 Cecilia P. Marin Oyarzún
Cecilia P. Marin Oyarzún Paula G. Heller
Paula G. Heller- Department of Hematology Research, National Scientific and Technical Research Council (CONICET), Institute of Medical Research (IDIM) “Dr. Alfredo Lanari”, University of Buenos Aires, Buenos Aires, Argentina
Chronic myeloproliferative neoplasms (MPN) are stem cell disorders driven by mutations in JAK2, CALR, or MPL genes and characterized by myeloid proliferation and increased blood cell counts. They encompass three closely related conditions, including essential thrombocythemia, polycythemia vera, and primary myelofibrosis. Elevated levels of cytokines released by clonal and non-clonal cells generate a chronic proinflammatory state that contributes to disease pathogenesis. Thrombosis represents the most common cause of morbidity and mortality in MPN, although paradoxically, patients may also present with a bleeding diathesis. The mechanisms leading to thrombosis are complex and multiple and include increased blood cells together with qualitative abnormalities of red cells, leukocytes, and platelets that favor a prothrombotic activated phenotype. The functional interplay between blood cells, the clotting cascade, and dysfunctional endothelium contributes to hypercoagulability and this process is perpetuated by the effect of inflammatory cytokines. In addition to their well-known function in hemostasis, platelets contribute to innate immunity and inflammation and play a key role in MPN thromboinflammatory state. In vivo platelet activation leads to platelet aggregate formation and exposure of adhesion molecules which favor their interaction with activated neutrophils and monocytes leading to circulating platelet-leukocyte heterotypic aggregates. Platelets are recruited to the activated endothelium further enhancing the reciprocal activation of both cell types. Crosstalk between activated cells drives cytokine production, further fuelling the self-reinforcing thromboinflammatory loop. In addition, MPN platelets provide a procoagulant scaffold which triggers the coagulation cascade and platelet-derived microparticles amplify this response. Markers of platelet, leukocyte, endothelial and coagulation activation are increased in MPN patients although prospective studies are required to determine the potential value of these parameters for identifying patients at increased thrombotic risk. Thrombosis remains the main complication of MPN patients, with a high risk of recurrence despite adequate cytoreductive and antithrombotic treatment. Deeper insight into the mechanism favoring thrombosis development in this setting may lead to novel therapeutic approaches for MPN thrombosis. Considering the critical role of inflammation in the vascular risk, concomitant targeting of inflammatory pathways could potentially impact on primary or secondary prevention strategies.
Introduction
Philadelphia-negative chronic myeloproliferative neoplasms (MPN) are clonal hematopoietic stem cell disorders characterized by excessive production of myeloid progenitors and mature blood cells. They comprise three closely related disorders, including essential thrombocythemia (ET), which is characterized by megakaryocyte proliferation and thrombocytosis, polycythemia vera (PV), which is defined by predominant erythroid expansion and increased red blood cells, frequently associated with high leukocyte and platelet counts, and primary myelofibrosis (PMF), featured by increased numbers of dysplastic megakaryocytes and granulocyte progenitors together with variable degrees of bone marrow fibrosis (1). Hyperactivation of JAK2-signaling is a common feature in MPN pathogenesis and is driven by mutations in three genes, including JAK2, CALR, and MPL. The JAK2V617F mutation is the most frequent molecular abnormality and may be found in over 95% of patients with PV and 50–60% of those with ET and PMF (1, 2). Defects in calreticulin (CALR) represent the second most frequent abnormality, which can be detected in 20–30% of ET and PMF patients (1, 2). Calreticulin mutants interact abnormally with the Mpl receptor leading to its activation and persistent JAK2 signaling (3). Finally, MPL mutations can be found in a low proportion (1–10%) of ET and PMF patients and generate constitutive receptor activation. None of the above-mentioned mutations are detected in 10–25% of ET and PMF cases, so-called triple-negative patients (1, 2).
Thrombosis is the main cause of morbidity and mortality in MPN and develops in around 20–35% of patients with PV, 15–30% in ET, and 10–15% in PMF (4, 5). Arterial thrombosis accounts for 60–70% of all vascular complications, and include stroke, cardiovascular events, and peripheral artery disease, whereas venous events include deep venous thrombosis and pulmonary embolism, but may also occur at unusual sites, such as the splachnic circulation. Indeed, MPN are the most frequent underlying disorders leading to Budd-Chiari syndrome and non-cirrhotic portal vein thrombosis, which may develop even in the absence of overt MPN (4, 5). A population-based study on the causes of death in MPN patients showed that cardiovascular and cerebrovascular disease accounted for high risk of death at all ages, particularly in younger patients. The most common cause of death was cardiovascular disease in patients with PV and ET, whereas patients with PMF had an increased probability of dying from hematologic malignancies (6). In addition to large-vessel thrombosis, transient platelet aggregates may clog small vessels and lead to microvascular disturbances, such as erythromelalgia and visual abnormalities, seen typically, but not exclusively, in ET (7). Paradoxically, MPN patients may also suffer from bleeding complications, which also substantially contribute to morbidity in these disorders.
Thrombotic Risk Factors
The main risk factors for thrombosis include age over 60 years and a previous history of thrombosis. According to the presence or absence of these factors, patients are stratified into low- or high-risk groups in order to guide treatment recommendations and the use of cytoreductive therapy (1). More recently, the IPSET-thrombosis model, which includes cardiovascular risk factors and the JAK2V617F mutation, has been proposed to better predict the thrombotic outcome in ET (8), although this score has not been yet incorporated into clinical practice. The influence of the JAK2V617F mutation in the thrombotic risk has been established by several studies (1, 9) and confirmed by a meta-analysis, which revealed a two-fold increase in vascular events (10). Interestingly, individuals harboring JAK2V617F-positive clonal hematopoiesis of indeterminate potential, in the absence of overt MPN, have an increased thrombotic risk, highlighting the relevance of the JAK2 mutation in the thrombotic predisposition (11, 12). Conversely, CALR-positive patients are at lower risk of thrombosis (1).
Pathogenesis of MPN Thrombosis
The pathogenesis of thrombosis in MPN is multifactorial and results from the complex interplay among blood cells, the endothelium and the clotting system. Increased numbers of red cells, leukocytes and platelets coupled to qualitative abnormalities that favor a prothrombotic phenotype contribute to the hypercoagulable state (4, 5). Hyperviscosity due to increased red cell mass clearly plays a role in the thrombotic predisposition of PV and, moreover, PV red cells display enhanced adhesion to endothelial laminin (13). In addition, high hematocrit favors platelet margination and accumulation at sites of vascular injury (14). In recent years, growing evidence has highlighted the key role of leukocytes in the prothrombotic state and leukocytosis has been shown to be an independent risk factor for thrombosis (15). In addition to increased numbers, there is evidence of in vivo neutrophil activation, as revealed by CD11b expression and elevated elastase and myeloperoxidase in circulation (16, 17), and of monocyte activation, as shown by elevated CD25 (18). Another element contributing to the thrombotic tendency involves endothelial dysfunction, which renders a pro-adhesive and proinflammatory surface, favoring leukocyte and platelet tethering and activation (4, 5).
It is currently well-established that the MPN clone induces a systemic inflammatory response, reflected by elevated levels of a wide spectrum of proinflammatory cytokines, such as IL-6, IL-1, IL-8, and TNFα (19). Inflammation and hemostasis are closely connected processes and, recently, the link between the innate immune system and coagulation as a host defense strategy against pathogens has led to the concept of immunothrombosis (20). Dysregulation of this mechanism may drive vascular disease and contribute to arterial and venous thrombosis in several disease conditions (20). Emerging work highlights the contribution of chronic inflammation to MPN hypercoagulable state, as demonstrated by the association of elevated C-reactive protein and thrombosis (5, 21). Inflammatory mediators favor the activation of both malignant and non-malignant blood cells, induce microparticle generation and elicit vascular damage, fuelling the thrombotic process (5).
Role of Platelets in MPN Thrombosis
Platelet Activation
Platelets are essential players in MPN thrombosis. Their role in this process and interplay with other elements of the procoagulant network represents the focus of this review. Both increased platelet numbers and in vivo activation may be involved in the prothrombotic phenotype. However, considerable controversy exists regarding the role of thrombocytosis in the thrombotic risk, as no correlation has been shown between platelet counts and vascular complications (22). Furthermore, extreme thrombocytosis (>1,000–1,500 × 109/L) is associated with increased bleeding rather than thrombosis. Indeed, ET patients with extreme thrombocytosis, in the absence of leucocytosis, carry a lower thrombotic risk (23). Nonetheless, reduction in platelet counts by cytoreductive therapy is useful at preventing thrombotic and bleeding complications (24–26) and current recommendations suggest that the goal of cytoreduction is to target the platelet count at <400 × 109/L (27). Notwithstanding controversy regarding the role of high platelet counts, there is unequivocal evidence supporting the contribution of platelet activation to the procoagulant state. In vivo platelet activation has been demonstrated in ET, PV and also in PMF (17, 28–31), as revealed by the finding of platelet activation markers, such as surface and soluble P-selectin and CD40L (17, 28, 29), raised β-thromboglobulin, platelet factor 4 and PDGF in plasma (32, 33) and urinary TXB2 metabolite as a reflection of thromboxane biosynthesis (30).
The mechanisms leading to platelet activation involve both intrinsic platelet abnormalities derived from disturbed hematopoietic stem cell function and linked to driver mutations that lead to hyperactive JAK2-dependent signaling (34), and extrinsic factors, such as cellular interaction with activated leukocytes, endothelial cells and soluble mediators, which may trigger activation of platelets derived not only from clonal cells, but also from megakaryocytes not involved in the malignant clone (Figure 1). In addition, increased platelet turnover leading to higher proportion of newly-formed platelets, which are known to display enhanced platelet reactivity, may also contribute to the hyperactivated state (29, 35). Previous data suggest that changes in megakaryocyte gene expression profile might give rise to circulating platelets with an altered hemostatic or inflammatory function in infectious or inflammatory conditions (36). Whether changes in megakaryocyte transcriptome may be associated with a similar phenotype in MPN platelets remains an intriguing possibility. In this regard, downregulation of several genes involved in thrombin signaling and platelet activation has been demonstrated in CALR- vs. JAK2V617F-positive patient samples, correlating with lower thrombotic predisposition in the former (37).
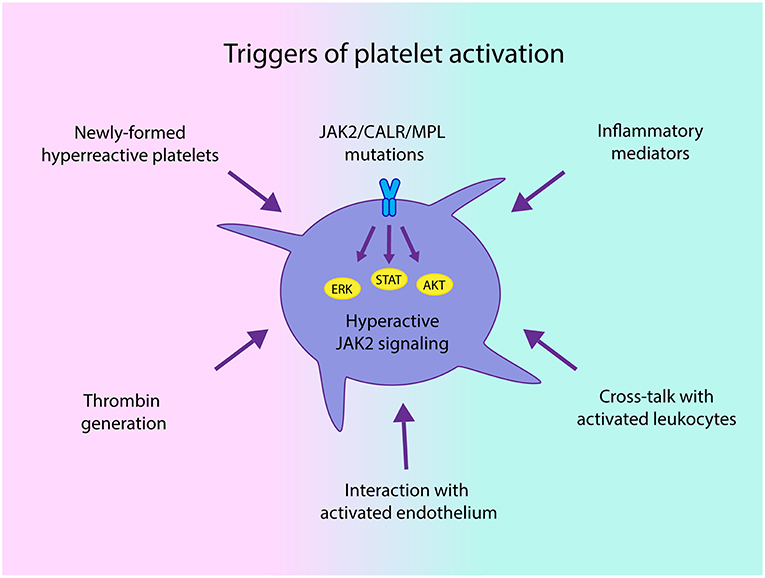
Figure 1. Triggers of platelet activation in chronic myeloproliferative neoplasms (MPN). Both intrinsic abnormalities derived from the MPN clone, such as JAK2-dependent hyperactivation of signaling pathways and hyperreactive newly-formed platelets, as well as extrinsic signals driven by enhanced interaction with activated leukocytes and endothelial cells and soluble mediators, including classical platelet agonists, such as thrombin generated by the hypercoagulable state and inflammatory factors may all converge to trigger platelet activation in MPN.
Among MPN disorders, platelet hemostatic abnormalities have been most thoroughly studied in ET. Despite the presence of basal platelet activation, conflicting results have been published regarding the ex vivo response to classical agonists, such as ADP and TRAP. Whereas, some studies showed normal or enhanced response to stimuli (17), others described an impaired hemostatic function, as shown by reduced P-selectin, CD63 expression and PAC-1 or fibrinogen binding triggered by classical agonists (38, 39). Similarly, light transmission aggregometry may reveal spontaneous platelet aggregation together with impaired response to different agonists, particularly ADP and epinephrine (40), although it has been suggested that the in vitro aggregation defect may be partly due to a laboratory artifact (41) or depend on analytical conditions (42). In vivo release of platelet granule contents due to spontaneous activation and secondary storage pool deficiency may explain the platelet function defect found ex vivo. Coexistence of platelet activation and dysfunction may contribute to the paradoxical occurrence of both thrombotic and bleeding complications. In addition, adsorption of high molecular weight multimers of von Willebrand factor (vWF) to platelet GPIbα leading to loss of large vWF multimers and acquired von Willebrand disease contributes to the bleeding diathesis (43).
Activated platelets mediate several functional responses which contribute to MPN prothrombotic state. In addition to the classical platelet hemostatic properties, growing body of evidence over the last decade highlights the relevance of alternative platelet functions, such as their role as effectors of inflammation and essential players in the innate immune response (44). The contribution of platelets as mediators of thromboinflammatory responses in MPN and their interplay with other components of the prothrombotic and proinflammatory circuit is discussed below and summarized in Figure 2.
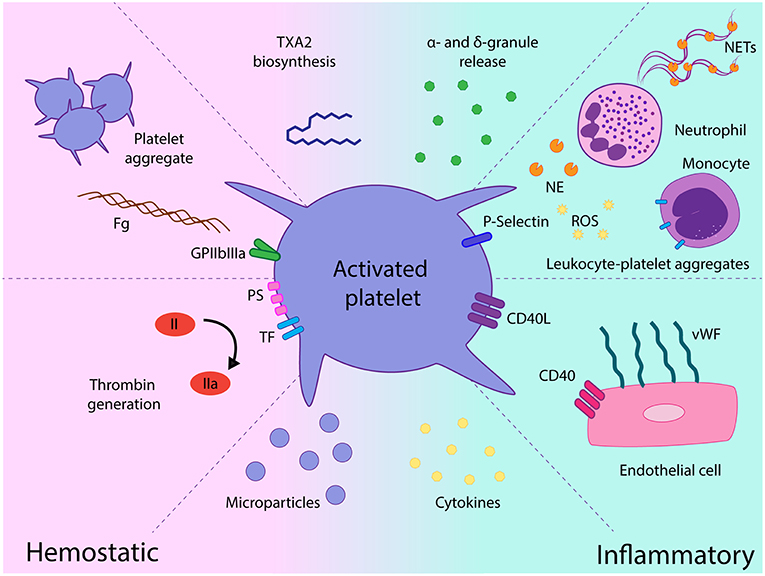
Figure 2. Role of platelets as mediators of hemostatic and proinflammatory responses in chronic myeloproliferative neoplasms (MPN). Platelet activation leads to increased GPIIbIIIa activation and platelet aggregate formation, platelet procoagulant response promotes thrombin generation on the platelet surface and platelet-derived microparticles futher fuel coagulation. Release of a miriad of α- and dense granule hemostatic and inflammatory mediators may contribute to the prothrombotic and proinflammatory loop. Enhanced interaction with leukocytes mediated by P-selectin leads to leukocyte-platelet heterotypic aggregates and may trigger several leukocyte responses, such as release of proteases, production of reactive oxygen species, and expression of tissue factor. Exposure of CD40L favors platelet recruitment and adhesion to the endothelium which, in turn, elicits endothelial cell activation, featured by vWF release from Weibel-Palade bodies. Elevated cytokines in the MPN milieu may promote platelet activation and, reciprocally, platelets may represent a potential source of inflammatory cytokines and chemokines. TXA2, thromboxane A2; Fg, fibrinogen; TF, tissue factor, PS phosphatidylserine; NE, neutrophil elastase; ROS, reactive oxygen species; NETs, neutrophil extracellular traps; vWF, von Willebrand factor, EC, endothelial cell.
Platelet-Leukocyte Cross-Talk
Platelet interaction with leukocytes is central to MPN prothrombotic scenario. Platelets bind to both neutrophils and monocytes through adhesion molecules such as P-selectin, which recognizes the PSGL-1 counterreceptor, and through GPIbα and GPIIbIIIa (via fibrinogen), both of which engage CD11b/CD18 (Mac-1). In accordance to elevated P-selectin, increased levels of platelet-neutrophil and platelet-monocyte aggregates have been shown in circulation in ET (17, 28), PV (28), and PMF (31). Crosstalk between platelets and leukocytes triggers the reciprocal activation of both cell types, contributing to the activated phenotype.
Several leukocyte functional responses are enhanced in MPN neutrophils, including the release of intracellular proteases, such as elastase and catepsin G, which activate platelets and trigger the clotting cascade by inactivating coagulation inhibitors (16). In addition, MPN neutrophils produce high levels of reactive oxygen species (45, 46), which lead to endothelial injury and modify coagulation factors. Besides these classical neutrophil functions, more recently, neutrophils have been shown to release extracellular traps (NETs), which are networks of DNA, histones and granular components which promote thrombus formation (47). Although deregulated NET formation underlies several prothrombotic conditions (47), the role of NETs in MPN remains controversial. Whereas, MPN neutrophils seldom undergo spontaneous NETosis ex vivo (11, 46), their response to stimuli was shown to vary according to the experimental setting. In this regard, enhanced response to ionomycin was shown in one study (11), while NETosis triggered by inflammatory cytokines and PMA was normal or impaired in another study (46), suggesting differential response to diverse NET inducers. Considering that activated platelets are a known trigger for NET formation, it might be relevant to study platelet-induced NETosis in these conditions. Likewise, platelet cross-talk with monocytes may prime monocyte functions, including tissue factor expression (48), which is increased at baseline in MPN (17), and cytokine synthesis (49), which has been found to be constitutively upregulated in PMF monocytes (50), thus perpetuating MPN prothrombotic and proinflammatory loop.
Platelet-Endothelial Interaction
The functional interplay between activated platelets and endothelial dysfunction plays an important role in the prothrombotic state. Evidence for endothelial activation in MPN is well-established, as reflected by elevated vWF antigen, soluble thrombomodulin and E-selectin (16, 29). Activated endothelial cells exhibit a pro-thrombotic phenotype which fosters platelet and leukocyte recruitment. Release of vWF from Weibel-Palade bodies tethers and activates platelets, leading to the surface translocation of platelet CD40 ligand (CD40L), which binds endothelial CD40. Cleavage of membrane CD40L generates a soluble fragment (sCD40L), which is increased in MPN plasma (29). Several factors contribute to endothelial activation in MPN, including interaction with blood cells, reactive oxygen species and inflammatory cytokines. Intriguingly, the JAK2V617F mutation has been detected in mature endothelial cells from selected organs, such as the spleen of PMF patients and the liver of PV patients with Budd-Chiari syndrome, suggesting the potential involvement of endothelial cells in the malignant clone (51, 52). Endothelial-like cells differentiated from MPN patient-derived induced pluripotent stem cells (53) and JAK2V617F-transduced HUVECs exhibit pro-adherent properties in vitro (54). Moreover, increased thrombus formation has been demonstrated in mouse models expressing JAK2V617F only in the endothelial compartment (54), overall suggesting that JAK2-mutant endothelial cells could contribute to the prothrombotic phenotype.
Procoagulant Potential of Platelets
MPN patients show several laboratory abnormalities indicative of chronic low-grade activation of the clotting system, such as elevated thrombin–antithrombin complexes, prothrombin fragment 1 + 2 and D-dimer levels (16, 29). Platelets are endowed with coagulation factors and activated platelets expose phosphatidylserine on their membrane, providing a catalytic substrate for the assembly of coagulation complexes and thrombin generation. Indeed, increased baseline phosphatidylserine expression on the platelet membrane has been shown in some MPN patients and overall platelet procoagulant potential was increased, as revealed by the finding of elevated platelet-induced thrombin generation (55). In addition, MPN platelets express higher surface levels of tissue factor, which represents the main initiator of blood coagulation, further enhancing the procoagulant activity (56).
Platelet-Derived Microparticles
Patients with ET harbor higher numbers of circulating microparticles of platelet, endothelial and leukocyte origin, with the former comprising the vast majority of the microparticle population (57). Remarkably, a subset of microparticles co-expressing platelet and endothelial markers were also detected in ET, suggesting their bilineage origin. Platelet-derived microparticles are rich in tissue factor and phospholipid-dependent procoagulant activity and may deliver platelet-derived cytokines and chemokines, thus amplifying platelet proinflammatory and procoagulant signals.
Platelets as Immune Cells
Platelets play a key role in innate immunity and inflammation through their interaction with other immune cells and the release of proinflammatory mediators (44) and thereby participate in several disease conditions characterized by acute or chronic inflammation, such as infection, autoimmune disorders, and atherosclerosis. MPN patients display raised levels of a broad array of cytokines and chemokines in circulation, which are aberrantly secreted by multiple cell populations, including monocytes, neutrophils and hematopoietic stem cells (58). Activated platelets release pro-inflammatory chemokines stored in their α-granules, such as RANTES (CCL5) and platelet factor 4 (PF4) (CXCL4) and may undergo de novo cytokine synthesis following agonist-triggered RNA splicing, as shown for IL-1β. The potential contribution of platelets as a source of cyto/chemokines and inflammatory mediators in MPN has not been explored. Alternatively, platelet interaction with monocytes could deliver signals that upregulate monocyte proinflammatory gene expression, thus contributing to elevated cytokine secretion (50). Reciprocally, elevated proinflammatory cytokines might contribute to platelet activation in MPN. In this regard, IL-1β has been shown to foster hemostatic responses in normal platelets (59).
Patients with MPN carry a significant risk of second malignancies, including both solid tumors and lymphomas (60). Platelets promote tumor growth and invasiveness through several mechanisms, including the release of growth factors, cytokines, and regulators of angiogenesis. Moreover, thrombocytosis in solid tumors is associated with inferior survival supporting the role of platelets in tumor progression. On this basis, it is tempting to consider the possibility that elevated platelet counts could contribute to tumorigenesis in the setting of MPN second cancers (61). Furthermore, although the contribution of platelets to innate immunity has been more extensively studied, platelets also influence adaptative immune responses through their interaction with T-cells, NK-cells and dendritic cells (44). Platelet-coated tumors may evade NK destruction by inhibiting NK cytotoxicity (62). In this setting, elevated platelet counts might also have implications in tumor immune surveillance and immunoregulation.
Relationship Between Platelet Activation and Clinical Features
Several factors account for the higher frequency of thrombosis observed in JAK2-positive ET patients, including higher hemoglobin and leukocyte counts, lower platelet counts and older age compared to CALR-positive and triple-negative patients. In addition, JAK2-positive patients display higher levels of platelet activation markers (17, 29, 56, 63), as well as leukocyte and endothelial activation and circulating microparticles (29, 56, 64), which may represent additional elements favoring thrombosis development in this subset. In addition, although sP-selectin and sCD40L were shown to correlate with a previous history of thrombosis in one study (29), currently, the value of platelet activation markers to estimate the vascular risk remains uncertain. Prospective studies assessing the role of platelet, together with leukocyte, endothelial and coagulation activation parameters in the same MPN cohort would be required to adequately address this issue and to establish whether one or more of these parameters might be useful to predict the risk of thrombosis in this setting.
On the other hand, despite the usefulness of cytoreductive therapy in preventing thrombosis development (24–26), controversy exists regarding its influence on platelet reactivity. Whereas, no difference in platelet activation was shown between patients with and without cytoreductive treatment in two different studies (16, 17), analysis of sequential samples of patients treated with hydroxyurea demonstrated a decrease in platelet-neutrophil aggregates (65) and this drug was shown to block P-selectin-triggered platelet-aggregate formation in vitro (65). Similarly, despite the relevance of JAK2-dependent signaling in MPN cellular abnormalities and the fact that the JAK1/2 inhibitor ruxolitinib reduces the thrombotic risk (66), no decrease in platelet activation markers was shown in patients treated with this drug (67). In another study, normalization of circulating platelet-derived microvesicles was noted in ruxolitinib spleen-responders (68), pointing out that the impact of JAK2 inhibitors in hemostatic parameters deserves further evaluation.
Antiplatelet Therapy
The efficacy of low-dose aspirin for the prevention of thrombotic complications in MPN highlights the key role of platelets in this setting (69). Patients with extreme thrombocytosis (>1,000 × 109/L) are at higher risk of bleeding under aspirin, mainly attributed to acquired von Willebrand disease. Screening for ristocetin cofactor activity and withholding aspirin therapy if <30% is suggested in this scenario (70). Current guidelines recommend aspirin for primary prevention in PV, high-risk ET and low-risk JAK2-mutated ET patients who have no contraindications for antiplatelet therapy (70). On the other hand, considering that aspirin treatment of CALR-positive low-risk ET patients does not reduce thrombosis and may actually increase bleeding, it is not routinely recommended for primary prophylaxis in this subgroup (71). Aspirin has also been shown to reduce thrombosis recurrence after both a first arterial or venous event (72). However, this risk remains high even after combined cytoreductive plus anticoagulant or antiplatelet therapy (73), highlighting the need for novel therapeutic antithrombotic strategies. Persistent thromboxane biosynthesis due to accelerated platelet production may lead to aspirin resistance in MPN (74). On this basis, a twice-daily schedule has been proposed for optimal platelet inhibition in patients with very high-risk features (70).
Statins have been shown to decrease the risk of both atherothrombotic events and venous thromboembolism. Among their pleiotropic actions, statins exert potent anti-inflammatory effects on the vascular wall and suppress platelet, endothelial and leukocyte activation (75). Due to these properties, statins might be relevant agents in MPN, although their role to prevent thrombotic events in this setting remains to be established (76).
Concluding Remarks
Thrombosis remains the main complication of MPN patients, with a high risk of recurrence despite adequate cytoreductive and antithrombotic treatment (73). Multiple and concomitant factors converge to increase the thrombotic risk in MPN, although the relative role of each factor may vary in the individual patient. Although progress has been made in unraveling the mechanisms underlying the thrombotic predisposition, the role of platelet and cellular activation markers in identifying patients at increased risk of thrombosis has not been established and prospective studies are needed to address this issue. Finally, the impact of new therapies on platelet and cellular activation and the potential development of novel therapeutic approaches to manage MPN coagulopathy could contribute to thrombosis prevention and result in improved patient outcome. Considering the critical role of inflammation in the vascular risk, simultaneous targeting of inflammatory pathways could potentially impact on primary or secondary prevention strategies.
Author Contributions
CM contributed to the writing of the article and prepared the figures. PH wrote the article. Both authors contributed to manuscript editing and final approval.
Funding
This work was supported by grants from the National Agency for Scientific and Technological Research (ANPCyT) and the Argentinian National Cancer Institute (INC).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rumi E, Cazzola M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood. (2017) 129:680–92. doi: 10.1182/blood-2016-10-695957
2. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. (2017) 129:667–79. doi: 10.1182/blood-2016-10-695940
3. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood. (2016) 127:1325–35. doi: 10.1182/blood-2015-11-681932
4. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. (2013) 122:2176–84. doi: 10.1182/blood-2013-03-460154
5. Falanga A, Marchetti M. Thrombosis in myeloproliferative neoplasms. Semin Thromb Hemost. (2014) 40:348–58. doi: 10.1055/s-0034-1370794
6. Hultcrantz M, Wilkes SR, Kristinsson SY, Andersson TM, Derolf ÅR, Eloranta S, et al. Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in Sweden between 1973 and 2005: a population-based study. J Clin Oncol. (2015) 33:2288–95. doi: 10.1200/JCO.2014.57.6652
7. Michiels JJ, Berneman Z, Van Bockstaele D, van der Planken M, De Raeve H, Schroyens W. Clinical and laboratory features, pathobiology of platelet-mediated thrombosis and bleeding complications, and the molecular etiology of essential thrombocythemia and polycythemia vera: therapeutic implications. Semin Thromb Hemost. (2006) 32:174–207. doi: 10.1055/s-2006-939431
8. Barbui T, Finazzi G, Carobbio A, Thiele J, Passamonti F, Rumi E, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood. (2012) 120:5128–33. doi: 10.1182/blood-2012-07-444067
9. Heller PG, Lev PR, Salim JP, Kornblihtt LI, Goette NP, Chazarreta CD, et al. JAK2V617F mutation in platelets from essential thrombocythemia patients: correlation with clinical features and analysis of STAT5 phosphorylation status. Eur J Haematol. (2006) 77:210–6. doi: 10.1111/j.1600-0609.2006.00688.x
10. Ziakas PD. Effect of JAK2 V617F on thrombotic risk in patients with essential thrombocythemia: measuring the uncertain. Haematologica. (2008) 93:1412–4. doi: 10.3324/haematol.12970
11. Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. (2018) 10:eaan8292. doi: 10.1126/scitranslmed.aan8292
12. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
13. Wautier MP, El Nemer W, Gane P, Rain JD, Cartron JP, Colin Y, et al. Increased adhesion to endothelial cells of erythrocytes from patients with polycythemia vera is mediated by laminin alpha5 chain and Lu/BCAM. Blood. (2007) 110:894–901. doi: 10.1182/blood-2006-10-048298
14. Walton BL, Lehmann M, Skorczewski T, Holle LA, Beckman JD, Cribb JA, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. (2017) 129:2537–46. doi: 10.1182/blood-2016-10-746479
15. Carobbio A, Thiele J, Passamonti F, Rumi E, Ruggeri M, Rodeghiero F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. (2011) 117:5857–9. doi: 10.1182/blood-2011-02-339002
16. Falanga A, Marchetti M, Evangelista V, Vignoli A, Licini M, Balicco M, et al. Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood. (2000) 96:4261–6.
17. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Villamor N, Colomer D, Cervantes F. Increased platelet and leukocyte activation as contributing mechanisms for thrombosis in essential thrombocythemia and correlation with the JAK2 mutational status. Haematologica. (2006) 91:169–75.
18. Goette NP, Lev PR, Heller PG, Kornblihtt LI, Korin L, Molinas FC, et al. Monocyte IL-2Ralpha expression is associated with thrombosis and the JAK2V617F mutation in myeloproliferative neoplasms. Cytokine. (2010) 51:67–72. doi: 10.1016/j.cyto.2010.04.011
19. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. (2011) 29:1356–63. doi: 10.1200/JCO.2010.32.9490
20. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
21. Barbui T, Carobbio A, Finazzi G, Vannucchi AM, Barosi G, Antonioli E, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. (2011) 96:315–8. doi: 10.3324/haematol.2010.031070
22. Campbell PJ, MacLean C, Beer PA, Buck G, Wheatley K, Kiladjian JJ, et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood. (2012) 120:1409–11. doi: 10.1182/blood-2012-04-424911
23. Carobbio A, Finazzi G, Antonioli E, Guglielmelli P, Vannucchi AM, Delaini F, et al. Thrombocytosis and leukocytosis interaction in vascular complications of essential thrombocythemia. Blood. (2008) 112:3135–7. doi: 10.1182/blood-2008-04-153783
24. Cortelazzo S, Finazzi G, Ruggeri M, Vestri O, Galli M, Rodeghiero F, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. (1995) 332:1132–6. doi: 10.1056/NEJM199504273321704
25. Mela Osorio MJ, Ferrari L, Goette NP, Gutierrez MI, Glembotsky AC, Maldonado AC, et al. Long-term follow-up of essential thrombocythemia patients treated with anagrelide: subgroup analysis according to JAK2/CALR/MPL mutational status. Eur J Haematol. (2016) 96:435–42. doi: 10.1111/ejh.12614
26. Gowin K, Thapaliya P, Samuelson J, Harrison C, Radia D, Andreasson B, et al. Experience with pegylated interferon α-2a in advanced myeloproliferative neoplasms in an international cohort of 118 patients. Haematologica. (2012) 97:1570–3. doi: 10.3324/haematol.2011.061390
27. Barosi G, Mesa R, Finazzi G, Harrison C, Kiladjian JJ, Lengfelder E, et al. Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood. (2013) 121:4778–81. doi: 10.1182/blood-2013-01-478891
28. Falanga A, Marchetti M, Vignoli A, Balducci D, Barbui T. Leukocyte-platelet interaction in patients with essential thrombocythemia and polycythemia vera. Exp Hematol. (2005) 33:523–30. doi: 10.1016/j.exphem.2005.01.015
29. Arellano-Rodrigo E, Alvarez-Larrán A, Reverter JC, Colomer D, Villamor N, Bellosillo B, et al. Platelet turnover, coagulation factors, and soluble markers of platelet and endothelial activation in essential thrombocythemia: relationship with thrombosis occurrence and JAK2 V617F allele burden. Am J Hematol. (2009) 84:102–8. doi: 10.1002/ajh.21338
30. Laguna MS, Kornblihtt LI, Marta RF, Molinas FC. Thromboxane B2 and platelet derived growth factor in essential thrombocythemia treated with anagrelide. Medicina. (2000) 60:448–52.
31. Alvarez-Larrán A, Arellano-Rodrigo E, Reverter JC, Domingo A, Villamor N, Colomer D, et al. Increased platelet, leukocyte, and coagulation activation in primary myelofibrosis. Ann Hematol. (2008) 87:269–76. doi: 10.1007/s00277-007-0386-3
32. Lev PR, Marta RF, Vassallu P, Molinas FC. Variation of PDGF, TGFbeta, and bFGF levels in essential thrombocythemia patients treated with anagrelide. Am J Hematol. (2002) 70:85–91. doi: 10.1002/ajh.10091
33. van Genderen PJ, Lucas IS, van Strik R, Vuzevski VD, Prins FJ, van Vliet HH, et al. Erythromelalgia in essential thrombocythemia is characterized by platelet activation and endothelial cell damage but not by thrombin generation. Thromb Haemost. (1996) 76:333–8. doi: 10.1055/s-0038-1650579
34. Rodriguez-Linares B, Watson SP. Thrombopoietin potentiates activation of human platelets in association with JAK2 and TYK2 phosphorylation. Biochem J. (1996) 316:93–8. doi: 10.1042/bj3160093
35. Panova-Noeva M, Marchetti M, Buoro S, Russo L, Leuzzi A, Finazzi G, et al. JAK2V617F mutation and hydroxyurea treatment as determinants of immature platelet parameters in essential thrombocythemia and polycythemia vera patients. Blood. (2011) 118:2599–601. doi: 10.1182/blood-2011-02-339655
36. Rondina MT, Weyrich AS. Regulation of the genetic code in megakaryocytes and platelets. J Thromb Haemost. (2015) 13 (Suppl 1):S26–32. doi: 10.1111/jth.12965
37. Zini R, Guglielmelli P, Pietra D, Rumi E, Rossi C, Rontauroli S, et al. CALR mutational status identifies different disease subtypes of essential thrombocythemia showing distinct expression profiles. Blood Cancer J. (2017) 7:638. doi: 10.1038/s41408-017-0010-2
38. Jensen MK, de Nully Brown P, Lund BV, Nielsen OJ, Hasselbalch HC. Increased platelet activation and abnormal membrane glycoprotein content and redistribution in myeloproliferative disorders. Br J Haematol. (2000) 110:116–24. doi: 10.1046/j.1365-2141.2000.02030.x
39. Moore SF, Hunter RW, Harper MT, Savage JS, Siddiq S, Westbury SK, et al. Dysfunction of the PI3 kinase/Rap1/integrin α(IIb)β(3) pathway underlies ex vivo platelet hypoactivity in essential thrombocythemia. Blood. (2013) 121:1209–19. doi: 10.1182/blood-2012-05-431288
40. Avram S, Lupu A, Angelescu S, Olteanu N, Mut-Popescu D. Abnormalities of platelet aggregation in chronic myeloproliferative disorders. J Cell Mol Med. (2001) 5:79–87. doi: 10.1111/j.1582-4934.2001.tb00140.x
41. Grignani C, Noris P, Tinelli C, Barosi G, Balduini CL. In vitro platelet aggregation defects in patients with myeloproliferative disorders and high platelet counts: are they laboratory artefacts? Platelets. (2009) 20:131–4. doi: 10.1080/09537100802691544
42. Lussana F, Femia EA, Pugliano M, Podda G, Razzari C, Maugeri N, et al. Evaluation of platelet function in essential thrombocythemia under different analytical conditions. Platelets. (2019) 1–8. doi: 10.1080/09537104.2019.1584668. [Epub ahead of print].
43. Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HH. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. (2006) 32:589–604. doi: 10.1055/s-2006-949664
44. Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. (2018) 122:337–51. doi: 10.1161/CIRCRESAHA.117.310795
45. Hurtado-Nedelec M, Csillag-Grange MJ, Boussetta T, Belambri SA, Fay M, Cassinat B, et al. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica. (2013) 98:1517–24. doi: 10.3324/haematol.2012.082560
46. Marin Oyarzún CP, Carestia A, Lev PR, Glembotsky AC, Castro Ríos MA, Moiraghi B, et al. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci Rep. (2016) 6:38738. doi: 10.1038/srep38738
47. Pfeiler S, Stark K, Massberg S, Engelmann B. Propagation of thrombosis by neutrophils and extracellular nucleosome networks. Haematologica. (2017) 102:206–13. doi: 10.3324/haematol.2016.142471
48. Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci USA. (1994) 91:8767–71. doi: 10.1073/pnas.91.19.8767
49. Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, et al. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest. (1996) 97:1525–34. doi: 10.1172/JCI118575
50. Fisher DAC, Miner CA, Engle EK, Hu H, Collins TB, Zhou A, et al. Cytokine production in myelofibrosis exhibits differential responsiveness to JAK-STAT, MAP kinase, and NFκB signaling. Leukemia. (2019). doi: 10.1038/s41375-019-0379-y. [Epub ahead of print].
51. Rosti V, Villani L, Riboni R, Poletto V, Bonetti E, Tozzi L, et al. Spleen endothelial cells from patients with myelofibrosis harbor the JAK2V617F mutation. Blood. (2013) 121:360–8. doi: 10.1182/blood-2012-01-404889
52. Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. (2009) 113:5246–9. doi: 10.1182/blood-2008-11-191544
53. Guadall A, Lesteven E, Letort G, Awan Toor S, Delord M, Pognant D, et al. Endothelial cells harbouring the JAK2V617F mutation display pro-adherent and pro-thrombotic features. Thromb Haemost. (2018) 118:1586–99. doi: 10.1055/s-0038-1667015
54. Guy A, Gourdou-Latyszenok V, Le Lay N, Peghaire C, Kilani B, Dias JV, et al. Vascular endothelial cell expression of JAK2V617F is sufficient to promote a pro-thrombotic state due to increased P-selectin expression. Haematologica. (2019) 104:70–81. doi: 10.3324/haematol.2018.195321
55. Panova-Noeva M, Marchetti M, Spronk HM, Russo L, Diani E, Finazzi G, et al. Platelet-induced thrombin generation by the calibrated automated thrombogram assay is increased in patients with essential thrombocythemia and polycythemia vera. Am J Hematol. (2011) 86:337–42. doi: 10.1002/ajh.21974
56. Falanga A, Marchetti M, Vignoli A, Balducci D, Russo L, Guerini V, et al. V617F JAK-2 mutation in patients with essential thrombocythemia: relation to platelet, granulocyte, and plasma hemostatic and inflammatory molecules. Exp Hematol. (2007) 35:702–11. doi: 10.1016/j.exphem.2007.01.053
57. Trappenburg MC, van Schilfgaarde M, Marchetti M, Spronk HM, ten Cate H, Leyte A, et al. Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia. Haematologica. (2009) 94:911–8. doi: 10.3324/haematol.13774
58. Kleppe M, Koche R, Zou L, van Galen P, Hill CE, Dong L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. (2018) 33:29–43.e7. doi: 10.1016/j.ccell.2018.03.024
59. Beaulieu LM, Lin E, Mick E, Koupenova M, Weinberg EO, Kramer CD, et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol. (2014) 34:552–64. doi: 10.1161/ATVBAHA.113.302700
60. Landtblom AR, Bower H, Andersson TM, Dickman PW, Samuelsson J, Björkholm M, et al. Second malignancies in patients with myeloproliferative neoplasms: a population-based cohort study of 9379 patients. Leukemia. (2018) 32:2203–10. doi: 10.1038/s41375-018-0027-y
61. Hasselbalch HC. The platelet-cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk Res. (2014) 38:1230–6. doi: 10.1016/j.leukres.2014.07.006
62. Kopp HG, Placke T, Salih HR. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. (2009) 69:7775–83. doi: 10.1158/0008-5472.CAN-09-2123
63. Torregrosa JM, Ferrer-Marín F, Lozano ML, Moreno MJ, Martinez C, Anton AI, et al. Impaired leucocyte activation is underlining the lower thrombotic risk of essential thrombocythaemia patients with CALR mutations as compared with those with the JAK2 mutation. Br J Haematol. (2016) 172:813–5. doi: 10.1111/bjh.13539
64. Charpentier A, Lebreton A, Rauch A, Bauters A, Trillot N, Nibourel O, et al. Microparticle phenotypes are associated with driver mutations and distinct thrombotic risks in essential thrombocythemia. Haematologica. (2016) 101:e365–8. doi: 10.3324/haematol.2016.144279
65. Maugeri N, Giordano G, Petrilli MP, Fraticelli V, de Gaetano G, Cerletti C, et al. Inhibition of tissue factor expression by hydroxyurea in polymorphonuclear leukocytes from patients with myeloproliferative disorders: a new effect for an old drug? J Thromb Haemost. (2006) 4:2593–8. doi: 10.1111/j.1538-7836.2006.02194.x
66. Verstovsek S, Vannucchi AM, Griesshammer M, Masszi T, Durrant S, Passamonti F, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. (2016) 101:821–9. doi: 10.3324/haematol.2016.143644
67. Keohane C, McLornan DP, Sanchez K, Connor C, Radia D, Harrison CN. The effects of JAK inhibitor therapy upon novel markers of thrombosis in myeloproliferative neoplasms. Haematologica. (2015) 100:e348–50. doi: 10.3324/haematol.2015.128918
68. Barone M, Ricci F, Sollazzo D, Ottaviani E, Romano M, Auteri G, et al. Circulating megakaryocyte and platelet microvesicles correlate with response to ruxolitinib and distinct disease severity in patients with myelofibrosis. Br J Haematol. (2018). 185:987–91 doi: 10.1111/bjh.15682
69. Landolfi R, Marchioli R, Kutti J, Gisslinger H, Tognoni G, Patrono C, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. (2004) 350:114–24. doi: 10.1056/NEJMoa035572
70. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am J Hematol. (2019) 9:133–43. doi: 10.1002/ajh.25303
71. Alvarez-Larrán A, Pereira A, Guglielmelli P, Hernández-Boluda JC, Arellano-Rodrigo E, Ferrer-Marín F, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica. (2016) 101:926–31. doi: 10.3324/haematol.2016.146654
72. De Stefano V, Za T, Rossi E, Vannucchi AM, Ruggeri M, Elli E, et al. Recurrent thrombosis in patients with polycythemia vera and essential thrombocythemia: incidence, risk factors, and effect of treatments. Haematologica. (2008) 93:372–80. doi: 10.3324/haematol.12053
73. De Stefano V, Ruggeri M, Cervantes F, Alvarez-Larrán A, Iurlo A, Randi ML, et al. High rate of recurrent venous thromboembolism in patients with myeloproliferative neoplasms and effect of prophylaxis with vitamin K antagonists. Leukemia. (2016) 30:2032–8. doi: 10.1038/leu.2016.85
74. Pascale S, Petrucci G, Dragani A, Habib A, Zaccardi F, Pagliaccia F, et al. Aspirin-insensitive thromboxane biosynthesis in essential thrombocythemia is explained by accelerated renewal of the drug target. Blood. (2012) 119:3595–603. doi: 10.1182/blood-2011-06-359224
75. Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C. Statins as anti-inflammatory agents in atherogenesis: molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des. (2012) 18:1519–30. doi: 10.2174/138161212799504803
Keywords: chronic myeloproliferative neoplasms, platelets, JAK2, thrombosis, inflammation, innate immunity, bleeding
Citation: Marin Oyarzún CP and Heller PG (2019) Platelets as Mediators of Thromboinflammation in Chronic Myeloproliferative Neoplasms. Front. Immunol. 10:1373. doi: 10.3389/fimmu.2019.01373
Received: 10 April 2019; Accepted: 30 May 2019;
Published: 14 June 2019.
Edited by:
Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Hans Carl Hasselbalch, Zealand University Hospital, DenmarkBo Shen, UT Southwestern Medical Center, United States
Copyright © 2019 Marin Oyarzún and Heller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paula G. Heller, cGF1bGFoZWxsZXJAaG90bWFpbC5jb20=