 Jana Meiners1
Jana Meiners1 Vittoria Palmieri1
Vittoria Palmieri1 Robert Klopfleisch2
Robert Klopfleisch2 Jana-Fabienne Ebel1Lukasz Japtok3Fabian Schumacher3,4Ayan Mohamud Yusuf5
Jana-Fabienne Ebel1Lukasz Japtok3Fabian Schumacher3,4Ayan Mohamud Yusuf5 Katrin A. Becker4Julia Zöller1
Katrin A. Becker4Julia Zöller1 Matthias Hose1Burkhard Kleuser3
Matthias Hose1Burkhard Kleuser3 Dirk M. Hermann5Richard N. Kolesnick6
Dirk M. Hermann5Richard N. Kolesnick6 Jan Buer1
Jan Buer1 Wiebke Hansen1
Wiebke Hansen1 Astrid M. Westendorf1*
Astrid M. Westendorf1*- 1Institute of Medical Microbiology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany
- 2Institute of Veterinary Pathology, Free University of Berlin, Berlin, Germany
- 3Department of Toxicology, Institute of Nutritional Science, University of Potsdam, Potsdam, Germany
- 4Department of Molecular Biology, University of Duisburg-Essen, Essen, Germany
- 5Department of Neurology, University Hospital Essen, University of Duisburg-Essen, Essen, Germany
- 6Laboratory of Signal Transduction, Memorial Sloan-Kettering Cancer Center, New York, NY, United States
Inflammatory diseases of the gastrointestinal tract are emerging as a global problem with increased evidence and prevalence in numerous countries. A dysregulated sphingolipid metabolism occurs in patients with ulcerative colitis and is discussed to contribute to its pathogenesis. In the present study, we determined the impact of acid sphingomyelinase (Asm), which catalyzes the hydrolysis of sphingomyelin to ceramide, on the course of Citrobacter (C.) rodentium-driven colitis. C. rodentium is an enteric pathogen and induces colonic inflammation very similar to the pathology in patients with ulcerative colitis. We found that mice with Asm deficiency or Asm inhibition were strongly susceptible to C. rodentium infection. These mice showed increased levels of C. rodentium in the feces and were prone to bacterial spreading to the systemic organs. In addition, mice lacking Asm activity showed an uncontrolled inflammatory Th1 and Th17 response, which was accompanied by a stronger colonic pathology compared to infected wild type mice. These findings identified Asm as an essential regulator of mucosal immunity to the enteric pathogen C. rodentium.
Introduction
Inflammatory bowel diseases (IBD), such as ulcerative colitis and Crohn's disease, are characterized by chronic, relapsing inflammatory conditions, resulting from a dysregulation of the mucosal immune system in the gastrointestinal tract (1). The exact mechanism underlying the pathogenesis of IBD is unknown; however, it is widely accepted that immunological abnormalities, genetic and environmental factors, as well as infections are important determinants of IBD (2).
Sphingolipids are a family of metabolic lipids that are ubiquitous in cellular membranes and include a bioactive subset that regulates various cellular mechanisms and biologic processes such as cell survival, growth, differentiation, and apoptosis (3). Interestingly, sphingolipids are also essential structural components of intestinal membranes, providing protection and integrity to the intestinal mucosa and regulating intestinal absorption processes (4, 5). Studies using common acute and chronic epithelial injury colitis models have shown that bioactive sphingolipids, particularly ceramide and sphingosine-1-phosphate, are important regulators of inflammation in IBD (6–8). The acid sphingomyelinase (Asm) is a relevant enzyme in this context, as it catalyzes the hydrolysis of sphingomyelin to ceramide. Asm is ubiquitously expressed and activated by a range of cellular stresses, including inflammatory cytokines and pathogens (9). The importance of Asm for cell functions was first recognized in Niemann-Pick disease type A and B, a genetic disorder with a severe accumulation of sphingomyelin in many organs (10). Recent studies implicated that Asm activity is also strongly involved in inflammatory processes (11). For example, Asm inhibition was shown to suppress the lipopolysaccharide (LPS) mediated release of inflammatory cytokines and to protect against disease pathology in chemical induced colitis in mice (12, 13). Furthermore, blockade of Asm bioactivity limited the in vitro differentiation of T helper cells derived from healthy volunteers and patients with Crohn's disease (14). These results implicate Asm inhibition as an innovative and effective immunoregulatory strategy for the treatment of IBD (12, 13, 15).
Nevertheless, the etiology of IBD is diverse and influenced by numerous factors (2). In this context, several enteropathogens have been implicated in the development of IBD (16), although to date, a causative bacterial agent for IBD has not been identified. Thus, further studies are needed to clarify the function of Asm under infectious and non-infectious conditions, as broad immunosuppression can increase the risk of infectious complications (17).
In the present study, we determined the impact of Asm activity on the course of Citrobacter (C.) rodentium induced colitis. In contrast to the protective effect of Asm inhibition in common acute and chronic epithelial injury colitis models, Asm inhibition or Asm deficiency strongly enhanced the susceptibility to enteric C. rodentium infection. Mice lacking Asm activity showed higher colon pathology, were prone to bacterial dissemination to the systemic organs, and showed an uncontrolled inflammatory Th1 and Th17 response compared to infected wild type mice. These findings identified Asm as a critical regulator of mucosal immunity to the enteric pathogen C. rodentium.
Materials and Methods
Mice
C57BL/6 mice were purchased from ENVIGO, Netherlands. To inhibit acid sphingomyelinase activity, amitriptyline or imipramine was administered to C57BL/6 mice at 180 mg/l via drinking water for 14 days prior to bacterial challenge, and for further 10 days of infection. Acid sphingomyelinase-deficient (Smpd1−/−) mice (18) were bred at the animal facility of the University Hospital Essen. All animals used in this study were 8–12 week old male or female mice kept in the animal experimental unit of the University Hospital Essen in individually ventilated cages and pathogen free conditions.
Asm Activity
The activity of Asm in colonic tissue was quantified using BODIPY-labeled sphingomyelin as a substrate. After colonic tissue was pulverized and lysed in 250 mM sodium acetate (pH 5) and 1% NP-40, 2 μg protein was incubated with 100 pmol BODIPY-labeled sphingomyelin (Thermo Fisher Scientific, Germany) for 1 h at 37°C in 250 mM sodium actetate (pH 5) and 0.1% NP-40. The reaction was terminated by the addition of chloroform:methanol (2:1, v:v) to extract the lipids. Subsequently, the lower phase containing lipids was collected, dried in a SpeedVac at 37°C, dissolved in 20 μl chloroform:methanol (2:1, v:v) and transferred onto a thin layer chromatography (TLC) plate. Product and uncleaved substrate were separated using chloroform:methanol (80:20, v/v). After separation, spots were imaged using a Typhoon FLA 9500 and quantified with ImageQuant software.
Ceramide and Sphingomyelin Quantification
Ceramide and sphingomyelin concentrations were quantified by rapid resolution liquid chromatography/mass spectrometry. Short lipids were extracted from colon biopsies with C17-ceramide and C16-d31sphingomyelin as internal standards, after homogenization of colonic tissue. Subsequently, samples were analyzed by rapid-resolution liquid chromatography-MS/MS using a Q-TOF 6530 mass spectrometer (Agilent Technologies, Waldbronn, Germany) operating in the positive ESI mode. The subsequent quantification was performed using Mass Hunter Software, and the resulting sphingolipid quantities were normalized to the actual protein content of the homogenate.
C. rodentium Infection Model
C. rodentium ICC169 strain was cultured overnight in Luria-Bertani (LB) medium at 37°C, centrifuged and washed with PBS. Mice were infected by oral gavage with ~2 × 109 colony forming units (CFUs) of C. rodentium. After gavage, an aliquot of the bacteria was plated in serial dilutions on MacConkey agar. Bacterial numbers in the feces were determined at indicated time points after infection. Mice were analyzed at various time points after infection (d.p. infection).
Intestinal Permeability-Assay
For in vivo analysis of the intestinal permeability fluorescein isothiocyanate-conjugated (FITC)-dextran beads have been used. Briefly, food and water were withdrawn for 2 h and mice were orally administrated with permeability tracer (60 mg/100 g body weight of FITC-labeled dextran, MW 4000; FD4, Sigma-Aldrich, St. Louis, USA). Serum was collected 4 h later and fluorescence intensity was determined (excitation, 492 nm; emission, 525 nm; BioTek). FITC-dextran concentrations were determined using a standard curve generated by serial dilution of FITC-dextran.
Isolation of Splenocytes and Mesenteric Lymph Node Cells
Spleens were rinsed with an erythrocyte lysis buffer [containing 0.15 M NH4Cl, 10 mM KHCO3, and 0.5 M ethylenediaminetetraacetic acid (EDTA)], meshed through a 100-μm cell strainer, and washed with PBS containing 2 mM EDTA and 2% fetal calf serum (FCS). Mesenteric lymph nodes (mLN) were meshed through a 100-μm cell strainer and washed with PBS containing 2 mM EDTA and 2% FCS.
Isolation of Lamina Propria Lymphocytes From the Colon
Lamina propria (LP) lymphocytes were isolated as described previously (19). In brief, colons were flushed with PBS, opened longitudinally, and cut into 1-cm pieces. Tissue pieces were washed twice in PBS containing 3 mM EDTA for 10 min at 37°C and twice in Roswell Park Memorial Institute (RPMI) medium containing 1% FCS, 1 mM EGTA, and 1.5 mM MgCl2 for 15 min at 37°C. Colon pieces were intensively vortexed, washed with phosphate-buffered saline (PBS), and digested in RPMI containing 20% FCS and 100 U/mL collagenase (Clostridium histolyticum; Sigma-Aldrich, St. Louis, MO) for 60 min at 37°C. Cell suspension was passed through a 40-μm cell strainer and washed with culture medium.
Macroscopic and Histopathologic Assessment of Colitis
Colonic damage was assessed based on two main characteristics: colon length and colon weight. Colons were embedded in paraffin, and tissue sections (4 μm) were prepared for histological scoring in a blinded manner. The colon was divided into three equal portions (oral, middle, and rectal) and assessed for inflammatory cell infiltrates, epithelial damage, neutrophil infiltration, crypt abscesses, and crypt hyperplasia as describe before (20). Crypt heights were measured by micrometry; 30 measurements were taken in the distal colon for each mouse.
Cytokine Detection
Cytokines in serum samples were quantified using a Procarta Cytokine assay kit, according to the manufacturer's guidelines. The assay was run on a Luminex 200 system and cytokine levels were quantified using the Luminex IS software (Luminex Corporation, Austin, TX).
Antibodies and Flow Cytometry
Cells were stained with fluorochrome-labeled anti-mouse CD4 (RM4-5), CD11b (M1/70), F4/80 (BM8), FoxP3 (FJK-16s), IFNγ (XMG1.2), IL-17 (TC11-18H10.1), and I-A/I-E (M5/114.15.2) antibodies and analyzed by flow cytometry on an LSR II instrument using DIVA software (BD Biosciences).
Statistical Analysis
Normality of data was tested using D'Agostino & Pearson normality test and Shapiro–Wilk normality test. Statistical analysis was performed using Student's t-test, one-way ANOVA or two-way ANOVA followed by Tukey's multiple comparisons test, Dunn's multiple comparisons test or Bonferroni's multiple comparisons test. P-values were set at a level of p < 0.05. Statistical analyzes were performed using GraphPad Prism software version 7.
Ethics Statement
This study was carried out in accordance with the recommendations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocol was approved by the North Rhine-Westphalia State Agency for Nature, Environment and Consumer Protection (LANUF), Germany.
Results
Alterations of the Sphingolipid Profile During C. rodentium Infection
Sphingolipids have been identified as important players to control intestinal inflammation. There is increasing evidence that a dyregulaton of several sphingolipid molecules occurs along with IBD and contributes to the pathogenesis and maintenance of the disease (21). To analyze the impact of the sphingolipid metabolism on pathogen-driven intestinal inflammation, C57BL/6 wild type (WT) mice were infected via oral gavage with ~2 × 109 CFUs C. rodentium, and the sphingomyelin and ceramide concentrations were determined in the colon at indicated time points post infection by mass spectrometry. Interestingly, sphingomyelin as well as ceramide concentrations in the colon decreased during the course of infection in comparison to non-infected WT mice (Figures 1A,B), suggesting an involvement of the sphingolipid metabolism in intestinal inflammation.
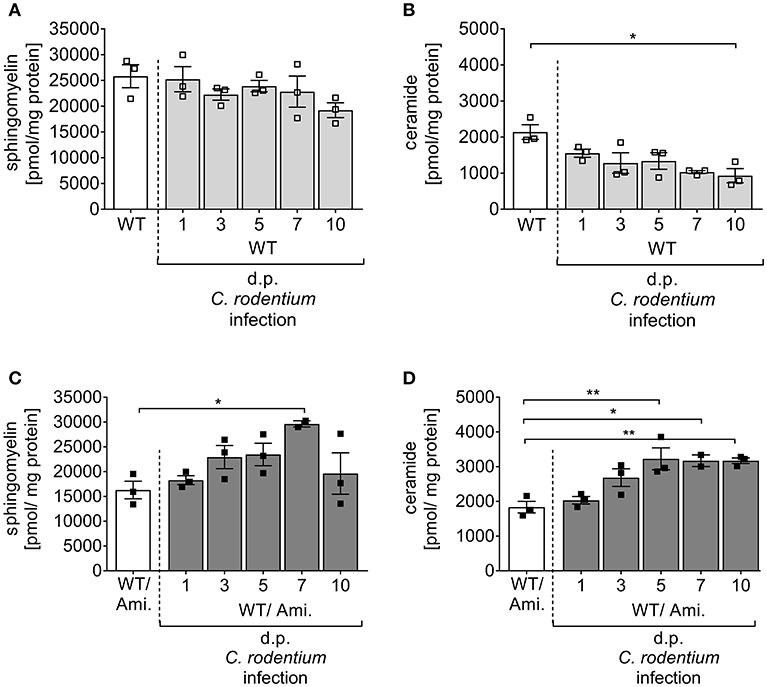
Figure 1. Alterations of sphingolipid concentrations during C. rodentium infection. C57BL/6 mice were either left untreated (WT) or pre-treated with 180 mg/l amitriptyline in drinking water 2 weeks prior to infection (WT/Ami) and during the course of infection. Mice were orally gavaged with PBS or with ~2 × 109 colony forming units (CFUs) of C. rodentium. At indicated days post (dp) infection colons were excised. In rectal colonic tissue, the (A,C) sphingomyelin and (B,D) ceramide contents were analyzed using rapid resolution liquid chromatography/mass spectrometry. Data are presented as the concentration of ceramide and sphingomyelin in pmol/mg protein. All data are presented as mean ± SEM. Statistics were performed using the parametric one-way ANOVA test with Tukey's multiple comparison test (*p < 0.05; **p < 0.01).
Oral Amitriptyline Pre-treatment Inhibits the Colonic Asm Activity During C. rodentium Infection
Acid sphingomyelinase (Asm) metabolizes sphingomyelin into ceramide, and this enzyme can be pharmacologically inhibited by amitriptyline (Ami) (22, 23). Amitriptyline accumulates in the acid compartments of the lysosomes, interferes with the translocation of Asm to the outer leaflet of the membrane and inhibits the activation of Asm (9). As sphingomyelin and ceramide were downregulated during the course of C. rodentium infection, we tested if the inhibition of Asm protects from pathogen-driven intestinal inflammation. For the specific in vivo inhibition of Asm in the gastrointestinal tract, WT mice were provided with amitriptyline in the drinking water for 14 days before and during a 10 day course of infection (WT/Ami). To check for the inhibition of the Asm activity, colonic tissues of WT/Ami mice, infected and non-infected, were analyzed for their sphingolipid profile by mass spectroscopy during the course of infection. Although no differences in the sphingomyelin and ceramide concentration were discovered under homeostasis, we detected an accumulation of sphingomyelin and ceramide in the colon of infected WT/Ami mice with constant increase during the course of infection (Figures 1C,D). This data clearly demonstrated a modulation of the sphingolipid pathway in the colonic tissue by amitriptyline treatment via the drinking water.
Asm Inhibition Increases the Susceptibility to C. rodentium Infection
To further elucidate the physiological effect of Asm inhibition on the course of infection, we characterized the C. rodentium induced inflammatory response in WT and WT/Ami mice in detail. First, we determined the body weight loss of the mice as indicator for diarrhea. Surprisingly, C. rodentium infected WT/Ami mice lost significantly more body weight within 5 and 10 days post infection compared to infected wild type mice (Figure 2A). Determination of the bacterial burden in the feces showed that bacterial loads were similar in WT and WT/Ami infected mice on day 3 post infection with 107 CFUs/g of feces. However, C. rodentium infected WT/Ami mice exhibited significantly higher bacterial numbers at day 5 and 7 post infection than did infected WT mice (Figure 2B), suggesting an impairment in the clearance of the pathogen. Interestingly, reduced bacterial eradication in WT/Ami mice was associated with exaggerated inflammation, characterized phenotypically by higher spleen weights (Figure 2C) and significantly higher colon weight-to-length ratios (Figure 2D) than in infected WT mice 10 days post infection. Well in line, histological analysis of the colon showed that C. rodentium-infected WT/Ami mice exhibited more severe crypt elongation and crypt hyperplasia (Figures 2E,F) and a higher inflammatory score compared to infected WT mice (Figure 2G). Of note, the same phenotype was obtained when infected mice were treated daily with amitriptyline via intraperitoneal injection (data not shown). These findings indicate that Asm activity is not only important for the eradication of C. rodentium but also for the control of infection-associated inflammation. Therefore, inhibition of Asm is not protective in pathogen-driven intestinal inflammation.
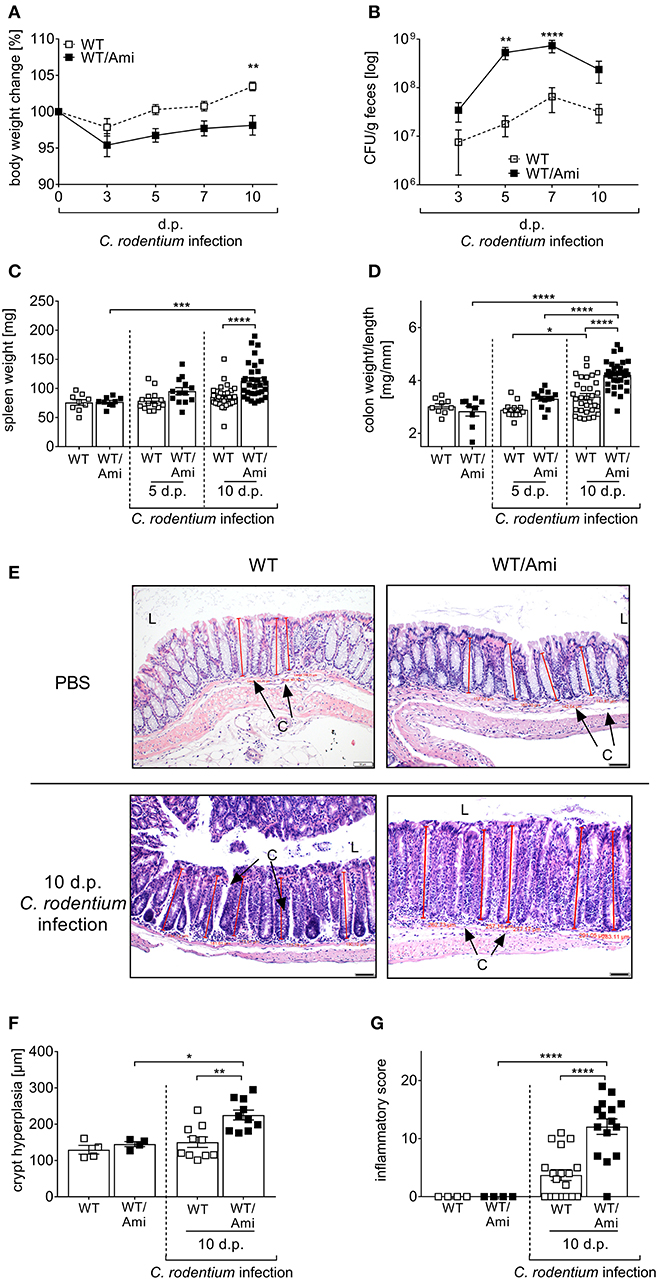
Figure 2. Amitriptyline pre-treatment increases the susceptibility to C. rodentium challenge. C57BL/6 mice were treated as described in Figure 1. (A,B) At indicated time points body weight and CFUs in feces in WT mice (white squares; n = 32–33) and WT/Ami mice (black squares; n = 32–35) were assessed. Statistics were performed using the Mann–Whitney test. (C) Spleen weight (n = 9–32) and (D) colon weight-to-length ratio were determined in uninfected, untreated and uninfected amitriptyline treated mice 5 and 10 dp C. rodentium infection (n = 9–32). (E) Representative H&E staining of colon sections from PBS or C. rodentium infected WT or WT/Ami mice 10 dp challenge. [Red lines indicate the crypt length. (L) lumen, (C) crypt. Length of scale bar is 50 μm]. (F) Measured crypt length in colons of uninfected WT or WT/Ami mice, and infected WT and WT/Ami 10 dp C. rodentium infection (n = 4–18). (G) Histopathology score of uninfected WT and WT/Ami mice, and WT and WT/Ami mice 10 dp C. rodentium infection (n = 4–18). All data are presented as mean ± SEM. Statistics were performed using the two-way ANOVA test followed by Tukey's multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Asm Provides Host Resistance to Bacterial Dissemination
A consequence of certain enteric bacterial infections is a breakdown of the intestinal barrier, allowing pathogen spreading from the intestine to the systemic organs of a host. Therefore, we tested whether Asm inhibition affects the intestinal permeability in vivo. Non-infected and infected WT and WT/Ami mice were orally gavaged with FITC-labeled dextran beads and 4 h later the intestinal permeability was assessed as relative concentration of serum FITC-dextran. Interestingly, the FITC concentrations in the serum of WT/Ami mice were significantly increased when compared to non-infected WT/Ami controls (Figure 3B). In contrast, only a slight but not significant increase in serum FITC levels was detected in infected WT mice compared to non-infected WT mice (Figure 3A).
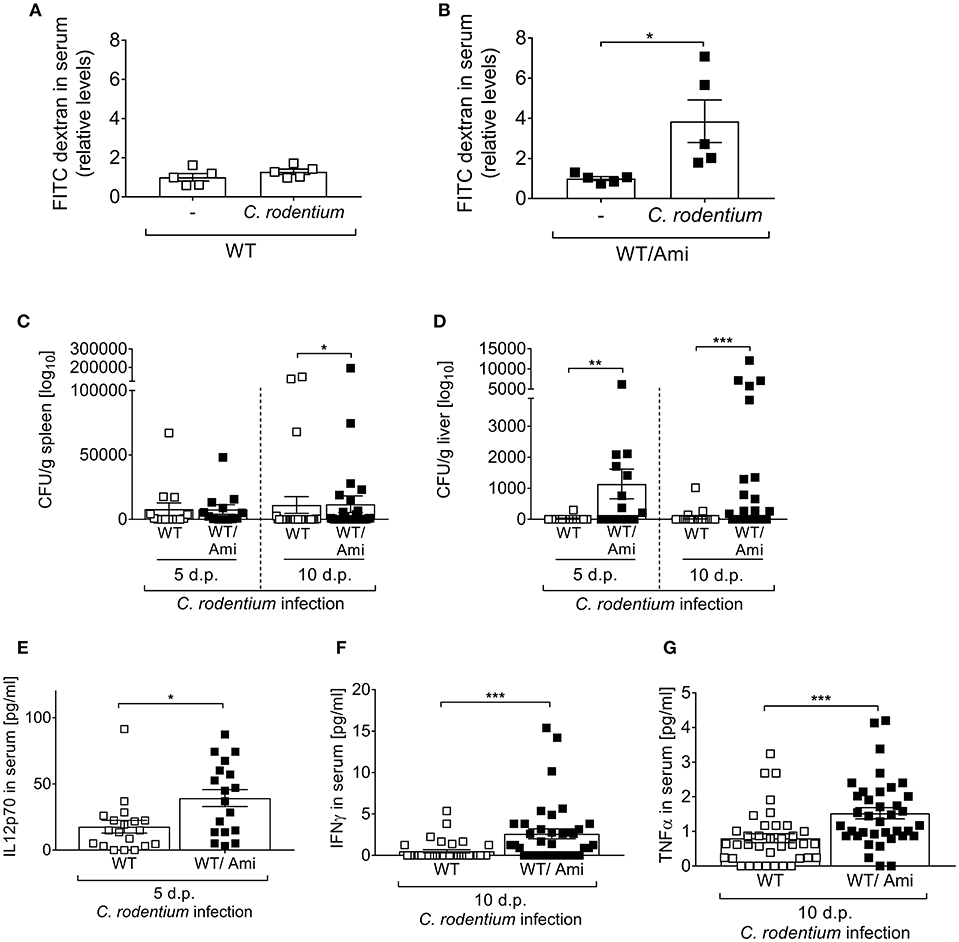
Figure 3. Enhanced systemic distribution of C. rodentium in mice pre-treated with amitriptyline. C57BL/6 mice were treated and infected as described in Figure 1. (A,B) Mice were orally administrated with FITC-labeled dextran beads. Serum was collected 4 h later, and fluorescence intensity was determined (n = 5). Serum FITC concentration in infected mice was normalized to serum FITC concentration of the respective control. (C,D) 5 and 10 dp C. rodentium infection spleen and liver were isolated and CFUs of C. rodentium were assessed (n = 13–27). (E–G) Concentration of the cytokines IL12p70, IFNγ, and TNFα in serum of WT and WT/Ami mice 10 dp C. rodentium infection were measured using Luminex technologies (n = 18–37). All data are presented as mean ± SEM. Statistics were performed using the Mann–Whitney test or Student's t-test (*p < 0.05; **p < 0.01; ***p < 0.001).
These results led us to hypothesize that bacteria, once invaded into the gastrointestinal tract, are spread to the systemic organs of infected WT/Ami mice. Thus, we infected WT and WT/Ami mice via oral gavage with ~2 × 109 CFUs C. rodentium per mouse, harvested the spleen and liver 5 and 10 days post-infection and analyzed the presence of viable bacteria. Well in line with the permeability assay, a higher bacterial burden was observed in the livers and spleens of infected WT/Ami mice compared to infected WT mice (Figures 3C,D).
The production of protective cytokines is a hallmark of immune responses being mounted toward the infection. To assess if the systemic distribution of C. rodentium in WT/Ami mice altered the level of cytokines that have been shown to orchestrate the immune response against C. rodentium infection (24), we infected WT and WT/Ami mice and measured the cytokine levels of IL-12p70, IFNγ, and TNFα in the sera. Of note, we found significantly elevated levels of IL12p70, INFγ, and TNFα in the sera of infected WT/Ami mice on day 10 post infection compared to infected WT mice (Figures 3E–G). Hence, Asm inhibition did not impair the production of protective pro-inflammatory cytokines. The high levels of certain pro-inflammatory cytokines in infected WT/Ami mice could be a consequence of the increased number of bacteria in their colon and liver tissues.
Colonic Infiltration of Macrophages Is Not Impaired by Asm Inhibition
Intestinal macrophages and macrophage-derived IL-12 are required for the initiation of adaptive immunity in response to C. rodentium (25). To investigate the role of Asm in mucosal immunity against C. rodentium infection, we measured the frequency of macrophages in the spleen, mesenteric lymph nodes, and the colonic lamina propria of WT and WT/Ami mice infected with C. rodentium. At day 5 and day 10 post infection the frequencies of macrophages were enhanced in the spleen and the LP, but no differences were found between infected WT and infected WT/Ami mice (Figure 4A). Furthermore, the expression of MHC II, which is essential for the activation of CD4+ T cells, was not impaired in infected WT/Ami mice compared to infected WT mice (Figure 4B). These data suggest that the function of macrophages to initiate adaptive immunity is independent of Asm during C. rodentium infection.
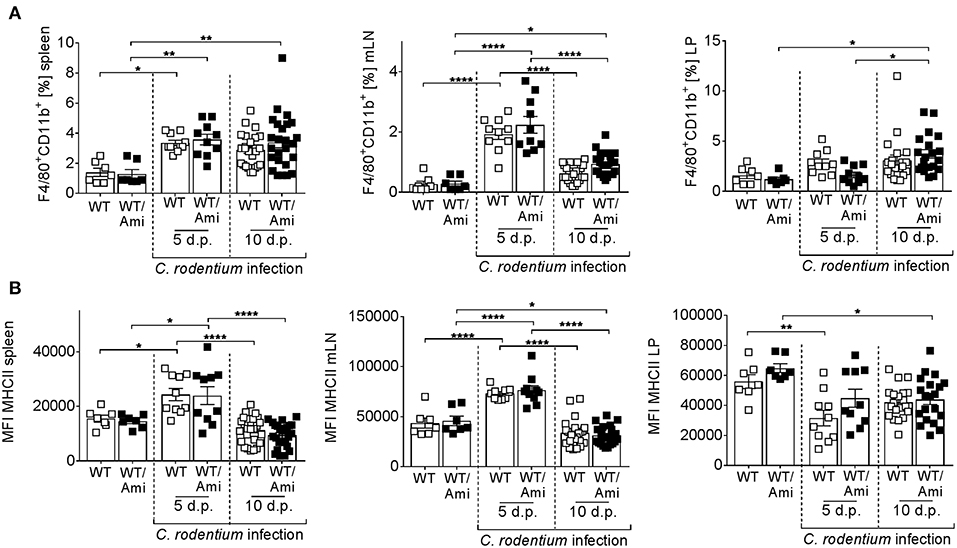
Figure 4. Infiltration of macrophages into colonic tissue is not altered in amitriptyline pre-treated mice after C. rodentium challenge. C57BL/6 mice were treated as described in Figure 2. Cells from the spleen, mesenteric lymph nodes (mLNs), and the lamina propria (LP) were isolated from uninfected WT and WT/Ami mice, 5 and 10 days post C. rodentium infection and analyzed for the frequency for macrophages by flow cytometry. (A) Percentages of macrophages from living cells are displayed (n = 7–25). (B) MFI of MHCII from macrophages is displayed (n = 7–25). All data are presented as mean ± SEM. Statistics were performed using the two-way ANOVA test with Tukey's multiple comparison test (*p < 0.05; **p < 0.01; ****p < 0.0001).
Asm Inhibition Leads to an Uncontrolled Expansion of Th1 and Th17 Cells
Infection with C. rodentium is associated with the induction of Th1 and Th17 adaptive immune responses (26–31). To measure the CD4+ T cell response during infection of WT and WT/Ami mice, splenocytes, mLN cells, and LP cells were restimulated ex vivo and analyzed for IFNγ and IL-17 production via flow cytometry. Comparable frequencies of Th1 and Th17 cells were detected in uninfected mice treated with or without amitriptyline. In contrast, C. rodentium infected mice exhibited enhanced frequencies of IFNγ- and IL-17-producing cells at day 10 post infection. Compared with infected WT controls, WT/Ami mice displayed an overall increase in activated T cells, with highest frequencies of Th1 and Th17 cells at day 10 post infection (Figures 5A,B).
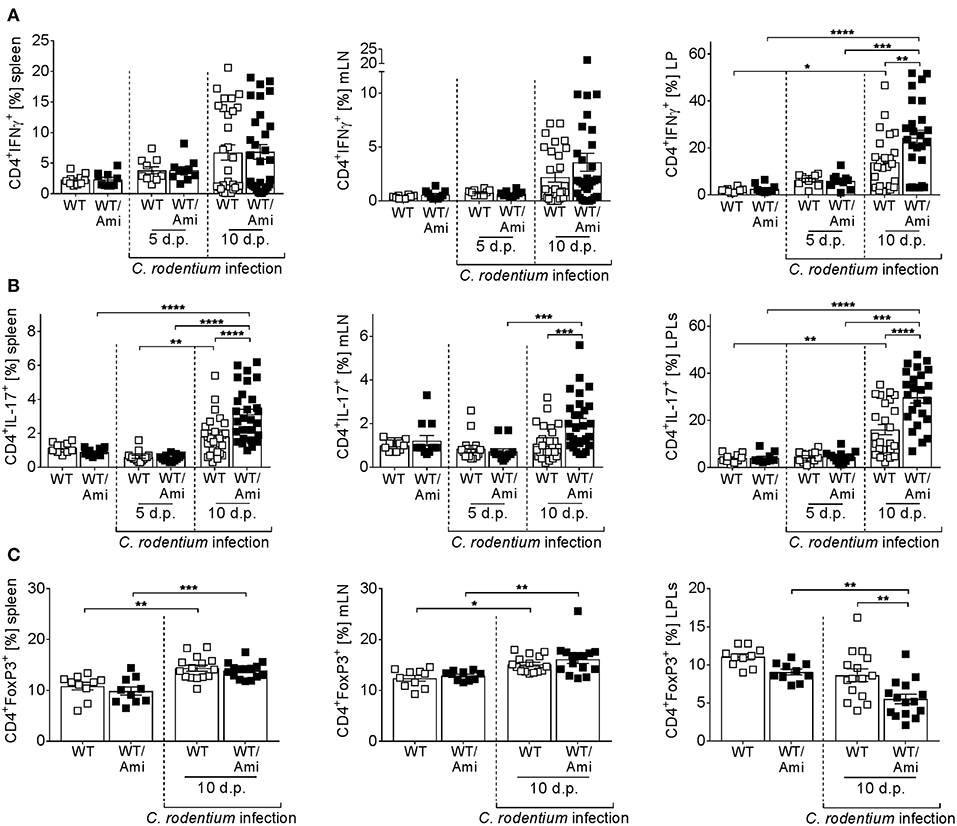
Figure 5. Enhanced proportions of Th1 and Th17 cells in colonic tissue of amitriptyline treated mice. C57BL/6 mice were treated as described in Figure 2. Single cells from spleen, mesenteric lymph nodes (mLNs), and lamina propria (LP) were prepared from uninfected WT or WT/Ami mice, 5 and 10 days post C. rodentium infection, and analyzed for (A) CD4+IFNγ+ cells (Th1), (B) CD4+IL-17+ cells (Th17), or (C) CD4+FoxP3+ cells (Tregs) by flow cytometry. Percentages of Th1, Th17, and Tregs from CD4+ T cells are displayed. All data are presented as mean ± SEM. Statistics were performed using the two-way ANOVA test with Tukey's multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Regulatory T cells (Tregs) are important to counterbalance effector T cell responses and to protect from severe pathology (32). Recently, it was shown that deficiency of Tregs enhances the susceptibility to C. rodentium infection (32). Therefore, we assessed the frequencies of Tregs in the spleen, mLN, and LP of C. rodentium infected WT and WT/Ami mice. No differences were observed in the spleens and mLN of non-infected and infected WT mice compared to infected WT/Ami mice. Nonetheless, the percentage of CD4+Foxp3+ Tregs was significantly decreased in the LP of infected WT/Ami mice compared to infected WT mice (Figure 5C). In summary, Asm inhibition enhances the colonic frequencies of Th1 and Th17 cells in C. rodentium infected mice, which seems to be a consequence of increased numbers of bacteria in the colon and a disturbed counterbalanced induction of Tregs.
Asm Knock-Out Mice Are Strongly Susceptible to C. rodentium Infection
Amitriptyline was initially introduced in order to treat major depressive disorders (33). However, nowadays the use of amitriptyline has expanded to numerous types of pain and other symptoms (34–38). In addition, anti-inflammatory and anti-microbial properties of the drug have been reported as well (39, 40). To exclude eventual drug unspecific effects, we used a second pharmacological Asm inhibitor, imipramine (Imi) within the same experimental set up. Well in line with the results from amitriptyline treated mice, imipramine treated mice are more susceptible to C. rodentium induced colitis, as shown by increased bacterial burden in the feces, enhanced colon-weight-to-length ratios and more severe pathology compared to infected WT mice (Figures 6A–E).
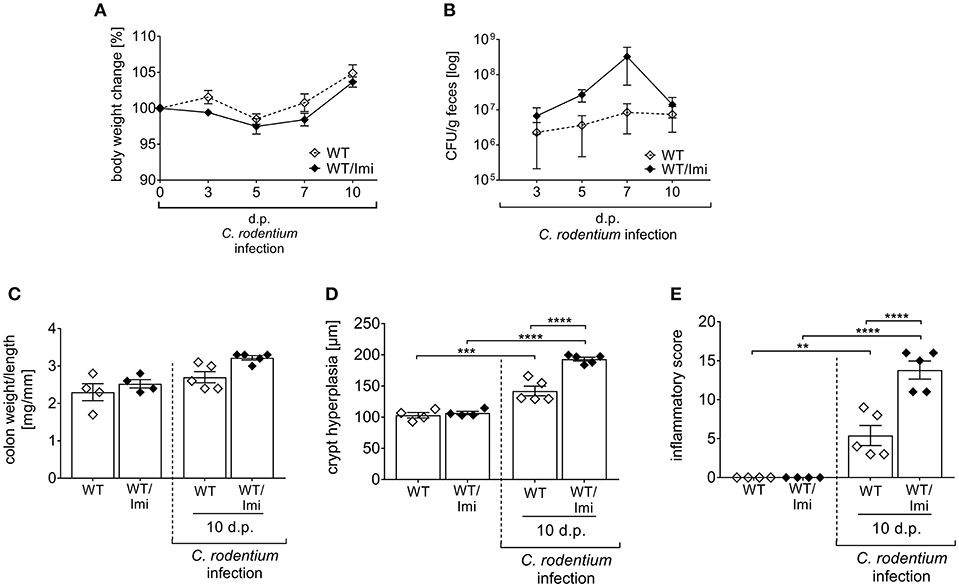
Figure 6. Imipramine pre-treatment increases the susceptibility to C. rodentium challenge. C57BL/6 mice were either left untreated (WT) or pre-treated with 180 mg/l imipramine in drinking water 2 weeks prior to infection (WT/Imi) and during the course of infection. Mice were orally gavaged with PBS or with ~2 × 109 colony forming units (CFUs) of C. rodentium. (A,B) At indicated time points body weight and CFUs in feces in WT mice (white diamonds; n = 4–5) and WT/Imi mice (black diamonds; n = 4–5) were assessed. Statistics were performed using the Mann–Whitney test. (C) Colon weight-to-length ratio were determined in uninfected, untreated, and uninfected imipramine treated mice 10 dp C. rodentium infection (n = 4–5). Crypt hyperplasia (D) and inflammation score (E) of uninfected WT and WT/Imi mice, and WT and WT/Imi mice 10 dp C. rodentium infection (n = 4–5). All data are presented as mean ± SEM. Statistics were performed using the one-way ANOVA test followed by Tukey's multiple comparison test (**p < 0.01; ***p < 0.001; ****p < 0.0001).
To prove that the effect in amitriptyline and imipramine treated animals on C. rodentium induced inflammation is specifically due to the inhibition of Asm, we repeated the infection experiments in Asm wild type (Asm WT) and Asm knock-out (Asm KO) mice. First, we confirmed the specific depletion of Asm in Asm KO mice under homeostasis and in infected animals. Therefore, the Asm activity as well as sphingomyelin and ceramide concentrations were determined in the colon. As expected, we observed no Asm activity accompanied with a strong accumulation of sphingomyelin in non-infected and infected Asm KO mice and only slight changes in the ceramide concentrations (Figures 7A–C). To approve the physiological effect of Asm deficiency in comparison to amitriptyline mediated inhibition of Asm, we infected Asm WT and Asm KO mice via oral gavage with ~2 × 109 CFUs C. rodentium per mouse and assessed the body weight, bacterial burden, spleen weight, colon weight-to-length ratio, and the histopathological score. Importantly, non-infected Asm WT mice and Asm KO mice exhibited no difference regarding the analyzed parameters (Figures 7D–F). In contrast, we observed an enhanced loss of body weight at day 10 post infection in Asm KO mice compared to Asm WT mice (Figure 7D). Determination of the bacterial burden in the feces showed that bacterial loads were higher in infected Asm KO mice on day 5 and 10 post infection compared to infected Asm WT mice (Figure 7G). In addition, a tendency of systemic dissemination in the liver and the spleen was observed in infected Asm KO mice but not in infected WT mice (Figures 7H,I). Well in line, also spleen weights and colon weight-to-length ratios were higher in infected Asm KO mice than in Asm WT mice (Figures 7E,F). Finally, the histopathological analysis of colon tissues revealed significantly elevated crypt hyperplasia and inflammatory scores in infected Asm KO mice compared to infected Asm WT mice (Figures 7J–L). In summary, Asm KO mice are strongly susceptible to C. rodentium infection and show the same histopathological phenotype as infected amitriptyline treated animals.
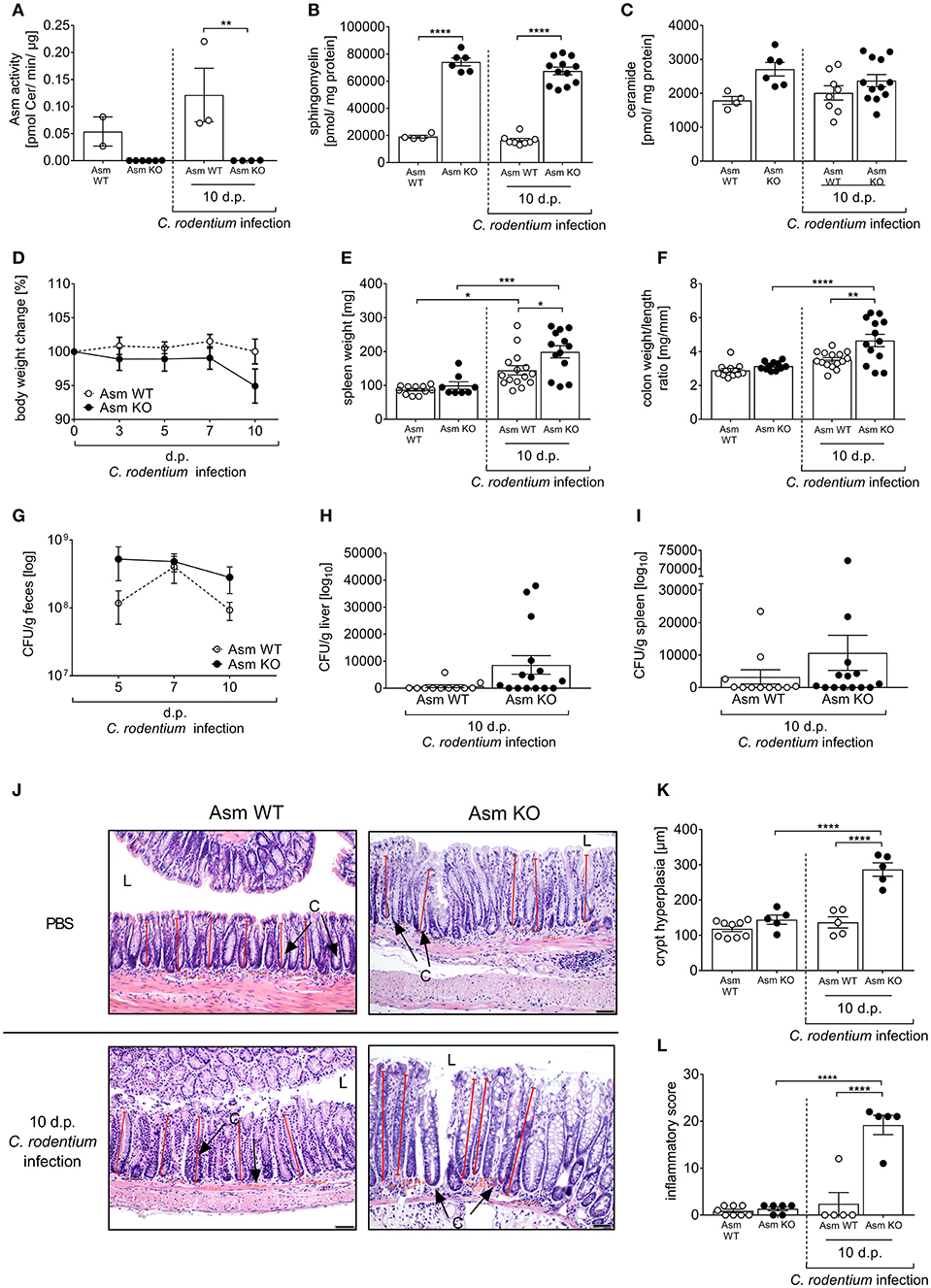
Figure 7. Deficiency of acid sphingomyelinase (Asm) increases susceptibility to C. rodentium infection. Asm wildtype (Asm WT) and Asm knock-out (Asm KO) mice were orally gavaged with PBS or ~2 × 109 colony forming units (CFUs) of C. rodentium. Colons were excised from uninfected Asm WT and Asm KO mice and 10 dp infection. (A) Asm activity was analyzed in rectal colonic tissue (n = 2–6) by rapid resolution liquid chromatography/mass spectrometry. (B) Sphingomyelin (n = 4–12) (C) and ceramide (n = 4–12) concentrations are displayed. (D) At indicated time points body weight was assessed in Asm WT mice (n = 16–17) and Asm KO mice (n = 14–20). Statistics were performed using the Mann–Whitney test or the Student's t-test. (E) Spleen weight (n = 8–15) and (F) colon weight-to-length ratio were determined in uninfected, untreated, and uninfected amitriptyline treated mice 5 and 10 dp C. rodentium infection (n = 11–15). (G–I) At indicated time points CFUs in feces, liver, and spleen of Asm WT mice (n = 11–12) and Asm KO mice (n = 14–15) were assessed. Statistics were performed using the Mann–Whitney test or the Student's t-test. (J) Representative H&E staining of colon sections from PBS or C. rodentium infected Asm WT or Asm KO mice 10 dp infection. [Red lines indicate the crypt length. (L) lumen, (C) crypt. Length of scale bar−50 μm]. (K) Crypt length in colons of uninfected Asm WT or Asm KO mice, and infected Asm WT and Asm KO 10 dp C. rodentium infection (n = 5–9). (L) Histopathology score of the colon of uninfected Asm WT and Asm KO mice, and Asm WT and Asm KO mice 10 dp C. rodentium infection (n = 5–8). All data are presented as mean ± SEM. Statistics were performed using the two-way ANOVA test followed by Tukey's multiple comparison test (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Discussion
The gastrointestinal tract is the largest mucosal surface in the human body, fulfilling the pivotal role of nutrition and water absorption. Pathogens preferentially invade the host through the gastrointestinal tract forcing it to distinguish between harmless and beneficial bacteria. Thus, the gastrointestinal tract adapted to these unique circumstances by limiting direct bacterial contact to the epithelial cell surface, rapid detection and killing of invading bacteria, and minimizing the exposure of commensal bacteria to the immune system (41). Dysregulation of this uniquely balanced system can lead to chronic inflammation, resulting in IBD (42). Although the exact etiology of IBD remains unclear, the role of sphingolipids in contributing to the inflammatory process is evident (4, 5, 43–47). Asm is of particular interest, as it catalyzes the hydrolysis of sphingomyelin to ceramide, which is the central molecule in the sphingolipid metabolism. Thus, research on therapeutic agents able to modulate Asm and tissue-specific delivery systems or application routes is mandatory (47).
The importance of Asm was first recognized in Niemann-Pick disease type A and B, also called Acid Sphingomyelinase Deficiency. Thus, sphingomyelin cannot be metabolized properly and is accumulated within cells, eventually causing cell death and the malfunction of major organ systems (10). Of note, recent studies point out that patients with Niemann-Pick disease are susceptible to pathogen infections (14, 48), indicating the association between deficiency and aberrant immune responses. Well in line, in the present study we identified Asm as a critical regulator of mucosal immunity to the C. rodentium. Asm inhibition or Asm deficiency strongly enhanced the susceptibility to enteric C. rodentium infection.
Interestingly, the impact of Asm and ceramide on IBD is intensively discussed. Evidence that Asm may be a therapeutic target in colitis has been demonstrated by a study using the Asm inhibitor SMA-7. SMA-7 inhibited LPS-induced activation of NFκB and release of pro-inflammatory cytokines TNFα, IL-1β, and IL-6 in macrophages (12). This was correlated with decreased ceramide production. In a chemically induced mouse model of colitis, oral administration of SMA-7 resulted in decreased cytokine levels in the colon and lower severity of colonic injury. Meanwhile, also desipramine and amitriptyline treatment was shown to inhibit Asm and to decrease pathology in common acute and chronic epithelial injury colitis models (13, 49). In all three studies, Asm inhibition resulted in a decreased pro-inflammatory cytokine production and impaired lymphocyte infiltration. Importantly, broad immunosuppression increases the risk of infectious complications (17) and several enteropathogens have been implicated in the development of gastrointestinal diseases (50, 51). Infection with Salmonella (S.) typhimurium results in severe gastroenteritis (52, 53). The pathogen infects the host by invading macrophages in Peyer's patches. Intracellular S. typhimurium survives within the lysosomal compartment by preventing lysosomal maturation of the phagosomes (54). Asm activity is required for the release of the reactive oxygen species that are necessary for the killing of intracellular S. typhimurium (55). Therefore, Asm deficiency strongly enhance the susceptible to S. typhimurium infection (56). Although we also observed a strong susceptibility of Asm inhibited and Asm deficient mice to C. rodentium infection, we could not observe any changes in the frequency and phenotype of macrophages in infected amitriptyline treated mice compared to infected wild type mice. However, C. rodentium is a noninvasive, attaching-effacing enteric bacterial pathogen, which does not infect macrophages but directly interacts with the intestinal epithelial layer (57). Therefore, it is more likely that the intestinal barrier is impaired in amitriptyline treated animals, which is well in line with the enhanced systemic bacterial distribution of C. rodentium in amitriptyline treated mice. Indeed, intestinal sphingolipids provide a non-specific barrier. In a porcine model, inhibition of de novo ceramide synthesis impaired the proliferation and barrier function of intestinal epithelial cells, which in turn led to the induction of inflammation (45). Furthermore, intestinal deletion of serine palmitoyltransferase (SPT), which is the rate-limiting enzyme for sphingolipid biosynthesis, significantly decreased the ceramide and sphingomyelin levels in the plasma membrane of gut cells and promoted intestinal cell apoptosis with the impairment of gut barrier function (58). However, further investigation is required to fully understand the impact of Asm on the barrier function in infectious and non-infectious colitis.
Th1 and Th17 cells, as part of the adaptive immunity, mediate the host defense against C. rodentium via the production of their signature cytokines IFNγ and IL-17 (26, 59), and mice depleted of either cell type have an impaired ability to clear the infection (29, 60). However, an uncontrolled CD4+ T cell response leads to severe immunopathology (20). Interestingly, we observed a significant increase of Th1 and Th17 cells accompanied with severe immunopathology in infected mice with Asm inhibition compared to infected wild type mice. Well in line, the regulation of murine Th1 differentiation by ceramide has been reported. A ceramide analog was shown to enhance IL-12 induced Th1 differentiation with increased T-bet expression and IFNγ production (61). In contrast, Asm inhibition in human CD4+ T cells abrogates Th1 cell differentiation (62). In addition, inhibition of Asm bioactivity by imipramine, which blocks the generation of ceramide, was shown to impair Th17 generation by blocking both mTor and Stat3 (63). Interestingly, we showed an enhanced frequency of Th17 cells in amitriptyline treated infected mice. Of note, amitriptyline also inhibits in part acid ceramidase, which metabolizes ceramide to sphingosine. Consequently, infected Asm inhibited mice displayed increased ceramide concentrations in the colon compared to infected wild type mice, which seems to further boost the Th17 response. During C. rodentium infection, Th1 and Th17 immunity is counterbalanced by regulatory T cells to inhibit severe immunopathology. Asm was recently described as negative regulator of Treg development (64). In comparison to wild type mice, Asm deficient mice have a higher number of systemic Treg cells under homeostasis. Furthermore, inhibition of Asm and supplementation of IL-2 result in augmentation of Foxp3 expression as well as induction of Treg differentiation in vitro (64, 65). Surprisingly, we identified lower frequencies of Tregs in the colon of infected Asm inhibited mice compared to infected wild type mice suggesting differences in the polarization process under homeostasis and during infection. Finally, in most of the in vivo studies the Asm inhibitor was applied intraperitoneally (66–68), whereas we chose the oral route via the drinking water to specifically target the gastrointestinal tract. Hence, we cannot exclude that the specific immunosuppressive micro-milieu in the intestine interferes with the effects induced by Asm inhibition.
In summary, in contrast to the protective effect of Asm inhibition in common acute and chronic epithelial injury colitis models, Asm inhibition or Asm deficiency strongly enhanced the susceptibility to enteric C. rodentium infection. Therefore, understanding the sphingolipid enzymes and metabolic pathways involved in regulating intestinal inflammation under infectious and non-infectious conditions is the prerequisite for the development of new therapeutic strategies, which target the sphingolipid metabolism.
Data Availability
All datasets generated for this study are included in the manuscript and/or the supplementary files.
Ethics Statement
This study was carried out in accordance with the recommendations of the Society for Laboratory Animal Science (GV-SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). The protocol was approved by the North Rhine-Westphalia State Agency for Nature, Environment and Consumer Protection (LANUF), Germany.
Author Contributions
JM and AW: conceived and designed the experiments and wrote the paper. JM, VP, RK, J-FE, FS, LJ, AY, KB, and JZ: performed the experiments. JM, VP, RK, J-FE, LJ, WH, and AW: analyzed the data and reviewed and edited the manuscript. MH, DH, BK, RNK, JB, and WH: contributed reagents, materials, analysis tools.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (DFG—GRK2098 to AW, KB, JB, and WH and GRK1949 to AW, JB, and WH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We kindly thank Mechthild Hemmler-Roloff and Christina Liebig for excellent technical assistance. We are grateful to Erich Gulbins for providing us with Asm KO mice.
References
1. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. (2007) 369:1641–57. doi: 10.1016/S0140-6736(07)60751-X
2. Fiocchi C. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. (1998) 115:182–205. doi: 10.1016/S0016-5085(98)70381-6
3. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. (2008) 9:139–50. doi: 10.1038/nrm2329
4. Oertel S, Scholich K, Weigert A, Thomas D, Schmetzer J, Trautmann S, et al. Ceramide synthase 2 deficiency aggravates AOM-DSS-induced colitis in mice: role of colon barrier integrity. Cell Mol Life Sci. (2017) 74:3039–55. doi: 10.1007/s00018-017-2518-9
5. Bock J, Liebisch G, Schweimer J, Schmitz G, Rogler G. Exogenous sphingomyelinase causes impaired intestinal epithelial barrier function. World J Gastroenterol. (2007) 13:5217–25. doi: 10.3748/wjg.v13.i39.5217
6. Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, et al. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. (2009) 23:143–52. doi: 10.1096/fj.08-118109
7. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. (2013) 23:107–20. doi: 10.1016/j.ccr.2012.11.013
8. Huang WC, Liang J, Nagahashi M, Avni D, Yamada A, Maceyka M, et al. Sphingosine-1-phosphate phosphatase 2 promotes disruption of mucosal integrity, and contributes to ulcerative colitis in mice and humans. FASEB J. (2016) 30:2945–58. doi: 10.1096/fj.201600394R
9. Beckmann N, Sharma D, Gulbins E, Becker KA, Edelmann B. Inhibition of acid sphingomyelinase by tricyclic antidepressants and analogons. Front Physiol. (2014) 5:331. doi: 10.3389/fphys.2014.00331
10. Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. (2008) 14:1247–55. doi: 10.1038/nm.1876
11. Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. (2014) 510:58–67. doi: 10.1038/nature13475
12. Sakata A, Ochiai T, Shimeno H, Hikishima S, Yokomatsu T, Shibuya S, et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology. (2007) 122:54–64. doi: 10.1111/j.1365-2567.2007.02612.x
13. Xiong Y, Zhu XD, Wan P, Ren YP, Wang C, Yan RW, et al. Inhibition of ASM activity ameliorates DSS-induced colitis in mice. Prostaglandins Other Lipid Mediat. (2018) 140:26–30. doi: 10.1016/j.prostaglandins.2018.12.002
14. Bai A, Guo Y. Acid sphingomyelinase mediates human CD4(+) T-cell signaling: potential roles in T-cell responses and diseases. Cell Death Dis. (2017) 8:e2963. doi: 10.1038/cddis.2017.360
15. Rahimi HR, Shiri M, Razmi A. Antidepressants can treat inflammatory bowel disease through regulation of the nuclear factor-kappaB/nitric oxide pathway and inhibition of cytokine production: a hypothesis. World J Gastrointest Pharmacol Ther. (2012) 3:83–5. doi: 10.4292/wjgpt.v3.i6.83
16. Campieri M, Gionchetti P. Bacteria as the cause of ulcerative colitis. Gut. (2001) 48:132–5. doi: 10.1136/gut.48.1.132
17. Orlicka K, Barnes E, Culver EL. Prevention of infection caused by immunosuppressive drugs in gastroenterology. Ther Adv Chronic Dis. (2013) 4:167–85. doi: 10.1177/2040622313485275
18. Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. (1995) 10:288–93. doi: 10.1038/ng0795-288
19. Pastille E, Bardini K, Fleissner D, Adamczyk A, Frede A, Wadwa M, et al. Transient ablation of regulatory T cells improves antitumor immunity in colitis-associated colon cancer. Cancer Res. (2014) 74:4258–69. doi: 10.1158/0008-5472.CAN-13-3065
20. Seiffart V, Zoeller J, Klopfleisch R, Wadwa M, Hansen W, Buer J, et al. IL10-Deficiency in CD4(+) T cells exacerbates the IFNγ and IL17 response during bacteria induced colitis. Cell Physiol Biochem. (2015) 36:1259–73. doi: 10.1159/000430295
21. Duan RD, Nilsson A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res. (2009) 48:62–72. doi: 10.1016/j.plipres.2008.04.003
22. Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. (2010) 26:9–20. doi: 10.1159/000315101
23. Kornhuber J, Tripal P, Gulbins E, Muehlbacher M. Functional inhibitors of acid sphingomyelinase (FIASMAs). Handb Exp Pharmacol. (2013) 169–86. doi: 10.1007/978-3-7091-1368-4_9
24. Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, et al. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol. (2014) 12:612–23. doi: 10.1038/nrmicro3315
25. Schreiber HA, Loschko J, Karssemeijer RA, Escolano A, Meredith MM, Mucida D, et al. Intestinal monocytes and macrophages are required for T cell polarization in response to Citrobacter rodentium. J Exp Med. (2013) 210:2025–39. doi: 10.1084/jem.20130903
26. Higgins LM, Frankel G, Douce G, Dougan G, Macdonald TT. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun. (1999) 67:3031–9.
27. Bry L, Brenner MB. Critical role of T cell-dependent serum antibody, but not the gut-associated lymphoid tissue, for surviving acute mucosal infection with Citrobacter rodentium, an attaching and effacing pathogen. J Immunol. (2004) 172:433–41. doi: 10.4049/jimmunol.172.1.433
28. Bry L, Brigl M, Brenner MB. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium. Infect Immun. (2006) 74:673–81. doi: 10.1128/IAI.74.1.673-681.2006
29. Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, et al. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol. (2002) 168:1804–12. doi: 10.4049/jimmunol.168.4.1804
30. Mundy R, Macdonald TT, Dougan G, Frankel G, Wiles S. Citrobacter rodentium of mice and man. Cell Microbiol. (2005) 7:1697–706. doi: 10.1111/j.1462-5822.2005.00625.x
31. Mangan PR, Harrington LE, O'quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. (2006) 441:231–4. doi: 10.1038/nature04754
32. Wang Z, Friedrich C, Hagemann SC, Korte WH, Goharani N, Cording S, et al. Regulatory T cells promote a protective Th17-associated immune response to intestinal bacterial infection with C. rodentium. Mucosal Immunol. (2014) 7:1290–301. doi: 10.1038/mi.2014.17
33. Merck Sharp AD. Amitriptyline Hydrochloride: An Antidepressive Agent: Résumé of Essential Information. Rahway, NJ: Merck & CO (1961).
34. Carette S, McCain GA, Bell DA, Fam AG. Evaluation of amitriptyline in primary fibrositis. A double-blind, placebo-controlled study. Arthritis Rheum. (1986) 29:655–9. doi: 10.1002/art.1780290510
35. Couch JR, Ziegler DK, Hassanein R. Amitriptyline in the prophylaxis of migraine. Effectiveness and relationship of antimigraine and antidepressant effects. Neurology. (1976) 26:121–7. doi: 10.1212/WNL.26.2.121
36. Egbunike IG, Chaffee BJ. Antidepressants in the management of chronic pain syndromes. Pharmacotherapy. (1990) 10:262–70.
37. Mishra PC, Agarwal VK, Rahman H. Therapeutic trial of amitryptiline in the treatment of nocturnal enuresis–a controlled study. Indian Pediatr. (1980) 17:279–85.
38. Friedman G. Treatment of the irritable bowel syndrome. Gastroenterol Clin North Am. (1991) 20:325–33.
39. Mandal A, Sinha C, Kumar Jena A, Ghosh S, Samanta A. An investigation on in vitro and in vivo antimicrobial properties of the antidepressant: amitriptyline hydrochloride. Braz J Microbiol. (2010) 41:635–45. doi: 10.1590/S1517-83822010000300014
40. Roumestan C, Michel A, Bichon F, Portet K, Detoc M, Henriquet C, et al. Anti-inflammatory properties of desipramine and fluoxetine. Respir Res. (2007) 8:35. doi: 10.1186/1465-9921-8-35
41. Shale M, Schiering C, Powrie F. CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev. (2013) 252:164–82. doi: 10.1111/imr.12039
42. Matricon J, Barnich N, Ardid D. Immunopathogenesis of inflammatory bowel disease. Self Nonself. (2010) 1:299–309. doi: 10.4161/self.1.4.13560
43. Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. (2014) 20:91–9. doi: 10.3748/wjg.v20.i1.91
44. Sandborn WJ, Feagan BG, Wolf DC, D'haens G, Vermeire S, Hanauer SB, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. (2016) 374:1754–62. doi: 10.1056/NEJMoa1513248
45. Bouhet S, Hourcade E, Loiseau N, Fikry A, Martinez S, Roselli M, et al. The mycotoxin fumonisin B1 alters the proliferation and the barrier function of porcine intestinal epithelial cells. Toxicol Sci. (2004) 77:165–71. doi: 10.1093/toxsci/kfh006
46. Tafazoli F, Magnusson KE, Zheng L. Disruption of epithelial barrier integrity by Salmonella enterica serovar typhimurium requires geranylgeranylated proteins. Infect Immun. (2003) 71:872–81. doi: 10.1128/IAI.71.2.872-881.2003
47. Bryan P-F, Karla C, Edgar Alejandro M-T, Sara Elva E-P, Gemma F, Luz C. Sphingolipids as mediators in the crosstalk between microbiota and intestinal cells: implications for inflammatory bowel disease. Mediat Inflamm. (2016) 2016:9890141. doi: 10.1155/2016/9890141
48. Gulhan B, Ozcelik U, Gurakan F, Gucer S, Orhan D, Cinel G, et al. Different features of lung involvement in Niemann-Pick disease and Gaucher disease. Respir Med. (2012) 106:1278–85. doi: 10.1016/j.rmed.2012.06.014
49. Fattahian E, Hajhashemi V, Rabbani M, Minaiyan M, Mahzouni P. Anti-inflammatory effect of amitriptyline on ulcerative colitis in normal and reserpine-induced depressed rats. Iran J Pharm Res. (2016) 15(Suppl.):125–37. doi: 10.22037/ijpr.2016.1813
50. Garcia Rodriguez LA, Ruigomez A, Panes J. Acute gastroenteritis is followed by an increased risk of inflammatory bowel disease. Gastroenterology. (2006) 130:1588–94. doi: 10.1053/j.gastro.2006.02.004
51. Sartor RB, Rath HC, Sellon RK. Microbial factors in chronic intestinal inflammation. Curr Opin Gastroenterol. (1996) 12:327–33. doi: 10.1097/00001574-199607000-00003
52. Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. (2012) 379:2489–99. doi: 10.1016/S0140-6736(11)61752-2
53. Ramachandran G, Panda A, Higginson EE, Ateh E, Lipsky MM, Sen S, et al. Virulence of invasive Salmonella Typhimurium ST313 in animal models of infection. PLoS Negl Trop Dis. (2017) 11:e0005697. doi: 10.1371/journal.pntd.0005697
54. McCollister BD, Bourret TJ, Gill R, Jones-Carson J, Vazquez-Torres A. Repression of SPI2 transcription by nitric oxide-producing, IFNgamma-activated macrophages promotes maturation of Salmonella phagosomes. J Exp Med. (2005) 202:625–35. doi: 10.1084/jem.20050246
55. McCollister BD, Myers JT, Jones-Carson J, Voelker DR, Vazquez-Torres A. Constitutive acid sphingomyelinase enhances early and late macrophage killing of Salmonella enterica serovar Typhimurium. Infect Immun. (2007) 75:5346–52. doi: 10.1128/IAI.00689-07
56. Utermohlen O, Karow U, Lohler J, Kronke M. Severe impairment in early host defense against Listeria monocytogenes in mice deficient in acid sphingomyelinase. J Immunol. (2003) 170:2621–8. doi: 10.4049/jimmunol.170.5.2621
57. Bergstrom KSB, Guttman JA, Rumi M, Ma C, Bouzari S, Khan MA, et al. Modulation of intestinal goblet cell function during infection by an attaching and effacing bacterial pathogen. Infect Immun. (2008) 76:796–811. doi: 10.1128/IAI.00093-07
58. Li Z, Kabir I, Tietelman G, Huan C, Fan J, Worgall T, et al. Sphingolipid de novo biosynthesis is essential for intestine cell survival and barrier function. Cell Death Dis. (2018) 9:173. doi: 10.1038/s41419-017-0214-1
59. Symonds EL, Riedel CU, O'mahony D, Lapthorne S, O'mahony L, Shanahan F. Involvement of T helper type 17 and regulatory T cell activity in Citrobacter rodentium invasion and inflammatory damage. Clin Exp Immunol. (2009) 157:148–54. doi: 10.1111/j.1365-2249.2009.03934.x
60. Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, et al. Differential roles of interleukin-17A and−17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. (2009) 30:108–19. doi: 10.1016/j.immuni.2008.11.009
61. Kue CS, Jung MY, Cho D, Kim TS. C6-ceramide enhances Interleukin-12-mediated T helper type 1 cell responses through a cyclooxygenase-2-dependent pathway. Immunobiology. (2012) 217:601–9. doi: 10.1016/j.imbio.2011.10.021
62. Bai A, Kokkotou E, Zheng Y, Robson SC. Role of acid sphingomyelinase bioactivity in human CD4+ T-cell activation and immune responses. Cell Death Dis. (2015) 6:e1828. doi: 10.1038/cddis.2015.178
63. Bai A, Moss A, Kokkotou E, Usheva A, Sun X, Cheifetz A, et al. CD39 and CD161 modulate Th17 responses in Crohn's disease. J Immunol. (2014) 193:3366–77. doi: 10.4049/jimmunol.1400346
64. Zhou Y, Salker MS, Walker B, Munzer P, Borst O, Gawaz M, et al. Acid Sphingomyelinase (ASM) is a negative regulator of regulatory T Cell (Treg) development. Cell Physiol Biochem. (2016) 39:985–95. doi: 10.1159/000447806
65. Schneider-Schaulies J, Beyersdorf N. CD4+ Foxp3+ regulatory T cell-mediated immunomodulation by anti-depressants inhibiting acid sphingomyelinase. Biol Chem. (2018) 399:1175–82. doi: 10.1515/hsz-2018-0159
66. Peng H, Li C, Kadow S, Henry BD, Steinmann J, Becker KA, et al. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. J Mol Med. (2015) 93:675–89. doi: 10.1007/s00109-014-1246-y
67. Johnson BL III, Rice TC, Xia BT, Boone KI, Green EA, Gulbins E, et al. Amitriptyline usage exacerbates the immune suppression following burn injury. Shock (Augusta Ga.). (2016) 46:541–8. doi: 10.1097/SHK.0000000000000648
Keywords: Citrobacter rodentium, colitis, acid sphingomyelinase, amitriptyline, Th1, Th17
Citation: Meiners J, Palmieri V, Klopfleisch R, Ebel J-F, Japtok L, Schumacher F, Yusuf AM, Becker KA, Zöller J, Hose M, Kleuser B, Hermann DM, Kolesnick RN, Buer J, Hansen W and Westendorf AM (2019) Intestinal Acid Sphingomyelinase Protects From Severe Pathogen-Driven Colitis. Front. Immunol. 10:1386. doi: 10.3389/fimmu.2019.01386
Received: 05 March 2019; Accepted: 03 June 2019;
Published: 19 June 2019.
Edited by:
Sibylle Schneider-Schaulies, University of Wuerzburg, GermanyReviewed by:
Sabine Grösch, Goethe-Universität Frankfurt am Main, GermanyMarkus M. Heimesaat, Charité Medical University of Berlin, Germany
Copyright © 2019 Meiners, Palmieri, Klopfleisch, Ebel, Japtok, Schumacher, Yusuf, Becker, Zöller, Hose, Kleuser, Hermann, Kolesnick, Buer, Hansen and Westendorf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Astrid M. Westendorf, YXN0cmlkLndlc3RlbmRvcmZAdWstZXNzZW4uZGU=