenge with LPS. Other reports reveal the impact of VPA and TSA on macrophage activity. Host anti-bacterial responses are inhibited via multiple mechanisms: phagocytic receptors are downregulated and release of reactive oxygen species and nitric oxide is reduced. Bacterial killing, demonstrated in mice with E. coli and S. aureus, is significantly impeded (123).
Roger et al. demonstrated a significant reduction in mortality in mouse models of toxic shock induced by Pam3CSK4 and cecal ligation and puncture when HDACi were given (64). TSA negatively regulated the expression of several pattern recognition receptors involved in microbial antigen detection. In addition, they observed that treatment with TSA, Vorinostat, and VPA all repressed cytokine release following TLR stimulation. A recent study exploring the effects of the HDAC6 inhibitor Tubastatin A in a cecal ligation and puncture model of sepsis demonstrated strong therapeutic efficacy. Survival was greater in Tubastatin A-treated mice vs. controls, and pro-inflammatory TNF-α and IL-6 were significantly reduced in peritoneal fluid and plasma of treated animals (106). In addition, a significant reduction in lung injury and bacterial load in the spleen 24 h after cecal ligation and puncture was observed (23 h after HDAC6 inhibitor treatment) (106).
Histone deacetylase inhibitors have been used synergistically with other epigenetic modifiers to ameliorate endothelial integrity and prevent lung injury. Prophylactic inhibition of histone deacetylation alone or combined with inhibition of histone methylation reduced capillary leak and pulmonary oedema in endothelium in vivo and substantially minimized lung histopathology (109).
Evidence from the clinic suggests that HDACi could be useful in attenuating the deleterious pro-inflammatory responses seen in sepsis as there is already a precedent for using HDACi in inflammatory disorders. Vorinostat and another HDACi, Givinostat, are licensed therapies for autoimmune inflammatory disorders graft-vs.-host disease and systemic onset juvenile idiopathic arthritis (SOJIA) (juvenile onset Still's disease), respectively (124, 125).
Considerations and Limitations of HDACi Use
A potential caveat of HDACi treatment is the associated increased risk of subsequent infection that accompanies a reduction in pro-inflammatory responses. Some phase I and II trials of HDACi as cancer treatments noted an increase in severe infections (126, 127) although trials of Vorinostat in graft-vs.-host disease and Givinostat in systemic onset juvenile idiopathic arthritis (SOJIA) have not reported such findings.
It should be noted that the effects of HDACi on the inflammatory response may not be restricted to alterations to the epigenome. The exact effect of HDACi on histone and non-histone acetylation is difficult to characterize, particularly for pan-inhibitors where alterations are likely to be widespread. This impairs our understanding of the exact mechanism driving potentially beneficial effects, in turn hampering the improvement of therapeutic specificity. HDACs have a degree of functional redundancy, therefore knockdown of a given enzyme is frequently compensated for by another of the same class (128). In addition, several HDAC enzymes are known to form multiple complexes, each of which targets a different histone substrate (128).
In several studies of the effects of HDACi on sepsis outcomes treatment was given either prophylactically or very soon following sepsis induction (within an hour), well ahead of the development of symptoms (in CLP models the first symptoms generally appear around 6–12 h post-induction) (Table 1). Therefore, the impact of HDACi treatment in a clinical setting when administered during symptomatic disease is currently unclear.
Histone deacetylase inhibitors may not be appropriate for use in individuals with latent infections. Vorinostat has been proven to reactivate transcription of the HIV reservoir in infected CD4+ T-cells (129). To this end, it has been heavily investigated as part of the “shock and kill” strategy for HIV reservoir eradication (130). Several latent infections such as Epson-Barr virus (EBV) and other herpesviruses also enter lytic replication following HDACi treatment (131). Given the ubiquity of herpesviruses and the seriousness of HIV, reactivation of latent infections during or after a septic episode could be highly detrimental. Whilst HDACi treatment in the context of sepsis is unlikely to be administered long-term, more detailed understanding of HDACi effects on viral latency and reactivation is critical for safe usage.
The Role of HATs Inhibitors in Inflammation
Given the role of acetylation in sepsis, there is a surprising paucity of data examining the role of HATs activity and inhibition. Whilst less well-characterized than that of HDACs, several studies suggest that HATs inhibition could also elicit anti-inflammatory effects. Several HATs inhibitors including delphinidin, gallic acid, epigallocatechin-3-gallate, diferuloylmethane, and cerulenin have all been shown to reduce pro-inflammatory cytokine release by regulating NF-κB acetylation (132–136). In animal models of acute respiratory distress syndrome and renal injury, elevated HATs activity associated with worsened tissue injury suggesting these inhibitors could have therapeutic benefits in cases of sepsis (137, 138). However, contradictory findings have been reported with some suggesting HATs inhibition has either no effect on pro-inflammatory responses or could in fact exaggerate cytokine release (139, 140). This discordance demonstrates the highly context-specific effect of these drugs. Further, exploration of their role in vivo and in sepsis pathophysiology would be welcome.
Concluding Remarks
Sepsis has a worldwide clinical burden with significant associated morbidity and mortality. Whilst our understanding of the underlying immunopathology has improved over the last 30 years, this has yet to inform effective therapeutic strategies. In this review, we have collated evidence from a large number of studies that highlight the epigenetic mechanisms underlying some of the major aspects of sepsis pathology. Together these reveal the importance of epigenetic changes at transcriptional promotors or enhancers in driving many pathological adaptions. It is key to note that the cell-specific context and stage of sepsis in which these changes occur is important for determining phenotypic effect. The potential use of HDACi as therapeutics in inflammatory disorders has garnered interest over the past decade. These drugs have proven tolerability and are already used in the treatment of a number of cancers. Their mechanism of action is incompletely understood and there are legitimate concerns about off-target effects. Histone deacetylase enzymes are involved in modulating thousands of genes and there are likely to be numerous non-histone targets within the cell that are also affected by their activity. Therefore, detailed exploration of enzyme selectivity and development of more targeted inhibitors are vital next steps in the clinical development of HDACi for use in inflammatory disorders.
We are only just beginning to understand the full scale of epigenetic influence on immune function (141). A critical question to address is the longevity of these adaptions. A limitation of many of the above studies is the relatively short time frame in which epigenetic changes are reported. Results from Mitroulis et al. which demonstrate sustained epigenetic modulation in myeloid progenitors now need to be expanded and built upon (98). A more comprehensive description of both the nature of epigenetic changes and the retention of them is needed to fully understand epigenetic contribution to sepsis pathology and outcome.
Author Contributions
DC wrote the first draft on the manuscript. RD contributed significantly to the discussion of the clinical presentation of sepsis. RD and DC produced the figures. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
The authors would like to acknowledge support from the Bill and Melinda Gates Foundation and the Wellcome Trust. JH and RD gratefully acknowledge support from the George and Susan Brownlee Fellowship at Linacre College and the Medical Research Council, respectively. The authors acknowledge the support of the National Institute for Health Research (NIHR), Oxford Biomedical Research Center, and the NIHR Thames Valley, and South Midlands Clinical Research Network. AP is an NIHR Senior Investigator. The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors kindly acknowledge Tatjana Petrinic for her assistance with the initial literature searches.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2. Marik PE. Don't miss the diagnosis of sepsis! Crit Care. (2014) 18:529. doi: 10.1186/s13054-014-0529-6
3. Nunez Lopez O, Cambiaso-Daniel J, Branski LK, Norbury WB, Herndon DN. Predicting and managing sepsis in burn patients: current perspectives. Ther Clin Risk Manag. (2017) 13:1107–17. doi: 10.2147/TCRM.S119938
4. Vincent J-L. The clinical challenge of sepsis identification and monitoring. PLoS Med. (2016) 13:17–8. doi: 10.1371/journal.pmed.1002022
5. Henning DJ, Carey JR, Oedorf K, Day DE, Redfield CS, Huguenel CJ, et al. The absence of fever is associated with higher mortality and decreased antibiotic and Iv fluid administration in emergency department patients with suspected septic shock. Crit Care Med. (2017) 45:575–82. doi: 10.1097/CCM.0000000000002311
6. Young PJ, Bellomo R. Fever in sepsis: is it cool to be hot? Crit Care. (2014) 18:109. doi: 10.1186/cc13726
7. Clifford KM, Dy-Boarman EA, Haase KK, Maxvill K, Pass S, Alvarez CA. Challenges with diagnosing and managing sepsis in older adults. Expert Rev Anti Infect Ther. (2016) 14:231–41. doi: 10.1586/14787210.2016.1135052
8. Warnock C, Totterdell P, Tod AM, Mead R, Gynn JL, Hancock B. The role of temperature in the detection and diagnosis of neutropenic sepsis in adult solid tumour cancer patients receiving chemotherapy. Eur J Oncol Nurs. (2018) 37:12–8. doi: 10.1016/j.ejon.2018.10.001.
9. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. (2017) 17:407–20. doi: 10.1038/nri.2017.36
10. Inada-Kim M, Nsutebu E. NEWS 2: an opportunity to standardise the management of deterioration and sepsis. BMJ. (2018) 360:k1260. doi: 10.1136/bmj.k1260
11. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Intensive Care Med. (2017) 43:304–77. doi: 10.1007/s00134-017-4683-6
12. Sepsis Trust. Screening and Action tools. UK Sepsis Trust (2019). Available online at: https://sepsistrust.org/professional-resources/clinical/
13. Vincent J, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. on behalf of the working group on sepsis-related problems of the European society of intensive care medicine. Intensive Care Med. (1996) 22:707–10. doi: 10.1007/BF01709751
14. Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. (2006) 34:1589–96. doi: 10.1097/01.CCM.0000217961.75225.E9
15. Dolin HH, Papadimos TJ, Stepkowski S, Chen X, Pan ZK. A novel combination of biomarkers to herald the onset of sepsis prior to the manifestation of symptoms. Shock. (2018) 49:364–70. doi: 10.1097/SHK.0000000000001010
16. Larsen FF, Petersen JA. Novel biomarkers for sepsis: a narrative review. Eur J Intern Med. (2017) 45:46–50. doi: 10.1016/j.ejim.2017.09.030
17. Vijayan AL, Vanimaya Ravindran S, Saikant R, Lakshmi S, Kartik R, et al. Procalcitonin: a promising diagnostic marker for sepsis and antibiotic therapy. J Intensive Care. (2017) 5:51. doi: 10.1186/s40560-017-0246-8
18. Zhou Y, Zhang Y, Johnson A, Venable A, Griswold J, Pappas D. Combined CD25, CD64, and CD69 biomarker panel for flow cytometry diagnosis of sepsis. Talanta. (2019) 191:216–21. doi: 10.1016/j.talanta.2018.08.058
19. Kumar S, Tripathy S, Jyoti A, Singh SG. Recent advances in biosensors for diagnosis and detection of sepsis: a comprehensive review. Biosens Bioelectron. (2019) 124–125:205–15. doi: 10.1016/j.bios.2018.10.034
20. Ruan L, Chen G-Y, Liu Z, Zhao Y, Xu G-Y, Li S-F, et al. The combination of procalcitonin and C-reactive protein or presepsin alone improves the accuracy of diagnosis of neonatal sepsis: a meta-analysis and systematic review. Crit Care Lond Engl. (2018) 22:316. doi: 10.1186/s13054-018-2236-1
21. Lendak DF, Mihajlović DM, Novakov-Mikić AS, Boban JM, Ubavić M, Brkić S V. APRIL and sTACI could be predictors of multiorgan dysfunction syndrome in sepsis. Virulence. (2018) 9:946–53. doi: 10.1080/21505594.2018.1462636
22. Wiersinga WJ. Current insights in sepsis: from pathogenesis to new treatment targets. Curr Opin Crit Care. (2011) 17:480–6. doi: 10.1097/MCC.0b013e32834a4aeb
23. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
24. Ricklin D, Hajishengallis G, Yang K, Lambris J. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 11:785–97. doi: 10.1038/ni.1923.
25. Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. (2012) 249:158–75. doi: 10.1111/j.1600-065X.2012.01146.x
26. Abraham E. Nuclear factor–kB and its role in sepsis–associated organ failure. J Infect Dis. 187:364–9. doi: 10.1086/374750
27. Leligdowicz A, Richard-Greenblatt M, Wright J, Crowley VM, Kain KC. Endothelial activation: the ang/tie axis in sepsis. Front Immunol. (2018) 9:838. doi: 10.3389/fimmu.2018.00838
28. De Backer D, Ortiz JA, Salgado D. Coupling microcirculation to systemic hemodynamics. Curr Opin Crit Care. (2010) 16:250. doi: 10.1097/MCC.0b013e3283383621
29. Kane BA, Bryant KJ, McNeil HP, Tedla NT. Termination of immune activation: an essential component of healthy host immune responses. J Innate Immun. (2014) 6:727–38. doi: 10.1159/000363449
30. Osuchowski MF, Welch K, Siddiqui J, Remick DG. Circulating cytokine/inhibitor profiles reshape the understanding of the SIRS/CARS continuum in sepsis and predict mortality. J Immunol Baltim Md 1950. (2006) 177:1967–74. doi: 10.4049/jimmunol.177.3.1967
31. Mira JC, Brakenridge SC, Moldawer LL, Moore FA. Persistent inflammation, immunosuppression and catabolism syndrome. Crit Care Clin. (2017) 33:245–58. doi: 10.1016/j.ccc.2016.12.001
32. Hawkins RB, Raymond SL, Stortz JA, Horiguchi H, Brakenridge SC, Gardner A, et al. Chronic critical illness and the persistent inflammation, immunosuppression, and catabolism syndrome. Front Immunol. (2018) 9:1511. doi: 10.3389/fimmu.2018.01511
33. Denstaedt SJ, Singer BH, Standiford TJ. Sepsis and nosocomial infection: patient characteristics, mechanisms, and modulation. Front Immunol. (2018) 9:2446. doi: 10.3389/fimmu.2018.02446
34. Walton AH, Muenzer JT, Rasche D, Boomer JS, Sato B, Brownstein BH, et al. Reactivation of multiple viruses in patients with sepsis. PLoS ONE. (2014) 9:e98819. doi: 10.1371/journal.pone.0098819
35. Chang DW, Tseng C-H, Shapiro MF. Rehospitalizations following sepsis: common and costly. Crit Care Med. (2015) 43:2085–93. doi: 10.1097/CCM.0000000000001159
36. Arens C, Bajwa SA, Koch C, Siegler BH, Schneck E, Hecker A, et al. Sepsis-induced long-term immune paralysis–results of a descriptive, explorative study. Crit Care Lond Engl. (2016) 20:93. doi: 10.1186/s13054-016-1233-5
37. Prescott HC, Angus DC. Enhancing recovery from sepsis: a review. JAMA. (2018) 319:62–75. doi: 10.1001/jama.2017.17687
38. Ou S-M, Chu H, Chao P-W, Lee Y-J, Kuo S-C, Chen T-J, et al. Long-term mortality and major adverse cardiovascular events in sepsis survivors. A nationwide population-based study. Am J Respir Crit Care Med. (2016) 194:209–17. doi: 10.1164/rccm.201510-2023OC
39. Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ. (2016) 353:i2375. doi: 10.1136/bmj.i2375
40. Dupont C, Armant R, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. (2009) 27:351–7. doi: 10.1055/s-0029-1237423.Epigenetics
41. Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet. (2009) 43:559–99. doi: 10.1146/annurev.genet.032608.103928
42. Schmidt F, Gasparoni N, Gasparoni G, Gianmoena K, Cadenas C, Polansky JK, et al. Combining transcription factor binding affinities with open-chromatin data for accurate gene expression prediction. Nucleic Acids Res. (2017) 45:54–66. doi: 10.1093/nar/gkw1061
43. Illingworth RS, Bird AP. CpG islands–“A rough guide.” FEBS Lett. (2009) 583:1713–20. doi: 10.1016/j.febslet.2009.04.012
44. Ho J, Chan H, Wong SH, Wang MHT, Yu J, Xiao Z, et al. The involvement of regulatory non-coding RNAs in sepsis: a systematic review. Crit Care. (2016) 20:383. doi: 10.1186/s13054-016-1555-3
45. Drury RE, O'Connor D, Pollard AJ. The clinical application of microRNAs in infectious disease. Front Immunol. (2017) 8:1182. doi: 10.3389/fimmu.2017.01182
46. Fernández-Morera JL, Calvanese V, Rodríguez-Rodero S, Menéndez-Torre E, Fraga MF. Epigenetic regulation of the immune system in health and disease. Tissue Antigens. (2010) 76:431–9. doi: 10.1111/j.1399-0039.2010.01587.x
47. Busslinger M, Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb Perspect Biol. (2014) 6:a019307. doi: 10.1101/cshperspect.a019307
48. Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. (2016) 9:436–47. doi: 10.1161/CIRCGENETICS.116.001506
49. Sapienza C, Issa J-P. Diet, nutrition, and cancer epigenetics. Annu Rev Nutr. (2016) 36:665–81. doi: 10.1146/annurev-nutr-121415-112634
50. Minarovits J, Niller HH. Patho-Epigenetics of Infectious Disease. 1st ed. New York, NY: Springer International Publishing (2016).
51. Ghizzoni M, Haisma HJ, Maarsingh H, Dekker FJ. Histone acetyltransferases are crucial regulators in NF-κB mediated inflammation. Drug Discov Today. (2011) 16:504–11. doi: 10.1016/j.drudis.2011.03.009
52. Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. (2005) 25:552–63. doi: 10.1183/09031936.05.00117504
53. Marmorstein R, Zhou M-M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol. (2014) 6:a018762. doi: 10.1101/cshperspect.a018762
54. Hull E, Montgomery M, Leyva K. HDAC inhibitors as epigenetic regulators of the immune system: impacts on cancer therapy and inflammatory diseases. BioMed Res Int. (2016) 2016:8797206. doi: 10.1155/2016/8797206
55. Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. (2009) 138:1019–31. doi: 10.1016/j.cell.2009.06.049
56. Zhang R, Erler J, Langowski J. Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys J. (2017) 112:450–9. doi: 10.1016/j.bpj.2016.11.015
57. Ince C, Mik EG. Microcirculatory and mitochondrial hypoxia in sepsis, shock, and resuscitation. J Appl Physiol. (2015) 120:226–35. doi: 10.1152/japplphysiol.00298.2015
58. Zhao Y, Yi W, Wan X, Wang J, Tao T, Li J, et al. Blockade of ICAM-1 improves the outcome of polymicrobial sepsis via modulating neutrophil migration and reversing immunosuppression. Mediators Inflamm. (2014) 2014:195290. doi: 10.1155/2014/195290
59. Leone M, Boutière B, Camoin-Jau L, Albanèse J, Horschowsky N, Mège J-L, et al. Systemic endothelial activation is greater in septic than in traumatic-hemorrhagic shock but does not correlate with endothelial activation in skin biopsies. Crit Care Med. (2002) 30:808–814.
60. Sônego F, Castanheira FV e S, Ferreira RG, Kanashiro A, Leite CAVG, Nascimento DC, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. (2016) 7:155. doi: 10.3389/fimmu.2016.00155
61. Bomsztyk K, Mar D, An D, Sharifian R, Mikula M, Gharib SA, et al. Experimental acute lung injury induces multi-organ epigenetic modifications in key angiogenic genes implicated in sepsis-associated endothelial dysfunction. Crit Care. (2015) 19:225. doi: 10.1186/s13054-015-0943-4
62. Bataille A, Galichon P, Ziliotis M-J, Sadia I, Hertig A. Epigenetic changes during sepsis: on your marks! Crit Care. (2015) 19:358. doi: 10.1186/s13054-015-1068-5
63. Zhang L, Jin S, Wang C, Jiang R, Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J Surg. (2010) 34:1676–83. doi: 10.1007/s00268-010-0493-5
64. Roger T, Lugrin J, Roy DL, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to toll-like receptor agonists and to infection. Blood. (2011) 117:1205–17. doi: 10.1182/blood-2010-05-284711
65. Goodwin AJ, Guo C, Cook JA, Wolf B, Halushka PV, Fan H. Plasma levels of microRNA are altered with the development of shock in human sepsis: an observational study. Crit Care. (2015) 19:440. doi: 10.1186/s13054-015-1162-8
66. Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, et al. MicroRNA-181b regulates NF-κB–mediated vascular inflammation. J Clin Invest. (2012) 122:1973–90. doi: 10.1172/JCI61495
67. An R, Feng J, Xi C, Xu J, Sun L. miR-146a attenuates sepsis-induced myocardial dysfunction by suppressing IRAK1 and TRAF6 via targeting ErbB4 expression. Oxid Med Cell Longev. (2018) 2018:1–9. doi: 10.1155/2018/7163057
68. Wang X, Yu Y. MiR-146b protect against sepsis induced mice myocardial injury through inhibition of Notch1. J Mol Histol. (2018) 49:411–7. doi: 10.1007/s10735-018-9781-4
69. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, et al. Attenuation of cardiac dysfunction in polymicrobial sepsis by microRNA-146a is mediated via targeting of IRAK1 and TRAF6 expression. J Immunol Baltim Md 1950. (2015) 195:672–82. doi: 10.4049/jimmunol.1403155
70. Christou NV, Meakins JL, Gordon J, Yee J, Hassan-Zahraee M, Nohr CW, et al. The delayed hypersensitivity response and host resistance in surgical patients. 20 years later. Ann Surg. (1995) 222:534–46. doi: 10.1097/00000658-199522240-00011
71. Mira JC, Gentile LF, Mathias BJ, Efron PA, Brakenridge SC, Mohr AM, et al. Sepsis pathophysiology, chronic critical illness and PICS. Crit Care Med. (2017) 45:253–62. doi: 10.1097/CCM.0000000000002074
72. Schrijver IT, Théroude C, Roger T. Myeloid-derived suppressor cells in sepsis. Front Immunol. (2019) 10:327. doi: 10.3389/fimmu.2019.00327
73. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
74. Uhel F, Azzaoui I, Grégoire M, Pangault C, Dulong J, Tadié J-M, et al. Early expansion of circulating granulocytic myeloid-derived suppressor cells predicts development of nosocomial infections in patients with sepsis. Am J Respir Crit Care Med. (2017) 196:315–27. doi: 10.1164/rccm.201606-1143OC
75. Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. (2005) 123:819–31. doi: 10.1016/j.cell.2005.09.023
76. McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, et al. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun. (2014) 82:3816–25. doi: 10.1128/IAI.01495-14
77. Chen J, Sahakian E, Powers JJ, LienlafMoreno M, Xing L, Deng S, et al. Histone deacetylase 11 (HDAC11) as a novel transcriptional regulator of C/EBP-β, in immature myeloid cell to myeloid derived suppressor cell transition. Blood. (2014) 124:225.
78. Tian J, Rui K, Tang X, Ma J, Wang Y, Tian X, et al. MicroRNA-9 regulates the differentiation and function of myeloid-derived suppressor cells via targeting runx1. J Immunol. (2015) 195:1301–11. doi: 10.4049/jimmunol.1500209
79. Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, et al. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci. (2009) 106:5282–7. doi: 10.1073/pnas.0810909106
80. Seeley JJ, Baker RG, Mohamed G, Bruns T, Hayden MS, Deshmukh SD, et al. Induction of innate immune memory via microRNA targeting of chromatin remodelling factors. Nature. (2018) 559:114. doi: 10.1038/s41586-018-0253-5
81. Foster S, Hargreaves D, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 447:972–8. doi: 10.1038/nature05836
82. Weiterer S, Uhle F, Lichtenstern C, Siegler BH, Bhuju S, Jarek M, et al. Sepsis induces specific changes in histone modification patterns in human monocytes. PLoS ONE. (2015) 10:e0121748. doi: 10.1371/journal.pone.0121748
83. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. (2007) 130:1083–94. doi: 10.1016/j.cell.2007.08.019
84. Santa FD, Narang V, Yap ZH, Tusi BK, Burgold T, Austenaa L, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. (2009) 28:3341–52. doi: 10.1038/emboj.2009.271
85. Calao M, Burny A, Quivy V, Dekoninck A, Lint CV. A pervasive role of histone acetyltransferases and deacetylases in an NF-κB-signaling code. Trends Biochem Sci. (2008) 33:339–49. doi: 10.1016/j.tibs.2008.04.015
86. Wei J, Dong S, Bowser RK, Khoo A, Zhang L, Jacko AM, et al. Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci Signal. (2017) 10:eaak9660. doi: 10.1126/scisignal.aak9660
87. Long C, Lai Y, Li J, Huang J, Zou C. LPS promotes HBO1 stability via USP25 to modulate inflammatory gene transcription in THP-1 cells. Biochim Biophys Acta BBA Gene Regul Mech. (2018) 1861:773–82. doi: 10.1016/j.bbagrm.2018.08.001
88. Liu TF, Yoza BK, Gazzar ME, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. (2011) 286:9856–64. doi: 10.1074/jbc.M110.196790
89. Kawahara T, Michishita E, Adler A, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-κB-dependent gene expression and organismal life span: cell. Cell. 136:62–74. doi: 10.1016/j.cell.2008.10.052
90. El Gazzar M, Yoza B, Chen X, Hu J, Hawkins G, McCall C. G9a and HP1 couple histone and DNA methylation to TNFα transcription silencing during endotoxin tolerance. J Biol Chem. 283:198–208. doi: 10.1074/jbc.M803446200
91. Netea MG, Quintin J, Van Der Meer JWM. Trained immunity: a memory for innate host defense. Cell Host Microbe. (2011) 9:355–61. doi: 10.1016/j.chom.2011.04.006
92. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, et al. Bacille calmette-guerin induces NOD2-dependent non-specific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci. (2012) 109:17537–42. doi: 10.1073/pnas.1202870109
93. Quintin J, Saeed S, Martens JH a, Giamarellos- EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. (2013) 12:223–32. doi: 10.1016/j.chom.2012.06.006.Candida
94. Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
95. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming during monocyte to macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
96. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
97. Novakovic B, Habibi E, Wang S-Y, Arts RJW, Davar R, Megchelenbrink W, et al. β-glucan reverses the epigenetic state of LPS-induced immunological tolerance. Cell. (2016) 167:1354–68.e14. doi: 10.1016/j.cell.2016.09.034
98. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. (2018) 172:147–61.e12. doi: 10.1016/j.cell.2017.11.034
99. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. (2018) 172:176–90.e19. doi: 10.1016/j.cell.2017.12.031
100. Song R, Yu D, Yoon J, Park J. Valproic acid attenuates the expression of pro-inflammatory cytokines lipopolysaccharide-treated canine peripheral blood mononuclear cells (in vitro) and in a canine endotoxemia model (in vivo). Vet Immunol Immunopathol. (2015) 166:132–7. doi: 10.1016/j.vetimm.2015.06.012
101. Zheng Q, Liu W, Liu Z, Zhao H, Han X, Zhao M. Valproic acid protects septic mice from renal injury by reducing the inflammatory response. J Surg Res. (2014) 192:163–9. doi: 10.1016/j.jss.2014.05.030
102. Liu Z, Li Y, Chong W, Deperalta DK, Duan X, Liu B, et al. Creating a prosurvival phenotype through a histone deacetylase inhibitor in a lethal two-hit model. Shock. (2014) 41:104. doi: 10.1097/SHK.0000000000000074
103. Takebe M, Oishi H, Taguchi K, Aoki Y, Takashina M, Tomita K, et al. Inhibition of histone deacetylases protects septic mice from lung and splenic apoptosis. J Surg Res. (2014) 187:559–70. doi: 10.1016/j.jss.2013.10.050
104. Wu J, Dong L, Zhang M, Jia M, Zhang G, Qiu L, et al. Class I histone deacetylase inhibitor valproic acid reverses cognitive deficits in a mouse model of septic encephalopathy. Neurochem Res. (2013) 38:2440–9. doi: 10.1007/s11064-013-1159-0
105. Cheng X, Liu Z, Liu B, Zhao T, Li Y, Alam HB. Selective histone deacetylase 6 inhibition prolongs survival in a lethal two-hit model. J Surg Res. (2015) 197:39–44. doi: 10.1016/j.jss.2015.02.070
106. Li Y, Zhao T, Liu B, Halaweish I, Mazitschek R, Duan X, et al. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J Trauma Acute Care Surg. (2015) 78:378–85. doi: 10.1097/TA.0000000000000510
107. Samanta S, Zhou Z, Rajasingh S, Panda A, Sampath V, Rajasingh J. DNMT and HDAC inhibitors together abrogate endotoxemia mediated macrophage death by STAT3-JMJD3 signaling. Int J Biochem Cell Biol. (2018) 102:117–27. doi: 10.1016/j.biocel.2018.07.002
108. Thangavel J, Samanta S, Rajasingh S, Barani B, Xuan Y-T, Dawn B, et al. Epigenetic modifiers reduce inflammation and modulate macrophage phenotype during endotoxemia-induced acute lung injury. J Cell Sci. (2015) 128:3094–105. doi: 10.1242/jcs.170258
109. Thangavel J, Malik AB, Elias HK, Rajasingh S, Simpson AD, Sundivakkam PK, et al. Combinatorial therapy with acetylation and methylation modifiers attenuates lung vascular hyperpermeability in endotoxemia-induced mouse inflammatory lung injury. Am J Pathol. (2014) 184:2237–49. doi: 10.1016/j.ajpath.2014.05.008
110. Fang J, Lian Y, Xie K, Cai S, Wen P. Epigenetic modulation of neuronal apoptosis and cognitive functions in sepsis-associated encephalopathy. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. (2014) 35:283–8. doi: 10.1007/s10072-013-1508-4
111. Alamdari N, Smith IJ, Aversa Z, Hasselgren P-O. Sepsis and glucocorticoids upregulate p300 and downregulate HDAC6 expression and activity in skeletal muscle. Am J Physiol–Regul Integr Comp Physiol. (2010) 299:R509–20. doi: 10.1152/ajpregu.00858.2009
112. Zhao T, Li Y, Liu B, Wu E, Sillesen M, Velmahos GC, et al. Histone deacetylase inhibitor treatment attenuates coagulation imbalance in a lethal murine model of sepsis. Surgery. (2014) 156:214–20. doi: 10.1016/j.surg.2014.04.022
113. Zhao T, Li Y, Liu B, Liu Z, Chong W, Duan X, et al. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surgery. (2013) 154:206–13. doi: 10.1016/j.surg.2013.04.003
114. Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, et al. Treatment with histone deacetylase inhibitor attenuates MAP kinase mediated liver injury in a lethal model of septic shock. J Surg Res. (2010) 163:146–54. doi: 10.1016/j.jss.2010.04.024
115. Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, et al. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. (2010) 148:246–54. doi: 10.1016/j.surg.2010.05.003
116. Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, et al. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. (2009) 32:517. doi: 10.1097/SHK.0b013e3181a44c79
117. Zhang L-T, Yao Y-M, Lu J-Q, Yan X-J, Yu Y, Sheng Z-Y. Sodium butyrate prevents lethality of severe sepsis in rats. Shock. (2007) 27:672. doi: 10.1097/SHK.0b013e31802e3f4c
118. Lugrin J, Ciarlo E, Santos A, Grandmaison G, Dos Santos I, Le Roy D, Roger T. The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim Biophys Acta BBA–Mol Cell Res. (2013) 1833:1498–510. doi: 10.1016/j.bbamcr.2013.03.004
119. Zhao T, Li Y, Liu B, Bronson RT, Halaweish I, Alam HB. Histone deacetylase III as a potential therapeutic target for the treatment of lethal sepsis. J Trauma Acute Care Surg. (2014) 77:913–9. doi: 10.1097/TA.0000000000000347
120. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. (2007) 12:1247–52. doi: 10.1634/theoncologist.12-10-1247
121. Raedler LA. Farydak (Panobinostat): first HDAC inhibitor approved for patients with relapsed multiple myeloma. Am Health Drug Benefits. (2016) 9:84–87.
122. Lee H. FDA approval: belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin Cancer Res. (2015) 11:1659–64. doi: 10.2217/fon.15.62.
123. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci. (2002) 99:2995–3000. doi: 10.1073/pnas.052702999
124. Choi SW, Braun T, Henig I, Gatza E, Magenau J, Parkin B, et al. Vorinostat plus tacrolimus/methotrexate to prevent GVHD following myeloablative conditioning unrelated donor HCT. Blood. (2017) 130:1760–7. doi: 10.1182/blood-2017-06-790469
125. Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med. (2011) 17:397–403. doi: 10.2119/molmed.2011.00030
126. Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N, et al. A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol. (2010) 89:185–90. doi: 10.1007/s00277-009-0793-8
127. Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. (2005) 23:3912–22. doi: 10.1200/JCO.2005.02.188
128. Seto E, Yoshida M. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol. (2014) 6:a018713. doi: 10.1101/cshperspect.a018713
129. Barton K, Hiener B, Winckelmann A, Rasmussen TA, Shao W, Byth K, et al. Broad activation of latent HIV-1 in vivo. Nat Commun. (2016) 7:12731. doi: 10.1038/ncomms12731
130. Ke R, Lewin SR, Elliott JH, Perelson AS. Modeling the effects of vorinostat in vivo reveals both transient and delayed HIV transcriptional activation and minimal killing of latently infected cells. PLoS Pathog. (2015) 11:e1005237. doi: 10.1371/journal.ppat.1005237
131. Ghosh SK, Perrine SP, Williams RM, Faller DV. Histone deacetylase inhibitors are potent inducers of gene expression in latent ebv and sensitize lymphoma cells to nucleoside antiviral agents. Blood. (2012) 119:1008–17. doi: 10.1182/blood-2011-06-362434
132. Seong A-R, Yoo J-Y, Choi K, Lee M-H, Lee Y-H, Lee J, et al. Delphinidin, a specific inhibitor of histone acetyltransferase, suppresses inflammatory signaling via prevention of NF-κB acetylation in fibroblast-like synoviocyte MH7A cells. Biochem Biophys Res Commun. (2011) 410:581–6. doi: 10.1016/j.bbrc.2011.06.029
133. Choi K-C, Jung MG, Lee Y-H, Yoon JC, Kwon SH, Kang H-B, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. (2009) 69:583–92. doi: 10.1158/0008-5472.CAN-08-2442
134. Kim M-J, Seong A-R, Yoo J-Y, Jin C-H, Lee Y-H, Kim YJ, et al. Gallic acid, a histone acetyltransferase inhibitor, suppresses β-amyloid neurotoxicity by inhibiting microglial-mediated neuroinflammation. Mol Nutr Food Res. (2011) 55:1798–808. doi: 10.1002/mnfr.201100262
135. Yuan Z, Syed MA, Panchal D, Rogers D, Joo M, Sadikot RT. Curcumin mediated epigenetic modulation inhibits TREM-1 expression in response to lipopolysaccharide. Int J Biochem Cell Biol. (2012) 44:2032–43. doi: 10.1016/j.biocel.2012.08.001
136. Lee HH, Sanada S, An SM, Ye BJ, Lee JH, Seo Y-K, et al. LPS-induced NFκB enhanceosome requires tonEBP/NFAT5 without DNA binding. Sci Rep. (2016) 6:24921. doi: 10.1038/srep24921
137. Huang J, Wan D, Li J, Chen H, Huang K, Zheng L. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics. (2015) 10:62–72. doi: 10.4161/15592294.2014.990780
138. Chen Y, Wang D, Zhao Y, Huang B, Cao H, Qi D. p300 promotes differentiation of Th17 cells via positive regulation of the nuclear transcription factor RORγt in acute respiratory distress syndrome. Immunol Lett. (2018) 202:8–15. doi: 10.1016/j.imlet.2018.07.004
139. Wang B, Lin L, Ai Q, Zeng T, Ge P, Zhang L. HAT inhibitor, garcinol, exacerbates lipopolysaccharide-induced inflammation in vitro and in vivo. Mol Med Rep. (2016) 13:5290–6. doi: 10.3892/mmr.2016.5189
140. Jing Y, Ai Q, Lin L, Dai J, Jia M, Zhou D, et al. Protective effects of garcinol in mice with lipopolysaccharide/D-galactosamine-induced apoptotic liver injury. Int Immunopharmacol. (2014) 19:373–80. doi: 10.1016/j.intimp.2014.02.012
Keywords: sepsis, epigenetics, immunosuppression, endothelial dysfunction, histone deacetylase inhibitors
Citation: Cross D, Drury R, Hill J and Pollard AJ (2019) Epigenetics in Sepsis: Understanding Its Role in Endothelial Dysfunction, Immunosuppression, and Potential Therapeutics. Front. Immunol. 10:1363. doi: 10.3389/fimmu.2019.01363
Received: 30 June 2018; Accepted: 29 May 2019;
Published: 18 June 2019.
Edited by:
Thierry Roger, Lausanne University Hospital (CHUV), SwitzerlandReviewed by:
Charles E. McCall, Wake Forest Baptist Medical Center, United StatesViswanathan Natarajan, University of Illinois at Chicago, United States
Copyright © 2019 Cross, Drury, Hill and Pollard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah Cross, ZGVib3JhaC5jcm9zczJAcGFlZGlhdHJpY3Mub3guYWMudWs=