 Franziska Hübner
Franziska Hübner Ewan A. Langan
Ewan A. Langan Andreas Recke
Andreas Recke- 1Department of Dermatology, University of Lübeck, Lübeck, Germany
- 2Dermatological Sciences, University of Manchester, Manchester, United Kingdom
- 3Lübeck Institute of Dermatological Research, University of Lübeck, Lübeck, Germany
Lichen planus pemphigoides (LPP) is a very rare autoimmune sub-epidermal blistering disease associated with lichenoid skin changes. Initially thought to be a mere variant of more common inflammatory dermatoses, particularly Bullous Pemphigoid (BP) or Lichen Planus (LP), a growing body of evidence suggests that it is a disease entity in its own right. In common with a range of autoimmune blistering diseases, including BP, pemphigoid gestationis (PG), mucous membrane pemphigoid (MMP) and linear IgA dermatosis (LAD), a key feature of the disease is the development of autoantibodies against type XVII collagen (COL17). However, accurately establishing the diagnosis is dependent on a careful correlation between the clinical, histological and immunological features of the disease. Therefore, we present an up to date summary of the epidemiology and etiopathogenesis of LPP, before illustrating the predisposing and precipitating factors implicated in the development of the disease. In addition to a selective literature search, we compare reports of potential drug-induced cases of LPP with pharmacovigilance data available via OpenVigil. We subsequently outline the cardinal clinical features, important differential diagnoses and current treatment options. We conclude by demonstrating that an improved understanding of LPP may not only lead to the development of novel treatment strategies for the disease itself, but may also shed new light on the pathophysiology of more common and treatment-refractory autoimmune blistering diseases.
Introduction
First described by Kaposi over a century ago (1), Lichen planus pemphigoides (LPP, syn. Lichen ruber pemphigoides) is commonly considered to be a variant of Lichen Planus (LP), characterized and complicated by the formation of tense blisters and bullae.
In addition to the clinical findings, including lichenoid plaques and tense blisters, the gold standard for the diagnosis of LPP is the demonstration of autoantibody deposition along the dermal-epidermal junctional zone in perilesional skin biopsies; first reported by Stingl and Holubar in 1975 (2). Almost two decades later, Tamada et al. established that the autoantigen in LPP is a 180 kDa protein expressed in the hemidesmosomes of the dermal-epidermal junction (3). Interestingly, the same autoantigen is responsible for the development of Bullous Pemphigoid (BP), namely type XVII collagen (COL17) (4).
Definition
Lichen planus pemphigoides can be best defined as an autoimmune dermatosis, the hallmarks of which are lichenoid and bullous skin lesions, which develop in the context of autoantibodies targeting type XVII collagen COL17.
Clinically, the diagnosis relies on carefully distinguishing the disease phenotype from that seen in bullous LP. Given the potential clinical, histological and immunological overlap between LPP, bullous LP and BP, the clinician must also determine which disease is present, although they may occasionally occur simultaneous, which may further complicate reaching the correct diagnosis(ses). Several clinical features can support reaching the correct diagnosis. For example, bullous LP classically describes the formation of blisters on pre-existing lichenoid plaques. In contrast to bullous LP, the blisters of LPP are typically located outside of LP lesions. However, several cases of LPP with blistering restricted to the lichenoid plaques have been reported, casting doubt on the utility of blister localization to clinically differentiate between LPP and Bullous LP with any degree of certainty (5, 6). The blisters in Bullous Pemphigoid tend to occur on urticated plaques and may evolve into erosions and crusts.
Moreover, there are other subtle differences in the clinical presentation of BP and LPP. For example, the typical age of onset of LPP is significantly younger than that in BP (7, 8). Furthermore, LPP lesions are predominantly found on the extremities, whereas in BP they are more often generalized (8) and associated with pruritus. The clinical course of LPP is usually less protracted and generally milder than that in BP.
Ultimately, the detection of autoantibodies to the dermal-epidermal junction is central to supporting and securing the diagnosis. In contrast to LPP, the skin changes seen in bullous LP develop in the absence of autoantibodies against structural proteins of the skin, especially COL17 (9). In terms of pathogenesis, given that in the majority of cases of LPP the development of lichenoid skin lesions precedes the formation of blisters, it has been hypothesized that lichenoid inflammation itself may actually promote the development of an autoimmune response, targeting proteins of the epidermal basement membrane (10–14).
In fact, Kromminga et al. identified subtle differences in the epitope specificity of autoantibodies in the sera of patients with LPP, BP, and mucous membrane pemphigoid (MMP) (15), using recombinant fragments of the NC16A subdomain of COL17. The sera of 12 patients with BP, 6 with gestational pemphigoid (PG), 10 with MMP and 4 with LPP were examined using nine overlapping dihydrofolate reductase-fused subfragments with a size of 13–18 amino acids of the NC16A (amino acids E490-L565 of COL17, Uniprot entry Q9UMD9) to evaluate the epitope binding pattern by immunoblotting (15). Here, most BP and PG patient sera bound to fragments representing amino acids E490-G532; MMP patient sera preferentially bound to fragments E490-R507 and D514-L565; while LPP sera generally lacked binding to E490-R507 but showed reactivity with fragments comprising D514-L565. Kromminga et al. did not perform any statistical evaluation as to whether these differences are significant. However, on the basis of the published data it was possible to carry out a statistical analysis1. We found that the differences between BP and LPP (p = 0.0032), MMP and LPP (p = 1.07 × 10−13) and LPP and PG (p = 3.08 × 10−43) were actually highly significant. In addition, BP and PG (p = 3.82 × 10−13) and MMP and PG (p = 1.95 × 10−24) had significantly different binding patterns, while MMP and BP did not.
Epidemiology
The exact prevalence of LPP is unknown. Only 4 cases of LPP were identified in a cohort of 68 patients with blistering diseases from Kuwait; equivalent to an incidence of 0.3/1,000,000 inhabitants (16). A study from India reported 3 patients with LPP in a series of 268 cases with autoimmune blistering dermatoses (17). In contrast, epidemiological studies in patients with blistering dermatoses, based in France, Germany, Greece, Serbia, and Singapore, with patient numbers ranging from 41 to 1,161, did not identify any cases of LPP (18–23). Based on ICD10 classification data from health insurance providers in Germany, the reported prevalence of L12.8 (other pemphigoid diseases) was 4.7 per million patients and 259 per million patients for BP (L12.0) (7). Unfortunately, the LPP ICD10 code L43.1, was not specifically evaluated. However, the epidemiological data analysis based upon ICD10 codes is complicated by the fact that the ICD10 code L43.1 is shared between LPP and bullous LP. Nevertheless, based on the available data the prevalence may be estimated at about 1 per 1,000,000 patients.
The sex ratio (male/female) is described to be roughly 0.8/1 in adults and 3.3/1 in children and adolescents (8), failing to support a specific predilection according to sex. The mean age of onset is approximately 46 years (range between 4 and 85), which is well below the typical age of onset of BP (7). Interestingly, it is not exceptionally rare for LPP to affect children and adolescents. Indeed, in a case report collection with 78 patients, 13 (~16%) were children or adolescents (8).
Etiopathogenesis
LPP is characterized by autoantibodies against type XVII collagen (COL17, BPAG2), a structural protein that resides in hemidesmosomes at the dermal-epidermal junction (4, 24, 25). Similarly to BP, autoantibodies in LPP may also bind to the 230 kDa BPAG1 (3). In most cases, the COL17-specific autoantibodies in LPP react with the membrane-proximal NC16A subdomain (amino acid residues 490–565 of UniProt entry Q9UMD9) (4, 24). In addition, the C-terminal portion of COL17 and desmoglein 1 have been identified as epitopes and antigens, respectively, in LPP (26). Other autoantibodies against unidentified antigens with a molecular weight of 130 kDa (27) and 200 kDa (28) have also been described. The reported variability in autoantigen specificity may result in clinical variants of LPP which appear similar to BP, with autoantibodies against NC16A (24, 29), and MMP with mucosal lesions and autoantibodies against the C-terminal portion of COL17 (26, 30).
In fact, COL17 is a common autoantigen in a variety of autoimmune blistering dermatoses (31, 32), including LPP, BP, linear IgA dermatosis (33, 34), PG, MMP and paraneoplastic pemphigus (35). Autoantibodies against COL17 have been demonstrated to induce inflammation and blistering due to the effector functions of the Fc portion (36–38). Moreover, a deposition of complement factor C3 at the dermal-epidermal junction found in skin biopsies of LPP indicates an involvement of complement in the pathogenesis. In case of BP and epidermolysis bullosa acquisita, a similar subepidermal blistering disease, the activation of the complement system has been described as a crucial event in the pathogenesis (39, 40).
However, a growing evidence suggests that both complement-dependent and complement-independent mechanisms may both be relevant and effective in subepidermal blistering dermatoses (41–44). The amount of complement-activating IgG1 and non-activating IgG4 autoantibodies (45) is variable between patients. Cases with only IgG4 autoantibodies and without any complement deposition at the derma-epidermal junction exist, suggesting complement-independent mechanisms in blister formation (43). Binding of leporine autoantibodies to type XVII collagen was demonstrated to induce skin fragility, both in a complement-dependent and independent manner, in a murine model for bullous pemphigoid (44). This was confirmed in a similar mouse model, additionally indicating a disease mitigating effect of complement receptor C5aR2 (42).
Although not specifically demonstrated for all diseases, the pathogenic mechanisms ultimately resulting in subepidermal cleavage and macroscopic may be similar and/or shared between distinct autoimmune blistering dermatoses.
The development of autoantibodies against COL17 in LP appears to be a primary event in the development of LPP. This is supported by the fact that blistering almost exclusively follows the appearance of the typical lichenoid skin lesions (8).
In more than 40% of cases with vulvar LP, the NC16A domain of COL17 has been demonstrated to be a target for circulating T cells with rapid effector function (10). The authors used an ELISpot assay that detected IFNγ-producing autoreactive T cells, i.e., the only determined Th1 responses. The T-cell responses in bullous pemphigoid have been investigated with more detail: it appears that most patients have a Th2 or mixed Th1/Th2 response to the complete extracellular portion of BP180 (46). The situation of T cell responses in LPP is unknown. One may speculate that the Th1 response in LP could protect from conversion to a blistering disease, and that a dysregulated Th2 response to NC16A might be necessary for the development of LPP.
In addition to T cell responses, several authors have reported the presence of circulating autoantibodies against COL17 in patients with LP, especially oral or genital LP (10–14). A clinical course similar to LPP was observed in LP-type chronic graft-vs.-host disease, where the patient developed autoantibodies against COL17 and mucous membrane pemphigoid (47). In summary, the development of autoantibodies against COL17 in LPP appears to be linked to the T cell-mediated lichenoid inflammation.
Clinical Features and Establishment of Diagnosis
Diagnosis is based on careful correlation of the clinical, histopathological and immunopathological features.
In LPP, two discrete primary skin lesions occur: lichenoid papules/plaques and tense blisters. Cases of LPP exclusively restricted to mucous membranes have also been reported (48, 49). The nail apparatus may also be affected by LPP, resulting in nail atrophy or even loss of the nail plate (50). This suggests that LPP is in fact a very heterogeneous disease, whose clinical symptoms and immunologic markers can mimic both BP and mucous membrane pemphigoid (49).
The lichenoid eruption consists of pruritic violaceous polygonal papules and plaques with a shiny surface (Figures 1A,B). On the mucosa, patterned white streaks may be found, most prominently on the buccal and the outer genital mucosa (48, 51). Blisters and erosions typically appear after the development of the lichenoid skin changes and classically on previously unaffected skin. Histopathology of a bullous lesion shows the typical feature of BP (2, 31): subepidermal separation with multiple eosinophils in the blister fluid and an eosinophilic infiltrate, whereas histopathology of a lichenoid lesion shows the typical features of lichen planus with focal hyperkeratosis, hypergranulosis, subepidermal band-like lymphocytic infiltrate, and Interface dermatitis with vacuolar change at the dermal-epidermal junction and apoptotic keratinocytes (so called Civatte bodies) (2, 51, 52).
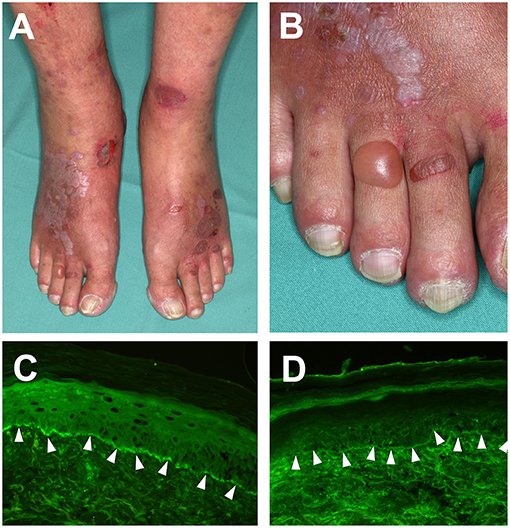
Figure 1. Clinical features of Lichen planus pemphigoides. (A) Violaceous plaques with polygonal configuration affecting the dorsal aspects of the feet, with tense blisters and erosions on non-lesional skin. (B) Close-up view of the right foot. (C) detection of complement factor C3 deposition and, (D) IgG deposition at the dermal-epidermal junction in a punch biopsy from perilesional skin, using direct immunofluorescence microscopy (magnification 200×).
IgG and complement factor C3 deposition at the dermal-epidermal junction zone can be detected using direct immunofluorescence of perilesional biopsies (2) (Figures 1C,D). In contrast, direct immunofluorescence studies of lichenoid lesions may show typical features of LP: irregular band of fibrinogen at the dermal-epidermal junction and colloid bodies in the papillary dermis (53). Circulating autoantibodies in patient sera that bind to the NC16A or C-terminal domains of COL17 or desmogleins may be detected using routine methods including indirect immunofluorescence on monkey esophagus or human neonatal foreskin (54), ELISA and immunoblotting of human keratinocyte extracts (32, 55). In addition to this, circulating autoantibodies will bind to the roof of the artificial blister in indirect immunofluorescence on 1M NaCl-salt split skin (56).
The gold standard for the diagnosis of LPP is the combination of the typical clinical features and the demonstration of autoantibodies binding to the dermal-epidermal junction.
Differential Diagnosis
In addition to BP and bullous LP, several other dermatoses form the differential diagnosis. For example, erythema multiforme has morpho- and histo-logical features that resemble LPP, most notably a lichenoid or interface dermatitis and blistering (57).
Atypical subacute cutaneous lupus erythematosus may present with similar clinical features as LPP (57, 58), underscoring the importance of a complete immunologic work-up to demonstrate autoantibodies against structural proteins of the skin, and especially the NC16A domain of BP180. Furthermore, in case of atypical clinical presentation, antinuclear antibody and dsDNA antibody testing is recommended.
Paraneoplastic pemphigus may present with similar skin changes and immunological features are those in LPP (59, 60), including autoantibodies against BP180. However, paraneoplastic pemphigus serum autoantibodies typically cause an intercellular binding pattern in the epidermis and can bind to rat bladder epithelia and to envoplakin and periplakin (61).
During the course of disease, LPP may even evolve into other types of autoimmune blistering skin diseases, such as pemphigoid nodularis (62), also known as non-bullous pemphigoid.
Drugs and Conditions Associated with LPP
Several reports describe an association between the development of LPP and medication and/or and pre-existing medical conditions.
LPP and Drugs
Relying solely on observational evidence, several drugs have been associated with the development of LPP (Table 1). A causal association is at least conceivable when the LPP developed shortly after the intake of new medication and fully resolved after cessation of the suspected drug-trigger. Perhaps the strongest evidence for drug-induced LPP is in the context angiotensin-converting enzyme (ACE) inhibitor use. There are four reports of an association of LPP with different angiotensin-converting enzyme (ACE) inhibitors (64, 67, 68, 74). Nevertheless, it should be acknowledged that in the majority of published reports of medication-induced LPP, drug re-challenge was either not performed or not associated with disease recurrence.
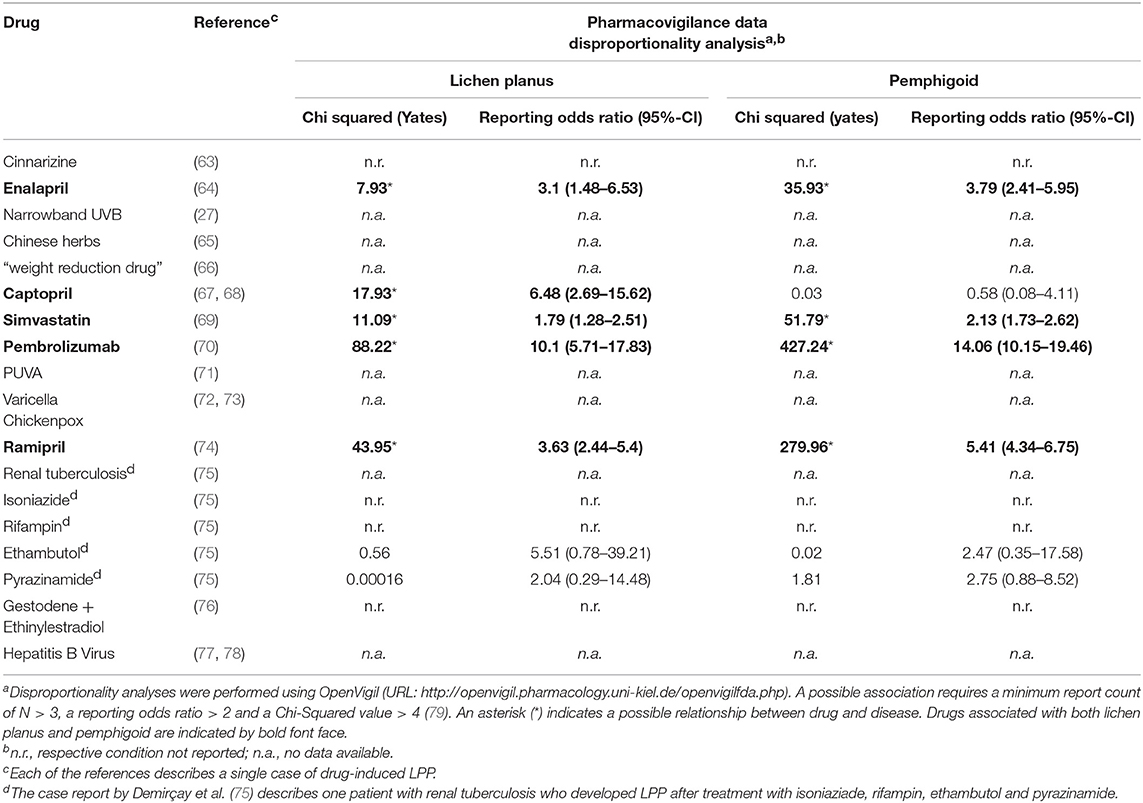
Table 1. Drugs and conditions that were reported to be associated with LPP.
LPP and Infections
LPP has also been reported to be a complication of infection, particularly viral infections (Table 1), for example varicella (72, 73) and hepatitis B (77, 78). In fact, all of the published cases of LPP and viral hepatitis to date have been in association with hepatitis B (77, 78) and not hepatitis C infection. This is in contrast to LP, in which only hepatitis C has been reported as a trigger factor. The odds ratio to develop LP, especially oral LP, is described to be 2.5–4.5 for individual who are seropositive for HCV (80). Vice versa, a meta-analysis showed that the odds ratio to have a positive serology of HCV was 2.73–13.48 in patients with oral LP (81).
LPP and Cancer
An association of LPP with colon adenocarcinoma points raises the possibility of a paraneoplastic variant of the disease (82).
Medication as a Trigger of Both LP and BP
If one considers the possibility that LPP occurs as a complication of LP, it is conceivable that drugs that trigger the more prevalent disease LP also increase the risk of developing LPP. In fact, a large number of drugs have been described as possibly related to the development of LP, including ACE inhibitors. Several drugs are reportedly associated with the development of LP, including antimalarials, thiazide diuretics, NSAIDs, quinidine, beta-blockers, gold compounds, and tumor necrosis factor alpha inhibitors (51). The list of drugs that have been reported to induce BP is more extensive than the equivalent list for LP. It includes antibiotics, Calcium channel antagonists, ACE inhibitors, beta-blockers, angiotensin 1 antagonists, vaccines, NSAIDs, diuretics, Gliptins, TNF-alpha inhibitors, D-penicillamine, and tiobutarid (52). An induction of BP by PUVA has been reported only anecdotally (83, 84).
Comparison of Case Report Data With Pharmacovigilance Information
Pharmacovigilance databases provide another valuable resource to identify drugs that may trigger the development of LPP. However, one major drawback is that LPP is not specified as a disease in the WHO list of adverse events. Instead, the terms “lichen planus” and “pemphigoid” were used to analyze pharmacovigilance data using the OpenVigil database tool (URL: http://openvigil.pharmacology.uni-kiel.de/openvigilfda.php). Pharmacovigilance report analyses are compared with case reports in Table 1, supported the observation of LPP cases that were caused by ACE inhibitors (enalapril, capropril, and ramipril), but also simvastatin and pembrolizumab. With the exception of captopril, both LP and pemphigoid disease were found to be putative side effects of these drugs. The reported association of anti-tuberculosis drugs with LPP could not be confirmed by pharmacovigilance data. For all other drugs and conditions that were suspected to be associated with LPP, no data could be found.
Identification of Putative Triggers for LPP, LP, and BP From Pharmacovigilance Information
In addition, pharmacovigilance data also allows the identification of drugs that are not yet reported as possible triggers for LPP but are putative triggers for LP and/or pemphigoid (Table 2). Drugs that were reported as possible triggers for both LP and pemphigoid were hydrochlorothiazide, candesartan, sitagliptine, amlodipine, losartan, fluoxetine, and terbinafine. In addition, vildagliptine has been identified as a potent trigger for the development of BP (90), though OpenVigil did not contain any entries with this drug.
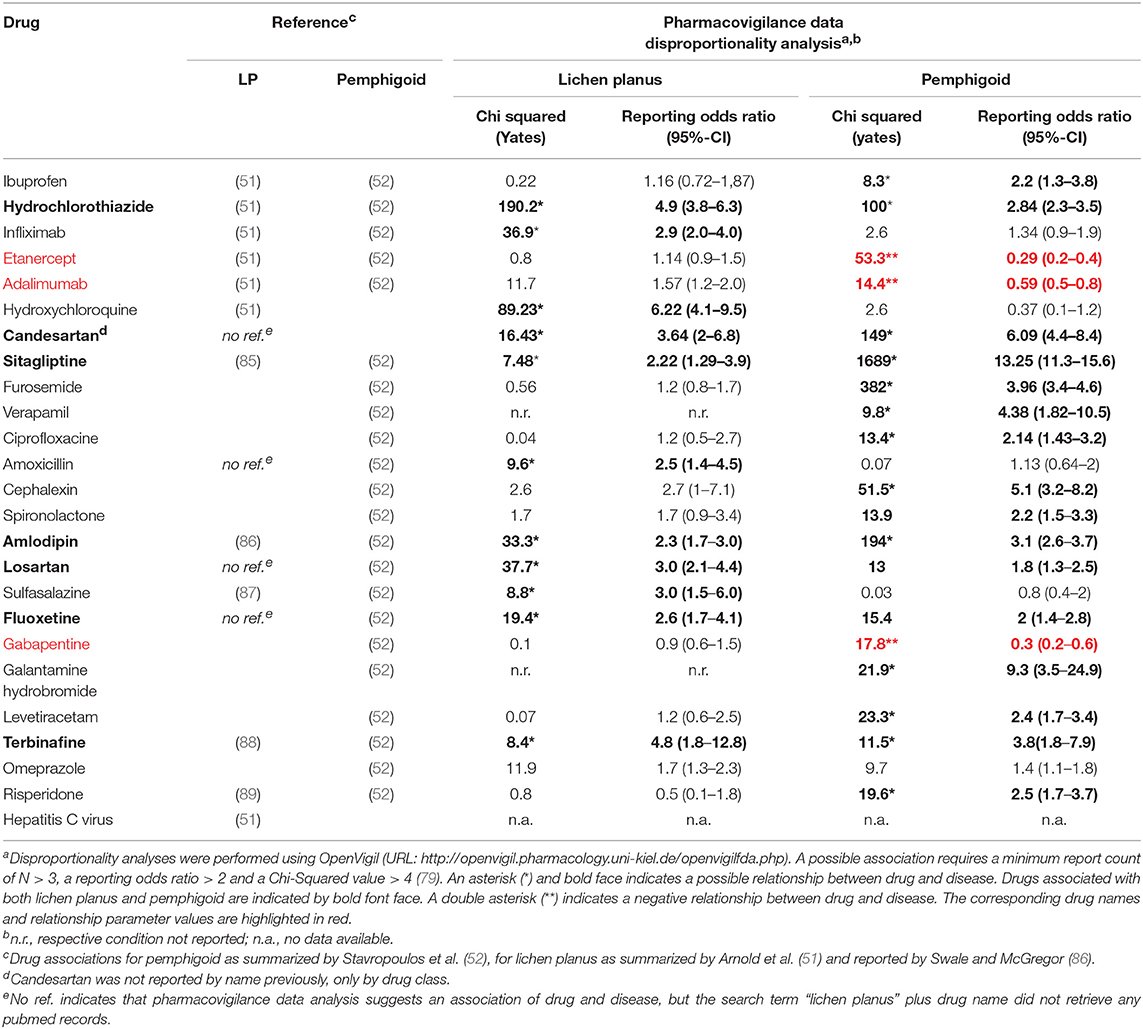
Table 2. Drugs and conditions not yet reported to be associated with LPP, but with LP or pemphigoid.
For some drugs, pharmacovigilance report analyses do not overlap or even contradict the published literature (Table 2). In case of Ibuprofen, the association with pemphigoid could be confirmed, but not in case of LP. The same holds true for sulfasalazine and risperidone. In case of gabapentin, a negative association with pemphigoid was found. For TNFα inhibitors, pharmacovigilance data indicates a negative association of etanercept and adalimumab with pemphigoid and no association with LP. A negative association (odds ratio below 1) may indicate here a protective effect of the respective drug regarding pemphigoid, although gabapentin, etanercept and adalimumab are no established treatment options for this disease. Alternatively, this negative association may be the result of a statistical phenomenon where patients who tend to develop pemphigoid are less likely to be treated with the aforementioned drugs. In the other hand, infliximab seems to be positively associated with LP, but not with pemphigoid.
It has to be noted that pharmacovigilance data is based on physician-initiated reports about possible adverse events and not on a prospective data collection. Common belief about possible relationships between drugs and adverse events may lead to further skewing of data. Moreover, it is difficult to control for confounders or interactions. If a certain drug is used to treat a disease that predisposes for LP, BP or LPP, the disproportionality analysis cannot distinguish this from a drug-related predisposition. Furthermore, it is not possible to distinguish whether a certain drug truly causes LPP or whether it just increases the probability for a clinical manifestation in patients that are otherwise predisposed to LPP.
Treatment
In a selective PubMed literature review of case reports published from the year 2000 onward, we were able to find treatment information from N = 53 patients in N = 43 articles (Table 3). Most reports (N = 42) describe the use of corticosteroids (mainly oral prednisolone) in various doses, from very low dose in some Japanese cases up to 2 mg/kg body weight. High doses of prednisolone are used for treatment of LPP in children. Topical corticosteroids were used in N = 20 cases, followed by dapsone in N = 15 cases.
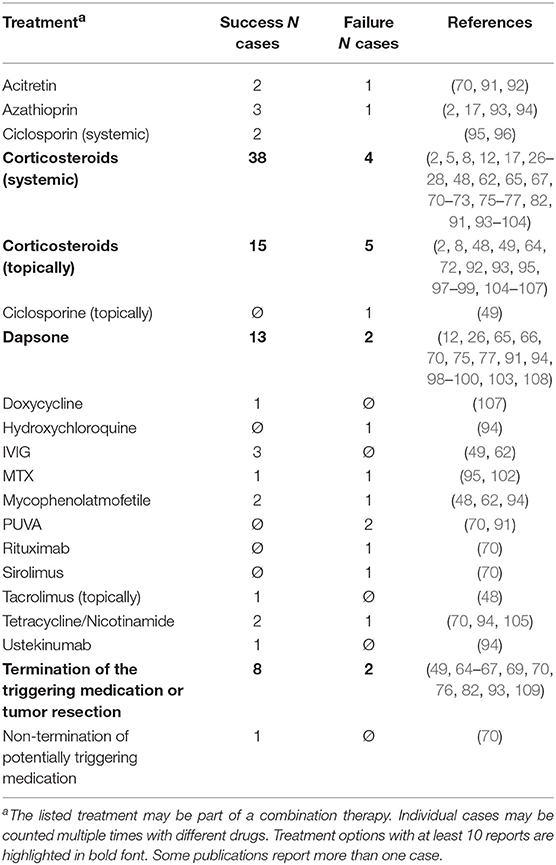
Table 3. Reported treatment options for LPP since 2000.
In a case of oral lichen planus pemphigoides, topical gel ointment containing fluocinonid or dexamethasone was reportedly effective (48).
Although in most of the reported cases, patients were successfully treated with systemic corticosteroids, this might not be the best option given the side-effect profile. Other treatment options include topical corticosteroids, dapsone, and acitretin, whose use may be associated with fewer side-effects. This situation may be compared with that in BP. Here, systemic oral corticosteroids had been considered as standard treatment for decades (110) until Joly et al. effected a paradigmatic shift in treatment by demonstrating the equivalent efficacy of highly potent topical steroids, with fewer side-effects (111). On the other hand, given that patients with LPP tend to be younger, with fewer co-morbidities, one could argue that systemic treatment would be less hazardous and may result in speedier disease resolution.
Another interesting observation made during the review of the case reports is the very low doses of corticosteroids that were used for treatment in some Japanese cases (71, 97). Here, a dose of 15 mg, irrespective of body weight, was reportedly effective. In contrast, corticosteroid doses in cases from other countries are often between 0.5 and 1 mg/kg body weight, i.e., 2 to 4-fold higher.
From our experience (112), a combination of topical dexamethasone, prednisolone pulse therapy (100 mg/day for 3 days, initially every 3rd week) and acitretin (20 mg/day p.o.) is often sufficient to induce remission of blistering within 3 months and disappearance of LP lesions within 1 year. Similar observations have been reported in the literature (91).
Alternatives or additives to corticosteroid treatment could include dapsone (100 mg/daily), tetracycline (2 × 500 mg/daily) in combination with nicotinamide (2 × 500 mg/daily), mycophenolate mofetil (1000–1500 mg/daily in two doses), cyclosporine A (2 mg/kg body weight in 2 doses/day), methotrexate (7.5–20 mg/week or 0.5 mg/kg body weight/week in children).
Our PubMed literature search also retrieved the use of hypnotic suggestion for the treatment of LPP (113), though this article was published in 1959.
In 10 cases, a triggering medication or condition such as a underlying malignancy was identified and treated, resulting in resolution of skin lesions in 8 cases. One report describes the management of LPP associated with pembrolizumab therapy in a patient with malignant melanoma (70). In this patient, dapsone led to resolution of LPP, allowing the treatment with pembrolizumab to be continued. In this context, pembrolizumab was not necessarily the direct trigger of LPP per se, but may have been an important co-factor for the clinical manifestation of a subclinical disease, potentially related to its mode of action.
Conclusion
LPP is a very rare disease entity that belongs to a larger family of diseases characterized by autoantibodies against COL17. It is a heterogenic disease, but shared common clinical features with other autoimmune blistering diseases, including BP and MMP. Its pathognomonic feature is the association with LP, supported by the demonstration of IgG and complement factor C3 deposition at the dermal-epidermal junction. Several case reports indicate an association with ACE inhibitors, simvastatin and checkpoint inhibitors, but also with HBV infection. There is no current consensus on the optimal treatment regime for LPP. However, combinations of systemic with topical corticosteroids, potentially in combination with dapsone or acitretin are worth considering. An improved understanding of the pathophysiology of LPP may help to shed new light on the mechanisms that lead to the development of autoantibodies against COL17 and subsequent blister formation.
Author Contributions
FH, EL and AR discussed data and wrote the manuscript. AR identified the patient, performed literature search, and pharmacovigilance data analyses.
Funding
We acknowledge financial support by Land Schleswig-Holstein within the funding programme Open Access Publikationsfonds.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all people not mentioned here who were involved in patient care and routine laboratory diagnostics.
Footnote
1. ^Using multinomial regression (R open source statistical software, packages nnet for the function multinom and car for the function Anova) and the Benjamini-Hochberg correction for multiple testing.
References
2. Stingl G, Holubar K. Coexistence of lichen planus and bullous pemphigoid. A immunopathological study. Br J Dermatol. (1975) 93:313–20. doi: 10.1111/j.1365-2133.1975.tb06497.x
3. Tamada Y, Yokochi K, Nitta Y, Ikeya T, Hara K, Owaribe K. Lichen planus pemphigoides: identification of 180 kd hemidesmosome antigen. J Am Acad Dermatol. (1995) 32:883–7. doi: 10.1016/0190-9622(95)91554-0
4. Zillikens D, Caux F, Mascaro JM, Wesselmann U, Schmidt E, Prost C, et al. Autoantibodies in lichen planus pemphigoides react with a novel epitope within the C-terminal NC16A domain of BP180. J Invest Dermatol. (1999) 113:117–21. doi: 10.1046/j.1523-1747.1999.00618.x
5. Matos-Pires E, Campos S, Lencastre A, João A, Mendes-Bastos P. Lichen planus pemphigoides. J Dtsch Dermatol Ges J Ger Soc Dermatol. (2018) 16:335–7. doi: 10.1111/ddg.13434_g
6. Archer CB, Cronin E, Smith NP. Diagnosis of lichen planus pemphigoides in the absence of bullae on normal-appearing skin. Clin Exp Dermatol. (1992) 17:433–6. doi: 10.1111/j.1365-2230.1992.tb00253.x
7. Hübner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–8. doi: 10.1016/j.jid.2016.07.013
8. Zaraa I, Mahfoudh A, Sellami MK, Chelly I, El Euch D, Zitouna M, et al. Lichen planus pemphigoides: four new cases and a review of the literature. Int J Dermatol. (2013) 52:406–12. doi: 10.1111/j.1365-4632.2012.05693.x
9. Liakopoulou A, Rallis E. Bullous lichen planus - a review. J Dermatol Case Rep. (2017) 11:1–4. doi: 10.3315/jdcr.2017.1239
10. Baldo M, Bailey A, Bhogal B, Groves RW, Ogg G, Wojnarowska F. T cells reactive with the NC16A domain of BP180 are present in vulval lichen sclerosus and lichen planus. J Eur Acad Dermatol Venereol. (2010) 24:186–90. doi: 10.1111/j.1468-3083.2009.03375.x
11. Buijsrogge JJA, Hagel C, Duske U, Kromminga A, Vissink A, Kloosterhuis AJ, et al. IgG antibodies to BP180 in a subset of oral lichen planus patients. J Dermatol Sci. (2007) 47:256–8. doi: 10.1016/j.jdermsci.2007.05.011
12. Barnadas MA, Roé E, Dalmau J, Alomar A, Martínez L, Gelpí C. Lichen planus pemphigoides: detection of anti-BP 180 antibodies by ELISA and immunoblotting tests. J Eur Acad Dermatol Venereol. (2010) 24:1360–1. doi: 10.1111/j.1468-3083.2010.03632.x
13. Herrero-González JE, Parera Amer E, Segura S, Mas Bosch V, Pujol RM, Martínez Escala ME. Epithelial antigenic specificities of circulating autoantibodies in mucosal lichen planus. Int J Dermatol. (2016) 55:634–9. doi: 10.1111/ijd.12990
14. Shipman AR, Cooper S, Wojnarowska F. Autoreactivity to bullous pemphigoid 180: is this the link between subepidermal blistering diseases and oral lichen planus? Clin Exp Dermatol. (2011) 36:267–9. doi: 10.1111/j.1365-2230.2010.03878.x
15. Kromminga A, Sitaru C, Meyer J, Arndt R, Schmidt E, Christophers E, et al. Cicatricial pemphigoid differs from bullous pemphigoid and pemphigoid gestationis regarding the fine specificity of autoantibodies to the BP180 NC16A domain. J Dermatol Sci. (2002) 28:68–75. doi: 10.1016/S0923-1811(01)00144-X
16. Nanda A, Dvorak R, Al-Saeed K, Al-Sabah H, Alsaleh QA. Spectrum of autoimmune bullous diseases in Kuwait. Int J Dermatol. (2004) 43:876–81. doi: 10.1111/j.1365-4632.2004.02292.x
17. De D, Khullar G, Handa S, Saikia UN, Radotra BD, Saikia B, et al. Clinical, demographic and immunopathological spectrum of subepidermal autoimmune bullous diseases at a tertiary center: A 1-year audit. Indian J Dermatol Venereol Leprol. (2016) 82:358. doi: 10.4103/0378-6323.175928
18. Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year review. Br J Dermatol. (2002) 147:476–80. doi: 10.1046/j.1365-2133.2002.04919.x
19. Bertram F, Bröcker E-B, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges J Ger Soc Dermatol. (2009) 7:434–40. doi: 10.1111/j.1610-0387.2008.06976.x
20. Zillikens D, Wever S, Roth A, Weidenthaler-Barth B, Hashimoto T, Bröcker EB. Incidence of autoimmune subepidermal blistering dermatoses in a region of central Germany. Arch Dermatol. (1995) 131:957–8. doi: 10.1001/archderm.131.8.957
21. Bernard P, Vaillant L, Labeille B, Bedane C, Arbeille B, Denoeux JP, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. bullous diseases French study group. Arch Dermatol. (1995) 131:48–52. doi: 10.1001/archderm.1995.01690130050009
22. Patsatsi A, Lamprou F, Kokolios M, Stylianidou D, Trigoni A, Kalampalikis D, et al. Spectrum of autoimmune bullous diseases in Northern Greece. A 4-year retrospective study and review of the literature. Acta Dermatovenerol Croat. (2017) 25:195–201.
23. Milinković MV, Janković S, Medenica L, Nikolić M, Reljić V, Popadić S, et al. Incidence of autoimmune bullous diseases in Serbia: a 20-year retrospective study: Incidence of autoimmune bullous diseases. J Dtsch Dermatol Ges. (2016) 14:995–1005. doi: 10.1111/ddg.13081
24. Skaria M, Salomon D, Jaunin F, Friedli A, Saurat JH, Borradori L. IgG autoantibodies from a lichen planus pemphigoides patient recognize the NC16A domain of the bullous pemphigoid antigen 180. Dermatol Basel Switz. (1999) 199:253–5. doi: 10.1159/000018257
25. Hsu S, Ghohestani RF, Uitto J. Lichen planus pemphigoides with IgG autoantibodies to the 180 kd bullous pemphigoid antigen (type XVII collagen). J Am Acad Dermatol. (2000) 42:136–41. doi: 10.1016/S0190-9622(00)90024-0
26. Sekiya A, Kodera M, Yamaoka T, Iwata Y, Usuda T, Ohzono A, et al. A case of lichen planus pemphigoides with autoantibodies to the NC16a and C-terminal domains of BP180 and to desmoglein-1. Br J Dermatol. (2014) 171:1230–5. doi: 10.1111/bjd.13097
27. Mandy Chan WM, Lee JSS, Thiam Theng CS, Chua SH, Boon Oon HH. Narrowband UVB-induced lichen planus pemphigoide. Dermatol Rep. (2011) 3:e43. doi: 10.4081/dr.2011.e43
28. Yoon KH, Kim SC, Kang DS, Lee IJ. Lichen planus pemphigoides with circulating autoantibodies against 200 and 180 kDa epidermal antigens. Eur J Dermatol. (2000) 10:212–4.
29. Giudice GJ, Emery DJ, Zelickson BD, Anhalt GJ, Liu Z, Diaz LA. Bullous pemphigoid and herpes gestationis autoantibodies recognize a common non-collagenous site on the BP180 ectodomain. J Immunol. (1993) 151:5742–50. doi: 10.1016/0923-1811(93)90940-Q
30. Lee JB, Liu Y, Hashimoto T. Cicatricial pemphigoid sera specifically react with the most C-terminal portion of BP180. J Dermatol Sci. (2003) 32:59–64. doi: 10.1016/S0923-1811(03)00035-5
31. Witte M, Zillikens D, Schmidt E. Diagnosis of autoimmune blistering diseases. Front Med. (2018) 5:296. doi: 10.3389/fmed.2018.00296
32. van Beek N, Zillikens D, Schmidt E. Diagnosis of autoimmune bullous diseases. J Dtsch Dermatol Ges. (2018) 16:1077–91. doi: 10.1111/ddg.13637
33. Pas HH, Kloosterhuis GJ, Heeres K, van der Meer JB, Jonkman MF. Bullous pemphigoid and linear IgA dermatosis sera recognize a similar 120-kDa keratinocyte collagenous glycoprotein with antigenic cross-reactivity to BP180. J Invest Dermatol. (1997) 108:423–9. doi: 10.1111/1523-1747.ep12289703
34. Hashimoto T, Ishii N, Tsuruta D. Production of neoepitopes by dynamic structural changes on BP180/type XVII collagen. J Invest Dermatol. (2017) 137:2462–4. doi: 10.1016/j.jid.2017.09.001
35. Yashiro M, Nakano T, Taniguchi T, Katsuoka K, Tadera N, Miyazaki K, et al. IgA paraneoplastic pemphigus in angioimmunoblastic T-cell lymphoma with antibodies to desmocollin 1, type VII collagen and laminin 332. Acta Derm Venereol. (2014) 94:235–6. doi: 10.2340/00015555-1660
36. Sitaru C, Schmidt E, Petermann S, Munteanu LS, Bröcker E-B, Zillikens D. Autoantibodies to bullous pemphigoid antigen 180 induce dermal-epidermal separation in cryosections of human skin. J Invest Dermatol. (2002) 118:664–71. doi: 10.1046/j.1523-1747.2002.01720.x
37. Shimanovich I, Mihai S, Oostingh GJ, Ilenchuk TT, Bröcker E-B, Opdenakker G, et al. Granulocyte-derived elastase and gelatinase B are required for dermal-epidermal separation induced by autoantibodies from patients with epidermolysis bullosa acquisita and bullous pemphigoid. J Pathol. (2004) 204:519–27. doi: 10.1002/path.1674
38. Hirose M, Recke A, Beckmann T, Shimizu A, Ishiko A, Bieber K, et al. Repetitive immunization breaks tolerance to type XVII collagen and leads to bullous pemphigoid in mice. J Immunol. (2011) 187:1176–83. doi: 10.4049/jimmunol.1100596
39. Liu Z, Giudice GJ, Swartz SJ, Fairley JA, Till GO, Troy JL, et al. The role of complement in experimental bullous pemphigoid. J Clin Invest. (1995) 95:1539–44. doi: 10.1172/JCI117826
40. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. (2007) 178:6514–21. doi: 10.4049/jimmunol.178.10.6514
41. Iwata H, Ujiie H. Complement-independent blistering mechanisms in bullous pemphigoid. Exp Dermatol. (2017) 26:1235–9. doi: 10.1111/exd.13367
42. Karsten CM, Beckmann T, Holtsche MM, Tillmann J, Tofern S, Schulze FS, et al. Tissue destruction in bullous pemphigoid can be complement independent and may be mitigated by C5aR2. Front Immunol. (2018) 9:488. doi: 10.3389/fimmu.2018.00488
43. Dainichi T, Nishie W, Yamagami Y, Sonobe H, Ujiie H, Kaku Y, et al. Bullous pemphigoid suggestive of complement-independent blister formation with anti-BP180 IgG4 autoantibodies. Br J Dermatol. (2016) 175:187–90. doi: 10.1111/bjd.14411
44. Natsuga K, Nishie W, Shinkuma S, Ujiie H, Nishimura M, Sawamura D, et al. Antibodies to pathogenic epitopes on type XVII collagen cause skin fragility in a complement-dependent and -independent manner. J Immunol. (2012) 188:5792–9. doi: 10.4049/jimmunol.1003402
45. Recke A, Sitaru C, Vidarsson G, Evensen M, Chiriac MT, Ludwig RJ, et al. Pathogenicity of IgG subclass autoantibodies to type VII collagen: induction of dermal-epidermal separation. J Autoimmun. (2010) 34:435–44. doi: 10.1016/j.jaut.2009.11.003
46. Büdinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest. (1998) 102:2082–9. doi: 10.1172/JCI3335
47. Hanafusa T, Azukizawa H, Nishioka M, Tanemura A, Murota H, Yoshida H, et al. Lichen planus-type chronic graft-versus-host disease complicated by mucous membrane pemphigoid with positive anti-BP180/230 and scleroderma-related autoantibodies followed by reduced regulatory T cell frequency. Eur J Dermatol. (2012) 22:140–2. doi: 10.1684/ejd.2011.1587
48. Sultan A, Stojanov IJ, Lerman MA, Kabani S, Haber J, Freedman J, et al. Oral lichen planus pemphigoides: a series of four cases. Oral Surg Oral Med Oral Pathol Oral Radiol. (2015) 120:58–68. doi: 10.1016/j.oooo.2015.03.012
49. Mignogna MD, Fortuna G, Leuci S, Stasio L, Mezza E, Ruoppo E. Lichen planus pemphigoides, a possible example of epitope spreading. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (2010) 109:837–43. doi: 10.1016/j.tripleo.2009.12.044
50. Khullar G, Handa S, De D, Saikia UN. Bullous lichen planus of the nails. JAMA Dermatol. (2015) 151:674–5. doi: 10.1001/jamadermatol.2014.5701
51. Arnold DL, Krishnamurthy K. Lichen, Planus. In: StatPearls. Treasure Island, FL: StatPearls Publishing. (2019). Available online at: http://www.ncbi.nlm.nih.gov/books/NBK526126/
52. Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. (2014) 28:1133–40. doi: 10.1111/jdv.12366
53. Yamanaka Y, Yamashita M, Innocentini LMA, Macedo LD, Chahud F, Ribeiro-Silva A, et al. Direct immunofluorescence as a helpful tool for the differential diagnosis of oral lichen planus and oral lichenoid lesions. Am J Dermatopathol. (2018) 40:491–7. doi: 10.1097/DAD.0000000000001071
54. Emtenani S, Yuan H, Lin C, Pan M, Hundt JE, Schmidt E, et al. Normal human skin is superior to monkey oesophagus substrate for detection of circulating BP180-NC16A-specific IgG antibodies in bullous pemphigoid. Br J Dermatol. (2018) 180:1099–1106. doi: 10.1111/bjd.17313
55. van Beek N, Dähnrich C, Johannsen N, Lemcke S, Goletz S, Hübner F, et al. Prospective studies on the routine use of a novel multivariant enzyme-linked immunosorbent assay for the diagnosis of autoimmune bullous diseases. J Am Acad Dermatol. (2017) 76:889–94.e5. doi: 10.1016/j.jaad.2016.11.002
56. Zillikens D, Kawahara Y, Ishiko A, Shimizu H, Mayer J, Rank CV, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol. (1996) 106:1333–8. doi: 10.1111/1523-1747.ep12349283
57. Gru AA, Salavaggione AL. Lichenoid and interface dermatoses. Semin Diagn Pathol. (2017) 34:237–49. doi: 10.1053/j.semdp.2017.03.001
58. Inoue Y, Adachi A, Ueno M, Fukumoto T, Nishitani N, Fujiwara N, et al. Atypical subacute cutaneous lupus erythematosus presenting as lichen planus pemphigoides with autoantibodies to C-terminus of BP180, desmoglein 1 and SS-A/Ro antigen. J Dermatol. (2012) 39:960–2. doi: 10.1111/j.1346-8138.2012.01536.x
59. Stevens SR, Griffiths CE, Anhalt GJ, Cooper KD. Paraneoplastic pemphigus presenting as a lichen planus pemphigoides-like eruption. Arch Dermatol. (1993) 129:866–9. doi: 10.1001/archderm.1993.01680280054010
60. Hsiao CJ, Hsu MM, Lee JY, Chen WC, Hsieh WC. Paraneoplastic pemphigus in association with a retroperitoneal Castleman's disease presenting with a lichen planus pemphigoides-like eruption. A case report and review of literature. Br J Dermatol. (2001) 144:372–6. doi: 10.1046/j.1365-2133.2001.04030.x
61. Probst C, Schlumberger W, Stöcker W, Recke A, Schmidt E, Hashimoto T, et al. Development of ELISA for the specific determination of autoantibodies against envoplakin and periplakin in paraneoplastic pemphigus. Clin Chim Acta. (2009) 410:13–8. doi: 10.1016/j.cca.2009.08.022
62. Sakuma-Oyama Y, Powell AM, Albert S, Oyama N, Bhogal BS, Black MM. Lichen planus pemphigoides evolving into pemphigoid nodularis. Clin Exp Dermatol. (2003) 28:613–6. doi: 10.1046/j.1365-2230.2003.01401.x
63. Miyagawa S, Ohi H, Muramatsu T, Okuchi T, Shirai T, Sakamoto K. Lichen planus pemphigoides-like lesions induced by cinnarizine. Br J Dermatol. (1985) 112:607–13. doi: 10.1111/j.1365-2133.1985.tb15272.x
64. Onprasert W, Chanprapaph K. Lichen planus pemphigoides induced by enalapril: a case report and a review of literature. Case Rep Dermatol. (2017) 9:217–24. doi: 10.1159/000481449
65. Xu H-H, Xiao T, He C-D, Jin G-Y, Wang Y-K, Gao X-H, et al. Lichen planus pemphigoides associated with Chinese herbs. Clin Exp Dermatol. (2009) 34:329–32. doi: 10.1111/j.1365-2230.2008.02900.x
66. Rosmaninho A, Sanches M, Oliveira A, Alves R, Selores M. Lichen planus pemphigoides induced by a weight reduction drug. Cutan Ocul Toxicol. (2011) 30:306–8. doi: 10.3109/15569527.2011.566234
67. Ben Salem C, Chenguel L, Ghariani N, Denguezli M, Hmouda H, Bouraoui K. Captopril-induced lichen planus pemphigoides. Pharmacoepidemiol Drug Saf. (2008) 17:722–4. doi: 10.1002/pds.1618
68. Flageul B, Foldes C, Wallach D, Vignon-Pennamen MD, Cottenot F. Captopril-induced lichen planus pemphigoides with pemphigus-like features. A case report. Dermatologica. (1986) 173:248–55. doi: 10.1159/000249262
69. Stoebner P-E, Michot C, Ligeron C, Durand L, Meynadier J, Meunier L. [Simvastatin-induced lichen planus pemphigoides]. Ann Dermatol Venereol. (2003) 130:187–90.
70. Schmidgen MI, Butsch F, Schadmand-Fischer S, Steinbrink K, Grabbe S, Weidenthaler-Barth B, et al. Pembrolizumab-induced lichen planus pemphigoides in a patient with metastatic melanoma. J Dtsch Dermatol Ges J Ger Soc Dermatol. (2017) 15:742–5. doi: 10.1111/ddg.13272
71. Kuramoto N, Kishimoto S, Shibagaki R, Yasuno H. PUVA-induced lichen planus pemphigoides. Br J Dermatol. (2000) 142:509–12. doi: 10.1046/j.1365-2133.2000.03366.x
72. Ilknur T, Akarsu S, Uzun S, Özer E, Fetil E. Heterogeneous disease: a child case of lichen planus pemphigoides triggered by varicella. J Dermatol. (2011) 38:707–10. doi: 10.1111/j.1346-8138.2011.01220.x
73. Mohanarao TS, Kumar GA, Chennamsetty K, Priyadarshini T. Childhood lichen planus pemphigoides triggered by chickenpox. Indian Dermatol Online J. (2014) 5:S98–100. doi: 10.4103/2229-5178.146169
74. Ogg GS, Bhogal BS, Hashimoto T, Coleman R, Barker JN. Ramipril-associated lichen planus pemphigoides. Br J Dermatol. (1997) 136:412–4. doi: 10.1111/j.1365-2133.1997.tb14956.x
75. Demirçay Z, Baykal C, Demirkesen C. Lichen planus pemphigoides: report of two cases. Int J Dermatol. (2001) 40:757–9. doi: 10.1046/j.1365-4362.2001.01334.x
76. Laureano A, Rafael M, Marques Pinto G, Cardoso J. Lichen planus pemphigoides possibly induced by hormone therapy. Eur J Dermatol. (2013) 23:903–4. doi: 10.1684/ejd.2013.2180
77. Jang SH, Yun SJ, Lee SC, Lee JB. Lichen planus pemphigoides associated with chronic hepatitis B virus infection. Clin Exp Dermatol. (2015) 40:868–71. doi: 10.1111/ced.12530
78. Flageul B, Hassan F, Pinquier L, Blanchet-Bardon C, Dubertret L. [Lichen pemphigoid associated with developing hepatitis B in a child]. Ann Dermatol Venereol. (1999) 126:604–7.
79. Evans SJ, Waller PC, Davis S. Use of proportional reporting ratios (PRRs) for signal generation from spontaneous adverse drug reaction reports. Pharmacoepidemiol Drug Saf. (2001) 10:483–6. doi: 10.1002/pds.677
80. Le Cleach L, Chosidow O. Clinical practice. Lichen planus. N Engl J Med. (2012) 366:723–32. doi: 10.1056/NEJMcp1103641
81. Alaizari NA, Al-Maweri SA, Al-Shamiri HM, Tarakji B, Shugaa-Addin B. Hepatitis C virus infections in oral lichen planus: a systematic review and meta-analysis. Aust Dent J. (2016) 61:282–7. doi: 10.1111/adj.12382
82. Hamada T, Fujimoto W, Okazaki F, Asagoe K, Arata J, Iwatsuki K. Lichen planus pemphigoides and multiple keratoacanthomas associated with colon adenocarcinoma. Br J Dermatol. (2004) 151:252–4. doi: 10.1111/j.1365-2133.2004.06074.x
83. George PM. Bullous pemphigoid possibly induced by psoralen plus ultraviolet A therapy. Photodermatol Photoimmunol Photomed. (1996) 11:185–7. doi: 10.1111/j.1600-0781.1995.tb00166.x
84. Lutowiecka-Wranicz A, Sysa-Jedrzejowska A, Skwarczynska-Banyś E. [Effect of drugs and ultraviolet rays on the development of pemphigoid]. Przegl Dermatol. (1980) 67:641–6.
85. Ohtani A, Takenaka Y, Ishiguro N, Kawashima M. Case of lichen planus induced by sitagliptin phosphate hydrate. J Dermatol. (2017) 44:1081–2. doi: 10.1111/1346-8138.13624
86. Swale VJ, McGregor JM. Amlodipine-associated lichen planus. Br J Dermatol. (2001) 144:920–1. doi: 10.1046/j.1365-2133.2001.04172.x
87. Ghosh S, Jain VK, Chaudhuri S, Mathur SK. Sulfasalazine induced lichen planus in a patient of rheumatoid arthritis. Indian J Dermatol Venereol Leprol. (2013) 79:541–4. doi: 10.4103/0378-6323.113103
88. Zheng Y, Zhang J, Chen H, Lai W, Maibach HI. Terbinafine-induced lichenoid drug eruption. Cutan Ocul Toxicol. (2017) 36:101–3. doi: 10.3109/15569527.2016.1160101
89. Woo V, Bonks J, Borukhova L, Zegarelli D. Oral lichenoid drug eruption: a report of a pediatric case and review of the literature. Pediatr Dermatol. (2009) 26:458–64. doi: 10.1111/j.1525-1470.2009.00953.x
90. Varpuluoma O, Försti A-K, Jokelainen J, Turpeinen M, Timonen M, Huilaja L, et al. Vildagliptin significantly increases the risk of bullous pemphigoid: a finnish nationwide registry study. J Invest Dermatol. (2018) 138:1659–61. doi: 10.1016/j.jid.2018.01.027
91. Kolb-Mäurer A, Sitaru C, Rose C, Bröcker E-B, Goebeler M, Zillikens D. [Treatment of lichen planus pemphigoides with acitretin and pulsed corticosteroids]. Hautarzt. (2003) 54:268–73. doi: 10.1007/s00105-002-0459-0
92. Hackländer K, Lehmann P, Hofmann SC. Successful treatment of lichen planus pemphigoides using acitretin as monotherapy: Clinical Letter. J Dtsch Dermatol Ges. (2014) 12:818–9. doi: 10.1111/ddg.12380
93. Davis AL, Bhocal BS, Whitehead P, Frith P, Murdoch ME, Leigh IM, et al. Lichen planus pemphigoides: its relationship to bullous pemphigoid. Br J Dermatol. (1991) 125:263–71. doi: 10.1111/j.1365-2133.1991.tb14753.x
94. Knisley RR, Petropolis AA, Mackey VT. Lichen Planus Pemphigoides Treated With Ustekinumab. Cutis. (2017) 100:415–418.
95. Malakar S, Saha A. Successful treatment of resistant lichen planus pemphigoides with cyclosporine: a new hope. Indian J Dermatol. (2016) 61:112–4. doi: 10.4103/0019-5154.174067
96. Washio K, Nakamura A, Fukuda S, Hashimoto T, Horikawa T. A case of lichen planus pemphigoides successfully treated with a combination of cyclosporine A and prednisolone. Case Rep Dermatol. (2013) 5:84–87. doi: 10.1159/000350285
97. Kabuto M, Fujimoto N, Yamaguchi A, Tanaka T. Evaluation of mononuclear cells in lichen planus pemphigoides. Acta Derm Venereol. (2016) 96:276–8. doi: 10.2340/00015555-2191
98. Cohen DM, Ben-Amitai D, Feinmesser M, Zvulunov A. Childhood lichen planus pemphigoides: a case report and review of the literature. Pediatr Dermatol. (2009) 26:569–74. doi: 10.1111/j.1525-1470.2009.00988.x
99. Goldscheider I, Herzinger T, Varga R, Eming R, Ruzicka T, Flaig MJ, et al. Childhood lichen planus pemphigoides: report of two cases treated successfully with systemic glucocorticoids and dapsone. Pediatr Dermatol. (2014) 31:751–3. doi: 10.1111/pde.12214
100. Conde Fernandes I, Pinto Almeida T, Mendes I, Cunha Velho G, Alves R, Selores M. Lichen planus pemphigoides in a child. Eur J Dermatol. (2012) 22:570–1. doi: 10.1684/ejd.2012.1764
101. von Stebut E, Steinbrink K, Bräuninger W. Lichen ruber pemphigoides. Aktuelle Dermatol. (2004) 30:73–6. doi: 10.1055/s-2004-814476
102. Duong B, Marks S, Sami N, Theos A. Lichen planus pemphigoides in a 2-year-old girl: response to treatment with methotrexate. J Am Acad Dermatol. (2012) 67:e154–6. doi: 10.1016/j.jaad.2011.12.024
103. Skvara H, Stingl G. Lichenoid eruption with single plantar blisters: a very rare case of lichen planus pemphigoides. J Eur Acad Dermatol Venereol. (2009) 23:596–7. doi: 10.1111/j.1468-3083.2008.02982.x
104. Loyal J, Rashtak S. Vulvar lichen planus pemphigoides. Int J Womens Dermatol. (2017) 3:225–7. doi: 10.1016/j.ijwd.2017.07.002
105. Shimada H, Shono T, Sakai T, Ishikawa K, Takeo N, Hatano Y, et al. Lichen planus pemphigoides concomitant with rectal adenocarcinoma: fortuitous or a true association? Eur J Dermatol. (2015) 25:501–3. doi: 10.1684/ejd.2015.2619
106. Jensen AØ, Steiniche T, Veien NK, Deleuran MS. Lichen planus pemphigoides in a 6-year-old child. Acta Paediatr. (2009) 98:2–3. doi: 10.1111/j.1651-2227.2008.01069.x
107. Solomon LW, Helm TN, Stevens C, Neiders ME, Kumar V. Clinical and immunopathologic findings in oral lichen planus pemphigoides. Oral Surg Oral Med Oral Pathol Oral Radiol Endodontol. (2007) 103:808–13. doi: 10.1016/j.tripleo.2006.03.020
108. Harjai B, Mendiratta V, Kakkar S, Koranne R. Childhood lichen planus pemphigoides - a rare entity. J Eur Acad Dermatol Venereol. (2006) 20:117–8. doi: 10.1111/j.1468-3083.2005.01346.x
109. Zhu YI, Fitzpatrick JE, Kornfeld BW, Fitzpatrick JE. Lichen planus pemphigoides associated with ramipril. Int J Dermatol. (2006) 45:1453–5. doi: 10.1111/j.1365-4632.2006.02711.x
110. Fine JD. Management of acquired bullous skin diseases. N Engl J Med. (1995) 333:1475–84. doi: 10.1056/NEJM199511303332207
111. Joly P, Roujeau J-C, Benichou J, Picard C, Dreno B, Delaporte E, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. (2002) 346:321–7. doi: 10.1056/NEJMoa011592
112. Recke A, Schmidt E, Zillikens D, Shimanovich I. Lichen planus pemphigoides. J. Deusch. Dermatol. Gesellschaft 9(Suppl. 1):251–5. doi: 10.1111/j.1610-0387.2011.07633.x
Keywords: Lichen planus pemphigoides, autoantibodies, Bullous Pemphigoid, Lichen Planus, BP180, BP230
Citation: Hübner F, Langan EA and Recke A (2019) Lichen Planus Pemphigoides: From Lichenoid Inflammation to Autoantibody-Mediated Blistering. Front. Immunol. 10:1389. doi: 10.3389/fimmu.2019.01389
Received: 26 March 2019; Accepted: 03 June 2019;
Published: 02 July 2019.
Edited by:
Karin Loser, University of Münster, GermanyReviewed by:
Chika Ohata, Kurume University School of Medicine, JapanHideyuki Ujiie, Hokkaido University Graduate School of Medicine, Japan
Zhi Liu, University of North Carolina at Chapel Hill, United States
Copyright © 2019 Hübner, Langan and Recke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Recke, QW5kcmVhcy5SZWNrZUB1a3NoLmRl
†These authors have contributed equally to this work